

Abstract
A recent breakthrough in the classification of soft tissue tumors (STT) has been a significant expansion in the number of neoplasms associated with NTRK and other kinase related fusions. This is important not only for diagnostic purposes, but also because it opens new avenues for targeted therapy. Indeed, recent clincal trials have shown significant benefit across multiple tumor-types, prompting approval of NTRK inhibitors for clinical use in the setting of advanced/metastatic NTRK-rearranged neoplasms. Despite these therapeutic oportunities, diagnostic challenges have transpired in recognizing these emerging new histologic subtypes of kinase fusions positive-STT prospectively. This, in part, is attributable to their wide morphologic spectrum, variable risk of malignancy, and non-specific immunoprofile. As such, recommendations for pathologic criteria and immunohistochemical testing are needed to improve classification and streamline the small subset of potential candidates for further molecular validation. This overview summarizes the key histologic features of various STT associated with NTRK and other kinase fusions, which appear to share a similar morphologic spectrum. Immunohistochemically, many of these tumors, regardless of the kinase fusion type, notably show co-expression of S100 and CD34; issues related to the utility of pan-NTRK and NTRK1 immunostaining are therefore summarized. Finally, I discuss the role of confirmatory molecular testing and how, in some instances, this may also be of prognostic value. This review is intended as a critical summary of the current literature to emphasize pathologic criteria for improving recognition of this emerging and complex group of kinase fusion associated STT.
Keywords: kinase, fusion, sarcoma, lipofibromatosis, fibrosarcoma, MPNST, NTRK, BRAF, RAF1, RET
INTRODUCTION
Structural gene rearrangements resulting in gene fusions are frequent events in solid tumors, and the list of known oncogenic fusions reported in oncology continues to grow with the availability of next generation sequencing (NGS) in clinical practice. Current evidence indicates that many of these fusions lead to a state of oncogene addiction and, therefore, targeted therapies directed at constitutively activated oncogenic tyrosine kinases have proven remarkably effective against cancers with fusions involving ALK, ROS1, or PDGFB. The efficacy of this approach was recently extended to malignancies driven by RET, NTRK1/2/3, and BRAF fusions, with a number of tyrosine kinase inhibitors (TKI) available in clinical trials or FDA-approved, such as cabozantinib, larotrectinib, vemurafenib, respectively1.
Gene fusions resulting in oncogenic activation of various protein kinases, including NTRK1-3, BRAF, RAF1, and RET, have been also increasingly recognized in soft tissue tumors (STT); and, these neoplasms are associated with a wide spectrum of morphologies and grades. As morphologic-genotypic correlations continue to evolve, there remains uncertainty as to which types of STT should be evaluated for NTRK-rearrangement, and the optimal hierarchy for evaluation (i.e., immunohistochemistry and/or molecular). This heightened awareness is driven, in large part, from the recent approval of NTRK inhibitors, among other specific kinase inhibitors, in the treatment of clinically advanced kinase fusion positive STT. Well-defined pathologic criteria are not available as these entities are still evolving, and there remain limited publications on this topic to date. As a result, and often at the behest of clinicians, this has undoubtably resulted in over-testing of a wide range of sarcoma types, both immunohistochemically and molecularly. Further confusion relates to the natural history of kinase-fusion positive STT- a significant number of these tumors may follow a benign/indolent course, being surgically cured without a need for adjuvant therapy. Given concerns of inadvertently denying patients with susceptible tumors the benefit of targeted therapy – or conversely over investigating and/or over treating patients – there is currently a need for education on the morphologic features associated with these neoplasms, and a more rationalized approach to ancillary testing. This overview provides a critical review of the emerging literature on this subject and proposes practical recommendations for the diagnosis and prognosis of these neoplasms, which may assist pathologists and medical oncologists in identifying the candidates for molecular confirmation and subsequent targeted treatment.
DISCUSSION
When approaching STT pathologists typically form a diagnosis or a differential diagnosis based on histologic impressions, which is refined through ancillary immunohistochemical and molecular techniques. At times an unexpected diagnosis may be facilitated through a broad immunohistochemical or molecular (e.g., RNA-sequencing) panel. Finally, and rather disconcertingly, a diagnosis is occasionally prompted when a tumor is blindly interrogated in the absence of morphologic guidance (e.g., molecular testing in the context of a clinical trial). Given this increasingly pluralistic, yet interconnected, diagnostic environment this overview is structured to cover these various scenarios. The first part describes the primary histologic patterns associated with various gene fusions. In contrast, the second part offers a complementary approach discussing the association of individual kinase fusions with various histologic patterns and pathologic entities. I believe an increased awareness of these morphologic patterns, coroborated with routine immunohistochemical testing, may assist practicing pathologists in identifying tumors with a higher likelihood for harboring kinase fusions, which should then be further confirmed at the molecular level. In the third part, this review also provides a practical guide on the utility of NTRK immunohistochemistry as well as the various molecular methods that can be applied to confirm a diagnosis of kinase fusion positive STT.
I. Distinctive Morphologic Patterns Associated with Kinase Fusions STT
1. Lipofibromatosis-like neural tumor (LPF-NT).
Lipofibromatosis-like neural tumor is a newly described, morphologically distinct, soft tissue tumor. It is predominantly a superficial neoplasm characterized by mildly atypical spindle cells with a highly infiltrative pattern of growth in the subcutis. These tumors often occur in children, and less commonly in young adults, with predilection for the extremities and trunk2. A subset of cases has been reported in infants3,4, which may be confused clinically with infantile fibrosarcoma. In young children these lesions may closely resemble lipofibromatosis; however, they often display a more pronounced and spindle cell component (Fig 1). The spindle cells are typically positive for S100 and CD34. This peculiar immunoprofile makes identification of the line of differentiation challenging; CD34 expression raises the possibility of fibroblastic differentiation, while the presence of multifocal positivity for S100 protein suggests a possible neural or neuroectodermal lineage. In many of these cases the presence of cytologic atypia and an infiltrative growth pattern within adipose tissue prompts concern for a low grade malignant peripheral nerve sheath tumor (MPNST) (Fig 1). However, none of the patients reported to date have a clinical history of type 1 neurofibromatosis (NF1). Furthermore, SOX10 – a relatively more specific marker of neural crest and peripheral nerve sheath tissues – is consistently negative; conversely, H3K27me3 – a marker that is frequently lost in MPNST – expression is consistently retained.2 The majority of tumors with LPF-NT histology have been associated with NTRK1 fusions2, and a smaller subset, particularly in infants and young children, may harbor RET gene fusions4. Rare cases with ALK fusions have also been reported2. To date, these NTRK1-related translocations overwhelmingly appear to be intra-chromosomal, either interstitial deletions (LMNA-NTRK1), or unbalanced insertions (TPM3- or TPR-NTRK1 fusions)2. As a result, these lesions are often immunoreactive for both NTRK12 and pan-NTRK5; as one might expect, while RET-positive LPF-NT are negative with panNTRK immunomarkers. None of the patients with LPF-NT reported to date have developed metastases or died of disease. Local recurrence has been documented in patients with incomplete surgical resection. Although most patients with LPF-NT do not require intervention with targeted therapy, as these lesions are often small and surgically resectable, occasional patients with locally aggressive, inoperable lesions have been treated with larotrectinib, showing considerable shrinkage and subsequently become surgical candidates (CRA, personal communications).
Figure 1. Lipofibromatosis-like neural tumor (LPF-NT) – morphologic spectrum.
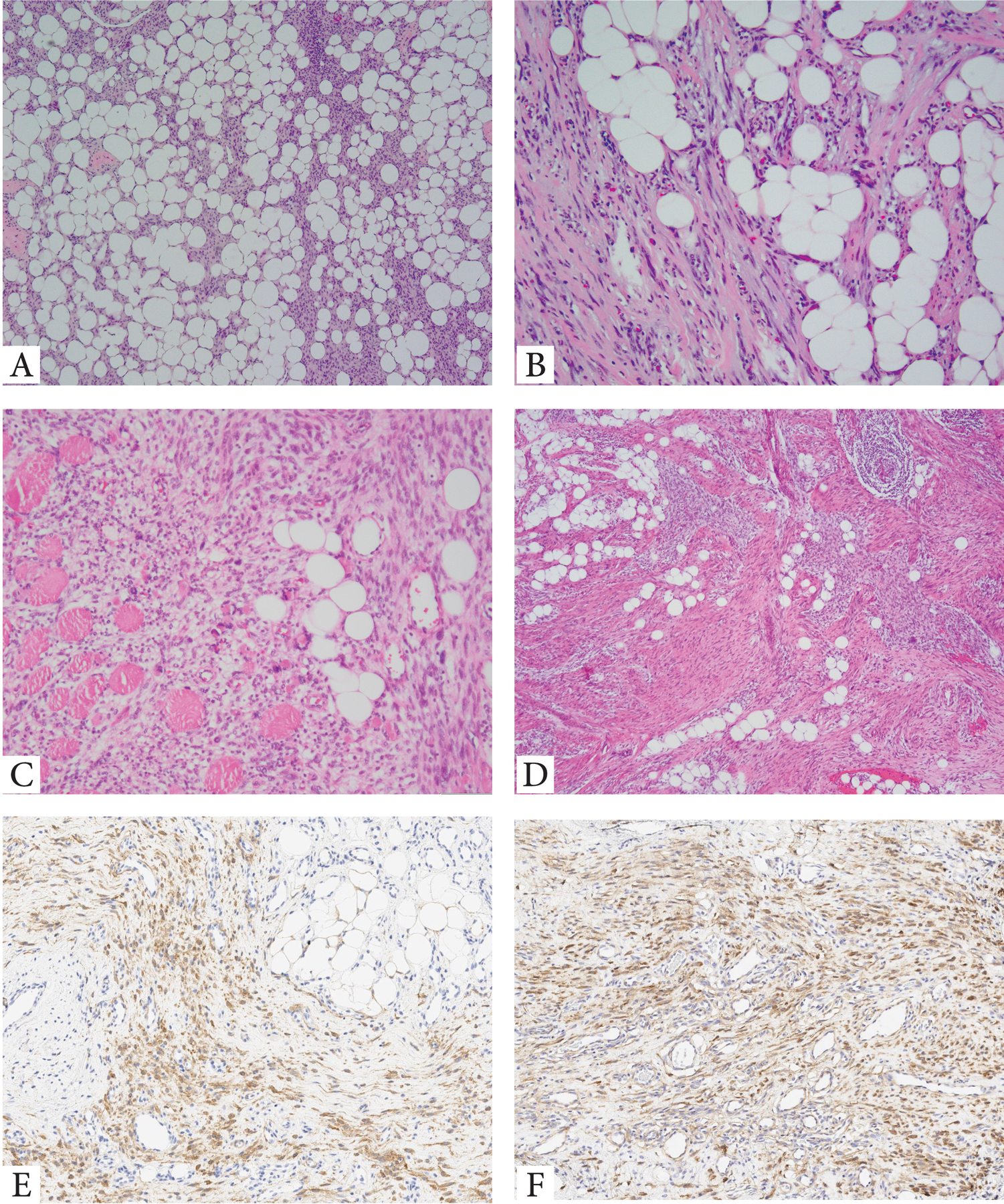
A. Highly infiltrative hypocellular neoplasm with thin strands of uniform spindle cells (locally recurrent, superficial soft tissue hand mass, 7/F, TPR-NTRK1 fusion); B. Delicate spindle cells arranged in short fascicles witin a fibrous stroma infiltrating surrounding adipose tissue (forearm, 64/M, CLIP1-ALK fusion). C. Deep-seated LPF-NT showing bland spindle cells arranged in solid sheets infiltratibe adipose tissue and skeletal muscle (buttock, 14/F, ALK gene rearrangement). D-F. Unusual LPF-NT with a distinctive triphasic growth pattern within the subcutis reminiscent of fibous hamartoma of infancy: compact nodules of primitive round to ovoid cells, admixed with long fascicles of fibrous spindle cells and mature adipose component. Tumor was positive for panNTRK (E) and NTRK1 (F)(forearm, 15/M, LMNA-NTRK1 fusion).
2. Spindle cell tumors with S100 and CD34 co-reactivity resembling malignant peripheral nerve sheath tumors (MPNST).
This recently identified group encompasses spindle cell tumors morphologically resembling low- to intermediate-grade MPNST. They predominate in children and young adults, and may occur at various anatomic sites, including bone, soft tissue, and viscera6. Microscopically, most tumors are characterized by a monomorphic spindle cell proliferation; they may be arranged in short fascicles or display a relatively patternless architecture (Fig 2). A distinctive feature of these neoplasms is the presence of band-like stromal, and ring-like perivascular, keloidal collagen. The cells have scant eosinophilic cytoplasm. The nuclei are ovoid and generally uniform and hyperchromatic; occasionally, scattered pleomorphic and/or multinucleated cells may be interspersed. Despite a morphologic resemblance to MPNST, most lesions show a low to intermediate mitotic count and lack necrosis. Although a small subset may show overlap with LPF-NT, an important distinguishing feature remains predominant solid and fascicular growth of primitive spindle cells, associated with stromal and perivascular hyalinization. Similar to LPF-NT, this group shows co-expression of S100 and CD34, which ranges from focal to diffuse. SOX10 is typically negative, while H3K27me3 expression is retained. At the molecular level, tumors harbor recurrent gene fusions with various kinases, such as RAF1, BRAF, NTRK1, and NTRK26. PanNTRK is typically positive in those tumors with NTRK-related fusions, but negative in tumors harboring RAF1 and BRAF fusions; additionally, NTRK1 is expressed only in tumors with NTRK1 gene rearrangement6. These tumors should be distinguished from conventional MPNST – which may be sporadic, occur in the setting of NF1, or prior radiation – as these patients may benefit from targeted therapy. Despite the limited number of cases reported to date, it is clear these tumors have malignant potential, and this appears to correlate with histologic grade. Among the 14 patients with available follow-up, two patients reportedly died of disease, two were alive with wide-spread metastases and one was alive with persistent local disease.6
Figure 2. Kinase fusion positive STT resembling MPNST and co-expression of S100 and CD34 immunoprofile.
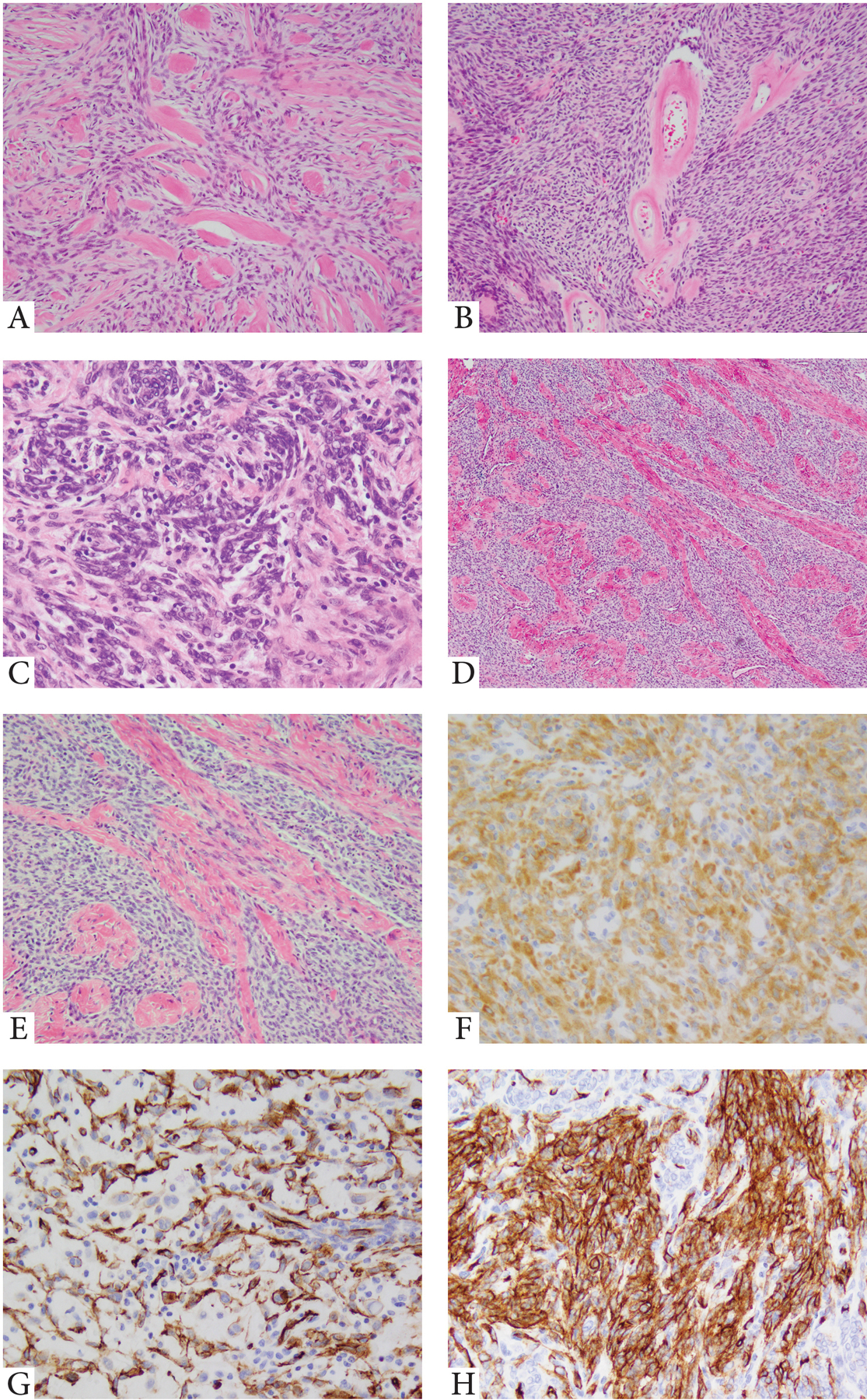
A. Variably cellular spindle cell neoplasm with abundant thick, band-like stromal hyalinization (paraspinal, 45/F, RAF1 gene rearrangement); B. Solid growth of primitive ovoid to spindle cells with scant cytoplasm, arranged in streaming or short fascicular patterns, with distinctive ring-like perivascular hyalinization (intra-abdominal, 67/F, PDZRN3-RAF1); C. Vague worling growth of primitive ovoid to short spindle cells within a fibrotic stroma (BRAF gene rearrangement); D-F. Spindle cell neoplasm resembling low-to intermediate grade MPNST, composed of primitive spindle cells separated by columns of thick, collagen bundles (foot, 31/M, NTRK1 gene rearrangement); F. Strong and diffuse NTRK1 immunoreactivity (cytoplasmic pattern). G,H. BRAF-fusion positive MPNST-like spindle cell neoplasm showing S100 and CD34 reactivity (18/F, thigh).
3. Infantile Fibrosarcoma (IFS) with cannonical ETV6-NTRK3 fusions.
IFS is a childhood sarcoma that typically presents at birth or in the first two years of life7. IFS has a variable anatomic distribution, with a predilection for the soft tissues of the extremities, and less commonly the trunk and head and neck area. Histologically, IFS is a monomorphic spindle cell sarcoma composed of spindle cells arranged in cellular sheets, fascicles and/or herringbone patterns, sometimes with a prominent lymphocytic infiltrate and hemangiopericytoma-like vasculature. The nuclei tend to be monomorphic, and mitotically active. The immunoprofile of IFS is nonspecific, with variable expression of SMA, desmin, S100 and CD34. IHC using a Pan-TRK rabbit monoclonal antibody (EPR17341) has proven to be a sensitive marker, showing nuclear and/or cytoplasmic staining in cases with ETV6-NTRK3 fusions8. IFS has a favorable outcome, with conservative surgical resection being the mainstay of therapy, particularly when aiming for limb salvage. Cytotoxic chemotherapy has shown activity in IFS, and its use is limited to selective cases that are unamenable to surgery. Recently, targeted therapy with Larotrectinib, a selective NTRK inhibitor, was shown to be highly beneficial in advanced stage IFS.9
4. IFS-like lesions with related fusion kinases.
IFS-like tumors represent a subset of childhood sarcomas, morphologically resembling IFS (Fig. 3); however, these are currently differentiated based on the presence of genetic abnormalities other than the cannonical ETV6-NTRK3 fusion. Most IFS-like sarcomas occur in children under age 2, but some present at an older age. These may show a predilection for intra-abdominal sites. Clinical outcome is less predictable, with some cases showing aggressive clinical behavior, including distant metastases. The alternate fusions may involve genes such as BRAF, NTRK110–12 and MET13; a minority of IFS-like tumors demonstrate compound intragenic BRAF deletions associated with tandem dupplication of exon 2, which may occur with or without the cannonical ETV6-NTRK3 fusion12. Their mechanism of activation is most likely similar to the BRAF-related fusions, resulting in loss of the N-terminal of BRAF protein containing the negative regulatory Ras-binding domain (BRD), with subsequent constitutive activation of BRAF. Although data on IFS-like lesions is still evolving, this provisional category will be included together with the more common IFS group in the 2020 WHO classification of bone and soft tissue tumors.
Figure 3. Kinase fusion positive STT with Fibrosarcoma and high grade MPNST phenotype.
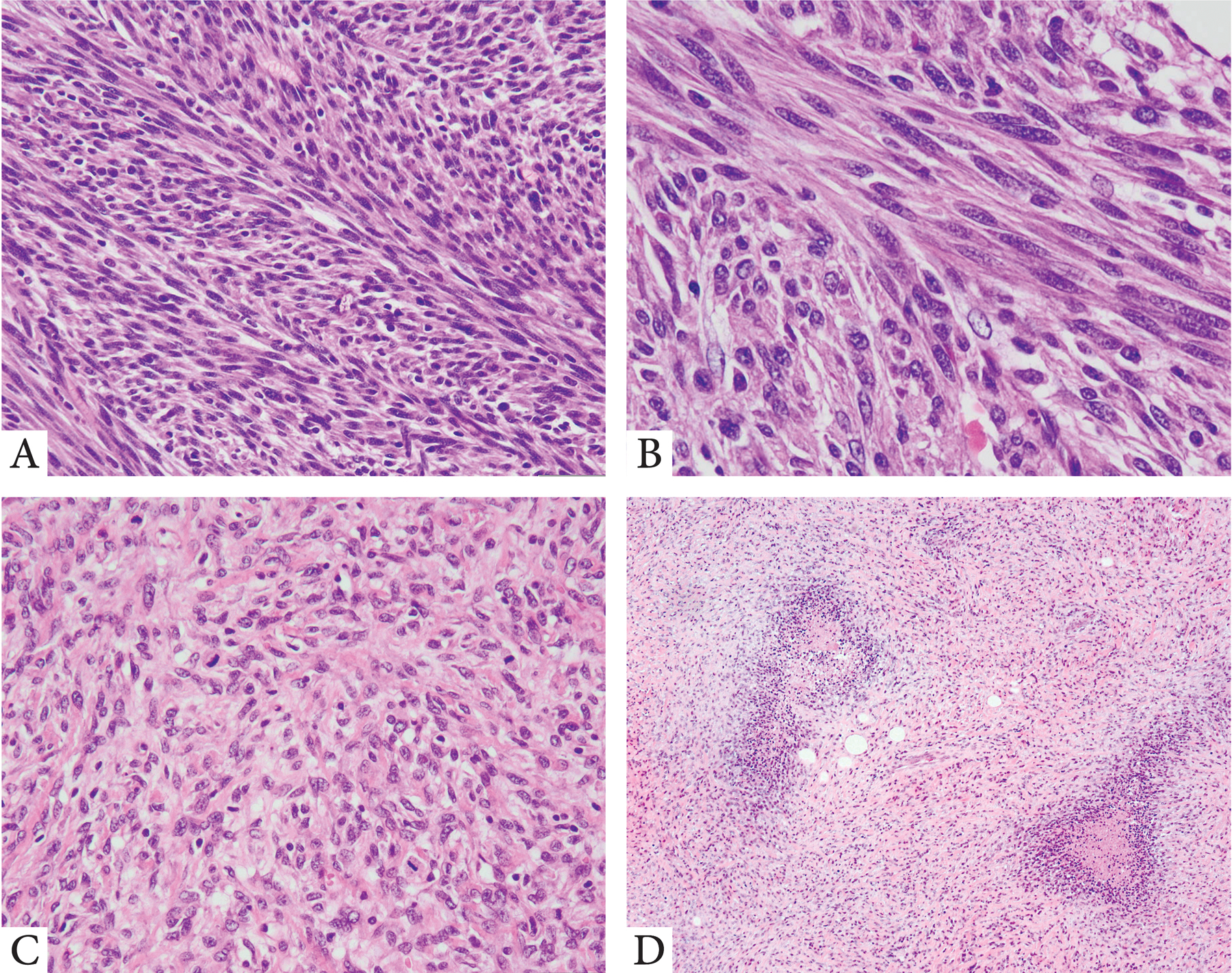
A,B. IFS-like morphology with monomorphic spindle cells with fibrillary eosinnophilic cytoplasm arranged in long, intersecting fascicles (superficial arm soft tissue mass, developed wide spread lung metastases, 3 years after diagnosis; 26/M, TPM3-NTRK1 fusion); C. High grade fibrosarcoma-like pattern (abdominal, 49/F, NTRK3 rearrangement); D. High grade spindle cell sarcoma with geographic areas of necrosis and focal S100 positivity, resembling high grade MPNST (buttock, 47/M, RET gene rearrangement).
5. Adult-type fibrosarcoma
A subset of kinase fusion-positive spindle cell sarcomas, with a fibrosarcoma herringbone growth pattern, have been reported in adults with a wide age range at diagnosis and various anatomic locations including soft tissue and viscera14. A number of these cases have been labeled in the past as adult-type fibrosarcomas (Fig. 3). A few cases were reported to express S100 protein; they may entrap glands, thereby mimicking adenosarcoma15. At the molecular level, STT with a fibrosarcoma-like morphology and ostensibly null immunoprofile have been reported with NTRK314 and RET gene fusions4. Similar lesions have also been described in the uterine cervix, as uterine sarcomas with features of fibrosarcoma16,17. A recent case report with a RET-SPECC1L fusion was also documented18. The relationship between this group and the more common IFS occurring in the pediatric age groups remains uncertain. As most of the reported cases to date appear histologically high grade, targeted therapy either with NTRK or RET inhibitors might be of significant clinical benefit in metastatic or locally advanced settings.
6. Spindle cell sarcomas with hemangiopericytic or myopericytoma-like pattern
A single study to date has described the association of spindle cell sarcomas with a hemangiopericytoma or myopericytoma phenotype and NTRK1 fusions. Haller et al described 4 patients (2 children and 2 adults) with soft tissue sarcomas with NTRK1 associated gene fusions. These tumors had a variable morphology and immunophenotype; their features could be likened to infantile hemangiopericytoma, myofibroblastic sarcoma, and myopericytoma. Mitotic activity in each of these cases was higher than 10 MF/ 10 high power fields. Immunohistochemically, two of the cases showed focal SMA staining and CD34 positivity. Molecularly there was a TPM3-NTRK1 fusion product in two cases, LMNA-NTRK1 fusion gene in one case, and the fourth lacked an identifiable gene partner19. As only one study to date has reported on this morphologic pattern, it remains uncertain the relationship of these lesions to other phenotypes described herein.
II. Distinctive Kinase Fusions are Associated with Recurring Morphologic Patterns in STTs
1. NTRK1 fusion-positive STTs are associated with a wide morphologic spectrum, predilection for children and benign behavior
NTRK1 gene fusions have been described in various pathologic entities occuring in children and adolescents, such as lipofibromatosis-like neural tumor, infantile fibrosarcoma and tumors resembling low grade malignant peripheral nerve sheath tumors (MPNST). NTRK1 rearrangement represents the most common genetic alteration among both lipofibromatosis-like neural tumors2 and spindle cell tumor resembling MPNST6 (Figs 1,2). In fact, a small subset of the latter was found to show intermixed foci of LPF-NT, suggesting the possibility of a morphologic spectrum6. In that study, although most of the patients with follow-up had an uneventful clinical course, two patients died of disease, one with documented lung metastases, at 375 and 20 months after diagnosis, respectively6. It is also worth noting the report of a myxoid spindle cell tumor with NTRK1-rearrangment with an aggressive clinical course, yet marked benefit with targeted therapy20.
2. NTRK3-fusion positive STTs are often malignant with a fibrosarcoma or MPNST-like phenotypes.
NTRK3 gene fusions (outside the canonical ETV6-NTRK3 fusions in IFS) have been described in sarcomas with either a fibrosarcoma phenotype (so-called FS-like sarcomas) or those resembling MPNST14,16,17,21,22 (Fig. 3). Among the 12 cases described to date, all except three were classified as high grade tumors, with most patients developing metastases and/or succumbing of disease. Two of these high grade fibrosarcoma-like tumors occurred in the uterine cervix16,17. The MPNST-like tumors show positivity for S100 and CD34, while all tumors expressed panNTRK, which can be used as a screening tool. Identification of high-grade NTRK3-rearranged sarcomas is clinically important, since the development of selective NTRK inhibitors has opened new avenues for targeted therapy.
3. RAF1-fusion positive spindle cell neoplasms
The study by Suurmeijer et al documenting eight spindle cell sarcomas with RAF1 gene fusions represents the first report of RAF1 gene fusions in soft tissue tumors6. RAF1 is a member of the RAF family of signaling kinases, downstream of RAS, which activate the MEK–ERK pathway that promotes cell proliferation and survival. RAF1 was fused to various gene partners, including PDZRN3, SLMAP, and TMF1, interestingly all of which are located on chromosome 3. Among the eight RAF1-positive tumors, two presented in children and six in adults, with no gender predilection. Five tumors were located on the trunk, one was intra-abdominal (involving stomach and pancreas), one visceral (rectum), and one occurred in the extremity. The tumors showed a predominantly bland spindle morphology with a patternless architecture and scant mitotic activity. A striking feature was the appearance of stromal collagen deposition, which included keloidal bands and perivascular rings (Fig 2). One of the cases showed low-grade areas that were mostly demarcated from higher grade areas with increased cellularity and fascicular growth. The latter component was composed of primitive monomorphic spindle cells with scant cytoplasm and fusiform nuclei, with scant collagenous stroma, which in a three-tier grading system would correspond to an intermediate histologic grade. Tumors lacked lipofibromatosis-like areas or nuclear pleomorphism. Among the 4 patients with follow-up only one behaved aggressively, with widespread metastatic disease to lung, liver, etc.
4. BRAF fusions positive STT
BRAF gene rearrangements have been associated with either IFS-like variants10 or with spindle cell sarcoma resembling MPNST6 (Fig 2). BRAF-positive IFS-like tumors were reported in three children, ranging from 6 months to 16 years old, with tumors occurring in the abdominal cavity (1 retroperitoneum and 2 pelvic)10. The morphologic appearance was characterized by primitive monomorphic spindle cells arranged in long, intersecting fascicles, associated with a variable hemangiopericytoma-like vascular pattern. The tumors showed no specific line of differentiation by immunohistochemistry, with only focal SMA reactivity in the two cases tested.
The two cases resembling MPNST with BRAF fusions showed a patternless growth of monomorphic spindle cells separated by “amianthoid-like” stromal collagen and perivascular rings6. One of the cases showed alternating areas of low cellularity with cellular components of primitive spindle cells arranged in intertwining fascicles. Focal areas with pleomorphic and multinucleate cells were present. Tumors were positive for S100 and CD34 (Fig 2).
5. RET fusion positive tumors
There are two series to date reporting recurrent RET fusions in STT4,23. Both studies highlight their strong predilection for children, including infants, and their diverse histologic spectrum, closely reproducing the phenotype observed in NTRK-fusion positive tumors. The morphologic patterns of RET-positive STT include lipofibromatosis-like neural tumors, IFS-like and MPNST-like tumors (Fig 3). Their histologic grade range from benign to highly malignant4. Immunohistochemically, tumors either co-express S100 and CD344,24, or display a non-specific immunoprofile. These results suggest that RET-rearranged neoplasms may share a similar phenotypic spectrum with the NTRK positive tumors, displaying either fibroblastic or neural-like differentiation, and open new avenues for targeted therapy with RET inhibitors available in clinical trials.
III. Ancillary Testing Recommendations.
1. NTRK Immunohistochemistry.
Pan-NTRK immunohistochemistry is recommended as a first-line screening for NTRK-related fusions in any STT with the histologic patterns described above, particularly if they show co-expression of S100 and CD34 immunostaining. The pattern of staining is quite variable and appears to be related to the type of underlying genetic abnormality5. Thus, IFS with canonical ETV6-NTRK3 fusion display nuclear positivity, while LPF-NT and MPNST-like spindle cell tumors harboring NTRK1 fusions have a cytoplasmic staining pattern2,6. Tumors with positive staining will then be streamlined for molecular validation. Additional NTRK1 immunoreactivity, especially in the setting of LPF-NT or MPNST-like spindle cell tumors, likely indicates the presence of NTRK1 gene abnormalities (Fig 1). However, a pan-NTRK immunostaining alone is insufficient in our opinion to confidently confirm a diagnosis of NTRK-fusion positive STT. In contrast to most epithelial malignancies with NTRK fusions, where this antibody was proven reliable and used as a surrogate for this genetic alteration25, the specificity and sensitivity of pan-NTRK immunostaining among sarcomas have been poor25 or has not been well investigated across various STT subtypes26. Importantly, STT with non-NTRK fusions, such as BRAF, RAF1, RET, MET, etc, are typically negative for pan-NTRK immunostaining, and should be further investigated by molecular analysis6.
2. Molecular Methods
As kinase fusion positive STT can involve a handful of genes (NTRK1/3, RAF1, BRAF, RET, MET, ALK, etc), a variety of potential fusion partners, and a few possible breakpoints at which different exons of the tyrosine kinase would join the fusion partner, screening for kinase fusions remains complex. Historically, gene fusions, such as ETV6-NTRK3 detected in most IFS, have been assayed by fluorescence in situ hybridization (FISH) and reverse transcriptase (RT)-PCR. However, given the multitude of 5’ partners involved in NTRK1/2/3, BRAF, and RET fusion genes in other STTs, assays that allow for the detection of multiple variants in a single test, including next-generation sequencing (NGS)-based RNA and DNA approaches, have been widely used. FISH analysis remains an option in cases with limited material or when no access to an NGS platform is available. FISH interrogates one gene rearrangement at the time and can be applied targeting the likely abnormality, such as NTRK3 in IFS and NTRK1 or RET in LPF-NT, etc. One caveat in applying FISH for detecting NTRK1 gene fusions relates to the high incidence of intra-chromosomal fusions, either small interstitial deletions or inversions, which can be easily missed by unexperienced labs2.
DNA-based sequencing, such as the targeted DNA-based next-generation sequencing panel (MSK-IMPACT) at our institution, shows an overall high specificity for the detection of NTRK fusions (99%), but a lower sensitivity (81%), when compared to RNA-based sequencing25. False negative results typically occur when breakpoint fusions are not covered by the assay. Novel structural variants involving gene kinases need to be confirmed at the RNA level to exclude random or non-transcribed events. In this respect, one recent study identified NTRK fusions by DNA-based NGS in rare osteosarcoma cases, however, these abnormalities lacked functional impact, suggesting that treatment approaches using pan-NTRK TKIs in this setting are unlikely to benefit these patients, as these fusions likely represent randomly occurring passenger alterations.27 Moreover, based on accumulated sequencing data, sarcomas characterized by complex genomics are less likely to harbor kinase gene fusions as driver events28.
Among the RNA-based approaches available, Archer panel, a next generation sequencing platform that uses anchored multiplex PCR, has emerged as a sensitive, robust and most widely applied platform for fusion detection in clinical practice, requiring a relatively limited amount of tissue and a 7–10 days turn-around time29. The panel covers fusions involving the kinase domains of the following genes: ALK, BRAF, MET, NTRK1, NTRK2, NTRK3, and RET.
SUMMARY
Recurrent fusions involving genes encoding receptor tyrosine or cytoplasmic kinases have been described in distinct STT with spindle cell morphology, and fibroblastic or neural-like differentiation. The clinical importance of the oncogenic gene fusions involving kinases in these tumors is twofold. First, they may serve as molecular diagnostic markers in the classification of spindle cell sarcomas with overlapping morphology and a nonspecific immunoprofile. Second, several of these oncogenic kinases have been shown to be therapeutically targetable, which translates into better treatment strategies and improved patient outcome. The correlation between various kinase fusions and different histologic subtypes is still evolving in soft tissue neoplasia and their specificity remains uncertain. A number of key points regarding diagnostic criteria and immunoprofile of kinase positive STT are summarized in Table 1. These pathologic guidelines may assist practicing pathologists in better recognizing these morphologic patterns and select only a small subset of likely candidates for a more definitive molecular confirmation.
Table 1.
Summary of Key Clinical, Pathologic and Molecular Features in Kinase Fusion positive STT
|
Disclosures:
Supported in part by: P50 CA140146 (CRA), P50 CA217694 (CRA), P30 CA008748
Footnotes
Data Availability Statement: No data is used or analyzed in the study.
Conflict of Interest: none
REFERENCES
- 1.Schram AM, Chang MT, Jonsson P, Drilon A. Fusions in solid tumours: diagnostic strategies, targeted therapy, and acquired resistance. Nat Rev Clin Oncol. 2017;14:735–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agaram NP, Zhang L, Sung YS, et al. Recurrent NTRK1 Gene Fusions Define a Novel Subset of Locally Aggressive Lipofibromatosis-like Neural Tumors. Am J Surg Pathol. 2016;40:1407–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartenstein DW, Coe TM, Gordon SC, et al. Lipofibromatosis-like neural tumor: Case report of a unique infantile presentation. JAAD Case Rep. 2018;4:185–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antonescu CR, Dickson BC, Swanson D, et al. Spindle Cell Tumors With RET Gene Fusions Exhibit a Morphologic Spectrum Akin to Tumors With NTRK Gene Fusions. Am J Surg Pathol. 2019;43:1384–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hung YP, Fletcher CDM, Hornick JL. Evaluation of pan-TRK immunohistochemistry in infantile fibrosarcoma, lipofibromatosis-like neural tumour and histological mimics. Histopathology. 2018;73:634–644. [DOI] [PubMed] [Google Scholar]
- 6.Suurmeijer AJH, Dickson BC, Swanson D, et al. A novel group of spindle cell tumors defined by S100 and CD34 co-expression shows recurrent fusions involving RAF1, BRAF, and NTRK1/2 genes. Genes Chromosomes Cancer. 2018;57:611–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bourgeois JM, Nagel K, Pearce E, Wright M, Barr RD, Tarnopolsky MA. Creatine monohydrate attenuates body fat accumulation in children with acute lymphoblastic leukemia during maintenance chemotherapy. Pediatric blood & cancer. 2008;51:183–187. [DOI] [PubMed] [Google Scholar]
- 8.Rudzinski ER, Lockwood CM, Stohr BA, et al. Pan-Trk Immunohistochemistry Identifies NTRK Rearrangements in Pediatric Mesenchymal Tumors. Am J Surg Pathol. 2018;42:927–935. [DOI] [PubMed] [Google Scholar]
- 9.Laetsch TW, DuBois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. The Lancet Oncology. 2018;19:705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kao YC, Fletcher CDM, Alaggio R, et al. Recurrent BRAF Gene Fusions in a Subset of Pediatric Spindle Cell Sarcomas: Expanding the Genetic Spectrum of Tumors With Overlapping Features With Infantile Fibrosarcoma. Am J Surg Pathol. 2018;42:28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pavlick D, Schrock AB, Malicki D, et al. Identification of NTRK fusions in pediatric mesenchymal tumors. Pediatric blood & cancer. 2017;64. [DOI] [PubMed] [Google Scholar]
- 12.Wegert J, Vokuhl C, Collord G, et al. Recurrent intragenic rearrangements of EGFR and BRAF in soft tissue tumors of infants. Nature communications. 2018;9:2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flucke U, van Noesel MM, Wijnen M, et al. TFG-MET fusion in an infantile spindle cell sarcoma with neural features. Genes Chromosomes Cancer. 2017;56:663–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suurmeijer AJ, Dickson BC, Swanson D, et al. The histologic spectrum of soft tissue spindle cell tumors with NTRK3 gene rearrangements. Genes Chromosomes Cancer. 2019;58:739–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rabban JT, Devine P, Sangoi AR, et al. NTRK Fusion Cervical Sarcoma: A Report of 3 Cases, Emphasizing Morphological and Immunohistochemical Distinction from Other Uterine Sarcomas, including Adenosarcoma. Histopathology. 2020. [DOI] [PubMed] [Google Scholar]
- 16.Chiang S, Cotzia P, Hyman DM, et al. NTRK Fusions Define a Novel Uterine Sarcoma Subtype With Features of Fibrosarcoma. Am J Surg Pathol. 2018;42:791–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Croce S, Hostein I, Longacre TA, et al. Uterine and vaginal sarcomas resembling fibrosarcoma: a clinicopathological and molecular analysis of 13 cases showing common NTRK-rearrangements and the description of a COL1A1-PDGFB fusion novel to uterine neoplasms. Mod Pathol. 2019;32:1008–1022. [DOI] [PubMed] [Google Scholar]
- 18.Weisman PS, Altinok M, Carballo EV, et al. Uterine Cervical Sarcoma With a Novel RET-SPECC1L Fusion in an Adult: A Case Which Expands the Homology Between RET-rearranged and NTRK-rearranged Tumors. Am J Surg Pathol. 2020. [DOI] [PubMed] [Google Scholar]
- 19.Haller F, Knoph J Ackermann A, et al. Paediatric and adult soft tissue sarcomas with NTRK1 gene fusions: a subset of spindle cell sarcomas unified by a prominent myopericytic/haemangiopericytic pattern. J Pathology. 2016; 238:700–10. [DOI] [PubMed] [Google Scholar]
- 20.So YK, Chow C, To KF, Chan JKC, Cheuk W. Myxoid Spindle Cell Sarcoma With LMNA-NTRK Fusion: Expanding the Morphologic Spectrum of NTRK-Rearranged Tumors. International journal of surgical pathology. 2020:1066896920905888. [DOI] [PubMed] [Google Scholar]
- 21.Olson N, Rouhi O, Zhang L, et al. A novel case of an aggressive superficial spindle cell sarcoma in an adult resembling fibrosarcomatous dermatofibrosarcoma protuberans and harboring an EML4-NTRK3 fusion. Journal of cutaneous pathology. 2018;45:933–939. [DOI] [PubMed] [Google Scholar]
- 22.Yamazaki F, Nakatani F, Asano N, et al. Novel NTRK3 Fusions in Fibrosarcomas of Adults. Am J Surg Pathol. 2019;43:523–530. [DOI] [PubMed] [Google Scholar]
- 23.Davis JL, Vargas SO, Rudzinski ER, et al. Recurrent RET Gene Fusions in Pediatric Spindle Mesenchymal Neoplasms. Histopathology. 2020. [DOI] [PubMed] [Google Scholar]
- 24.Michal M, Ptakova N, Martinek P, et al. S100 and CD34 positive spindle cell tumor with prominent perivascular hyalinization and a novel NCOA4-RET fusion. Genes Chromosomes Cancer. 2019;58:680–685. [DOI] [PubMed] [Google Scholar]
- 25.Solomon JP, Linkov I, Rosado A, et al. NTRK fusion detection across multiple assays and 33,997 cases: diagnostic implications and pitfalls. Mod Pathol. 2020;33:38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Solomon JP, Benayed R, Hechtman JF, Ladanyi M. Identifying patients with NTRK fusion cancer. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2019;30:viii16–viii22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ameline B, Saba KH, Kovac M, et al. NTRK fusions in osteosarcoma are rare and non-functional events. J Pathol Clin Res. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cancer Genome Atlas Research Network. Electronic address edsc, Cancer Genome Atlas Research N. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell. 2017;171:950–965 e928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng Z, Liebers M, Zhelyazkova B, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nature medicine. 2014;20:1479–1484. [DOI] [PubMed] [Google Scholar]