

Abstract
Purpose
To investigate the molecular mechanism underlying the Leber's hereditary optic neuropathy (LHON)-linked MT-ND1 3460G>A mutation.
Methods
Cybrid cell models were generated by fusing mitochondrial DNA-less ρ0 cells with enucleated cells from a patient carrying the m.3460G>A mutation and a control subject. The impact of m.3460G>A mutations on oxidative phosphorylation was evaluated using Blue Native gel electrophoresis, and measurements of oxygen consumption were made with an extracellular flux analyzer. Assessment of reactive oxygen species (ROS) production in cell lines was performed by flow cytometry with MitoSOX Red reagent. Assays for apoptosis and mitophagy were undertaken via immunofluorescence analysis.
Results
Nineteen Chinese Han pedigrees bearing the m.3460G>A mutation exhibited variable penetrance and expression of LHON. The m.3460G>A mutation altered the structure and function of MT-ND1, as evidenced by reduced MT-ND1 levels in mutant cybrids bearing the mutation. The instability of mutated MT-ND1 manifested as defects in the assembly and activity of complex I, respiratory deficiency, diminished mitochondrial adenosine triphosphate production, and decreased membrane potential, in addition to increased production of mitochondrial ROS in the mutant cybrids carrying the m.3460G>A mutation. The m.3460G>A mutation mediated apoptosis, as evidenced by the elevated release of cytochrome c into the cytosol and increasing levels of the apoptotic-associated proteins BAK, BAX, and PARP, as well as cleaved caspases 3, 7, and 9, in the mutant cybrids. The cybrids bearing the m.3460G>A mutation exhibited reduced levels of autophagy protein light chain 3, accumulation of autophagic substrate P62, and impaired PTEN-induced kinase 1/parkin-dependent mitophagy.
Conclusions
Our findings highlight the critical role of m.3460G>A mutation in the pathogenesis of LHON, manifested by mitochondrial dysfunction and alterations in apoptosis and mitophagy.
Keywords: Leber's hereditary optic neuropathy, complex I, mitochondrial dysfunction, apoptosis, mitophagy
Leber's hereditary optic neuropathy (LHON) is the most common example of maternal inheritance of eye disease, with the degeneration of retinal ganglion cells (RGCs) leading to the loss of central vision.1–3 A number of mutations in mitochondrial DNA (mtDNA) have been associated with LHON, contributing at varying degrees to pathogenesis.4–11 Of these, NADH dehydrogenase (ND subunit 1 [MT-ND1] 3460G>A, MT-ND4 11778G>A, and MT-ND6 14484T>C) mutations affecting essential subunits of NADH:ubiquinone oxidoreductase (complex I) have been found to be responsible for the majority of LHON cases worldwide.4–8,12–14 The primary defects in these LHON-linked mtDNA mutations appear to be a failure in the activity of complex I, thereby causing respiratory deficiency, diminishing adenosine triphosphate (ATP) synthesis, and increasing generation of reactive oxygen species (ROS).15–20 Subsequently, the energy failure and increasing oxidative stress may lead to degeneration of the retinal ganglion cells.1–4 The pathophysiology of these LHON-linked mtDNA mutations is not well understood; therefore, it is necessary to establish a functional link between LHON mitochondrial dysfunction and the associated causes and effects.
The m.3460G>A mutation changed a highly conserved alanine at position 52 with threonine (A52T) in MT-ND1, the essential subunit of complex I.21–25 Therefore, we hypothesized that the m.3460G>A mutation may alter both the structure and function of complex I, thereby causing mitochondrial dysfunction. Recently, we identified 19 unrelated Han Chinese families bearing the m.3460G>A mutation among a large cohort of 1793 genetically unrelated Han Chinese subjects.14,26,27 The occurrence of the m.3460G>A mutation in the genetically unrelated families of different ethnic backgrounds strongly supports the notion that this mutation is involved in the pathogenesis of LHON.8,23–27 In the present study, we performed clinical evaluations and Sanger sequence analyses of the mtDNA of these Chinese families bearing the m.3460G>A mutation. The functional significance of the m.3460G>A mutation was further investigated through the use of cybrid cell lines constructed by transferring mitochondria from lymphoblastoid cell lines derived from an affected matrilineal relative carrying the m.3460G>A mutation and from a control subject belonging to the same mtDNA haplogroup into human mtDNA-less (ρ0) cells.28,29 Using western blot and Blue Native polyacrylamide gel electrophoresis (BN-PAGE) analyses, we examined whether or not the m.3460G>A mutation influenced the stability of MT-ND1 and the assembly and activity of complex I. These cell lines were then assessed for effects of the mtDNA mutations on the enzymatic activities of respiratory chain complexes, the rate of O2 consumption, mitochondrial ATP production, mitochondrial membrane potential, and generation of ROS. These cell lines were further evaluated for the effect of these mtDNA mutations on apoptotic state and mitophagy.
Materials and Methods
Families and Subjects
A total of 19 unrelated Han Chinese families bearing the m.3460G>A mutation were recruited from eye clinics across China, as described previously.14,20,27 This study was in compliance with the tenets of the Declaration of Helsinki. Informed consent, blood samples, and clinical evaluations were obtained from all participating family members under protocols approved by the ethics committees of Zhejiang University School of Medicine and Wenzhou Medical University. Comprehensive histories were obtained and physical examinations were performed for these participating subjects to identify personal or family medical histories of visual impairment or other clinical abnormalities. The ophthalmic examinations of probands and other members of these families were conducted as detailed previously.30,31 The degree of visual impairment was defined as follows: normal, >0.3; mild, 0.3 to 0.1; moderate, <0.1to 0.05; severe, <0.05 to 0.02; and profound, <0.02. A total of 485 control DNA samples were obtained from the adult Han Chinese participants.
Sanger Sequence Analysis
Genomic DNA was isolated from whole blood of family members and control subjects using the QIAamp DNA Blood Mini Kit (51104; QIAGEN, Hilden, Germany). The entire mitochondrial genomes of family members were PCR amplified in 24 overlapping fragments using sets of light-strand and heavy-strand oligonucleotide primers; they were subsequently analyzed by direct sequencing as described elsewhere.32 These sequence results were compared with the updated Cambridge Reference Sequence (GenBank accession number: NC_012920).33
Cell Cultures and Culture Conditions
Lymphoblastoid cell lines were immortalized by transformation with the Epstein–Barr virus, as described elsewhere.34 Cell lines derived from the affected matrilineal relative (WZ83-III1) carrying the m.3460G>A mutation and one age- and sex-matched control subject (A70) lacking the mutation but belonging to the same mtDNA haplogroup D4 (Supplementary Fig. S2) were grown in the RPMI 1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS). The 143B.TK– cell line was grown in Dulbecco's modified Eagle's medium (containing 4.5 mg/mL glucose and 0.11 mg/mL pyruvate), supplemented with 100 µg/mL bromodeoxyuridine (BrdU) and 5% FBS. The mtDNA-less ρ0206 cell line, derived from 143B.TK–, was grown under the same conditions as the parental line, except for the addition of 50 µg/mL uridine.28 Transformation by cytoplasts of mtDNA-less ρ0206 cells was performed using immortalized lymphoblastoid cell lines, as detailed previously.28,29 The cybrids derived from each donor cell line were analyzed for the presence and level of the m.3460G>A mutation and mtDNA copy numbers as detailed elsewhere.11,27 Three cybrids derived from each donor cell line with homoplasmy of mtDNA mutations and similar mtDNA copy numbers were used for the following biochemical characterization. All cybrid cell lines were maintained in the same medium as the 143B.TK– cell line.
Western Blot Analysis
Western blot analysis was performed as detailed elsewhere.29 Cellular proteins (20 µg) were electrophoresed through 10% bis-Tris SDS-polyacrylamide gels and then transferred to a polyvinyl difluoride membrane. The following primary antibodies used for this experiment were obtained from Abcam (Cambridge, UK): anti-ND1 (ab74257), VDAC1 (ab14734), AFG3L2 (ab68023), TOM20 (ab56783), and anti-cytochrome c (ab13575). Anti-ND4 (sc-20499-R) was obtained from Santa Cruz Biotechnology (Dallas, TX, USA). Antibodies obtained from Cell Signaling Technology (Danvers, MA, USA) included CLPP (14181), SOD1 (4266T), SOD2 (13141T), catalase (12980T), LC3 (12741), P62 (8025), PINK1 (6946), parkin (4211), PARP (9542), caspase 3 (14420) caspase 7 (12827), caspase 9 (9508), BAK (12105T), BAX (5023T), cleaved PARP (5625), cleaved caspase 3 (9664), cleaved caspase 7 (8438), and cleaved caspase 9 (52873). Other antibodies included NDUFA3 (17257-1-AP; ProteinTech Group, Rosemont, IL, USA), NDUFA8 (A12118; ABclonal Technology, Woburn, MA, USA), and NDUFA13 (A5412; ABclonal Technology). Peroxidase AffiniPure Goat Anti-Mouse IgG and Peroxidase AffiniPure Goat Anti-Rabbit IgG (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) were used as secondary antibodies, and protein signals were detected using the ECL Prime Western Blotting System (MilliporeSigma, Burlington, MA, USA). Quantification of the density in each band was performed as detailed previously.29
BN-PAGE and In-Gel Activity Assays
BN-PAGE and in-gel activity assays were performed using mitochondrial proteins isolated from mutant and control cybrid cell lines, as detailed elsewhere.35,36 Samples containing 30 µg of mitochondrial proteins were separated on 3% to 11% Bis-Tris NativePAGE gel (Thermo Fisher Scientific). The primary antibody used for this experiment was the total human OXPHOS antibody cocktail (Abcam), with voltage-dependent anion channel as a loading control. Alkaline phosphatase-labeled goat anti-mouse IgG and goat anti-rabbit IgG (Beyotime, Jiangsu, China) were used as secondary antibodies, and protein signals were detected using the BCIP/NBT Alkaline Phosphatase Color Development Kit (Beyotime).
The in-gel activity assay was performed as detailed elsewhere.37 Briefly, the NativePAGE gels were prewashed in ice-cold water and then incubated with the substrates of complex I (1-mM Tris-HCl, pH 7.4; 1 mg/mL nitroblue tetrazolium [NBT]; 0.1 mg/mL NADH), complex II (84-mM sodium succinate; 50-mM phosphate buffer, pH 7.4; 2 mg/mL NBT; 0.2-mM phenazine methosulfate) at room temperature (RT), and complex V (35-mM Tris-HCl, pH 7.4; 14-mM magnesium sulfate; 270-mM glycine; 10-mM ATP; 0.2% lead nitrate). The gels were then incubated at 37°C overnight. After the reaction was stopped with 10% acetic acid, the gels were washed extensively in water and scanned to visualize the activities of the respiratory chain complexes.
Measurements of Oxygen Consumption Rates
The oxygen consumption rates (OCRs) in various cybrid cell lines were measured with a Seahorse XF96 extracellular flux analyzer (Seahorse Bioscience, North Billerica, MA, USA), as detailed previously.38 Cybrid cells were seeded at a density of 2 × 104 cells per well on the Seahorse XF96 polystyrene tissue culture plates. Inhibitors were used at the following concentrations: oligomycin (1.5 µM), carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP; 0.8 µM), antimycin A (1.5 µM), and rotenone (3 µM).
ATP Measurements
The CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI, USA) was used to measure cellular and mitochondrial ATP levels, modifying the manufacturer's instructions.29 Briefly, the assay buffer and substrate were equilibrated at RT and transferred to and gently mixed with the substrate to obtain a homogeneous solution. After a 30-minute equilibration of the cell plate at RT, 100 µL of the assay reagents was added to each well with 2 × 104 cells and the content was mixed for 2 minutes on an orbital shaker to induce cell lysis. After a 10-minute incubation at RT, the luminescence was read on a microplate reader (Synergy H1; BioTek, Winooski, VT, USA).
Assessment of Mitochondrial Membrane Potential
Mitochondrial membrane potential was measured with a JC-10 Mitochondrial Membrane Potential Assay Kit (Abcam), generally following the manufacturer's recommendations with some modifications, as detailed elsewhere.39,40 In brief, ∼2 × 106 cells of each cybrid cell line were harvested, resuspended in 200 µL 1× JC-10 assay buffer, and then incubated at 37°C for 30 minutes. Alternatively, harvested cells were preincubated with 10 µM of FCCP for 30 minutes at 37°C prior to staining with JC-10 dye. After they were washed twice with PBS, the cells were resuspended in 200 µL PBS. The fluorescent intensities for both J-aggregates and monomeric forms of JC-10 were measured at excitation (Ex)/emission (Em) = 490/530 and 490/590 nm with a BD Biosciences LSR II flow cytometer (Becton, Dickinson and Company, Franklin Lakes, NJ, USA).
Measurement of ROS Production
The levels of mitochondrial ROS generation were determined using the MitoSOX assay (Thermo Fisher Scientific) as detailed previously.41,42 Briefly, approximate 2 × 106 cells of each cell line were harvested, resuspended in PBS supplemented with 5 µM of MitoSOX, and then incubated at 37°C for 20 minutes. After they were washed twice with PBS, the cells were resuspended in PBS in the presence of 0.8-mM freshly prepared H2O2 and 2% FBS and then incubated at RT for another 45 minutes. Cells were further washed with PBS and resuspended with 1 mL of PBS with 0.5% paraformaldehyde. Samples with or without H2O2 stimulation were analyzed using the BD Biosciences LSR II flow cytometer, with excitation at 488 nm and emission at 529 nm. In each sample, 10,000 events were analyzed.
Tunel Assay
The TUNEL assay was carried out using the In Situ Cell Death Detection Kit (Sigma-Aldrich) according to the manufacturer's protocol. Briefly, cybrid cells were seeded at a density of 2 × 104 cells per well into a V-bottomed 96-well microplate overnight. After they were washed twice with PBS, the cells were incubated in the presence of 0.8-mM freshly prepared H2O2 at RT for 30 minutes, fixed with freshly prepared 4% paraformaldehyde in PBS for 60 minutes at RT, permeabilized with 0.1% Triton X-100 (Sigma-Aldrich) for 2 minutes on ice, and incubated in the TUNEL reaction mixture for 60 minutes at 37°C. Samples were analyzed under a fluorescence microscope (DMi8; Leica Microsystems, Wetzlar, Germany).
Immunofluorescence Analysis
Cells were cultured on glass coverslips (Thermo Fisher Scientific), fixed with 4% (w/v) formaldehyde (Sigma-Aldrich) in PBS for 15 minutes at RT, permeabilized with 0.2% Triton X-100 in PBS for 10 minutes, blocked with 5% BSA for 30 minutes, and incubated with primary antibodies (TOM20, cytochrome c, and LAMP1) at 4°C overnight. Samples were washed three times in PBS for 5 minutes each at RT, incubated with fluorescent-conjugated secondary antibodies—Alexa Fluor 488 Goat anti-Mouse IgG (H+L) and Alexa Fluor 594 Goat anti-Rabbit IgG (H+L) (Thermo Fisher Scientific)—for 1 hour at RT, stained with 4′,6-diamidino-2-phenylindole (DAPI; Thermo Fisher Scientific) for 15 minutes, and then mounted with a drop of Fluoromount-G (Sigma-Aldrich). Cells were examined using a confocal microscope (LSM710; Carl Zeiss Meditec, Oberkochen, Germany) with three lasers (Ex/Em = 492/520, 540/590, and 358/461 nm).
Statistical Analysis
Statistical analysis was carried out using Prism 8.0 (GraphPad Software, San Diego, CA, USA) and was conducted on data from three or more biologically independent experimental replicates. Comparisons between groups were planned before statistical testing, and target effect sizes were not predetermined. In all graphs, error bars displayed on graphs represent means ± SEM of at least three independent experiments. Statistical analysis included two-tailed Student's t-test (with 95% confidence interval) for two groups or ordinary one-way analysis of variance (ANOVA) with Dunnett's multiple comparison test for three or more groups. Differences were considered statistically significant at P < 0.05.
Results
Clinical Genetic Evaluation of 19 Chinese Families Carrying the m.3460G>A Mutation
Nineteen Han Chinese pedigrees bearing the m.3460 G>A mutation were identified in a large cohort of 1793 Chinese probands with LHON.14,20,27 All available members of these families underwent comprehensive physical and ophthalmologic examinations to identify personal or family medical histories of visual impairments and other clinical abnormalities. As shown in Supplemental Table S1 and Supplementary Figure S1, 42 (28 males/14 females) of 247 matrilineal relatives exhibited variable penetrance and expression of optic neuropathy among and within families. In particular, the severity of visual loss ranged from profound visual loss to normal vision. The age at onset of optic neuropathy of matrilineal relatives bearing the m.3460G>A mutation ranged from 5 to 29 years, with an average of 18.9 years. As shown in Supplementary Table S2, the penetrance of optical neuropathy among these pedigrees varied from 2.8% to 50%, with an average of 20.3%. Furthermore, all affected matrilineal relatives of these families revealed no other clinical abnormalities, including hearing loss, diabetes, and neurological diseases.
Sanger sequence analysis of whole mtDNAs among these Chinese pedigrees revealed the presence of the m.3460G>A mutation and distinct sets of mtDNA polymorphisms, including 269 known variants, as shown in Supplementary Table S3. As shown in Supplementary Figure S2 and Supplementary Table S4, the mtDNAs from 19 pedigrees resided at mtDNA haplogroups A, B5, C4a1, D, F, M12, M7, M8a2, R9, and H2.43 These mtDNA variants included 76 in the D-loop region, 11 in the 12S rRNA gene, 10 in the 16S rRNA gene, eight in the tRNA genes, and 114 silent and 50 missense variants in the genes encoding polypeptides. These variants in RNAs and polypeptides were further evaluated by phylogenetic analyses of the sequences from 17 other vertebrates, as shown in Supplementary Table S3. These variants were further assessed for their presence in 485 control subjects and potential structural and functional alterations. The lack of potential structural and functional alterations indicates that these mtDNA variants may not play an important role in the phenotypic manifestation of the m.3460G>A mutation.
Effect of the m.3460G>A Mutation on the Stability of MT-ND1
We evaluated the effect of the m.3460G>A (A52T) mutation on the structure and function of MT-ND1. Based on the cryo-electron microscopy (cryo-EM) structure of mammalian complex I (PDB entry: 5XTD and 6G2J),22,44 A52 forms hydrophobic interactions with P48, L55, and 56F in MT-ND1 (Fig. 1A). Hence, the substitution of non-polar hydrophobic alanine at position 52 with the hydrophilic threonine due to m.3460G>A mutation in MT-ND1 may destabilize these interactions inside MT-ND1, thereby perturbing the structure and stability of MT-ND1.
Figure 1.

Alterations in the structure and stability of MT-ND1. (A) Electrostatic interactions in A52 of MT-ND1. Based on the cryo-EM structure of complex I from Homo sapiens (PDB entry: 5XTD), the hydrophobic residue A52 interacts with 48P, 55L, and 56F. (B) Western blot analysis. Cellular proteins (20 µg) from various cell lines were electrophoresed through a denaturing polyacrylamide gel, electroblotted, and hybridized with MT-ND1, MT-ND4, NDUFA3, NDUFA8, NDUFA13, AFG3L2, and CLPP antibodies, with GAPDH as a loading control. (C) Quantification of MT-ND1. Average relative values of MT-ND1 were normalized to the average values of VDAC1 in various cell lines. The calculations were based on three independent determinations in each cell line. The error bars indicate 2 SEM. P indicates significance based on Student's t-test of the differences between mutant and control cells.
To test this hypothesis, we analyzed the levels of MT-ND1and MT-ND4 by western blot in these mutant and control cell lines. As shown in Figures 1B and 1C, the levels of MT-ND1 in three mutant cell lines ranged from 45.7% to 50.6%, with an average of 47.4% (P = 0.0010), relative to the mean value measured in three control cell lines, whereas the levels of MT-ND4 in the mutant cybrids were comparable with those in control cybrids.
Complex I of human and mice is composed of 45 subunits, including seven subunits encoded by mtDNA and 38 subunits encoded by nuclear genes.21 In fact, MT-ND1 interacts with NDUFA1, NDUFA3, NDUFA8, and NDUFA13, whereas MT-ND4 interacts with MT-ND5, NDFS2, NDUFB1, NDUFB4, DDUFB5, NDUFB8, and NDUFB11.22,44 To examine whether m.3460G>A mutation affects the expression of other subunits of complex I, we measured the levels of NDUFA3, NDUFA8, and NDUFA13 by western blot analysis among mutant and control cell lines. As shown in Figure 1B, the levels of these subunits in mutant cybrids were comparable to those in control cybrids.
To analyze whether the m.3460G>A mutation affects mitochondrial protein homeostasis, we measured the levels of the caseinolytic mitochondrial matrix peptidase proteolytic subunit (CLPP) involved in mitoribosome assembly45 and AFG3 like matrix AAA protease stress46 in the various cell lines. As shown in Figure 1B, there were no significant differences in the levels of AFG3L2 and CLPP between the mutant and control cybrids. These data indicate that the m.3460G>A mutation may not affect proteostasis stress.
Perturbed the Assembly and Activity of Complex I
To determine whether or not the mutated MT-ND1 affects the assembly of complex I, mitochondrial membrane proteins isolated from mutant and control cell lines were separated by BN-PAGE, electroblotting, and hybridizing, with NDUFA9, SDHA, UQCRC2, and CO2 (subunits of complexes I, II, III, and IV, respectively) and ATP5A (subunit of complex V) as loading controls. As shown in Figures 2A and 2B, the levels of complex I in the mutant cybrids were 67.2% relative to the average values in the control cybrids (P = 0.0267), whereas the levels of complexes II, III, and IV in the mutant cybrids were comparable to those in the control cybrids.
Figure 2.

Altered assembly and defective stability of complex I. (A) Respiratory complex I assembly. Whole cells from mutant and control cell lines were solubilized with digitonin and then subjected to BN-PAGE analysis. Blots were then hybridized with NDUFA9 (complex I, CI), SDHA (complex II, CII), UQCRC2 (complex III, CIII), and CO2 (complex IV, CIV) antibodies, with ATP5A (complex V, CV) as the internal control. (B) Quantification of complexes. The levels of four complexes in the mutant and control cell lines were determined based on three independent determinations in each cell line. (C) In-gel activity of complexes I, II, and V. Mitochondrial proteins (20 µg) from various mutant and control cell lines were used for BN-PAGE, and the activities of the complexes were measured in the presence of specific substrates (NADH and NBT for complex I; sodium succinate, phenazine methosulfate, and NBT for complex II; ATP and lead nitrate for complex V). (D) Quantification of in-gel activities of complexes I and II. The calculations were based on three independent determinations in each cell line. Graph details and symbols are explained in Figure 1.
We analyzed the potential consequence of m.3460G>A mutation on the stability and activity of complex I using the in-gel activity assay. Mitochondrial membrane proteins isolated from mutant and control cell lines were separated by BN-PAGE and stained with specific substrates of complexes I, II, and V (complex V as a loading control).36,37 As illustrated in Figures 2C and 2D, the activities of complex I in the mutant cell lines were 78.2% (P = 0.0023) relative to the average values of control cell lines. In contrast, the average in-gel activities of complex II in the mutants were comparable to those of the controls.
Respiration Deficiency
To further elucidate whether the m.3460G>A mutation affects cellular bioenergetics, we examined the OCRs of various mutant and control cybrid cell lines with the Seahorse XF96 extracellular flux analyzer.38 As shown in Figure 3, the basal OCR in the mutant cybrids carrying only m.3460G>A was 33.1% (P = 0.0098), relative to the mean value measured in the control cybrids. To assess which of the enzyme complexes in the mitochondrial respiratory chain was perturbed in the mutant cell lines, the OCR was measured after the sequential addition of oligomycin (to inhibit ATP synthase), FCCP (to uncouple the inner mitochondrial membrane and allow for maximum electron flux through the electron transport chain), rotenone (to inhibit complex I), and antimycin (to inhibit complex III). The difference between the basal OCR and the drug-insensitive OCR yielded the ATP-linked OCR, proton leak OCR, maximal OCR, reserve capacity, and nonmitochondrial OCR. As shown in Figure 3, the basal OCR, ATP-linked OCR, proton leak OCR, maximal OCR, reserve capacity, and nonmitochondrial OCR in mutant cell lines carrying the m.3460G>A mutation were 33.1%, 27.8%, 82.2%, 52.9%, 74.5%, and 110.9%, respectively, relative to the mean values measured in the control cybrids.
Figure 3.
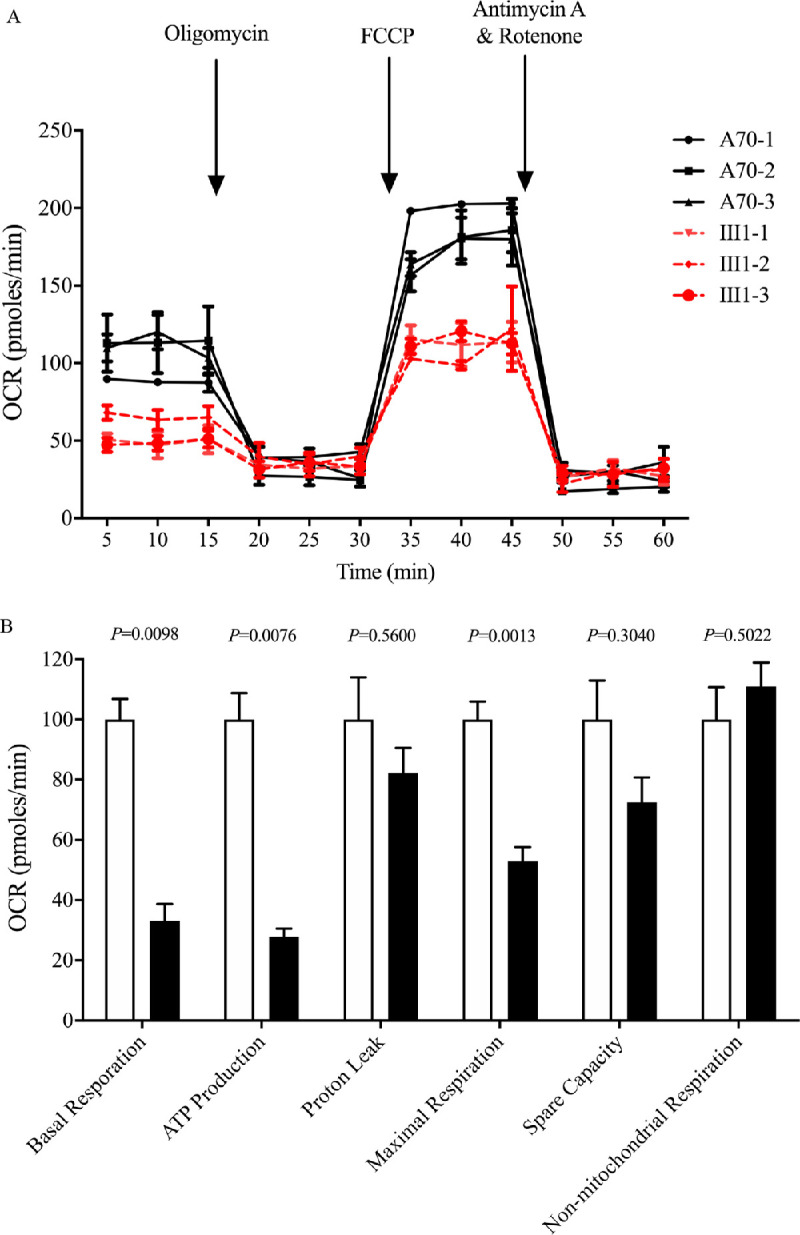
Respiration assays. (A) An analysis of O2 consumption in the various cell lines using different inhibitors. The OCRs were first measured on 1 × 104 cells of each cell line under basal conditions and then sequentially added to oligomycin (1.0 µM), FCCP (0.5 µM), rotenone (1.0 µM), and antimycin A (1.0 µM) at the indicated times to determine the different parameters of mitochondrial functions. (B) Graphs show the ATP-linked OCR, proton leak OCR, maximal OCR, reserve capacity, and non-mitochondrial OCR in the mutant and control cell lines. The non-mitochondrial OCR was determined as the OCR after rotenone/antimycin A treatment. The basal OCR was determined as the OCR before oligomycin minus the OCR after rotenone/antimycin A treatment. The ATP-linked OCR was determined as the OCR before oligomycin minus the OCR after oligomycin. The proton leak OCR was determined as the basal OCR minus the ATP-linked OCR. The maximal OCR was determined as the OCR after FCCP minus the non-mitochondrial OCR. Reserve capacity was defined as the difference between the maximal OCR after FCCP minus the basal OCR. The average values of four independent experiments for each cell line are shown. The horizontal dashed lines represent the average value for each group. Graph details and symbols are explained in Figure 1.
Impaired Mitochondrial ATP Synthesis
The capacity of oxidative phosphorylation in mutant and control cybrids was examined by measuring the levels of cellular ATP using a luciferin/luciferase assay. Populations of cells were incubated in media in the presence of glucose (total cellular ATP), and 2-deoxy-d-glucose (2-DG) with pyruvate to inhibit glycolysis (oxidative phosphorylation).29 As shown in Figure 4, the levels of mitochondrial ATP production in the mutant cybrids ranged from 73.3% to 87.5%, with an average of 81.1% relative to the mean values measured in the control cybrids (P = 0.0114), whereas there were no differences in the levels of total cellular ATP between mutant and control cybrids.
Figure 4.
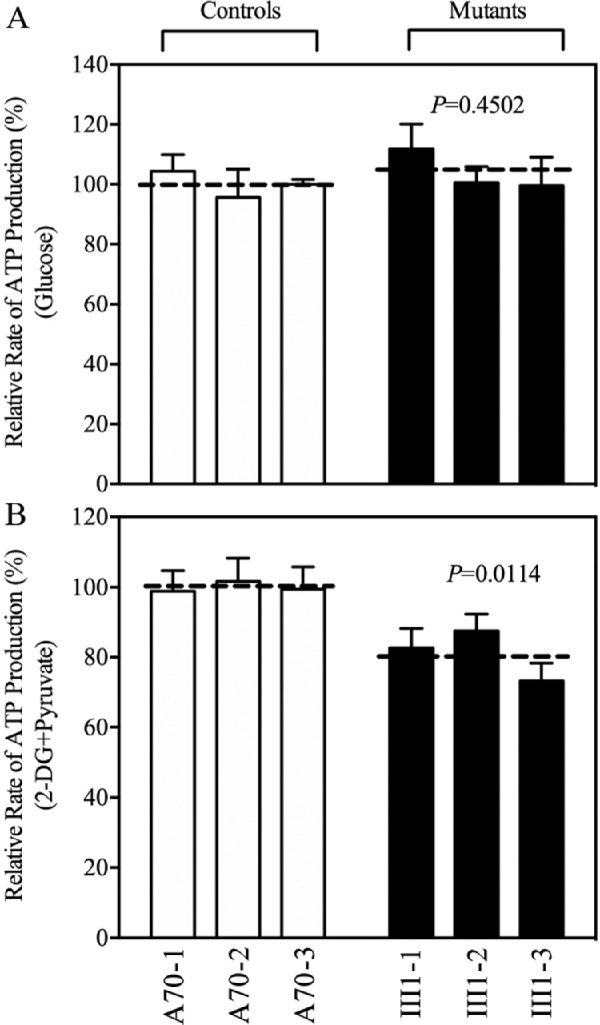
Measurement of cellular and mitochondrial ATP levels. ATP levels in the mutant and control cell lines were measured using a luciferin/luciferase assay. Cells were incubated with 10-mM glucose or 5-mM 2-DG plus 5-mM pyruvate to determine ATP generation under mitochondrial ATP synthesis. (A) Average ATP levels in total cells. (B) Average ATP levels in mitochondria. Four independent experiments were conducted for each cell line. Graph details and symbols are explained in Figure 1.
Decrease in Mitochondrial Membrane Potential
The JC-10 Mitochondrial Membrane Potential Assay Kit was used to measure the mitochondrial membrane potential (ΔΨm) in three mutant and three control cybrids.39,40 The ratios of fluorescence intensities Ex/Em = 490/590 and 490/525 nm (FL590/FL525) were recorded to determine the ΔΨm of each sample. The geometric means of the relative ratios of FL590/FL525 between mutant and control cybrids were calculated to represent the ΔΨm levels, as described elsewhere.39 As shown in Figure 5, the levels of ΔΨm in three mutant cybrids carrying the m.3460G>A mutation were decreased, ranging from 28.9% to 35.7%, with a mean value of 31.5% (P = 0.0006) measured in three control cybrids. By contrast, the levels of ΔΨm in mutant cybrids in the presence of FCCP were comparable to those measured in the control cybrids.
Figure 5.

Mitochondrial membrane potential analysis. (A, C) The mitochondrial membrane potential was measured in mutant and control cell lines using the BD Biosciences LSR II flow cytometer and JC-10 Mitochondrial Membrane Potential Assay Kit. The ratios of fluorescence—490/525 nm and 490/525 nm (FL590/FL525)—were recorded to determine the ΔΨm level of each sample. Representative flow cytometry images of mutant cell lines and control cell lines with and without 10 µM of FCCP are shown. (B, D) The geometric means of the FL590/FL525 relative ratios between mutant and control cell lines were calculated to reflect the level of ΔΨm. The averages of three to five determinations for each cell line are shown. Graph details and symbols are explained in Figure 1.
ROS Production Increase
The levels of ROS generation in the vital cells in three mutant cybrids carrying the m.3460G>A mutation and three control cybrids lacking this mutation were measured using the MitoSOX assay via flow cytometry under normal conditions and H2O2 stimulation.41,42 Geometric mean intensity was recorded to measure the rate of ROS in each sample. The ratio of geometric mean intensities between unstimulated and stimulated cell lines with H2O2 was calculated to determine the reaction to increasing levels of ROS under oxidative stress. As shown in Figures 6A and 6B, the levels of mitochondrial ROS in the three mutant cybrids varied from 123.5% to 135.2%, with an average of 129.3% (P = 0.0040) of the mean value measured in three control cybrids. To determine whether the m.3460G>A mutation affects antioxidant systems, we used western blot analysis to measure the levels of three antioxidant enzymes in the mutant and control cybrids: SOD2 in the mitochondrial matrix and SOD1 and catalase in the cytosol and mitochondrial intermembrane space.47 As shown in Figure 6C, the mutant cell lines revealed marked increases in the levels of SOD1 and catalase and mild increases in the levels of SOD2, as compared with those in the control cell lines.
Figure 6.

Assays for mitochondrial ROS production. (A) Ratios of geometric mean intensity between levels of ROS generation in the vital cells. The rates of ROS production from six cell lines were analyzed using the BD Biosciences LSR II flow cytometer system with MitoSOX (5 µM). (B) The relative ratios of intensity were calculated. The average values of three independent determinations for each cell line are shown. Graph details and symbols are explained in Figure 1. (C) Western blotting analysis of antioxidative enzymes SOD2, SOD1, and catalase in six cell lines, with actin as a loading control.
Promoting Intrinsic Apoptosis
Deficient activities of oxidative phosphorylation have been linked to protecting against certain apoptotic stimuli.11,18 To evaluate whether or not the m.3460G>A mutation affects the apoptotic process, we performed TUNEL assays to examine cell death in the mutant (III1-1) and control (A70-1) cybrids with or without H2O2 stimulation. As shown in Figure 7A, cell death in the mutant cybrids (III1-1) increased as compared with the control cybrids (A70-1). Without H2O2 stimulation, there were 36 cell deaths out of 3147 total cells (n = 10 sides) in the control cybrids (A70-1) and 43 cell deaths out of 2686 total cells (n = 10 sides) in the mutant cybrids (III-1). With H2O2 stimulation, the mutant cybrids (III1-1) had 243 cell deaths out of 2724 total cells (n = 10 sides), as compared with 81 cell deaths out of 2721 total cells (n = 10 slides) in the control cybrids (A70-1).
Figure 7.

Apoptosis assays. (A) TUNEL assays of mutant III1-1 and control A70-1 cybrids with or without H2O2 stimulation. Arrows indicate death cells. (B) The distribution of cytochrome c from mutant III1-1 and control A70-1 cybrids was visualized by immunofluorescent labeling with TOM20 antibody conjugated to Alexa Fluor 594 (red) and cytochrome c antibody conjugated to Alexa Fluor 488 (green) and analyzed by confocal microscopy. DAPI-stained nuclei were identified by their blue fluorescence. (C, E, G) Western blotting analysis. Cellular proteins (20 µg) from various cell lines were electrophoresed, electroblotted, and hybridized with several apoptosis-associated protein antibodies: cytochrome c, BAK, and BAX (C); cleaved caspases 3, 7, and 9 and PARP (E); and uncleaved caspases 3, 7, and 9 and PARP (G), with β-actin as a loading control. (D, F, H) Quantification of apoptosis-associated proteins: cytochrome c, BAK, and BAX (D); cleaved caspases 3, 7, and 9 and PARP (F); and uncleaved caspases 3, 7, and 9 and PARP (H). The levels of apoptosis-associated proteins in various cell lines were determined as described elsewhere.11 Three independent determinations were made in each cell line. Graph details and symbols are explained in Figure 1.
We then examined the apoptotic state of the cybrids by using immunocytostaining assays to determine the immunofluorescence pattern of cybrids that were double labeled with rabbit monoclonal antibody specific for cytochrome c and mouse monoclonal antibody to TOM20, a nuclear-encoded mitochondrial protein. As shown in Figure 7B, the mutant cybrids carrying the m.3460G>A mutation revealed markedly increased levels of cytochrome c, as compared with the control cybrids. The impact of the m.3460G>A mutation on the apoptotic process was confirmed with western blot analysis, which showed that the mutant cybrids exhibited 67.4%, 53.0%, and 51.8% increased levels of cytochrome c, BAX, and BAK, respectively, which are able to pierce the mitochondrial outer membrane to mediate cell death by apoptosis,48 as compared with the control cybrids (Figures 7C, 7D). Furthermore, we used western blot analysis to measure the levels of apoptosis-related proteins: uncleaved/cleaved PARP and uncleaved/cleaved caspases 3, 7, and 9 in mutant and control cell lines.49 As shown in Figures 7E and 7F, the average levels of cleaved PARP protein and caspases 3, 7, and 9 in the three mutant cell lines were 153.5%, 116.2%, 140.8%, and 114.5%, respectively, of the average values measured in the three control cell lines. However, there were no significant differences in the average levels of uncleaved PARP or caspases 3, 7, and 9 between control and mutant cybrids (Figs. 7G, 7H).
Alteration in Mitophagy
Mitophagy, an important mitochondrial quality control system, selectively degrades damaged mitochondria with autophagosomes and their subsequent catabolism by lysosomes.50 To investigate the effect of the m.3460G>A mutation on mitophagy, we evaluated the mitophagic states of mutant and control cell lines using immunoblotting and endogenous immunofluorescence assays. As shown in Figure 8A, mutant cell lines displayed ∼16% reduced levels of lysosome-associated membrane glycoprotein 1 (LAMP1), indicating that the m.3460G>A mutation affects the mitophagy process. The levels of autophagy in mutant and control cell lines were then examined using two markers: microtubule-associated protein 1A/1B light chain 3 (LC3) and Beclin-1.51 During autophagy, the cytoplasmic form (LC3-I) is processed into a cleaved and lipidated membrane-bound form (LC3-II), which is recruited to autophagosomal membranes. The amount of LC3-II is clearly correlated with the number of autophagosomes.52 The sequestosome 1/P62 protein, (SQSTM1, hereafter referred to P62), is an autophagy substrate that colocalizes with ubiquitinated protein aggregates in many neurodegenerative diseases.53 As shown in Figures 8B and 8D, reduced levels of LC3 and increased levels of P62 were observed in mutant cell lines carrying the m.3460G>A mutation, compared with controls. In particular, the average levels of LC3-II/I and P62 in the three mutant cell lines carrying the m.3460G>A mutation were 47.0% (P = 0.0140) and 137.5% (P = 0.0375) of the mean values measured in the three control cell lines, respectively. These data suggest that the m.3460G>A mutation impaired autophagy in the mutant cell lines.
Figure 8.
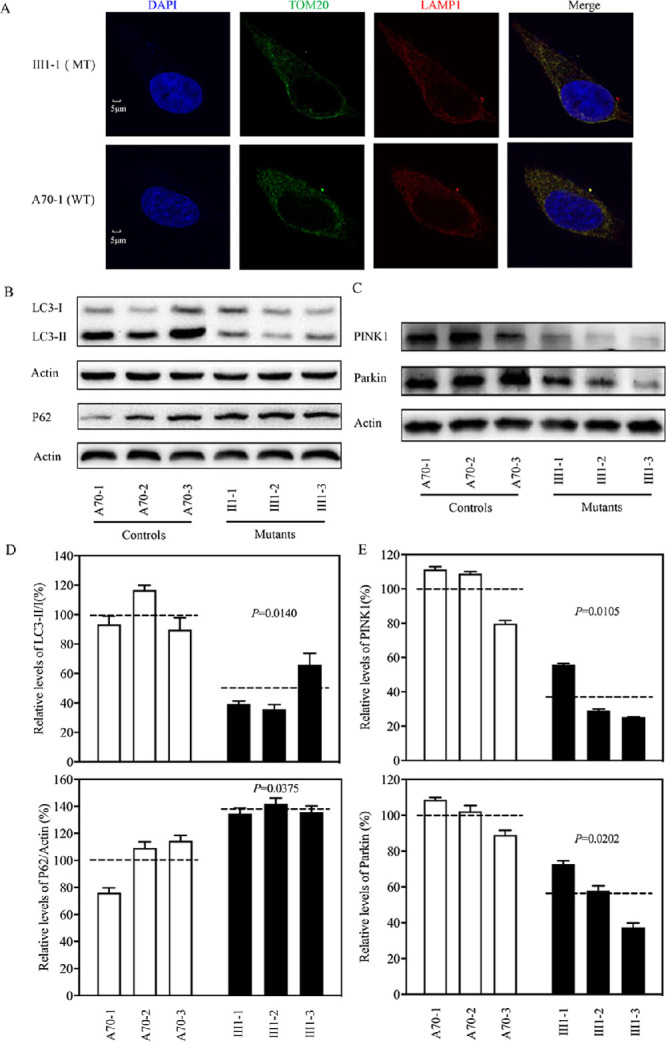
Analysis of mitophagy. (A) The distribution of LAMP1 from cybrids (III1-1 and A70-1) was visualized by immunofluorescent labeling with TOM20 antibody conjugated to Alexa Fluor 488 (green) and LAMP1 antibody conjugated to Alexa Fluor 594 (red) analyzed by confocal microscopy. DAPI-stained nuclei were revealed by blue fluorescence. (B, C) Western blot analysis. Cellular proteins (20 µg) from various cell lines were electrophoresed, electroblotted, and hybridized with LC3, P62, PINK1, and parkin antibodies, with β-actin as a loading control. (D) Quantification of autophagy markers LC3 and P62. (E) Quantification of mitophagy-associated proteins (PINK1 and parkin). Three independent determinations were made in each cell line. Graph details and symbols are explained in Figure 1.
Mitophagy promotes mitochondrial turnover and prevents accumulation of damaged mitochondria. It is regulated by the PINK1/parkin pathway. Damaged mitochondria are recognized by PINK1, which builds up on the outer mitochondrial membrane and recruits parkin. The accumulation of PINK1 and recruitment of parkin target mitochondria for degradation by lysosomes.50 To assess if the m.3460G>A mutation affects PINK1/parkin-mediated mitophagy, we evaluated the mitophagic states of mutant and control cell lines using immunoblotting. As shown in Figures 8C and 8E, reduced levels of PINK1 and parkin were observed in mutant cell lines carrying the m.3460G>A mutation compared with the controls. In particular, the average levels of PINK1 and parkin in the three mutant cell lines carrying the m.3460G>A mutation were 36.7% (P = 0.0105) and 56.0% (P = 0.0202), respectively, of the mean values measured in the three control cell lines lacking the mutation. These data reveal that the m.3460G>A mutation alters mitophagy.
Discussion
In the current study, we investigated the pathophysiology of the LHON-associated m.3460G>A mutation. Nineteen Han Chinese families bearing the m.3460G>A mutation were identified among a large cohort of Chinese patients with LHON.14,26,27 Optic neuropathy as a sole clinical phenotype was only present in the maternal lineage of these pedigrees, which showed a wide range of penetrance and expression of the optic neuropathy. The average age of onset of LHON ranged from 5 to 29 years, with an average of 18.9 years among the 19 Chinese families, whereas the average age of onset of optic neuropathy was 20 years in eight Caucasian families.54,55 The penetrance of optic neuropathy (affected matrilineal relatives/total matrilineal relatives) in the 19 Chinese pedigrees bearing the m.3460G>A mutation ranged from 2.8% to 50%, with an average of 20.3%; whereas, 134 affected matrilineal relatives out of 768 matrilineal relatives among 21 Caucasian families harboring the m.3469G>A mutation developed optic neuropathy.5,55 Furthermore, 28 males and 14 females of the 247 matrilineal relatives among the 19 Han Chinese families exhibited visual impairment, whereas the average ratio for affected male and female matrilineal relatives among 21 Caucasian families was 3.35.5,55 Moreover, the mtDNA haplogroups (A, B5, C4a1, D, F, M12, M7, M8a2, R9, and H2) of the 19 Chinese pedigrees and those (B4d1, F2, A5b, M12a, D4b2b, and D4b2) in six other Chinese families bearing the 3460G>A mutation differed from those (H, J, K, U, V, W, and X) in 11 Dutch and seven Italian families carrying the m.3460G>A mutation.56–58 The occurrence of the m.3460G>A mutation in these genetically unrelated families affected by optic neuropathy and that they differed considerably in their mtDNA haplogroups highlight the impact of the mutation on the pathogenesis of LHON. The phenotypic variability, including the penetrance and expression of the m.3460G>A mutation, may be due to the influence of mitochondrial haplotypes or nuclear modifier genes.19,26,27,30,58–60
The m.3460G>A mutation replaced the highly conserved alanine at position 52 with a threonine in MT-ND1, which is the essential subunit of complex I comprised of an additional six mitochondrion-encoding and 39 nucleus-encoding subunits.21 Based on the cryo-EM structure of mammalian complex I, the hydrophobic side chains of A52 contribute to the hydrophobic interactions with P48, L55, and 56F in MT-ND1.22 The substitution of nonpolar hydrophobic alanine at position 52 with the hydrophilic threonine by the m.3460G>A mutation in MT-ND1 may destabilize these interactions, thereby altering the structure and function of MT-ND1. The instability of mutated ND1 was evidenced by a 52.6% decrease in the levels of MT-ND1 observed in the cybrids harboring the m.3460G>A mutation. However, the m.3460G>A mutation did not affect the expression of NDUFA3, NDUFA8, or NDUFA13, which interact with MT-ND1.22 The mutated MT-ND1 altered the assembly of complex I, as is the case for m.3394T>C and m.3866T>C mutations.18,20 Alterations in the stability of MT-ND1 and the assembly of complex I were responsible for significant reductions in the activity of complex I observed in the cybrids in this study and lymphoblastoid cell lines carrying the m.3460G>A mutation.15 Furthermore, the m.3460G>A mutation gave rise to reduced basal OCRs, as well as reduced levels of ATP-linked OCR protons, leak OCRs, maximal OCRs, and reserve capacity in the mutant cybrids. These OXPHOS deficiencies diminished ATP synthesis, impaired the mitochondrial membrane potentials, and increased the production of oxidative reactive species in the mutant cell lines bearing the m.3460G>A mutation, as is the case for cells carrying the m.3394T>C and m.3866T>C mutations.18,20 These results indicate that defects in complex I in the mutant cell lines carrying the m.3460G>A mutation play a critical role in the mitochondrial dysfunction that leads to the development of LHON.
Mitochondrial dysfunction and overproduction of ROS caused by LHON-associated mtDNA mutations often affect apoptotic death and mitophagy.11,18,20,61–63 In the present investigation, we have shown that mitochondrial dysfunctions caused by the m.3460G>A mutation promote the apoptotic process. Both immunocytostaining assays and western blot analysis revealed the elevated releases of cytochrome c into cytosol in the mutant cybrids carrying the m.3460G>A mutation compared with the control cybrids lacking the mutation. Additional effects of the m.3460G>A mutation include the increased expression of BAK and BAX, which mediate cell death by apoptosis, and the elevated levels of apoptosis-activated proteins (cleaved caspases 3, 7, and 9 and PARP) in the mutant cybrids carrying the m.3460G>A mutation compared with the control cybrids.48,49 Mitophagy disposes of damaged mitochondria and maintains a healthy mitochondria population in cells.50 The effect of the m.3460G>A mutation on autophagy was first evidenced by reduced levels of LAMP1 in the mutant cell lines carrying the m.3460G>A mutation as compared with the control cell lines. The reductions in LC3 level in the mutant cell lines suggested a general decrease in the capacity of the mutant cells to generate autophagosomes, thereby perturbing the autophagic degradation of ubiquitinated proteins. Furthermore, the increased levels of p62 in cybrids carrying the m.3460G>A mutation indicate an accumulation of autophagic substrates, such as misfolded proteins, leading to the deleterious effects. Notably, the reduced levels of PINK1 and parkin observed in the mutant cell lines carrying the m.3460G>A mutation indicated that the m.3460G>A mutation altered the mitophagic removal of damaged mitochondria. Photoreceptor cells and RGCs account for the biggest demand for ATP by cells in the retina.37,64 Mitochondrial dysfunction may lead to the dysfunction or death of RGCs, thereby contributing to the development of optic neuropathy. Our findings highlight the critical role of the m.3460G>A mutation in the pathogenesis of LHON, manifested by mitochondrial dysfunction and alterations in apoptosis and mitophagy.
Supplementary Material
Acknowledgments
The authors thank the patients and their family members for their participation.
Supported by grants from the Natural Science Foundation of China and National Key R&D Program of China (82071007 and 31970557), Ministry of Science and Technology of China (2018YFC1004802 and 2016YFC0901504), Zhejiang Provincial Natural Science Foundation (LY20C060003), and Wenzhou Science and Technology Bureau (Y20190067).
Disclosure: J. Zhang, None; Y. Ji, None; J. Chen, None; M. Xu, None; G. Wang, None; X. Ci, None; B. Lin, None; J.Q. Mo, None; X. Zhou, None; M.-X. Guan, None
References
- 1. Wallace DC, Lott MT.. Leber hereditary optic neuropathy: exemplar of an mtDNA disease. Handb Exp Pharmacol . 2017; 240: 339–376. [DOI] [PubMed] [Google Scholar]
- 2. Carelli V, La Morga C, Valentino ML, Barboni P, Ross-Cisneros FN, Sadun AA. Retinal ganglion cell neurodegeneration in mitochondrial inherited disorders. Biochim Biophys Acta . 2009; 1787: 518–528. [DOI] [PubMed] [Google Scholar]
- 3. Sadun AA, La Morgia C, Carelli V. Leber's hereditary optic neuropathy. Curr Treat Options Neurol . 2011; 1391: 109–117. [DOI] [PubMed] [Google Scholar]
- 4. Wallace DC, Singh G, Lott MT, et al.. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science . 1988; 24: 1427–1430. [DOI] [PubMed] [Google Scholar]
- 5. Mackey DA, Oostra RJ, Rosenberg T, et al.. Primary pathogenic mtDNA mutations in multigeneration pedigrees with Leber hereditary optic neuropathy. Am J Hum Genet . 1996; 59: 481–485. [PMC free article] [PubMed] [Google Scholar]
- 6. Ruiz-Pesini E, Lott MT, Procaccio V, et al.. An enhanced mitomap with a global mtDNA mutational phylogeny. Nucleic Acids Res . 2007; 35: D823–D828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ji Y, Nie Z, Meng F, et al.. Mechanistic insights into mitochondrial tRNAAla 3′-end metabolism deficiency. J Biol Chem. 2021; 296: 100816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brown MD, Torroni A, Reckord CL, Wallace DC.. Phylogenetic analysis of Leber's hereditary optic neuropathy mitochondrial DNA's indicates multiple independent occurrences of the common mutations. Hum Mutat . 1995; 6: 311–325. [DOI] [PubMed] [Google Scholar]
- 9. Zhou X, Qian Y, Zhang J, et al.. Leber's hereditary optic neuropathy is associated with the T3866C mutation in mitochondrial ND1 gene in three Han Chinese families . Invest Ophthalmol Vis Sci . 2012; 53: 4586–4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang J, Jiang P, Jin X, et al.. Leber's hereditary optic neuropathy caused by the homoplasmic ND1 G3635A mutation in nine Han Chinese families. Mitochondrion. 2014; 18: 18–26. [DOI] [PubMed] [Google Scholar]
- 11. Zhang J, Ji Y, Lu Y, et al.. Leber's hereditary optic neuropathy (LHON)-associated ND5 12338T>C mutation altered the assembly and function of complex I, apoptosis and mitophagy. Hum Mol Genet. 2018; 27: 1999–2011. [DOI] [PubMed] [Google Scholar]
- 12. Jiang P, Liang M, Zhang J, et al.. Prevalence of mitochondrial ND4 mutations in 1281 Han Chinese subjects with Leber's hereditary optic neuropathy. Invest Ophthalmol Vis Sci . 2015; 56: 4778–4788. [DOI] [PubMed] [Google Scholar]
- 13. Liang M, Jiang P, Li F, et al.. Frequency and spectrum of mitochondrial ND6 mutations in 1218 Han Chinese subjects with Leber's hereditary optic neuropathy. Invest Ophthalmol Vis Sci . 2014; 55: 1321–1331. [DOI] [PubMed] [Google Scholar]
- 14. Ji Y, Liang M, Zhang J, et al.. Mitochondrial ND1 variants in 1281 Chinese subjects with Leber's hereditary optic neuropathy. Invest Ophthalmol Vis Sci . 2016; 56: 2377–2389. [DOI] [PubMed] [Google Scholar]
- 15. Brown MD, Trounce IA, Jun AS, Allen JC, Wallace DC.. Functional analysis of lymphoblast and cybrid mitochondria containing the 3460, 11778, or 14484 Leber's hereditary optic neuropathy mitochondrial DNA mutation. J Biol Chem . 2000; 275: 39831–39836. [DOI] [PubMed] [Google Scholar]
- 16. Hofhaus G, Johns DR, Hurkoi O, Attardi G, Chomyn A.. Respiration and growth defects in transmitochondrial cell lines carrying the 11778 mutation associated with Leber's hereditary optic neuropathy. J Biol Chem . 1996; 271: 13155–13161. [DOI] [PubMed] [Google Scholar]
- 17. Pello R, Martin MA, Carelli V, et al.. Mitochondrial DNA background modulates the assembly kinetics of OXPHOS complexes in a cellular model of mitochondrial disease. Hum Mol Genet . 2008; 17: 4001–4011. [DOI] [PubMed] [Google Scholar]
- 18. Ji Y, Zhang J, Lu Y, et al.. Complex I mutations synergize to worsen the phenotypic expression of Leber's hereditary optic neuropathy. J Biol Chem. 2020; 295: 13224–13238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jiang P, Jin, Peng Y, et al.. The exome sequencing identified the mutation in YARS2 encoding the mitochondrial tyrosyl-tRNA synthetase as a nuclear modifier for the phenotypic manifestation of Leber's hereditary optic neuropathy-associated mitochondrial DNA mutation. Hum Mol Genet . 2016; 25: 584–596. [DOI] [PubMed] [Google Scholar]
- 20. Ji Y, Zhang J, Yu J, et al.. Contribution of mitochondrial ND1 3394T>C mutation to the phenotypic manifestation of Leber's hereditary optic neuropathy. Hum Mol Genet . 2019; 28: 1515–1529. [DOI] [PubMed] [Google Scholar]
- 21. Scheffler IE. Mitochondrial disease associated with complex I (NADH-CoQ oxidoreductase) deficiency. J Inherit Metab Dis. 2015; 38: 405–415. [DOI] [PubMed] [Google Scholar]
- 22. Zhu J, Vinothkumar KR, Hirst J.. Structure of mammalian respiratory complex I. Nature. 2016; 536: 354–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huoponen K, Vilkki J, Aula P, Nikoskelainen EK, Savontaus ML.. A new mtDNA mutation associated with Leber hereditary optic neuroretinopathy. Am J Hum Genet . 1991; 48: 1147–1153. [PMC free article] [PubMed] [Google Scholar]
- 24. Howell N, Bindoff LA, McCullough DA, et al.. Leber hereditary optic neuropathy: identification of the same mitochondrial ND1 mutation in six pedigrees. Am J Hum Genet . 1991; 49: 939–950. [PMC free article] [PubMed] [Google Scholar]
- 25. Mashima Y, Yamada K, Wakakura M, et al.. Spectrum of pathogenic mitochondrial DNA mutations and clinical features in Japanese families with Leber's hereditary optic neuropathy. Curr Eye Res. 1998; 17: 403–408. [DOI] [PubMed] [Google Scholar]
- 26. Tong Y, Mao Y, Zhou X, et al.. The mitochondrial tRNAGlu A14693G mutation may influence the phenotypic manifestation of ND1 G3460A mutation in a Chinese family with Leber's hereditary optic neuropathy. Biochem Biophys Res Commun. 2007; 357: 524–530. [DOI] [PubMed] [Google Scholar]
- 27. Ji Y, Liang M, Zhang J, et al.. Mitochondrial haplotypes may modulate the phenotypic manifestation of the LHON-associated ND1 G3460A mutation in Chinese families. J Hum Genet . 2014; 59: 134–140. [DOI] [PubMed] [Google Scholar]
- 28. King MP, Attadi G.. Mitochondria-mediated transformation of human ρ0 cells. Methods Enzymol . 1996; 264: 313–334. [DOI] [PubMed] [Google Scholar]
- 29. Gong S, Peng Y, Jiang P, et al.. A deafness-associated tRNAHis mutation alters the mitochondrial function, ROS production and membrane potential. Nucleic Acids Res. 2014; 42: 8039–8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yu J, Liang X, Ji Y, et al.. PRICKLE3 linked to ATPase biogenesis manifested Leber's hereditary optic neuropathy. J Clin Invest. 2020; 130: 4935–4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Qu J, Li R, Zhou X, et al.. The novel A4435G mutation in the mitochondrial tRNAMet may modulate the phenotypic expression of the LHON-associated ND4 G11778A mutation in a Chinese family. Invest Ophthalmol Vis Sci. 2006; 47: 475–483. [DOI] [PubMed] [Google Scholar]
- 32. Rieder MJ, Taylor SL, VO Tobe, Nickerson DA.. Automating the identification of DNA variations using quality-based fluorescence re-sequencing: analysis of the human mitochondrial genome. Nucleic Acids Res. 1998; 26: 967–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N.. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet. 1999; 23: 147. [DOI] [PubMed] [Google Scholar]
- 34. Miller G, Lipman M.. Release of infectious Epstein-Barr virus by transformed marmoset leukocytes. Proc Natl Acad Sci USA. 1973; 70: 190–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jha P, Wang X, Auwerx J. Analysis of mitochondrial respiratory chain supercomplexes using blue native polyacrylamide gel electrophoresis (BN-PAGE). Curr Protoc Mouse Biol . 2016; 6: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wittig I, Braun HP, Schagger H.. Blue native PAGE. Nat Protoc . 2006; 1: 418–428. [DOI] [PubMed] [Google Scholar]
- 37. Jin X, Zhang Z, Nie Z, et al.. An animal model for mitochondrial tyrosyl-tRNA synthetase deficiency reveals links between oxidative phosphorylation and retinal function. J Biol Chem. 2021; 296: 100437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dranka BP, Benavides GA, Diers AR, et al.. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic Biol Med . 2011; 51: 1621–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Reers M, Smiley ST, Mottola-Hartshorn C, Chen A, Lin M, Chen LB. Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol. 1995; 260: 406–417. [DOI] [PubMed] [Google Scholar]
- 40. Zhou M, Xue L, Chen Y, et al.. A hypertension-associated mitochondrial DNA mutation introduces an m1G37 modification into tRNAMet, altering its structure and function. J Biol Chem. 2018; 293: 1425–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mahfouz R, Sharma R, Lackner J, Aziz N, Agarwal A.. Evaluation of chemiluminescence and flow cytometry as tools in assessing production of hydrogen peroxide and superoxide anion in human spermatozoa. Fertil Steril. 2009; 92: 819–827. [DOI] [PubMed] [Google Scholar]
- 42. Jiang P, Wang M, Xue L, et al.. A hypertension-associated tRNAAla mutation alters tRNA metabolism and mitochondrial function. Mol Cell Biol. 2016; 6: 1920–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kong QP, Bandelt HJ, Sun C, et al.. Updating the East Asian mtDNA phylogeny: a prerequisite for the identification of pathogenic mutations. Hum Mol Genet. 2006; 15: 2076–2086. [DOI] [PubMed] [Google Scholar]
- 44. Agip A-NA, Blaza JN, Bridges HR, et al.. Cryo-EM structures of complex I from mouse heart mitochondria in two biochemically defined states. Nat Struct Mol Biol. 2018; 25: 548–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Szczepanowska K, Maiti P, Kukat A, et al.. CLPP coordinates mitoribosomal assembly through the regulation of ERAL1 levels. EMBO J . 2016; 35: 2566–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nolden M, Ehses S, Koppen M, et al.. The m-AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in mitochondria. Cell . 2005; 123: 277–289. [DOI] [PubMed] [Google Scholar]
- 47. Gong S, Wang X, Meng F, et al.. Overexpression of mitochondrial histidyl-tRNA synthetase restores mitochondrial dysfunction caused by a deafness-associated tRNAHis mutation. J Biol Chem. 2020; 295: 940–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Westphal D, Dewson G, Czabotar PE, Kluck RM.. Molecular biology of Bax and Bak activation and action. Biochim Biophys Acta. 2011; 1813: 521–531. [DOI] [PubMed] [Google Scholar]
- 49. Wang C, Youle RJ. The role of mitochondria in apoptosis. Annu Rev Genet . 2009; 43: 95–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pickles S, Vigie P, Youle RJ.. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol . 2018; 28: R170–R185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sharma LK, Tiwari M, Rai NK, Bai Y.. Mitophagy activation repairs Leber's hereditary optic neuropathy-associated mitochondrial dysfunction and improves cell survival. Hum Mol Genet . 2019; 28: 422–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mizushima N, Yoshimori T.. How to interpret LC3 immunoblotting. Autophagy . 2007; 3: 542–545. [DOI] [PubMed] [Google Scholar]
- 53. Lamark T, Svenning S, Johansen T.. Regulation of selective autophagy: the p62/SQSTM1 paradigm. Essays Biochem . 2017; 61: 609–624. [DOI] [PubMed] [Google Scholar]
- 54. Johns DR, Smith KH, Miller NR.. Leber's hereditary optic neuropathy. Clinical manifestations of the 3460 mutation. Arch Ophthalmol. 1992; 110: 1577–1581. [DOI] [PubMed] [Google Scholar]
- 55. Harding AE, Sweeney MG, Govan GG, Riordan-Eva P.. Pedigree analysis in Leber hereditary optic neuropathy families with a pathogenic mtDNA mutation. Am J Hum Genet. 1995; 57: 77–86. [PMC free article] [PubMed] [Google Scholar]
- 56. Yu D, Jia X, Zhang AM, et al.. Molecular characterization of six Chinese families with m.3460G>A and Leber hereditary optic neuropathy. Neurogenetics . 2010; 11: 349–356. [DOI] [PubMed] [Google Scholar]
- 57. Howell N, Oostra R-J, Bolhuis PA, et al.. Sequence analysis of the mitochondrial genomes from Dutch pedigrees with Leber hereditary optic neuropathy. Am J Hum Genet. 2003; 72: 1460–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Torroni A, Petrozzi M, D'Urbano L, et al.. Haplotype and phylogenetic analyses suggest that one European-specific mtDNA background plays a role in the expression of Leber hereditary optic neuropathy by increasing the penetrance of the primary mutations 11778 and 14484. Am J Hum Genet. 1997; 60: 1107–1121. [PMC free article] [PubMed] [Google Scholar]
- 59. Ji Y, Zhang AM, Jia X, et al.. Mitochondrial DNA haplogroups M7b1'2 and M8a affect clinical expression of Leber hereditary optic neuropathy in Chinese families with the m.11778G–>a mutation. Am J Hum Genet. 2008; 83: 760–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jin X, Zhang J, Yi Q, et al.. Leber's hereditary optic neuropathy arising from the synergy between ND1 3635G>A mutation and mitochondrial YARS2 mutations. Invest Ophthalmol Vis Sci. 2021; 62: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Skeie JM, Nishimura DY, Wang CL, et al.. Mitophagy: an emerging target in ocular pathology. Invest Ophthalmol Vis Sci. 2021; 62: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ghelli A, Zanna C, Porcelli AM, et al.. Leber's hereditary optic neuropathy (LHON) pathogenic mutations induce mitochondrial-dependent apoptotic death in transmitochondrial cells incubated with galactose medium. J Biol Chem. 2003; 278: 4145–4150. [DOI] [PubMed] [Google Scholar]
- 63. Danielson SR, Wong A, Carelli V, Martinuzzi A, Schapira AH, Cortopassi GA.. Cells bearing mutations causing Leber's hereditary optic neuropathy are sensitized to Fas-induced apoptosis. J Biol Chem. 2002; 277: 5810–5815. [DOI] [PubMed] [Google Scholar]
- 64. Country MW. Retinal metabolism: a comparative look at energetics in the retina. Brain Res . 2017; 1672: 50–57. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.