

Abstract

Deoxyhypusine synthase (DHPS) is the primary enzyme responsible for the hypusine modification and, thereby, activation of the eukaryotic translation initiation factor 5A (eIF5A), which is key in regulating the protein translation processes associated with tumor proliferation. Although DHPS inhibitors could be a promising therapeutic option for treating cancer, only a few studies reported druglike compounds with this inhibition property. Thus, in this work, we designed and synthesized a new chemical series possessing fused ring scaffolds designed from high-throughput screening hit compounds, discovering a 5,6-dihydrothieno[2,3-c]pyridine derivative (26d) with potent inhibitory activity; furthermore, the X-ray crystallographic analysis of the DHPS complex with 26d demonstrated a distinct allosteric binding mode compared to a previously reported inhibitor. These findings could be significantly useful in the functional analysis of conformational changes in DHPS as well as the structure-based design of allosteric inhibitors.
Keywords: Deoxyhypusine synthase (DHPS), eukaryotic translation initiation factor 5A (eIF5A), nicotinamide adenine dinucleotide (NAD), spermidine, allosteric inhibitor
Deoxyhypusine synthase (DHPS) is an enzyme that activates via hypusine [Nε-(4-amino-2-hydroxybutyl)lysine] modification the eukaryotic translation initiation factor 5A (eIF5A), which is a regulator of the protein translation processes involved in tumor growth.1−5 This modification involves two enzymatic steps. DHPS catalyzes the first rate-limiting step, which is the cleavage of the polyamine spermidine: the 4-aminobutyl moiety is transferred to the ε-amino group of one specific lysine residue of the eIF5A precursor to form a deoxyhypusine intermediate.6−8 In the second step, deoxyhypusine hydroxylase (DOHH) converts the deoxyhypusine-containing intermediate into hypusine-containing mature eIF5A (Supporting Information). Blocking the biological activity of eIF5A by inhibiting the DHPS-mediated modification of its precursor could be an effective strategy in cancer therapy.9,10N1-Guanyl-1,7-diaminoheptane (GC-7) (1), a substrate (spermidine) mimetic compound, binds to DHPS similarly to spermidine11−13 and possesses inhibitory activity; nonetheless, the potential application of 1 is limited by its polyamine-like structure (Figure 1A).14−16 We recently reported a novel allosteric inhibitor (2) and its cocrystal structure with DHPS, which revealed the dynamic conformational change in the DHPS structure.17 However, its application for the further biological evaluation of DHPS is still limited due to the narrow structure–activity relationship (SAR) of its related compounds, although the structural analysis suggests that the allosteric pocket is druggable. Hence, more potent and druglike inhibitors are needed for detailed biologic studies on the therapeutic potential of targeting the DHPS-mediated hypusine synthesis. Here, we describe a new series of potent DHPS inhibitors discovered through SAR studies from hit compounds obtained via a high-throughput screening (HTS). Among them, the X-ray crystallographic analysis of a highly potent compound (26d, IC50 = 0.0092 μM) in complex with DHPS demonstrates a distinct binding mode compared to the previously reported 2.
Figure 1.

(A) Chemical structures of spermidine and reported DHPS inhibitors. (B) Structures of HTS hits (3 and 4), and a possible pharmacophore. The DHPS inhibitory activities were determined by an enzyme assay. IC50 values and 95% confidence intervals were calculated via the nonlinear regression analysis of the percent inhibition data.
To identify a new series of DHPS inhibitors, we conducted an HTS via an enzyme assay. Several hit compounds containing a 3-pyridyl group such as 3 and 4 (Figure 1B), whose chemical structures differ from those of other reported inhibitors (Figure 1A), showed inhibitory activities toward DHPS.17−20 An initial SAR study revealed that the 3-pyridine moiety is essential for their inhibitory activity (data not shown). Based on the structural similarity among these compounds, we presumed that a fused ring system with 3-pyridine is a pharmacophore of their series for inhibiting DHPS, as shown in Figure 1B; thus, we started chemical modifications according to the designed molecules maintaining the possible pharmacophore.
Benzyl derivatives with designed fused rings 6a–d were synthesized via benzylation of 5a–d as illustrated in Scheme 1. The precursors 5a–d were prepared as described in the Supporting Information.
Scheme 1. Synthesis of Benzyl Derivatives 6a–d.

Reagents and conditions: (a) benzyl bromide, NaH, DMF.
Scheme 2 illustrates the synthesis of 5,6-dihydrothieno[2,3-c]pyridine derivatives 9a–g from the intermediate 5d. Alkylation of 5d with methyl 2-bromo-2-phenylacetate gave the racemic ester 7a, which was successively reduced by NaBH4 in the presence of CaCl2 to yield the alcohol 8a. Conversion into the racemic amine 9a was performed via introduction of phthalimide by Mitsunobu reaction and subsequent treatment with hydrazine monohydrate at 70 °C in EtOH. The optical resolution of 9a via chiral HPLC afforded both its enantiomers: the S- (9b) and the R-form (9c). The absolute configuration of each enantiomer was determined through single-crystal X-ray analysis (Supporting Information). Using the same procedures for 9a, compounds 9d–e were synthesized from 5d and the corresponding bromides 10a–b, which were prepared as described in the Supporting Information. The Boc group of 9e was removed in the presence of K2CO3 at 80 °C in MeOH and H2O to obtain the indole derivative 9f. By treating 5d with the bromide 11, we formed the benzoyl derivative 12, which was hydrolyzed to yield the alcohol 13, which was then converted into the amine 9g similarly to 9a.
Scheme 2. Synthesis of Compounds 7a, 8a, and 9a–g.

Reagents and conditions: (a) methyl 2-bromo-2-phenylacetate, potassium tert-butoxide, THF, 90%; (b) NaBH4, CaCl2, MeOH; (c) (i) phthalimide, triphenylphosphine, diisopropyl azodicarboxylate, toluene; (ii) hydrazine monohydrate, EtOH, 70 °C; (d) 10a–b, potassium tert-butoxide, THF; (e) K2CO3, MeOH, H2O, 80 °C, 62%; (f) 11, potassium tert-butoxide, DMF, 52%; (g) 2 M NaOH, MeOH, 87%.
Scheme 3 describes an alternative method to obtain 5,6-dihydrothieno[2,3-c]pyridine derivatives. By using 3-hydroxythiophene 14 as a starting material, triflate 17 was synthesized via bromination at the 4-position, Suzuki coupling, and successive triflation of the resulting phenol 16. The introduction of an ethyl vinyl ether via Suzuki coupling followed by hydrolysis under an acidic condition yielded the aldehyde 18. The 5,6-dihydrothieno[2,3-c]pyridine scaffold was constructed via the reductive amination of 18 with the racemic amines 19a–b and the subsequent intramolecular cyclization in the presence of NaOMe in MeOH, producing the cyclized compounds 21a–b. The removal of the Boc group yielded the racemates 22a–b. In a similar way, the alcohols 25a–d were synthesized through the cyclization of the amines 24a–d, which were prepared from 18 and the commercially available R-form or racemic amines 23a–d. The application of the same reactions shown in Scheme 2 to alcohols 25a–d gave the amines 26a–d.
Scheme 3. Synthesis of Compounds 22a–b and 26a–d.

Reagents and conditions: (a) bromine, AcOH, 61%; (b) 3-pyridineboronic acid, Pd(dppf)Cl2·CH2Cl2, Cs2CO3, DME, H2O, 80 °C, 72%; (c) trifluoromethanesulfonic anhydride, pyridine; (d) (i) (E)-1-ethoxyethene-2-boronic acid pinacol ester, Pd(dppf)Cl2·CH2Cl2, Cs2CO3, DME, H2O, 80 °C; (ii) 12 M HCl, THF, 30% (two steps); (e) 19a–b, 2-picoline borane, MeOH, AcOH; (f) NaOMe, MeOH; (g) 4 M HCl in EtOAc; (h) 23a–d, 2-picoline borane, MeOH, AcOH; (j) (i) phthalimide, triphenylphosphine, diisopropyl azodicarboxylate, toluene; (ii) hydrazine monohydrate, EtOH, 70 °C.
To verify the potentials of each bicyclic scaffold having the 3-pyridyl moiety, the derivatives with a benzyl group derived from the hit compound 4 were evaluated in the enzyme assay (Table 1). The conversion of the thieno[3,2-d]pyrimidine ring in 4 to isoquinolone 6a improved the inhibitory activity (IC50 = 8.7 μM). Dihydroisoquinolone 6b also showed a comparable potency to 6a, indicating that the left ring of the bicyclic scaffold is not necessarily aromatic and the nitrogen atom on the 1-position of the thieno[3,2-d]pyrimidine (4) weakens the potency. A significant loss of potency was observed by changing the position of the 3-pyridine moiety (6c, IC50 > 100 μM), indicating that the orientation of the substituents from the scaffold is a key factor. Furthermore, the evaluation of scaffolds such as fused the 6,5- (6d) and 5,6-membered (27) bicyclic rings revealed the strong inhibitory activity of 5,6-dihydrothieno[2,3-c]pyridine 6d (IC50 = 6.3 μM).
Table 1. SAR for Fused Ring Scaffolds 6a–d and 27.
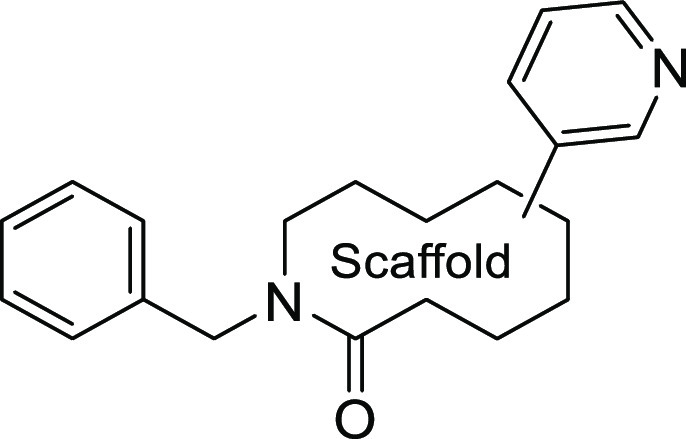

IC50 values were calculated via the nonlinear regression analysis of the percent inhibition data. 95% confidence intervals are shown in the Supporting Information.
Based on the above-mentioned SAR results, we further explored the substituents on the left side of the 5,6-dihydrothieno[2,3-c]pyridine scaffold (Table 2). Since the hit compound 3 bearing a hydroxymethyl group at the benzyl position showed inhibitory activity, we expected that the introduction of a polar functional group at the corresponding benzylic position of 6d would be tolerable. Indeed, introduction of the hydroxymethyl group (8a) significantly enhanced the potency although a methyl ester (7a) was unfavorable, indicating the necessity of a hydrogen donor for the inhibitory activity. The conversion of the primary alcohol to an amine further improved the potency, and the racemate amine 9a exhibited a strong IC50 value of 0.018 μM. Moreover, we found that R- form 9c is a eutomer after separation of each enantiomer of 9a. On the other hand, neither the extension of the alkyl chain (9g) nor the conformational restriction of the amine moiety via cyclization (22a) increased the potency compared to 9a.
Table 2. SAR for Substituents of 5,6-Dihydrothieno[2,3-c]pyridine-7(4H)-one Derivatives.


In a previous work, the allosteric inhibitor 2 showed nicotinamide adenine dinucleotide (NAD), a coenzyme of DHPS, competitive profile as a result of dramatic conformational changes of DHPS induced by its binding.17 We employed the radiometric assay using [3H]-spermidine to evaluate the inhibitory potency of compounds and the competitiveness against NAD or spermidine at each Km or higher concentrations although there is a limitation to analyze precise kinetic profiles due to 120 min incubation time needed in this assay. To analyze the inhibition mode of the newly discovered chemical series, the potent compound 9c was evaluated under the high NAD concentration (Table 3); its DHPS inhibition at 250 μM NAD was 29-fold lower than at 14 μM NAD, indicating a NAD competitive profile similar to that of 2.17 The X-ray cocrystal analysis of the new chemical series revealed its binding mode and a mechanism of the NAD competition (vide infra).
Table 3. Conversion of Benzene Ring to Other Hydrophobic Substituents.


To further improve the inhibition potency and identify a promising tool molecule, we successively converted the benzene ring on the left part into other hydrophobic substituents (Table 3). We used a DHPS concentration of 0.024 μM in the enzyme assay (Supporting Information). Since the tight-binding21 possibly caused an underestimation of the IC50 value of a highly potent compound, we analyzed the inhibitory activities also under high NAD concentrations to obtain weakened IC50 values for the SAR analysis of the NAD competitive inhibitors. The conversion of the benzene ring (9c) into bicyclic rings such as benzothiophene (9d) and indole (9f) decreased the inhibitory activity, while the compound with a cyclohexane substituent (26a) exhibited improved potency under high NAD concentration (IC50 = 0.17 μM). This result encouraged us to pursue other alkyl substituents. Although n-hexane derivative 22b also showed potent activity, its metabolic stability in mice liver microsome was diminished, probably due to high lipophilicity. Therefore, we examined shorter and/or branched alkyl chains (26b–d) to reduce the lipophilicity, resulting in the discovery of the isobutyl derivative 26d, which exhibited the most potent inhibitory activity among the 5,6-dihydrothieno[2,3-c]pyridine derivatives (IC50= 0.069 μM under high NAD condition). In addition, 26d showed orally available pharmacokinetic profiles in mice (Table 4). Further investigations are needed to verify the therapeutic potentials of the inhibitor, such as confirmation of effects in in vivo tumor xenograft models as well as in eIF5A-amplified cancer cells. However, we believe that the identified chemical series can be a good lead molecule.
Table 4. Pharmacokinetic Parameters of 26d in Micea.
| Vdssb (mL/kg) | CLtotalb (mL/h/kg) | Cmaxc (ng/mL) | AUC0–8hc (ng·h/mL) | MRTc (h) | Fd (%) |
|---|---|---|---|---|---|
| 8491 | 8965 | 4.8 | 13.1 | 2.46 | 11.7 |
Cassette dosing. Male Institute of Cancer Research mice (n = 3).
0.1 mg/kg, iv.
1 mg/kg, po.
Bioavailability.
To unravel the binding mode of the newly discovered compounds, we defined the X-ray crystal structure of the DHPS/26d complex via soaking experiments with a 2.15 Å resolution (Figure 2). The results revealed a tetramer of this binary complex similar to the previously reported structure of the DHPS/2 complex;1726d was bound to the allosteric pocket generated via an α-helix unfolding, which was induced by the inhibitor binding. Despite the fact that the binding site of 26d partially overlaps that of 2, their binding modes turned out to be quite different. In contrast to 2, Gly282 is located near a NAD binding site but neither flips nor seems to interrupt the NAD binding. This implies a different mechanism of competitive inhibition of 26d to NAD compared to 2. Instead of Gly282, upon the 26d binding, Lys287 moves to another NAD binding site and hinders its pocket. To our surprise, this Lys287 is overlapped with the spermidine binding site in the DHPS/2 complex structure (Figure 2B, C). Overall, these results indicate the versatility of the allosteric site as a druggable pocket, which can be utilized for exploring DHPS inhibitors.
Figure 2.

Complex crystal structure of compound 26d with DHPS (PDB ID 6WL6). All images were prepared using PyMOL.22 (A) Tetramer with each monomer indicated by a different color in cartoon model. Each 26d molecule is represented as a sphere. (B) 26d bound allosteric site. 26d is shown as a stick model in salmon. The protein is colored by chain and shown as a cartoon, while the two residues, Gly282 and Lys287, are shown as stick models. The black dashed circle indicates a loop structure that was conformationally changed from an α-helix in GC-7 bound structure (PDB ID 6P4V). NAD and GC-7 are shown in surface representation by superposition of the complex (PDB ID 6P4V) in pink and deep blue, respectively. Compound 2 is shown by the superposition of the complex (PDB ID 6PGR), which is shown as a stick model in magenta. (C) Schematic diagrams of 26d (PDB ID 6WL6) and 2 (PDB ID 6PGR) bound conformations in one dimer of a DHPS tetramer.
Additionally, this complex structure well explained the results of the SAR studies (Figure 3). Regarding the 3-pyridyl moiety, a hydrogen bonding was observed between a nitrogen atom of the pyridine ring and a side chain of Asp262 and/or a main chain of Ser233 via a water molecule, confirming that it is required for the potency. Furthermore, the carbonyl oxygen on the bicyclic scaffold formed a hydrogen bond with Lys287. The primary amine of 26d also formed a hydrogen bond with Asp243 (side chain) and Asp238 (main chain), resulting in high affinity in the pocket and strong inhibitory activity. The alkyl side chain occupies a hydrophobic pocket surrounded by Phe100, Ala270, Met278, and Val286; however, there might be other residues surrounding the alkyl side chain because the 288–298 and 316–334 residues are disordered in the 26d complex. As described in Table 1, the orientation of substituents on the fused ring scaffolds is important for the potency, which is reasonable to maintain or stabilize the interactions between inhibitor and DHPS.
Figure 3.

(A) Allosteric binding site of 26d. DHPS is shown at the surface (PDB ID 6WL6). 26d and a part of the surrounding residues and water molecules are represented as sticks and spheres, respectively. The colors are the same as in Figure 2B. The dashed lines indicate the hydrogen bonds between 26d and the protein/water molecules. (B) Schematic diagram of interactions in stabilizing 26d at the allosteric site.
In summary, we discovered a new class of potent allosteric DHPS inhibitors via SAR studies by initially exploring bicyclic scaffolds suggested by the pharmacophore of HTS hit compounds. These compounds were characterized as NAD competitive inhibitors, and the X-ray cocrystal analysis revealed that the potent inhibitor 26d binds to the allosteric pocket in a newly identified binding mode, distinct from that of the previously reported inhibitor 2. The identification of the novel binding mode with the compound possessing improved enzymatic inhibitory activity and favorable PK properties suggests the promise of using the allosteric site as a druggable pocket, and these results would pave the road for the structure-based design for further lead optimization of allosteric inhibitors targeting DHPS.
Acknowledgments
We thank all members of the DHPS Program Team at Takeda for their contribution. We thank Katsuaki Oda for assistance with chiral separation and Mitsuyoshi Nishitani for assistance with single-crystal X-ray study along with deposition of data to the Cambridge Crystallographic Data Centre. We also thank Weston Lane and Gyorgy Snell for diffraction data collection and Jaya Bhatnagar for assistance with crystallization. The GM/CA@APS has been funded in whole or in part with federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Glossary
Abbreviations
- DHPS
deoxyhypusine synthase
- eIF5A
eukaryotic translation initiation factor 5A
- DOHH
deoxyhypusine hydroxylase
- NAD
nicotinamide adenine dinucleotide
- SAR
structure–activity relationship
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00331.
Figure of the DHPS-eIF5A pathway and chemical reaction of DHPS, 95% confidence intervals of enzyme IC50, synthetic experimental details and characterization data, single-crystal X-ray structures of 9b and 9c, assay protocols, and cocrystal data of 9c and 26d (PDF)
Accession Codes
The atom coordinates and structure factors for the DHPS/9c and DHPS/26d complexes have been deposited in the Protein Data Bank with the accession codes 6WKZ, and 6WL6, respectively.
Author Contributions
All authors contributed equally and have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Park M. H.; Wolff E. C. Hypusine, a polyamine-derived amino acid critical for eukaryotic translation. J. Biol. Chem. 2018, 293, 18710–18718. 10.1074/jbc.TM118.003341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson A.; Hershey J. W. Eukaryotic translation initiation factor (eIF) 5A stimulates protein synthesis in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 6415–6419. 10.1073/pnas.1008150108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini P.; Eyler D. E.; Green R.; Dever T. E. Hypusine-containing protein eIF5A promotes translation elongation. Nature 2009, 459, 118–121. 10.1038/nature08034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan X. Y.; Fung J. M.; Ma N. F.; Lau S. H.; Tai L. S.; Xie D.; Zhang Y.; Hu L.; Wu Q. L.; Fang Y.; Sham J. S. Oncogenic role of eIF-5A2 in the development of ovarian cancer. Cancer Res. 2004, 64, 4197–4200. 10.1158/0008-5472.CAN-03-3747. [DOI] [PubMed] [Google Scholar]
- Clement P. M.; Johansson H. E.; Wolff E. C.; Park M. H. Differential expression of eIF5A-1 and eIF5A-2 in human cancer cells. FEBS J. 2006, 273, 1102–1114. 10.1111/j.1742-4658.2006.05135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greganova E.; Altmann M.; Butikofer P. Unique modification of translation elongation factors. FEBS J. 2011, 278, 2613–2624. 10.1111/j.1742-4658.2011.08199.x. [DOI] [PubMed] [Google Scholar]
- Park M. H.; Nishimura K.; Zanelli C. F.; Valentini S. R. Functional significance of eIF5A and its hypusine modification in eukaryotes. Amino Acids 2010, 38, 491–500. 10.1007/s00726-009-0408-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y.; Park I.; Hong B. S.; Nedyalkova L.; Tempel W.; Park H. W. Crystal structure of human eIF5A1: insight into functional similarity of human eIF5A1 and eIF5A2. Proteins: Struct., Funct., Genet. 2009, 75, 1040–1045. 10.1002/prot.22378. [DOI] [PubMed] [Google Scholar]
- Nishimura K.; Ohki Y.; Fukuchi-Shimogori T.; Sakata K.; Saiga K.; Beppu T.; Shirahata A.; Kashiwagi K.; Igarashi K. Inhibition of cell growth through inactivation of eukaryotic translation initiation factor 5A (eIF5A) by deoxyspergualin. Biochem. J. 2002, 363, 761–768. 10.1042/bj3630761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puleston D. J.; Buck M. D.; Klein Geltink R. I.; Kyle R. L.; Caputa G.; O’Sullivan D.; Cameron A. M.; Castoldi A.; Musa Y.; Kabat A. M.; Zhang Y.; Flachsmann L. J.; Field C. S.; Patterson A. E.; Scherer S.; Alfei F.; Baixauli F.; Austin S. K.; Kelly B.; Matsushita M.; Curtis J. D.; Grzes K. M.; Villa M.; Corrado M.; Sanin D. E.; Qiu J.; Pallman N.; Paz K.; Maccari M. E.; Blazar B. R.; Mittler G.; Buescher J. M.; Zehn D.; Rospert S.; Pearce E. J.; Balabanov S.; Pearce E. L. Polyamines and eIF5A hypusination modulate mitochondrial respiration and macrophage activation. Cell Metab. 2019, 30, 352–363. 10.1016/j.cmet.2019.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D. I.; Wolff E. C.; Park M. H.; Davies D. R. Crystal structure of the NAD complex of human deoxyhypusine synthase: an enzyme with a ball-and-chain mechanism for blocking the active site. Structure 1998, 6, 23–32. 10.1016/S0969-2126(98)00004-5. [DOI] [PubMed] [Google Scholar]
- Umland T. C.; Wolff E. C.; Park M. H.; Davies D. R. A new crystal structure of deoxyhypusine synthase reveals the configuration of the active enzyme and of an enzyme·NAD·inhibitor ternary complex. J. Biol. Chem. 2004, 279, 28697–28705. 10.1074/jbc.M404095200. [DOI] [PubMed] [Google Scholar]
- Wator E.; Wilk P.; Grudnik P. Half way to hypusine-structural basis for substrate recognition by human deoxyhypusine synthase. Biomolecules 2020, 10, 522. 10.3390/biom10040522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakus J.; Wolff E. C.; Park M. H.; Folk J. E. Features of the spermidine-binding site of deoxyhypusine synthase as derived from inhibition studies. J. Biol. Chem. 1993, 268, 13151–13159. [PubMed] [Google Scholar]
- Lee Y. B.; Park M. H.; Folk J. E. Diamine and triamine analogs and derivatives as inhibitors of deoxyhypusine synthase; synthesis and biological activity. J. Med. Chem. 1995, 38, 3053–3061. 10.1021/jm00016a008. [DOI] [PubMed] [Google Scholar]
- Marton L. J.; Pegg A. E. Polyamines as targets for therapeutic intervention. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 55–91. 10.1146/annurev.pa.35.040195.000415. [DOI] [PubMed] [Google Scholar]
- Tanaka Y.; Kurasawa O.; Yokota A.; Klein M. G.; Ono K.; Saito B.; Matsumoto S.; Okaniwa M.; Ambrus-Aikelin G.; Morishita D.; Kitazawa S.; Uchiyama N.; Ogawa K.; Kimura H.; Imamura S. Discovery of novel allosteric inhibitors of deoxyhypusine synthase. J. Med. Chem. 2020, 63, 3215–3226. 10.1021/acs.jmedchem.9b01979. [DOI] [PubMed] [Google Scholar]
- Lee Y. B.; Folk J. E. Branched-chain and unsaturated 1,7-diaminoheptane derivatives as deoxyhypusine synthase inhibitors. Bioorg. Med. Chem. 1998, 6, 253–270. 10.1016/S0968-0896(97)10030-X. [DOI] [PubMed] [Google Scholar]
- Ziegler P.; Chahoud T.; Wilhelm T.; Pallman N.; Braig M.; Wiehle V.; Ziegler S.; Schroder M.; Meier C.; Kolodzik A.; Rarey M.; Panse J.; Hauber J.; Balabanov S.; Brummendorf T. H. Evaluation of deoxyhypusine synthase inhibitors targeting BCR-ABL positive leukemias. Invest. New Drugs 2012, 30, 2274–2283. 10.1007/s10637-012-9810-1. [DOI] [PubMed] [Google Scholar]
- Schroeder M.; Kolodzik A.; Pfaff K.; Priyadarshini P.; Krepstakies M.; Hauber J.; Rarey M.; Meier C. In silico design, synthesis, and screening of novel deoxyhypusine synthase inhibitors targeting HIV-1 replication. ChemMedChem 2014, 9, 940–952. 10.1002/cmdc.201300481. [DOI] [PubMed] [Google Scholar]
- Murphy D. J. Determination of accurate KI values for tight-binding enzyme inhibitors: an in silico study of experimental error and assay design. Anal. Biochem. 2004, 327, 61–67. 10.1016/j.ab.2003.12.018. [DOI] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, Version 2.2.3; Schrödinger, LLC. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.