

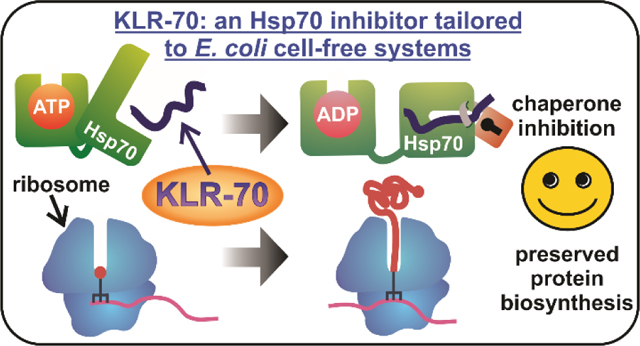
Abstract
The heat-shock factor Hsp70 and other molecular chaperones play a central role in nascent protein folding. Elucidating the task performed by individual chaperones within the complex cellular milieu, however, has been challenging. One strategy to address this goal has been to monitor protein biogenesis in the absence and presence of inhibitors of a specific chaperone, followed by analysis of folding outcomes under both conditions. In this way, the role of the chaperone of interest can be discerned. However, development of chaperone inhibitors, including well-known proline-rich antimicrobial peptides, has been fraught with undesirable side effects including decreased protein expression yields. Here, we introduce KLR-70, a rationally designed cationic inhibitor of the E. coli Hsp70 chaperone (also known as DnaK). KLR-70 is a 14-amino-acid peptide bearing naturally occurring residues and engineered to interact with the DnaK substrate-binding domain. The interaction of KLR-70 with DnaK is enantioselective and is characterized by high affinity in buffered solution. Importantly, KLR-70 does not significantly interact with the DnaJ and GroEL/ES chaperones, and it does not alter yields of nascent protein biosynthesis across a wide concentration range. Some attenuation of the anti-DnaK activity of KLR-70, however, has been observed in the complex E. coli cell-free environment. Interestingly, the D enantiomer D-KLR-70, unlike its all-L KLR-70 counterpart, does not bind the DnaK and DnaJ chaperones, yet it strongly inhibits translation. This outcome suggests that the two enantiomers (KLR-70 and D-KLR-70) may serve as orthogonal inhibitors of chaperone binding and translation. In summary, KLR-70 is a novel chaperone inhibitor with high affinity and selectivity for bacterial Hsp70 and with considerable potential to help parsing out the role of Hsp70 in nascent protein folding.
Keywords: Hsp70, DnaK, molecular chaperone, inhibitor, protein folding
TOC Graphic
Introduction
The heat-shock factor Hsp70 is a well-known molecular chaperone that plays a seminal role in key biological processes1 and helps maintain proteostasis2, 3 in living organisms. Hsp70 has been recently described as a molecular hub4 due to its ability to interact not only with its own co-chaperones but also with a variety of other chaperone networks that control intracellular activity. The function of Hsp70 spans from promoting the proper folding of nascent proteins to preventing and reversing protein aggregation, as well as to regulating protein degradation.1, 4–9
Upon interaction with its client proteins, Hsp70 undergoes allosterically regulated cycles of binding and release that ultimately result in proper protein folding. The most salient aspects of the Hsp70 chaperone cycle are illustrated in Figure 1 in the case of DnaK, i.e., the Hsp70 molecular chaperone of the E. coli bacterium. The DnaK chaperone cycle involves allosterically regulated conformational changes driven by the hydrolysis of nucleotide cofactors.10–13 These conformational changes, partly involving the closing and opening of an α-helical lid over a β-sheet binding cleft in the substrate binding domain (SBD) of DnaK, are responsible for regulating client-protein binding and release. The allosteric regulation of DnaK’s chaperone cycle is further mediated by its co-chaperones DnaJ and GrpE.14 DnaJ is responsible for improving the efficiency of client-protein binding by delivering some of the clients to DnaK and stimulating hydrolysis of DnaK-bound adenosine triphosphate (ATP), thus enhancing DnaK’s lid closure and affinity for its bound client.15–17 In contrast, GrpE stimulates the release of DnaK-bound adenosine diphosphate (ADP), which in turn allows the subsequent binding of a new ATP molecule, thus enhancing lid opening and encouraging client-protein release from DnaK.16, 17
Figure 1.

The DnaK chaperone cycle supports the proper folding of client proteins. The cycle involves interactions between unfolded, partially folded or misfolded client proteins (CP) and DnaK. These interactions are mediated by DnaK conformational changes that are allosterically regulated by nucleotide cofactors and the DnaK co-chaperones DnaJ and GrpE. The DnaK chaperone cycle can be summarized as consisting of three main steps: CP binding, nucleotide recycling and CP release. Once the client protein is released, DnaK is primed to accept new CPs for the next round of the chaperone cycle. While conformational heterogeneity of both CP and DnaK has been reported for each state of DnaK illustrated above, only the canonical conformation of each state is shown here, for simplicity.
DnaK is estimated to interact with approximately 25% of the E. coli cytosolic proteome4 by preferentially binding unfolded or partially-folded client-proteins that either fold slowly or are inherently thermodynamically unstable.18, 19 Despite its large interactome, however, DnaK is not rigorously required for cell survival under physiologically-relevant growth conditions (i.e., lacking an environmental stressor like high temperature, ≥ 42°C), given that other molecular chaperones share functional overlap with DnaK.4, 20, 21
An example of the above functional overlap involves DnaK acting in parallel with trigger factor (TF), a ribosome-binding molecular chaperone, to sequester nascent proteins as they become exposed outside the ribosomal exit tunnel during translation.20, 22 Other E. coli molecular chaperones known to coordinate directly with DnaK include Hsp9023, ClpB,24 and GroEL together with its co-chaperone GroES (GroEL/ES).25
To date, the functional overlap between bacterial Hsp70 and other molecular chaperones rendered it difficult to assess the independent effect of DnaK on protein folding, in the complex environment of the cell. Early attempts employed DnaK knockout strains.20, 26 Results were ambiguous, however, because elimination of DnaK causes upregulation of other heat-shock factors.4
Alternative approaches based on in vitro experiments employed fully reconstituted E.coli cell-free systems (denoted as PURE27) containing or lacking DnaK,28, 29 so that the identity and amounts of chaperones during nascent-protein biosynthesis and folding could be tightly controlled. However, one limitation of the PURE cell-free systems is that they are unable to mimic the natural complexity of the intracellular environment, given that they typically lack the majority of endogenous cytosolic components of living cells.30
An alternative in vitro method capturing the complexity of the E. coli cytoplasm relies on cell-free systems, which are based on extracts isolated from living E. coli cells. Cell-free systems lack endogenous DNA, hence they are incapable of up/down-regulating heat shock factors and other cellular components, following a perturbation.
Given that cell extracts retain most other soluble cytoplasmic components of the host organism, they are good proxies for the complex intracellular environment, and can easily be supplemented with plasmid DNA encoding the desired transcription/translation product. In addition, cell extracts are available from several organisms other than E. coli, including wheat germ,31 insects,32 rabbits,33 hamsters34 and humans,35 rendering the cell-free medium extremely versatile in biology.
In summary, understanding the role of individual chaperones in cell-like conditions is facilitated by environments lacking a homeostatic regulatory response, e.g., the cell-free systems and cell extracts described above, where selective chaperone inhibitors can be added and the impact of a missing or non-functional chaperone can be readily evaluated.
The major inhibitors developed so far against the bacterial Hsp70 chaperone DnaK include proline-rich antimicrobial peptides (prAMPs) like pyrrhocoricin, oncocin and related derivatives.22, 36–38 While several prAMPs with a variable spectrum of activity and selectivity against DnaK have been developed to date, 22, 37–39 it was recently reported that prAMPs interact with the ribosome,40–42 and hence directly inhibit translation.43, 44 This characteristic may be advantageous towards the potential antibacterial activity of prAMPs. However, the significant interaction of prAMPs with the ribosome renders them unsuitable for mechanistic studies targeting the role of DnaK in co- and immediately post-translational protein folding.
To overcome the above limitation, we developed a novel 14-residue peptide inhibitor of DnaK that, unlike prAMPs,22, 36–38 does not strongly impair nascent-protein expression across a wide concentration range. The peptide, named KLR-70 (pronounce: killer-70, i.e., an inactivator of Hsp70), displays high affinity (dissociation constant in the nM range) and selectivity for DnaK, and interacts with this chaperone stereoselectively. Conveniently, KLR-70 does not interact with the DnaJ co-chaperone, at the physiologically relevant pH values tested in this work.
In summary, KLR-70 is a novel inhibitor of bacterial Hsp70 with considerable potential to help parsing out the role of this chaperone in nascent protein folding.
Materials and Methods
Peptide synthesis.
Chemical synthesis of the KLR-70 peptide (RRGKKLLLLLKKKG), the KLR-70 enantiomer D-KLR-70, the AlexaFluor 488-conjugated KLR-70-C-AF488 peptide (RRGKKLLLLLKKKGC-AlexaFluor 488) and oncocin-112 (Onc112: VDKPPYLPRPRPPRrIYNr-NH2) was either carried out in-house at the UW-Madison Biotechnology Center or commissioned to a company (Biomatik, Genscript or Thermo Fisher Scientific). All non-fluorescent peptides were generated by solid-phase peptide synthesis via Fmoc chemistry and isolated to > 95% purity via reverse-phase HPLC followed by lyophilization. KLR-70-C-AF488 was prepared similarly, except for the additional thiol-reactive dye conjugation and the HPLC purification steps preceding lyophilization. Peptide identities were confirmed by either MALDI TOF/TOF or ESI mass spectrometry. Lyophilized peptides of known mass (typically > 10 mg to ensure sufficient mass accuracy) were dissolved in 100 mM HEPES-KOH adjusted to pH 7.0 prior to handling. Peptide stock solutions (1.5 mM) were generated as single-use aliquots and stored at – 80 °C. In addition, the concentration of some of the peptide stock solutions was confirmed via Bradford assay45 against a calibration curve comprised of several KLR-70 peptide solutions of accurately known concentration (assessed gravimetrically). The concentration of KLR-70-C-AF488 stock solutions was assessed via electronic absorption at 492 nm, using an extinction coefficient of 72,000 M−1 cm−1.46
Chaperone preparation.
DnaK, DnaJ and GrpE were prepared as described.47, 48 Single-use DnaK stock solutions (500 μM) were in 50 mM Tris/HCl pH 7.2, 5 mM MgCl2 and 50 mM KCl. DnaJ stick solutions (720 μM) were in 25 mM HEPES-KOH pH 7.0 and 100 mM KCl. GrpE stock solutions (345 μM) were in 30 mM HEPES pH 7.0, 100 mM KCl and 5% glycerol. Chaperone concentrations were assessed via electronic absorption at 280 nm using extinction coefficients of 15,800 M−1 cm−1, 14,000 M−1 cm−1 and 1,400 M−1 cm−1 for DnaK, DnaJ and GrpE, respectively.16, 49 GroEL and GroES were purchased (Sigma Aldrich) as a 1 : 1 mixture (wt : wt) of lyophilized chaperonins 60 and 10 (Tris/HCl, KCl, dithiothreitol and trehalose, at concentrations undisclosed by the company) and reconstituted in molecular-biology-grade water according to manufacturer’s specifications to final concentrations of 1.05 and 1.84 μM, respectively (assessed gravimetrically).
Cell-free expression.
Cell-free transcription-translation to generate the apoMb protein was carried out at 37 °C for 20 min as described,50 except for the following modifications. The in-house generated cell-free system included a plasmid composed of the pET-Blue1 vector harboring the apoMb gene (20 nM), a c.f. mix (see definition below), and S30 cell extracts from either wild-type or Δtig A19 E. coli cell strain. Here, Δtig denotes a strain lacking the gene encoding the trigger factor chaperone. The cell-free system also includes a fluorophore labeled aminoacylated tRNA, BODIPY 576/589-Met-tRNAfMet (0.1 μM), which was prepared as described,50 except for the fact that BODIPY 576/589 N-hydroxyl succinimidyl ester (i.e., BODIPY™ 576/589 NHS ester, ThermoFisher) was used as a reactive fluorophore in place of BODIPY™ FL NHS ester (ThermoFisher). The final solution pH was adjusted to 7.0 with dilute acetic acid. In addition, some of the transcription-translation mixtures also contained KLR-70, D-KLR-70 and Onc112 (see other sections for details). Protein expression was monitored by low-pH SDS PAGE.51 Fluorophore-labeled newly synthesized proteins were visualized on an FLA 9500 Typhoon gel imager (General Electric) equipped with an LPG (> 600 nm) longpass filter (Fuji-Film). Gel band intensities were quantified with the ImageJ software.52, 53
Determination of chaperone concentration in cell-free systems.
The intrinsic concentration of DnaK, DnaJ and GrpE in S30 cell extracts was assessed (either from wild-type or Δtig E. coli A19 strains) via Western blotting. Briefly, S30 cell extracts were subject to SDS-PAGE via two-layer gels comprising a stacking (130 mM Tris/Cl pH 8.8, 0.1% SDS, 5 % T and 3 % C) and a resolving (400 mM Tris/Cl pH 6.8, 0.1% SDS, 12% T and 3 % C) layer. T and C denote the percentage of acrylamide plus bis-acrylamide, and the percentage of bis-acrylamide alone, respectively. The running buffer was 250 mM Tris and 2 M glycine at pH 8.4 with 0.1% SDS. A PVDF membrane was used for the transferring step. The PVDF membrane was then treated with solution containing either mouse monoclonal anti-DnaK (Abcam), rabbit polyclonal anti-DnaJ (Stressgen) or rabbit polyclonal anti-GrpE (Stressgen) antibodies (1 : 2,000 dilution). Goat anti-rabbit or goat anti-mouse antibodies (ECL Amersham Western blotting kit, General Electric) were used as secondary antibodies. Protein bands were detected via chemiluminescence upon treatment with standard detection reagents (ECL Amersham Western blotting kit, General Electric), and quantified with ImageJ.55, 56 Chaperone concentrations were determined upon comparison with individual calibration curves comprising series of DnaK, DnaJ or GrpE standards of known concentration. Total concentrations of DnaK, DnaJ and GrpE in the wild-type S30 cell extract were found to be 3.8, 0.3 and 0.6 μM, respectively. Total concentrations of DnaK, DnaJ and GrpE in the Δtig S30 cell extract were found to be 4.0 μM, 0.2 μM and 0.4 μM, respectively.
Native gels involving DnaK.
The overall interactions of KLR-70 and D-KLR-70 for the ADP-state of DnaK were investigated via native gel electrophoresis by known procedures,47 with the following modifications. DnaK (5 μM) was incubated with an excess of either KLR-70 (50 μM) or D-KLR-70 (50 μM) in 50 mM Tris/HCl pH 7.0, 5 mM MgCl2, 50 mM KCl, 5 mM ADP and 15 mM dithiothreitol (DTT) for 1 hr at 21.5 ± 0.5 °C. Affinity of the peptides for ADP-DnaK was quantified via binding isotherms comprising multiple independent samples incubated as above, except that peptide concentration varied between 0 and 50 μM. Native polyacrylamide gel electrophoresis was then carried out on a vertical gel apparatus (Mini-PROTEAN 3, Bio-Rad) as follows. Briefly, gels consisted of 375 mM Tris-acetate at pH 7.8, 7.5 % w/v acrylamide and 0.2 % w/v bis-acrylamide (8 % T, 2.5 % C). T and C denote the percentage of acrylamide plus bis-acrylamide, and the percentage of bis-acrylamide alone, respectively. Tris-acetate (32 mM) at pH 7.5 was used as running buffer. Gels were run at constant voltage (200 V) for 5 hrs. Given the long run time and the tendency of the gel apparatus to overheat, temperature was controlled by immersing the device in a large container filled with either room-temperature water (effective water temp. during run: 21.5 – 24.5 ± 0.5 °C) or an ice/water mixture (effective water temp. during run: 15.0 – 24.5 ± 0.5 °C). Direct comparisons between the above conditions showed that slightly different gel running temperatures did not significantly affect the ability of KLR-70 and D-KLR-70 to bind DnaK. After electrophoresis, gels were fixed upon treatment with 40 % methanol and 10% acetic acid (fixing solution) for 15 min, and then stained with 0.025 % Brilliant Blue G-250 Coomassie dye (Fisher Scientific) in fixing solution for 1 hr, followed by destaining in fixing solution if/as needed. Gel images were collected on a Typhoon FLA 9500 laser scanner (General Electric) upon excitation at 635 nm with an LPR (> 700 nm) emission filter (Fuji-Film). As reference information, the expected isoelectric points of DnaK and the KLR-70 peptide are 5.1 and 12.04, respectively.
Native gels involving DnaJ.
The presence of interactions between DnaJ and the inhibitory peptides developed in this work was probed via native gel electrophoresis. Briefly, KLR-70 and D-KLR-70 (35 μM each) were independently incubated with and excess of DnaJ (200 μM) in 20 mM HEPES/KOH pH 7.2, 50 mM KCl, 5 mM DTT and 0.1 mM EDTA for 1 hr at 21.5 ± 0.5 °C, consistent with conditions known to be appropriate for this chaperone.54 Following incubation, native gel electrophoresis and gel staining/destaining were carried out at pH 7.5 under conditions similar to those employed for DnaK (see previous section), except for the following variations. Gels were run at constant voltage (120 V) for 3 hrs with reverse polarity. Under these conditions, the chaperone-peptide interactions had to be assessed so that the client peptides could be visualized, given that DnaJ did not enter the gel. Independent experiments performed at pH 6.8, where DnaJ enters gels, showed consistent results (Figure S1). As reference information, the expected isoelectric point of DnaJ is 8.5.
Luciferase refolding assays in buffered solution.
Luciferase denaturation and refolding was carried as described,55 with the following minor modifications. Pure luciferase (8 μM, Promega) was unfolded in unfolding buffer (25 mM HEPES/KOH pH 7.6, 5 mM MgCl2, 50 mM KCl, 6 M guanidine hydrochloride (GdnHCl), 5 mM β-mercaptoethanol (BME)) at room temperature for one hr. Refolding was carried out at 30 °C for 1 hr after 400-fold dilution into a refolding buffer (25 mM HEPES/KOH pH 7.6, 5 mM MgCl2, 50 mM KCl, 5 mM BME, 2 mM ATP) containing variable amounts of KLR-70 peptide (0 − 400 μM). In some instances (see specific figures), the refolding buffer also contained DnaK, DnaJ and GrpE (0.6 μM, 0.12 μM and 0.24 μM, respectively). After 1 hr incubation, the refolding mixtures were then diluted 33-fold into Steady-Glo solution (Promega), and samples were transferred into a 96-well plate, followed by luminescence detection via an EnVision 2100 or EnVision 2105 microplate reader (Perkin Elmer) equipped with a < 700 nm shortpass filter.
Luciferase refolding assays in cell-free systems.
Luciferase unfolding was carried out as listed in the previous section. Refolding was performed at 30 °C for 1 hr after 150-fold dilution into an E. coli cell-free system identical to the one employed for protein expression with the following modifications. The cell-free system employed in luciferase refolding experiments included 2.6 mM ATP, 0.4 mM of other NTPs (e.g. GTP, CTP and UTP) and lacked both T7 RNA polymerase and BODIPY 576/589-Met-tRNAfMet. When specified, the cell-free systems also included S30 extract at 1.5-fold higher concentration. Some of the assays included cell-free systems lacking S30 extracts. These solutions are denoted as cell-free mixtures (c.f. mix) in this work. Some of the refolding solutions also contained additional amounts of chaperones, including DnaK, DnaJ and GrpE (at 0.6 μM, 0.12 μM and 0.24 μM, respectively) or GroEL and GroES (at 0.5 μM and 1.0 μM, respectively). After 1 hr incubation, the refolding mixtures were diluted 33-fold into Steady-Glo solution (Promega), and samples were transferred into a 96-well plate, followed by luminescence detection via an EnVision 2100 or EnVision 2105 microplate reader (Perkin Elmer) equipped with a shortpass filter with 700 nm cut-off wavelength.
Data analysis of native gels to generate binding-isotherms.
Binding isotherms monitoring the affinity of KLR-70 for DnaK were generated as described in the section titled Native gels involving DnaK. Gel-band intensities were initially quantified with the ImageJ software.52, 53 A representative example is shown in panel A of Figure S2. Inspection of these intensity profiles highlights three challenging aspects of the ligand-binding data processing.
First, the peptide-bound and unbound states of DnaK are extremely poorly resolved on native gels even after extensive attempts of optimizing experimental conditions.
Second, the native gels exhibited subtle species-independent, random fluctuations in vertical band position.
Third, the ImageJ software is only able to produce discrete gel-band intensity values (e.g., see dotted traces in plot of panel A of Figure S2).
The above complications required the raw ImageJ traces to be carefully deconvoluted with continuous mathematical functions, and the data to correct for random vertical-height fluctuations. We achieved the above goals by a series of steps. First, we fit the raw ImageJ data to sums of Gaussians via the MATLAB software package (The MathWorks Inc. Natick, MA, v. R2016b). The Gaussian curves were used in all downstream analysis, in place of the ImageJ raw band intensities. Then, we carried out gel-band deconvolution followed by mathematical realignment. Representative examples of the above procedure are shown in panels B – D of Figure S2.
Gel band intensities of DnaK samples lacking KLR-70 were fit to an unconstrained sum of two Gaussians (see panel B of Figure S2) according to
| (1) |
where I(d)0 μM denotes the gel band intensity, which is a function of migration distance (d) for DnaK samples lacking KLR-70 (0 μM). In addition, the a, b and c terms denote amplitude, mean value and width of the Gaussian curves, respectively. The Gaussian curves represent intensity distributions of the top (distribution 1) and bottom (distribution 2) native-gel bands, likely representing different conformational states of DnaK. Gel band intensities of samples containing varied KLR-70 concentrations (X μM) were fit to constrained models which forced the bottom band Gaussians to have the same difference in mean migration distances as the samples lacking KLR-70 as shown in panel B of Figure S2, according to
| (2) |
The resulting Gaussians accurately represent gel-band intensity profiles for the peptide-unbound and -bound states of DnaK (r2 ≥ 0.98 for all samples). Next, the Gaussians were adjusted so that all curves within the same gel had their top and bottom Gaussians overlap, to account for lane-dependent small random migration differences through the native gel (see panel C of Figure S2), according to
| (3) |
Finally, the adjusted Gaussians had their total areas normalized to 1 to correct for small errors due to different total sample amounts loaded onto different gel lanes.
Once the above procedure was complete, the increase in the bottom DnaK-band (termed “DnaK band 2”) intensity upon addition of KLR-70 was plotted and fit to a mathematical model in Origin (OriginLab Corporation, v. 2018b) to determine their dissociation constant (see panel D of Figure S2).
The direct binding of KLR-70 to DnaK was modeled as a simple two-state process shown below,
| (4) |
where D, P and DP denote DnaK, KLR-70 peptide and DnaK-KLR-70 complex, respectively. The dissociation constant for the direct binding of KLR-70 to DnaK (KD1) can be written as
| (5) |
with Deq, Peq and DPeq,KLR−70 denoting the equilibrium concentrations of DnaK, KLR-70 and DnaK-KLR-70 complex. Equation 5 can be recast as
| (6) |
where D0 and P0 denote the total concentrations of DnaK and KLR-70 and fB1 denotes the fraction of KLR-70-bound directly to DnaK. Equation 6 can be further simplified to solve for fB1. The only physically meaningful solution of the resulting quadratic equation is
| (7) |
Equation 7 can be substituted directly into the following expression which describes how the observed maximum normalized gel band intensity of DnaK band 2 (Iobs) varies with fB1:
| (8) |
where I free and Ibound are the maximum normalized gel band intensities of DnaK band 2 under fully-free and fully-bound conditions.
Experimental data points were then fit to equation 8, after replacement of equation 7 into it,, with KD1, Ifree and Ibound serving as the adjustable parameters. Data are reported as an average KD1 from n = 3 independent experiments ± standard error.
Steady-state fluorescence anisotropy: data collection and analysis to generate binding isotherms.
Experiments monitoring the binding of the KLR-70 peptide for ADP-DnaK via fluorescence anisotropy were carried out either in direct-binding or competitive-inhibition modes. In both cases, the fluorescently labeled KLR-70-C-AF488 analog of the KLR-70 peptide was employed.
Direct binding experiments.
The direct-binding studies of KLR-70-C-AF488 to DnaK were carried out under conditions similar to those employed for binding isotherms assessed via native gels. Briefly, DnaK (0 – 100 μM) was incubated with KLR-70-C-AF488 (10 nM) in 50 mM Tris/HCl pH 7.0, 5 mM MgCl2, 50 mM KCl, 5 mM ADP and 15 mM dithiothreitol (DTT) for 1 hr at 21.0 – 23.5 ± 0.5 °C. The resulting changes in fluorescence anisotropy for KLR-70-C-AF488 in the presence or absence of DnaK were measured on a PC1 photon-counting steady-state fluorimeter (ISS Inc.). The excitation wavelength was 485 nM and the total fluorescence emission was recorded in the presence of a > 500 nm long-pass emission filter. Conveniently, fluorescence anisotropy depends linearly on the population of components i within a mixture, according to
| (9) |
where ri is the anisotropy of component i while αi is its fractional contribution to the total anisotropy (r). When the excitation source is vertically polarized, r becomes
| (10) |
with
| (11) |
where IVV and IVH denote the fluorescence intensity following excitation by vertically-polarized light and passing through either a vertical or horizontal emission polarizer, respectively. Similarly, IHV and IHH denote the sample fluorescence intensity following excitation by horizontally polarized light and passing through either a vertical or horizontal emission polarizer, respectively. All intensities were background-subtracted prior to further analysis. The background consisted of an independently-prepared equivalent sample lacking KLR-70-C-AF488. The changes in KLR-70-C-AF488 anisotropy as a function of total DnaK concentration were then plotted and fitted to the expressions below with Origin (OriginLab Corporation, v. 2020). Binding to DnaK was modeled as a simple two-state process (see eqns 4 and 5). Equation 6 can be recast as
| (12) |
Where fB2 denotes the fraction of KLR-70-C-AF488-bound to ADP-DnaK, P0,AF488 is the total concentration KLR-70-C-AF488, KD2 denoes the dissociation constant for KLR-70-C-AF488 binding to DnaK. Equation 12 was then inserted into the expression below, which describes how the observed anisotropy of KLR-70-C-AF488 (robs) varies with fB2
| (13) |
where rfree and rbound are anisotropies of KLR-70-C-AF488 under fully-chaperone-free and fully-chaperone-bound conditions. Q is an additional correction term which accounts for changes in the total fluorescence intensities of free KLR-70-C-AF488 upon binding to ADP-DnaK. Q is defined as
| (14) |
where Itotal,bound and Itotal,free are the total fluorescence intensities for the fully-DnaK-bound and fully-free states of KLR-70-C-AF488. Total intensities were obtained from anisotropy experiments according to
| (15) |
Importantly, no statistically significant changes in total fluorescence intensity of KLR-70-C-AF488 were observed in the presence of ADP-DnaK. Therefore Q was taken to be equal 1.0 in our experiments (Figure S3).
Experimental data points were then fit to equation 13, after replacement of equation 12 into it,, with KD2, rfree and rbound as adjustable parameters. Data are reported as an average KD2 from n = 4 independent experiments ± standard error.
Competitive binding experiments.
In order to quantify the affinity of unlabeled KLR-70 for ADP-DnaK, competitive binding experiments were carried out. These experiments were conducted under similar conditions to those employed in the direct binding experiments, except that KLR-70 (1 – 1000 μM) was co-incubated with DnaK (10 μM) and KLR-70-C-AF488 (10 nM) in 50 mM Tris/HCl pH 7.0, 5 mM MgCl2, 50 mM KCl, 5 mM ADP and 15 mM dithiothreitol (DTT) for 1 hr at 21.5 ± 0.5 °C.
The fraction of KLR-70-bound DnaK in the presence of KLR-70-C-AF488 (fB3) is56, 57
| (16) |
with
| (17) |
| (18) |
| (19) |
and
| (20) |
where P0,AF488 and KD2 are the total concentrations of KLR-70-C-AF488 peptide. The affinity of KLR-70-C-AF488 for ADP-DnaK was quantified as described in the previous section. Namely, relation 16 was replaced into the expression below, which describes the dependence of the observed anisotropy of KLR-70-C-AF488 (robs) on fB3
| (21) |
Experimental data points were then fit to equation 21, after replacement of equation 16 into it, with KD3, rfree and rbound serving as adjustable parameters. A comparisons of rfree values resulting from the fits in direct and competitive binding experiments strongly suggest that c.a. 25% of KLR-70-C-AF488 remains bound to ADP-DnaK even in the presence of excess KLR-70. This effect is potentially due to nonspecific binding of the fluorescently labeled, hence more hydrophobic, KLR-70-C-AF488. Data were reported as average KD3 for n = 4 independent experiments ± standard error.
Circular dichroism.
The far-UV circular dichroism spectra of the KLR-70 and D-KLR-70 peptides (20 μM) in 1.3 mM HEPES (pH 7.0) were recorded within the 190 – 260 nm spectral range (1 nm steps, 20 s / step) on a MOS-450 spectropolarimeter (Bio-Logic Science Instruments) at room temperature. Raw data were baseline corrected and converted to mean residue ellipticity (MRE) according to
| (22) |
where θ denotes the experimentally determined ellipticity (in mdeg), b is the cuvette pathlength (in cm), C is the peptide concentration (in molarity), and Nr denotes the number of amino acids, i.e., 14. Data were smoothed via a Savitzky-Golay algorithm (2nd order polynomials, 5 nm window size) with MATLAB (The MathWorks Inc. Natick, MA, v. R2016b). The fractional contributions of secondary structure elements to the overall smoothed spectra were assessed with CONTINLL58, 59 via the CDPro software.60
Determination of minimum inhibitory concentrations.
Single colonies of wild-type E. coli (ATCC 25922) cells were grown overnight shaking at 250 rpm and 37°C in Muller Hinton Broth (MHB) prior to dilution to a McFarland Standard 0.5 turbidity (the optical density at 600 nm was OD600 = 0.08–0.10). The culture was then diluted 1:300 (vol:vol) in MHB, followed by addition of the resulting suspension (200 μL per well) to a 96-well plate. Serially diluted stock solutions of peptide dissolved in dimethyl sulfoxide (DMSO) were then diluted 1:100 into each well prior to overnight growth for c.a. 18 hrs at 37°C. The OD600 of each well was then recorded with a Victor 3-V plate reader (Perkin Elmer). Finally, the minimum inhibitory concentration (MIC) was assessed as the lowest total concentration of peptide that displayed a baseline at OD600 similar to that of a negative control containing only MHB and DMSO.
Results and Discussion
Design criteria.
The bacterial Hsp70 DnaK has a well-known canonical binding site for client proteins and polypeptides. Not surprisingly, this site is located within the substrate-binding domain (SBD) of the chaperone61, 62 (Figure 2A), and lies between a β-sheet region and the α-helical lid of DnaK, as shown in Figure 2B. Our design of a peptide inhibitor of DnaK targeted a potential interaction with the above canonical binding site, and was based on the three major criteria outlined below.
Figure 2.

KLR-70 is predicted to interact with DnaK more strongly than known proline-rich peptide inhibitors. (A) Structure ADP-DnaK (PDB: 2KHO), showing the nucleotide- and substrate-binding domains, denoted as NBD and SBD, respectively. The client-binding cleft of DnaK is labeled. (B) Structure of the DnaK – pyrrhocoricin complex (PDB: 4EZN). Pyrrhocoricin is a known proline-rich peptide inhibitor of DnaK. (C) Known binding motif for the interaction of client peptides with DnaK. The motif includes a core of five nonpolar amino acids flanked by positively-charged residues. (D) Amino-acid sequence of the KLR-70 peptide investigated in this work, designed to mimic the DnaK substrate binding motif. (E) Amino-acid sequences of the DnaK-binding peptides KLR-70, pyrrhocoricin and oncocin. The underlined segments highlight the 13-residue binding motif predicted to have the highest affinity for DnaK (F) Computationally predicted binding scores for the highest-affinity DnaK binding sites of the peptides listed in panel E. Binding scores below −5 kJ/mol are regarded as diagnostic of high affinity for DnaK.
First, we exploited well established background knowledge on the amino acid sequence of peptides that are experimentally known to interact with the SBD of DnaK.63 Based on extensive screening of a large library of peptides encompassing the sequence of a wide variety of client proteins, Rüdiger et al. developed an algorithm for the in-silico prediction of the amino-acid sequence of peptides likely to interact with the SBD of DnaK.63 The result of these studies, which were later validated in other laboratories,64 highlighted the presence of a client-peptide binding motif consisting of 3–6 nonpolar amino acids flanked by residues with positively charged side chains.63 The key features of the canonical DnaK client binding motif based on the studies by Rüdiger et al. are illustrated in Figure 2C. It is worth noticing that the authors proposed that the client peptides developed in their work interact with DnaK via the binding cleft of the SBD (Figure 2A). This conclusion is corroborated by a number of systematic controls that had to be fulfilled by the screened peptides.63 Namely: (1) the bound peptides had to become fully dissociated upon addition of excess Mg2+ and ATP, (2) the bound peptides had to be displaced by competing peptides known to interact with the client-binding cleft,65 (3) the bound peptides had to show similar affinities for the isolated SBD of DnaK and for wild-type full-length DnaK, and (4) the bound peptides had to show no affinity for the isolated nucleotide binding domain of DnaK.63
We took advantage of the above information, and employed the algorithm by Rüdiger et al. to computationally screen a collection of 13 − 14 residue peptides. This procedure led us to select a peptide with a very favorable predicted DnaK binding score. An alternative recently developed algorithm,66 which complements the experimental data source employed in the algorithm by Rüdiger et al.63 with computational homology modeling,66 yielded equivalent results.
Second, an additional design criterion was applied to help improve peptide selectivity towards DnaK over its DnaJ co-chaperone. Namely, DnaJ shows high affinity for peptides enriched in aromatic residues while DnaK prefers aliphatic residues, most notably leucine.15, 63 Conversely, lysines are rarely found in DnaJ clients.15, 63 Thus, to optimize affinity for DnaK and selectivity for DnaK over DnaJ, we selected peptides bearing a leucine-rich core flanked by arginines and lysines.
Third, we considered the fact that peptides with a high net positive charge are known to often display undesired nonspecific binding to highly negatively charged cellular components, including membranes, nucleic acids and the ribosome. Hence we reduced the net charge density of our peptide candidates by including a few glycines, which bear no net charge and whose presence does not influence binding to either DnaK nor DnaJ.15, 63
Upon keeping all the above criteria in mind, the 14-residue RRGKKLLLLLKKKG polypeptide was ultimately chosen and denoted as KLR-70. A salient characteristic of this compound is that it is predicted to bind strongly to DnaK via the algorithm by Rüdiger et al.63 despite its extremely simple primary structure (Figure 2D). KLR-70 contains only naturally-occurring residues, it includes small clusters of arginines, lysines and leucines, and it and lacks N- and C-terminal modifications.
A comparison between the computationally predicted63 DnaK binding scores of KLR-70 and that of well-known proline-rich inhibitors of DnaK, including pyrrhocoricin36 and a sequence-optimized variant of oncocin22 (Figure 2E), was carried out. This side-by-side evaluation revealed that the predicted binding score of all three peptides is below − 5 kcal/mol, i.e., the nominal cutoff for interaction with DnaK (Figure 2F). Thus, all three peptides are expected to bind DnaK. Yet, the KLR-70 sequence was predicted to have by far the highest affinity for this chaperone.
KLR-70 and its enantiomer D-KLR-70 are mostly unstructured in solution.
The KLR-70 peptide was synthesized and its far-UV circular dichroism spectrum, shown in Figure 3, was recorded to experimentally assess secondary structure in solution. Inspection of the spectrum and data deconvolution via CONTINLL58, 59 revealed that KLR-70 is mostly unordered, and displays small amounts of α-helical, β-sheet and β-turn secondary structure (Figure 3 and Table 1).
Figure 3.

The KLR-70 and D-KLR-70 peptides are predominantly unstructured in solution. Far-UV circular dichroism spectra of KLR-70 and D-KLR-70 in 1.3 mM HEPES (pH 7.0) at room temperature. Each spectrum results from the average of n = 2 independent experiments. See Table 1 for secondary structure deconvolution via CONTINLL.
Table 1.
Fractional secondary structure of KLR-70 and D-KLR-70 deconvoluted via CONTINLL.
| Peptide | secondary structure | |||
|---|---|---|---|---|
| unordered | α-helical | β-sheet | β-turn | |
| KLR-70 | 0.49 | 0.21 | 0.10 | 0.21 |
| D-KLR-70 | 0.52 | 0.20 | 0.08 | 0.20 |
Interestingly, DnaK is known to bind client peptides stereoselectively, with preferential interaction with L-enantiomers. In contrast, DnaJ tends to lack this stereoselectivity, and typically interacts with both L- and D-enantiomers of client peptides to a comparable extent.15, 67
In order to probe the affinity of both peptide enantiomers for DnaK and DnaJ, we prepared both the all-L-amino-acid KLR-70 peptide and its enantiomer D-KLR-70, which carries all-D amino acids. Not surprisingly, far-UV circular dichroism analysis shows that D-KLR-70 is as poorly structured as KLR-70. The far-UV circular dichroism spectrum of D-KLR-70 is the exact mirror image of the KLR-70 spectrum, confirming the opposite chirality of the two polypeptides (Figure 3 and Table 1).
KLR-70 interacts with DnaK but not DnaJ, while D-KLR-70 does not interact with either chaperone.
Gel shift assays utilizing native gel electrophoresis at pH 7.5 were carried out to assess whether the KLR-70 and D-KLR-70 peptides interact with the DnaK (ADP-state of the chaperone, Figures 4A) or the DnaJ (Figure 4B) molecular chaperones. In the absence of client peptides, DnaK travels through the native gel as a diffuse band at low temperature (15.0 – 24.5 ± 0.5 °C, Figure 4A) and as two distinct bands at room temperature (21.5 – 24.5 ± 0.5 °C, Figure 4C), suggesting DnaK exhibits conformational heterogeneity in the absence of client proteins. This observation fully consistent with the literature.12, 13, 68 Addition of an excess of KLR-70 leads to a clear and reproducible variation in gel migration pattern. Namely, the diffuse or double band turns into a single well-defined sharper band within the same gel region. This result strongly suggests the presence of an interaction between KLR-70 and DnaK. The data are consistent with the scheme in Figure 4D, showing that DnaK has a conformationally heterogeneous unbound state that narrows into an apparent single conformation upon interaction with the KLR-70 client peptide. The KLR-70 bound state may well coincide with the closed-lid conformation previously observed upon client binding to DnaK,12, 13, 68 as suggested in the scheme of Figure 4D. In addition, our data show that the D-KLR-70 enantiomer does not interact with DnaK at either temperature.
Figure 4.

KLR-70 enantioselectively binds DnaK and does not bind DnaJ. (A) Representative native gel data, run under conventional polarity and on ice (15.0 – 24.5 ± 0.5 °C), for the binding of DnaK (5 μM) to either KLR-70 or D-KLR-70 (50 μM each). (B) Representative native gel data, run under reverse polarity and on ice (15.0 – 24.5 ± 0.5 °C), for the binding of DnaJ (200 μM) to either KLR-70 or D-KLR-70 (35 μM each). A one-tailed Student T-test assuming unequal variances shows that the gel band intensities for the KLR-70 (p = 0.32) and D-KLR-70 (p = 0.39) peptides in the presence of DnaJ are not statistically different. A 95 % confidence interval cutoff (p = 0.05) was used. Native PAGE analysis of these samples at pH 6.8, to allow DnaJ entering the gel, confirm the lack of interaction with DnaJ (Figure S1). (C) Representative native-gel data, run under conventional polarity at room temperature (21.5 ± 0.5 °C), for the binding of DnaK (5 μM) to either KLR-70 or D-KLR-70 (50 μM each). (D) Schematic cartoon representation of the DnaK-peptide binding data in panels A and C. Unbound DnaK is conformationally heterogeneous and it adopts a single apparent conformation upon interaction with KLR-70. In contrast, DnaK does not interact with D-KLR-70. (E) Schematic cartoon representation of the DnaJ-peptide binding data in panel B. Neither KLR-70 nor D-KLR-70 binds DnaJ. All native gel data are representative of n = 3 independent experiments. Native gel electrophoresis was carried out at pH 7.5 (see Material and Methods for additional information).
The above conclusions are supported by quantitative evaluation of the fraction of client-bound DnaK (fB). By applying equation 8 to the gel-shift data in Figure 4C, we get fB = 1.04 ± 0.04 in the presence of excess KLR-70, consistent with the binding isotherm in Figure 5A. In contrast, we find that fB = 0.00 ± 0.05 in the presence of excess of D-KLR-70.
Figure 5.

KLR-70 binds DnaK with high affinity. (A) Representative direct binding isotherm generated from the native gel analysis of ADP-DnaK (5 μM) preequilibrated in the presence of variable concentrations (0 – 50 μM) of KLR-70. Additional information on the generation and analysis of these data can be found in the Materials and Methods and Figure S2. (B) Representative direct-binding isotherm generated from changes in the anisotropy of KLR-70-A-AF488 (10 nM) preequilibrated in the presence of variable concentrations (0 – 100 μM) of ADP-DnaK. (C) Representative competitive binding isotherm generated from changes in the steady-state anisotropy of KLR-70-C-AF488 (10 nM) preequilibrated with ADP-DnaK (10 μM) and variable amounts of KLR-70 (1 – 1000 μM). Curve fitting yields dissociation constants of (A) 158 ± 70 nM, (B) 1.73 ± 0.37 μM and (C) 889 ± 386 nM. All dissociation constants are reported as avg ± standard error for (A) n = 3 or (B and C) n = 4 independent experiments.
Investigations on client binding to DnaJ at physiologically-relevant pH requires running native gels run with reverse polarity, due to the relatively high isoelectric point of DnaJ (pI = 8.5).69 However, given that DnaJ does not enter the gel at pH 7.5 even under reverse-polarity conditions, we had to move to pH 6.8 to enable explicit visualization of DnaJ bands (Figure S1). On the other hand, native gels at both pH 7.5 and 6.8 enabled prompt visualization of any interaction by inspection of the peptide gel band, in the presence of an excess of DnaJ chaperone. Student T-tests indicate that KLR-70 and D-KLR-70 concentrations remain constant in the absence and presence of DnaJ, at both pH 7.5 (Figure 4B) and pH 6.8 (Figure S1). P values can be found in the legends of Figure 4 and Figure S1. The above results are consistent with no detectable binding of either peptide to DnaJ, as illustrated in the scheme of Figure 4E.
The lack of binding of DnaJ by KLR-70 suggests that the design criteria discussed above (i.e. the enrichment of lysines and lack of aromatic amino acids) was effective at improving the binding selectivity of KLR-70 for DnaK over its DnaJ co-chaperone.
KLR-70 has high affinity for DnaK.
Since DnaK displayed observable changes in native gel migration patterns upon binding KLR-70 at room temperature in Figure 4C, we next performed gel shift assays aimed at quantifying the interaction of the ADP-state of DnaK with KLR-70 (Figure 5A). The gel-shift experiments yielded direct binding isotherms that led to a dissociation constant (KD) of 158 ± 73 nM for the DnaK-KLR-70 complex during electrophoresis at 21.5 – 24.5 ± 0.5 °C.
To further probe and validate the above results, we also explored the binding affinity of KLR-70 for DnaK by steady-state fluorescence anisotropy at room temperature (21.0 – 23.5 ± 0.5 °C), via direct-binding and competitive inhibition experiments. A fluorophore-labeled variant of KLR-70, denoted as KLR-70-C-AF488, was designed for this assay. KLR-70-C-AF488 was generated by conjugating an AlexaFluor 488 dye to the thiol group of a C-terminal cysteine added to the KLR-70 sequence. The direct binding of KLR-70-C-AF488 to ADP-DnaK yielded binding isotherms corresponding to a KD of 1.73 ± 0.37 μM (Figure 5B). A competitive binding assay then probed the displacement of KLR-70-C-AF488 from ADP-DnaK upon progressively increasing KLR-70 concentration (Figure 5C). The resulting competitive binding isotherms yielded to a KD of 889 ± 386 nM for KLR-70. These experimentally determined dissociation constants are summarized in Table 2. Importantly, two-tailed T-test analysis assuming unequal variances highlights that the dissociation constants determined for KLR-70 binding to DnaK either directly (via gel-shift assay) or via competitive displacement experiments (via fluorescence anisotropy) are statistically similar (p = 0.15). Taken together, the combination of the above data supports the conclusion that KLR-70 binds DnaK with high affinity.
Table 2.
Dissociation constants (KD) for DnaK binding to KLR-70 and KLR-70-C-AF488.
| Peptide | Method | KD |
|---|---|---|
| KLR-70 | Direct binding (native gels) | 158 ± 73 nM (*) |
| KLR-70 | Competitive binding (fluorescence anisotropy) | 890 ± 390 nM (*) |
| KLR-70-C-AF488 | Direct binding (fluorescence anisotropy) | 1.73 ± 0.37 μM |
Differences in KD determined for KLR-70 binding DnaK by gel-shift and anisotropy assays are not statistically significant (p = 0.15) when analyzed using a two-tailed T-test assuming unequal variances and using a 95% confidence interval cutoff.
Interestingly, the high affinity of KLR-70 for ADP-DnaK is somewhat larger than the corresponding affinity of oncocin37 and pyrrhocoricin,43 which have KDs of c.a. 1 μM and 5 μM, respectively. These results agree with their predicted binding affinities in Figure 2F (relative to KLR-70) at the qualitative level.
However, it is worth noting that many other prAMPs also exhibit high affinity for DnaK (c.a. 0.08–4 uM).62 Interestingly, prAMPs are known to bind ADP-DnaK via a different binding mode, relative to the canonical, non-proline-rich peptides employed by Rüdiger et al. Specifically, prAMPs typically exhibit highest affinity for DnaK’s D-E helical hinge region, which belong to the chaperone’s α-helical lid.70, 71 In addition, prAMPs also bind the canonical DnaK client-binding cleft, but only weakly so. In contrast, the peptides employed by Rüdiger et al. target primarily DnaK’s client-binding cleft. This observation suggests why the computationally predicted affinity of prAMPs is often underestimated by the Rüdiger et al. algorithm.
KLR-70 disrupts the chaperone activity of DnaK.
Next, we performed experiments to assess whether the binding of KLR-70 to DnaK disrupts chaperone activity. This question was probed by evaluating the effect of KLR-70 on the K/J/E-mediated refolding of chemically-denatured luciferase (20 nM luciferase, Figure 6A) according to a well-known assay.55 We achieved nearly quantitative inhibition of luciferase refolding upon addition of moderate concentrations of KLR-70 (≥ 60 μM) in buffered solution. The Student T-tests reported in Figure 6B confirms the statistical reliability of this conclusion. The moderate concentration of KLR-70 required to eliminate the chaperone activity of DnaK (tot. conc. 0.6 μM) is remarkable and convenient for practical applications. Not surprisingly, this result is consistent with the high affinity of KLR-70 for this chaperone. On the other hand, the presence of some unbound DnaK at lower (≤ 6 μM) total inhibitor concentration is expected, despite the high affinity of KLR-70 for DnaK. Figure S4A shows simulations probing the KLR-70-DnaK interaction prior to addition of luciferase. These simulations highlight the expected partially bound state of DnaK in the presence of low total KLR-70 concentration (≤ 6 μM). Hence, at [KLR-70]total ≤ 6 μM, a significant concentration (> 10 nM) of KLR-70-unbound DnaK is estimated to remain available to catalyze the refolding of luciferase (tot. conc. 20 nM, Figure S4B).
Figure 6.

KLR-70 inhibits the DnaK/DnaJ/GrpE (i.e., K/J/E)-mediated refolding of luciferase. (A) Plot illustrating the normalized luminescence of luciferase (20 nM) refolded in the presence of K/J/E (0.6 μM K, 0.12 μM J and 0.24 μM E) and variable amounts (0 – 400 μM) of KLR-70. The negative control (N.C.) denotes the normalized luminescence arising from luciferase refolding in the absence of K/J/E and KLR-70. (B) Table summarizing the p values pertaining to a comparison of the negative control (N.C.) with luciferase refolding carried out in the presence of K/J/E and variable amounts of the KLR-70 inhibitory peptide. A one-tailed Student T-test assuming unequal variances was performed. Statistical significance was judged using a 95 % confidence interval (p ≤ 0.05). All data represent avg ± std. err. for n = 3 independent experiments
The mechanism by which DnaK promotes protein folding is still a subject of active debate.14 While some reports propose that DnaK (in the presence of its cochaperones) plays an active role by serving as a folding catalyst that is even able to unfold misfolded aggregates,72–74 other studies show evidence for DnaK serves as a mere kinetic partitioning agent by establishing a new parallel chaperone-interaction route that kinetically competes with the folding and aggregation paths.47, 48, 75 Regardless of the exact mechanism by which DnaK refolds luciferase, our data are consistent with KLR-70 behaving as an effective DnaK inhibitor. While this is an exciting result per se, future work is necessary to probe the nature of the KLR-70’s behavior in further detail, e.g., establishing whether KLR-70 serves as a competitive or noncompetitive inhibitor.
KLR-70 does not significantly disrupt de novo protein biosynthesis.
Next, we characterized the behavior of KLR-70 within more complex cell-like environments. Specifically, understanding whether KLR-70 has any undesirable effects on protein production is important, given that other classes of DnaK inhibitory peptides, e.g., prAMPs, are known to interfere with the translation machinery.43, 44 Reported binding-affinity data for prAMPs37, 39, 44 suggest that the ribosome is the primary intracellular target of prAMPs, rendering the interactions with DnaK a non-exclusive feature of these peptides.
One notable example is Onc112, a prAMP variant of oncocin that inhibits de novo protein synthesis by binding the peptidyl transferase center of the ribosomal 50S subunit, and physically blocking the ribosomal exit tunnel.40, 44 The interaction between Onc112 and the ribosome is stereoselective, as an all-D enantiomer variant of Onc112 does not inhibit green-fluorescent-protein production.44 In addition, both enantiomers of Onc112 exhibit generic weak interactions with nucleic acids, with a ca. 100-fold lower efficiency than binding to the ribosome.44 Both types of interactions are thought to be in-part due to electrostatic interactions facilitated by the high positive net charge (+5) of Onc112 at physiological pH, and in part due to the ability of the proline-rich scaffold to effectively enter and block the ribosomal exit tunnel.40
KLR-70 carries a fairly high positive net charge (+7) at physiological pH, despite the fact that one of our inhibitor design criteria included engineering two glycines to decrease net positive charge density. In order to experimentally probe whether electrostatic attraction may decrease the selectivity of KLR-70 in the presence of cell-like environments, we allowed KLR-70 to interact with highly negative charged biomolecules, including ribosomes and other nucleic acids.
For these experiments, we expressed the model protein sperm whale apomyoglobin (apoMb), in a cell-free system including an S30 cell extract derived from TF-depleted (Δtig) E. coli. TF and DnaK known to associate with nascent proteins and assist their proper folding upon release from the ribosome.20
Cell-free expression was carried out in the presence of aminoacylated initiator tRNA labeled with BODIPY 576/589 at the methionine amino group, as described.50 This procedure enables the selective N-terminal cotranslational labeling of nascent apoMb, facilitating its in-gel fluorescence detection (Figure 7A). As shown in panels A-C of Figure 7, nascent protein synthesis is not disrupted within the 60 – 150 μM total KLR-70 concentration range. Partial inhibition of protein expression takes place at higher total KLR-70 concentrations (200 – 400 μM).
Figure 7.

KLR-70 does not inhibit nascent protein production in cell-free systems, at concentrations sufficient to inhibit DnaK. (A) Representative low-pH SDS-PAGE data with fluorescence detection, illustrating the cell-free expression of BODIPY 576/589-labeled apoMb in the presence of 0 – 400 μM KLR-70. (B) Quantitative analysis of the nascent protein-expression data of panel A. Nascent protein expression is reported relative to the average expression level in the absence of KLR-70. (C) Table summarizing the p values for apoMb expression in the presence (60 – 400 μM) or absence (0 μM) of KLR-70. P values were obtained from the one-tailed Student T-test assuming unequal variances. Statistical significance was assessed with a 95 % confidence-level cutoff (p ≤ 0.05). All data represent avg ± std. err. for n = 3 independent experiments. An E. coli cell-free system lacking the gene for the trigger factor chaperone (Δtig strain) was used. The total concentration of the K/J/E chaperones in the Δtig S30 cell extract were 0.7 μM DnaK, 0.03 μM DnaJ and 0.07 μM GrpE.
Additional expression tests were carried out to directly compare the behavior of KLR-70, D- KLR-70 or Onc112 at moderate concentration (150 μM each, Figure 8A). While Onc112 fully inhibits nascent apoMb expression, as expected,40, 44 KLR-70 does not have any influence on apoMb production (Figure 8B).
Figure 8.

KLR-70 and D-KLR-70 have a dramatically different effect on cell-free protein expression. (A) Representative low pH SDS-PAGE gel illustrating the cell-free expression of fluorophore-labeled apoMb in the absence and presence of the KLR-70, D-KLR-70 and Onc112 peptides. (B) Quantitative analysis of nascent-protein expression data of panel A, in the absence (N.C.; negative control) and presence of DnaK-binding peptides. Nascent protein expression is quantified relative to expression level of the N.C. sample. Data are shown as avg ± std. err. for n = 3 independent experiments carried out with an E. coli cell-free system lacking the gene for the trigger factor chaperone (Δtig strain). The total concentration of the K/J/E chaperones in the Δtig S30 cell extract were 0.7 μM DnaK, 0.03 μM DnaJ and 0.07 μM GrpE. Asterisks (*) denote samples where no observable (obs.) nascent-protein expression could be detected.
Surprisingly, we found that D-KLR-70 completely inhibits protein biosynthesis, similarly to Onc112. The origin of this unexpected behavior of D-KLR-70 is presently unknown and will be further investigated at a later time.
Taken together, the above data show that the DnaK inhibitory peptide featured in this work, KLR-70, does not disrupt cell-free protein expression at moderate concentrations, up to 150 μM. Hence KLR-70 is an effective DnaK inhibitor with no negative effects on protein biosynthesis, at concentrations lower than 150 μM.
KLR-70 selectively inhibits DnaK in cell-like environments.
As a compliment to the above studies, we further assessed whether KLR-70 remains an effective inhibitor of the DnaK chaperone under complex conditions that more accurately take the cellular environment into account, including the presence of ribosomes and nucleic acids in general. In order to investigate this question, we performed a series of luciferase refolding experiments under cell-like conditions and in the absence and presence of KLR-70 (Figure 9). The assay conditions are similar to those used to express nascent apoMb in Figures 7 and 8, and comprise various combinations of a cell-free mixture (c.f. mix), purified K/J/E chaperones, and an S30 cell extract generated from either a wild-type (WT) or Δtig E. coli cell strain.
Figure 9.

KLR-70 inhibits a significant portion of the chaperone activity of cell-like environments. (A) Normalized luminescence data, illustrating of the extent of luciferase (50 nM) refolding in a cell-like environment (c.f. mix, see components in Methods section) containing either purified K/J/E chaperones (i.e., DnaK, DnaJ and GrpE), or S30 cell extracts from a wild-type (WT) strain, or S30 extracts from a trigger factor-depleted (Δtig) strain. The total concentration of the purified K/J/E chaperones was: 1.1 μM DnaK, 0.2 μM DnaJ and 0.5 μM GrpE, respectively. KLR-70 was 400 μM in the experiments that included it. Bar-graph colors denote refolding in the absence (black) or presence of E. coli S30 extracts (dark grey: WT; light grey: Δtig). (B) Alternative plot of the data of panel A, showing relative normalized relative luminescence values. All data represent avg ± std. err. for n = 3 independent experiments.
Importantly, the c.f. mix only contains small molecules necessary for chaperone activity and transcription-translation (e.g. salts, nucleotides, amino acids) as well as DNA plasmids. The c.f. mix is a key component of cell-free nascent protein expression. In contrast, the S30 cell extract has a more complex composition, and it includes a variety of cellular macromolecules, i.e., ribosomes, most water-soluble cellular proteins (including molecular chaperones), tRNAs and more. The c.f. mix and the cell extract are typically used in combination, to carry out cell-free transcription-translation.
As shown in Figure 9A, luciferase refolding is less efficient in a c.f. mix supplemented with purified K/J/E than in a c.f. mix that has been supplemented with either a WT of Δtig S30 cell extract. This result highlights the role of multiple chaperones, including those that are known to cooperate with K/J/E to promote protein folding.76 The major E. coli chaperone networks that are known to assist protein folding include K/J/E55, K/J/E in combination with Hsp9023 or ClpB,24 and GroEL/ES.25
The increased luciferase refolding activity in the presence of Δtig E. coli S30 extract may seem surprising at first. However, this result can be rationalized to reflect the known upregulation of the above chaperones and chaperone networks to compensate for the loss of trigger factor.4
The effect of the c.f. mix on luciferase refolding is interesting. Considering that we separately established that 400 μM KLR-70 fully inhibits K/J/E-mediated luciferase refolding under simple buffer conditions (Figure 6), the data of Figure 9 (which also include 400 μM KLR-70) suggest that c.f.-mix components, including nucleotides and nucleic acids, likely reduce KLR-70’s ability to inhibit DnaK. This observation agrees with the model proposed in the previous section where KLR-70 promiscuously engages with a variety of targets, especially negatively-charged nucleic acids and ribosomes, yet it retains high DnaK inhibitory activity.
The binding promiscuity of KLR-70 in cell-like environments is not inherently problematic for its use as a tool to address the effect of DnaK on nascent protein folding as long as it only inhibits molecular chaperone networks dependent on DnaK for function. For instance, Hsp90 and ClpB are two molecular chaperones known to promote the folding of luciferase. However, their chaperone actions also require K/J/E.23, 24
Given that GroEL/ES is known to refold luciferase independently of DnaK, we separately probed whether GroEL/ES activity is inhibited by KLR-70. As expected, 25 the addition of purified GroEL/ES facilitates luciferase refolding in a c.f. mix (Figure S5). However, addition of KLR-70 only weakly reduces the GroEL/ES-mediated refolding of luciferase. This result implies that the GroEL/ES chaperone is not a major biological target of the KLR-70 peptide.
KLR-70 is a useful biochemical tool for investigating the specific role of DnaK in nascent-protein folding.
Taken together, the data of Figures 6, 9 and Figure S5 show that the DnaK chaperone network remains the main target of KLR-70 within the complex cell-like environment. While interactions with nucleic acids and potentially other small molecules attenuate KLR-70’s inhibitory activity in complex cell-like environments (Figure 9), the overall effectiveness of this inhibitor is remarkably high.
Conveniently, KLR-70 exhibits high affinity for DnaK in the absence of any chemical modifications (e.g. C-terminal amidation) or unnatural amino acids (e.g. cyclohexylalanine) which are often featured in engineered prAMPs.62
Most importantly, KLR-70 is effective at inhibiting the bacterial Hsp70 chaperone at concentrations that do not interfere with protein biosynthesis, as highlighted in Figures 7 and 8.
D-KLR-70 inhibits bacterial growth more effectively than KLR-70.
Finally, we collected pilot data to probe the sensitivity of live wild-type E. coli cells towards KLR-70 and D-KLR-70. We also compared the results with corresponding experiments performed on the known DnaK inhibitor oncocin 112 (Onc112), a representative prAMP. The results are shown in Table 3. Onc112 exhibits a minimum inhibitory concentration (MIC) of 5 μg/mL, consistent with the literature sampling other E. coli strains.77 The MIC values of KLR-70 and D-KLR-70 are 41 – 83 and 2 – 5 μg/mL, respectively. Interestingly, D-KLR-70 has a lower MIC than KLR-70 by about one order of magnitude. Hence D-KLR shows more promise as an antimicrobial peptide against wild-type E. coli than KLR-70.
Table 3.
Minimum inhibitory concentrations (MICs) of KLR-70, D-KLR-70 and Onc112 against wild-type E. coli cells.
| Peptide | MIC (μg/mL)* |
|---|---|
| KLR-70 | 41 – 83 |
| D-KLR-70 | 2 – 5 |
| Onc112 | 5 |
Data report the range of MICs observed across n = 3 independent experiments.
This results raises two interesting questions that stand as interesting topics for future investigations. First, it is not clear whether the high MIC of KLR-70 in E. coli cells is due to lack of cellular penetration or to orthogonal stress response pathways that are inactive in cell-free systems.
Second, the promising MIC value of D-KLR-70 suggests that this peptide either acts upon interfering with the cell membrane, as many antimicrobial peptides, or by penetrating live cells and subsequently inhibiting translation. Future targeted studies are necessary to shed light on the above intriguing topics.
Conclusions
In summary, we developed KLR-70, a 14-residue peptide inhibitor of DnaK whose sequence was designed to match an optimal binding motif of DnaK for protein substrates.63 KLR-70 comprises only natural amino acids, is rich in lysines, leucines and arginines, and has no N- or C-terminal covalent modifications (e.g., acetylation or amidation). KLR-70 displays high affinity for DnaK, and inhibits the Hsp70 chaperone cycle by selectively targeting DnaK, and not the DnaJ co-chaperone. The interaction between KLR-70 and DnaK is highly stereoselective, as evidenced by the fact that DnaK fails to bind the D enantiomer of KLR-70. Importantly, the binding of KLR-70 to DnaK does not impair nascent-protein translation at [KLR-70]total ≤ 150 μM.
In all, KLR-70 is a useful DnaK inhibitor that bears promise to serve as a valuable tool in mechanistic studies focusing on the role of the DnaK molecular chaperone in buffered solutions and cell-free systems. Finally, the D enantiomer D-KLR-70 does not bind DnaK or DnaJ chaperones, yet it strongly inhibits translation. This result suggests that KLR-70 and its enantiomer D-KLR-70 may be used orthogonally when inhibition of DnaK/DnaJ chaperones and(or) inhibition of translation are desired.
Supplementary Material
Acknowledgments
We thank Alex Staikos, Collin Goebel and Natalie Feider for a gift of purified DnaK and DnaJ, Hanming Yang for his guidance on Savitsky-Golay filters in MatLab, and Kevin England for his help with spectral deconvolution of circular dichroism data. In addition, we are grateful to Gene Ananiev at the University of Wisconsin-Madison Small Molecule Screening Facility for his assistance in running the MIC assays reported in this work.
Funding
This work was supported by the National Science Foundation (NSF) grants MCB-1616459 and CBET-1912259 (to S.C.) and by the University of Wisconsin Carbone Cancer Center Support Grant P30 CA014520.
Footnotes
Associated Content
Supporting Information
The Supporting Information includes additional DnaJ native-gels, a detailed pictorial illustration of the procedures employed to quantify gel-band intensities to generate binding-isotherm plots, total fluorescence data analysis for KLR-70-C-AF488 binding DnaK, simulations estimating KLR-70’s effects on the fraction of bound DnaK and free DnaK concentrations for the data in Figure 6 and additional luciferase refolding data.
Accession IDs
DnaK (UniProtKB P0A6Y8), DnaJ (UniProtKB P08622), GrpE (UniProtKB P09372), GroEL (UniProtKB P0A6F5), GroES (UniProtKB P0A6F9), Firefly luciferase (UniProtKB P08659)
Notes
The authors declare no competing financial interest.
References
- (1).Clerico EM, Tilitsky JM, Meng W, and Gierasch LM (2015) How Hsp70 molecular machines interact with their substrates to mediate diverse physiological functions, J. Mol. Biol. 427, 1575–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Cho Y, Zhang X, Pobre Kristine Faye R., Liu Y, Powers David L., Kelly Jeffery W., Gierasch Lila M., and Powers Evan T. (2015) Individual and Collective Contributions of Chaperoning and Degradation to Protein Homeostasis in E. coli, Cell Reports 11, 321–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Balch WE, Morimoto RI, Dillin A, and Kelly JW (2008) Adapting Proteostasis for Disease Intervention, Science 319, 916–919. [DOI] [PubMed] [Google Scholar]
- (4).Calloni G, Chen T, Schermann Sonya M., Chang, Genevaux H.-c., Agostini P, Tartaglia F, Gian G, Hayer-Hartl M, and Hartl FU (2012) DnaK Functions as a Central Hub in the E. coli Chaperone Network, Cell Rep. 1, 251–264. [DOI] [PubMed] [Google Scholar]
- (5).Mayer MP, and Bukau B. (2005) Hsp70 chaperones: cellular functions and molecular mechanism, Cell. Mol. Life Sci. 62, 670–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Sharma SK, Christen P, and Goloubinoff P. (2009) Disaggregating Chaperones: An Unfolding Story, Curr. Protein Pept. Sci. 10, 432–446. [DOI] [PubMed] [Google Scholar]
- (7).Fernández-Fernández MR, Gragera M, Ochoa-Ibarrola L, Quintana-Gallardo L, and Valpuesta JM (2017) Hsp70 – a master regulator in protein degradation, FEBS Lett. 591, 2648–2660. [DOI] [PubMed] [Google Scholar]
- (8).Balchin D, Hayer-Hartl M, and Hartl FU (2016) In vivo aspects of protein folding and quality control, Science 353, 1–12. [DOI] [PubMed] [Google Scholar]
- (9).Nunes JM, Mayer-Hartl M, Hartl FU, and Müller DJ (2015) Action of the Hsp70 chaperone system observed with single proteins, Nat. Comm. 6, 1–9. [DOI] [PubMed] [Google Scholar]
- (10).Bertelsen EB, Chang L, Gestwicki JE, and Zuiderweg ERP (2009) Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate, Proc. Natl. Acad. Sci. U.S.A 106, 8471–8476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kityk R, Kopp J, Sinning I, and Mayer MP (2012) Structure and dynamics of the ATP-bound open conformation of Hsp70 chaperones, Mol. Cell 48, 863–874. [DOI] [PubMed] [Google Scholar]
- (12).Banerjee R, Jayaraj GG, Peter JJ, Kumar V, and Mapa K. (2016) Monitoring conformational heterogeneity of the lid of DnaK substrate-binding domain during its chaperone cycle, FEBS J. 283, 2853–2868. [DOI] [PubMed] [Google Scholar]
- (13).Lai AL, Clerico EM, Blackburn ME, Patel NA, Robinson CV, Borbat PP, Freed JH, and Gierasch LM (2017) Key features of an Hsp70 chaperone allosteric landscape revealed by ion-mobility native mass spectrometry and double electron-electron resonance, J. Biol. Chem. 292, 8773–8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Mayer MP, and Gierasch LM (2019) Recent advances in the structural and mechanistic aspects of Hsp70 molecular chaperones, J. Biol. Chem. 294, 2085–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Rüdiger S, Schneider-Mergener J, and Bukau B. (2001) Its substrate specificity characterizes the DnaJ co-chaperone as a scanning factor for the DnaK chaperone, EMBO J. 20, 1042–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Russell R, Karzai AW, Mehl AF, and McMacken R. (1999) DnaJ Dramatically Stimulates ATP Hydrolysis by DnaK: Insight into Targeting of Hsp70 Proteins to Polypeptide Substrates, Biochemistry 38, 4165–4176. [DOI] [PubMed] [Google Scholar]
- (17).Liberek K, Marszalek J, Ang D, Georgopoulos C, and Zylicz M. (1991) Escherichia coli DnaJ and GrpE heat shock proteins jointly stimulate ATPase activity of DnaK, Proc. Natl. Acad. Sci. U. S. A. 88, 2874–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sekhar A, Lam HN, and Cavagnero S. (2012) Protein folding rates and thermodynamic stability are key determinants for interaction with the Hsp70 chaperone system, Protein Sci. 21, 1489–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Mashaghi A, Bezrukavnikov S, Minde DP, Wentink AS, Kityk R, Zachmann-Brand B, Mayer MP, Kramer G, Bukau B, and Tans SJ (2016) Alternative modes of client binding enable functional plasticity of Hsp70, Nature 539, 448–451. [DOI] [PubMed] [Google Scholar]
- (20).Deuerling E, Schulze-Specking A, Tomoyasu T, Mogk A, and Bukau B. (1999) Trigger factor and DnaK cooperate in folding of newly synthesized proteins, Nature 400, 693–696. [DOI] [PubMed] [Google Scholar]
- (21).Deuerling E, Patzelt H, Vorderwulbecke S, Rauch T, Kramer G, Schaffitzel E, Mogk A, Schulze-Specking A, Langen H, and Bukau B. (2003) Trigger Factor and DnaK possess overlapping substrate pools and binding specificities, Mol. Microbiol. 47, 1317–1328. [DOI] [PubMed] [Google Scholar]
- (22).Knappe D, Piantavigna S, Hansen A, Mechler A, Binas A, Nolte O, Martin LL, and Hoffmann R. (2010) Oncocin (VDKPPYLPRPRPPRRIYNR-NH2): A Novel Antibacterial Peptide Optimized against Gram-Negative Human Pathogens, J. Med. Chem. 53, 5240–5247. [DOI] [PubMed] [Google Scholar]
- (23).Genest O, Hoskins JR, Camberg JL, Doyle SM, and Wickner S. (2011) Heat shock protein 90 from Escherichia coli collaborates with the DnaK chaperone system in client protein remodeling, Proc. Natl. Acad. Sci. U. S. A. 108, 8206–8211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zolkiewski M. (1999) ClpB cooperates with DnaK, DnaJ, and GrpE in suppressing protein aggregation. A novel multi-chaperone system from Escherichia coli, J. Biol. Chem. 274, 28083–28086. [DOI] [PubMed] [Google Scholar]
- (25).Buchberger A, Schroder H, Hesterkamp T, Schonfeld HJ, and Bukau B. (1996) Substrate shuttling between the DnaK and GroEL systems indicates a chaperone network promoting protein folding, J. Mol. Biol. 261, 328–333. [DOI] [PubMed] [Google Scholar]
- (26).Hesterkamp T, and Bukau B. (1998) Role of the DnaK and HscA homologs of Hsp70 chaperones in protein folding in E.coli, EMBO J. 17, 4818–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Shimizu Y, Kanamori T, and Ueda T. (2005) Protein synthesis by pure translation systems, Methods 36, 299–304. [DOI] [PubMed] [Google Scholar]
- (28).Niwa T, Ying B-W, Saito K, Jin W, Takada S, Ueda T, and Taguchi H. (2009) Bimodal protein solubility distribution revealed by an aggregation analysis of the entire ensemble of Escherichia coli proteins, Proc. Natl. Acad. Sci. U.S.A 106, 4201–4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Niwa T, Kanamori T, Ueda T, and Taguchi H. (2012) Global analysis of chaperone effects using a reconstituted cell-free translation system, Proc. Natl. Acad. Sci. U. S. A. 109, 8937–8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kramer G, Kudlicki W, Hardesty B. (1999) Cell-free coupled transcription-translation systems from Escherichia coli, In Protein Expression : A Practical Approach, pp 201–223, Oxford University Press. [Google Scholar]
- (31).Endo Y, and Sawasaki T. (2004) High-throughput, genome-scale protein production method based on the wheat germ cell-free expression system, J. Struct. Funct. Genomics 5, 45–57. [DOI] [PubMed] [Google Scholar]
- (32).Tarui H, Imanishi S, and Hara T. (2000) A novel cell-free translation/glycosylation system prepared from insect cells, J. Biosci. Bioeng. 90, 508–514. [PubMed] [Google Scholar]
- (33).Craig D, Howell MT, Gibbs CL, Hunt T, and Jackson RJ (1992) Plasmid cDNA-directed protein synthesis in a coupled eukaryotic in vitro transcription-translation system, Nucleic Acids Res. 20, 4987–4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Brodel AK, Sonnabend A, and Kubick S. (2014) Cell-free protein expression based on extracts from CHO cells, Biotechnol. Bioeng. 111, 25–36. [DOI] [PubMed] [Google Scholar]
- (35).Burgenson D, Gurramkonda C, Pilli M, Ge X, Andar A, Kostov Y, Tolosa L, and Rao G. (2018) Rapid recombinant protein expression in cell-free extracts from human blood, Sci. Rep. 8, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Cociancich S, Dupont A, Hegy G, Lanot R, Holder F, Hetru C, Hoffmann JA, and Bulet P. (1994) Novel inducible antibacterial peptides from a hemipteran insect, the sap-sucking bug Pyrrhocoris apterus, Biochem. J. 300, 567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Knappe D, Zahn M, Sauer U, Schiffer G, Sträter N, and Hoffmann R. (2011) Rational Design of Oncocin Derivatives with Superior Protease Stabilities and Antibacterial Activities Based on the High-Resolution Structure of the Oncocin-DnaK Complex, ChemBioChem 12, 874–876. [DOI] [PubMed] [Google Scholar]
- (38).Liebscher M, Haupt K, Yu C, Jahreis G, Lücke C, and Schiene‐Fischer C. (2010) Rational Design of Novel Peptidic DnaK Ligands, ChemBioChem 11, 1727–1737. [DOI] [PubMed] [Google Scholar]
- (39).Czihal P, Knappe D, Fritsche S, Zahn M, Berthold N, Piantavigna S, Müller U, Van Dorpe S, Herth N, Binas A, Köhler G, De Spiegeleer B, Martin LL, Nolte O, Sträter N, Alber G, and Hoffmann R. (2012) Api88 Is a Novel Antibacterial Designer Peptide To Treat Systemic Infections with Multidrug-Resistant Gram-Negative Pathogens, ACS Chem. Biol. 7, 1281–1291. [DOI] [PubMed] [Google Scholar]
- (40).Roy RN, Lomakin IB, Gagnon MG, and Steitz TA (2015) The mechanism of inhibition of protein synthesis by the proline-rich peptide oncocin, Nat. Struct. Mol. Biol. 22, 466–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Graf M, Mardirossian M, Nguyen F, Seefeldt AC, Guichard G, Scocchi M, Innis CA, and Wilson DN (2017) Proline-rich antimicrobial peptides targeting protein synthesis, Nat. Prod. Rep. 34, 702–711. [DOI] [PubMed] [Google Scholar]
- (42).Gagnon G, M., N. Roy R, Lomakin I, Florin T, Mankin A, and A. Steitz T. (2016) Structures of proline-rich peptides bound to the ribosome reveal a common mechanism of protein synthesis inhibition, Nucleic Acids Res. 44, 2439–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Taniguchi M, Ochiai A, Kondo H, Fukuda S, Ishiyama Y, Saitoh E, Kato T, and Tanaka T. (2016) Pyrrhocoricin, a proline-rich antimicrobial peptide derived from insect, inhibits the translation process in the cell-free Escherichia coli protein synthesis system, J. Biosci. Bioeng. 121, 591–598. [DOI] [PubMed] [Google Scholar]
- (44).Krizsan A, Volke D, Weinert S, Strater N, Knappe D, and Hoffmann R. (2014) Insect-derived proline-rich antimicrobial peptides kill bacteria by inhibiting bacterial protein translation at the 70S ribosome, Angew. Chem. Int. Ed. Engl. 53, 12236–12239. [DOI] [PubMed] [Google Scholar]
- (45).Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding, Anal. Biochem. 72, 248–254. [DOI] [PubMed] [Google Scholar]
- (46).Rusinova E, Tretyachenko-Ladokhina V, Vele OE, Senear DF, and Alexander Ross JB (2002) Alexa and Oregon Green dyes as fluorescence anisotropy probes for measuring protein-protein and protein-nucleic acid interactions, Anal. Biochem. 308, 18–25. [DOI] [PubMed] [Google Scholar]
- (47).Lee JH, Zhang D, Hughes C, Okuno Y, Sekhar A, and Cavagnero S. (2015) Heterogeneous binding of the SH3 client protein to the DnaK molecular chaperone, Proc. Natl. Acad. Sci. U. S. A. 112, 4206–4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Sekhar A, Santiago M, Lam HN, Lee JH, and Cavagnero S. (2012) Transient interactions of a slow-folding protein with the Hsp70 chaperone machinery, Protein Sci. 21, 1042–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Silberg JJ, Hoff KG, and Vickery LE (1998) The Hsc66-Hsc20 chaperone system in Escherichia coli: chaperone activity and interactions with the DnaK-DnaJ-grpE system, J. Bacteriol. 180, 6617–6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Ellis JP, Bakke CK, Kirchdoerfer RN, Jungbauer LM, and Cavagnero S. (2008) Chain dynamics of nascent polypeptides emerging from the ribosome, ACS Chem. Biol. 3, 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Kirchdoerfer RN, Huang JJT, Isola MK, and Cavagnero S. (2007) Fluorescence-based analysis of aminoacyl- and peptidyl-tRNA by low-pH sodium dodecyl sulfate–polyacrylamide gel electrophoresis, Anal. Biochem. 364, 92–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Schneider CA, Rasband WS, and Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis, Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Abràmoff MD, Magalhães PJ & Ram SJ (2004) Image processing with ImageJ., Biophotonics Int 11, 36–42. [Google Scholar]
- (54).Cuellar J, Perales-Calvo J, Muga A, Valpuesta JM, and Moro F. (2013) Structural insights into the chaperone activity of the 40-kDa heat shock protein DnaJ: binding and remodeling of a native substrate, J. Biol. Chem. 288, 15065–15074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Szabo A, Langer T, Schröder H, Flanagan J, Bukau B, and Hartl FU (1994) The ATP hydrolysis-dependent reaction cycle of the Escherichia coli Hsp70 system DnaK, DnaJ, and GrpE, Proc. Natl. Acad. Sci. U. S. A. 91, 10345–10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Wang ZX (1995) An exact mathematical expression for describing competitive binding of two different ligands to a protein molecule, FEBS Lett. 360, 111–114. [DOI] [PubMed] [Google Scholar]
- (57).Roehrl MH, Wang JY, and Wagner G. (2004) A general framework for development and data analysis of competitive high-throughput screens for small-molecule inhibitors of protein-protein interactions by fluorescence polarization, Biochemistry 43, 16056–16066. [DOI] [PubMed] [Google Scholar]
- (58).Provencher SW, and Glockner J. (1981) Estimation of globular protein secondary structure from circular dichroism, Biochemistry 20, 33–37. [DOI] [PubMed] [Google Scholar]
- (59).Sreerama N, and Woody RW (2000) Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set, Anal. Biochem. 287, 252–260. [DOI] [PubMed] [Google Scholar]
- (60).CDPro: A Software Package for Analyzing Protein CD Spectra. https://sites.bmb.colostate.edu/sreeram/CDPro/ (accessed August, 31 2019)
- (61).Zhu X, Zhao X, Burkholder WF, Gragerov A, Ogata CM, Gottesman ME, and Hendrickson WA (1996) Structural analysis of substrate binding by the molecular chaperone DnaK, Science 272, 1606–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Zahn M, Berthold N, Kieslich B, Knappe D, Hoffmann R, and Sträter N. (2013) Structural Studies on the Forward and Reverse Binding Modes of Peptides to the Chaperone DnaK, J. Mol. Biol. 425, 2463–2479. [DOI] [PubMed] [Google Scholar]
- (63).Rüdiger S, Germeroth L, Schneider-Mergener J, and Bukau B. (1997) Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries, EMBO J. 16, 1501–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Vega CA, Kurt N, Chen Z, Rüdiger S, and Cavagnero S. (2006) Binding specificity of an alpha-helical protein sequence to a full-length Hsp70 chaperone and its minimal substrate-binding domain, Biochemistry 45, 13835–13846. [DOI] [PubMed] [Google Scholar]
- (65).McCarty JS, R üdiger S, Sch önfeld H-J, Schneider-Mergener J, Nakahigashi K, Yura T, and Bukau B. (1996) Regulatory Region C of the E. coli Heat Shock Transcription Factor, σ32, Constitutes a DnaK Binding Site and is Conserved Among Eubacteria, J. Mol. Biol. 256, 829–837. [DOI] [PubMed] [Google Scholar]
- (66).Van Durme J, Maurer-Stroh S, Gallardo R, Wilkinson H, Rousseau F, and Schymkowitz J. (2009) Accurate prediction of DnaK-peptide binding via homology modelling and experimental data, PLoS Comput. Biol. 5, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Feifel B, Schönfeld H-J, and Christen P. (1998) d-Peptide Ligands for the Co-chaperone DnaJ, J. Biol. Chem. 273, 11999–12002. [DOI] [PubMed] [Google Scholar]
- (68).Mapa K, Sikor M, Kudryavtsev V, Waegemann K, Kalinin S, Seidel CAM, Neupert W, Lamb DC, and Mokranjac D. (2010) The Conformational Dynamics of the Mitochondrial Hsp70 Chaperone, Mol. Cell 38, 89–100. [DOI] [PubMed] [Google Scholar]
- (69).Zylicz M, Yamamoto T, McKittrick N, Sell S, and Georgopoulos C. (1985) Purification and properties of the dnaJ replication protein of Escherichia coli, J. Biol. Chem. 260, 7591–7598. [PubMed] [Google Scholar]
- (70).Kragol G, Lovas S, Varadi G, Condie BA, Hoffmann R, and Otvos L Jr. (2001) The antibacterial peptide pyrrhocoricin inhibits the ATPase actions of DnaK and prevents chaperone-assisted protein folding, Biochemistry 40, 3016–3026. [DOI] [PubMed] [Google Scholar]
- (71).Kragol G, Hoffmann R, Chattergoon MA, Lovas S, Cudic M, Bulet P, Condie BA, Rosengren KJ, Montaner LJ, and Otvos L. (2002) Identification of crucial residues for the antibacterial activity of the proline-rich peptide, pyrrhocoricin, Eur. J. Biochem. 269, 4226–4237. [DOI] [PubMed] [Google Scholar]
- (72).Sharma SK, De los Rios P, Christen P, Lustig A, and Goloubinoff P. (2010) The kinetic parameters and energy cost of the Hsp70 chaperone as a polypeptide unfoldase, Nat. Chem. Biol. 6, 914–920. [DOI] [PubMed] [Google Scholar]
- (73).Sharma SK, De Los Rios P, and Goloubinoff P. (2011) Probing the different chaperone activities of the bacterial HSP70-HSP40 system using a thermolabile luciferase substrate, Proteins 79, 1991–1998. [DOI] [PubMed] [Google Scholar]
- (74).Priya S, Sharma SK, and Goloubinoff P. (2013) Molecular chaperones as enzymes that catalytically unfold misfolded polypeptides, FEBS Lett. 587, 1981–1987. [DOI] [PubMed] [Google Scholar]
- (75).Seckler R, and Jaenicke R. (1992) Protein folding and protein refolding, FASEB J. 6, 2545–2552. [DOI] [PubMed] [Google Scholar]
- (76).Hiller S, and Burmann BM (2018) Chaperone–client complexes: A dynamic liaison, J. Magn. Reson. 289, 142–155. [DOI] [PubMed] [Google Scholar]
- (77).Krizsan A, Knappe D, and Hoffmann R. (2015) Influence of the yjiL-mdtM Gene Cluster on the Antibacterial Activity of Proline-Rich Antimicrobial Peptides Overcoming Escherichia coli Resistance Induced by the Missing SbmA Transporter System, Antimicrob. Agents Chemother. 59, 5992–5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.