

Abstract
Background
The disease course of adult immunoglobulin A (IgA) vasculitis (IgAV; Henoch–Schönlein purpura) has not been well defined.
Methods
In a retrospective survey, we studied 85 adult IgAV patients with extended follow-up (median 43 months) for 67 patients.
Results
Only 33 of 67 (49%) achieved complete remission. Ongoing renal disease was the most common persistent organ manifestation, but extra-renal disease activity was also present in >50% of patients not achieving complete remission. Twenty-nine of 67 (43%) had relapsing disease, with 18/67 (27%) experiencing several relapses. Skin disease was the most common feature in relapsing patients, followed by nephritis. At 4 years of follow-up, 6 of 29 (21%) experienced progressive disease and 10/29 (34%) relapsing disease. Five of 67 (7%) developed nephritis after diagnosis, within the first 6 months of follow-up. At final follow-up, 10 of 67 (15%) had chronic kidney disease Stage ≥G3a, 18 (27%) haematuria and 13 (19%) proteinuria. No therapy appeared particularly effective and only 6/17 patients treated with mycophenolate mofetil experienced a good response.
Conclusions
The disease course of adult IgAV is different from that seen in children, with higher frequency of persisting and relapsing disease. Renal disease is the main determinant of ongoing disease activity, but extra-renal features were seen in >50% of patients with chronic disease activity. No clear conclusions on use or choice of immunosuppressive agent could be made based on our experience.
Keywords: extra-renal outcome, Henoch–Schönlein purpura, IgA vasculitis, initial presentation, renal outcome, nephritis
INTRODUCTION
Immunoglobulin A (IgA) vasculitis (IgAV), formerly Henoch–Schönlein purpura, is a systemic small-vessel leukocytoclastic vasculitis, caused by the deposition of IgA-containing immune complexes. It is the most frequent systemic childhood vasculitis (annual incidence of 6.2–24/100 000 children [1]), but is less common in adults, with an estimated annual incidence of 0.8–1.8/100 000 patients [2], although this may be an underestimate: Hočevar et al. [3] reported an annual incidence of 5.1/100 000 adults with biopsy-proven disease. There are no established diagnostic criteria for IgAV. The American College of Rheumatology (ACR) 1990 classification criteria [4] (Table 1) have less utility in adults and do not differentiate well from other small vessel vasculitides. The European League Against Rheumatism, Paediatric Rheumatology International Trials Organisation and Paediatric Rheumatology European Society (EULAR/PRINTO/PRES) classification criteria [5] (Table 1) provide classification criteria only for childhood IgAV.
Table 1.
Classification criteria for IgAV
| Criterion | Definition |
|---|---|
| ACR classification criteriaa | |
| Palpable purpura | Slightly raised ‘palpable’ haemorrhagic skin lesions, not related to thrombocytopaenia |
| Age ≤20 years at disease onset | Patient 20 years or younger at onset of first symptoms |
| Bowel angina | Diffuse abdominal pain, worse after meals or the diagnosis of ischaemia, usually including bloody diarrhoea |
| Wall granulocytes on biopsy | Histologic changes showing granulocytes in the walls of arterioles or venules |
| EULAR/PRINTO/PRES classification criteriab | |
| Purpura (mandatory) | Purpura (commonly palpable and in crops) or petechiae, with lower limb predominance, not related to thrombocytopaeniac |
| Abdominal pain | Diffuse abdominal colicky pain with acute onset assessed by history and physical examination; may include intussusception and gastrointestinal bleeding |
| Histopathology | Typically leukocytoclastic vasculitis with predominant IgA deposits or proliferative glomerulonephritis with predominant IgA deposits |
| Arthritis or arthralgias | Arthritis of acute onset defined as joint swelling or joint pain with limitation on motion; arthralgia of acute onset defined as joint pain without joint swelling or limitation on motion |
| Renal involvement | Proteinuria >0.3 g/24 h or >30 mg/mmol of urine albumin/creatinine ratio on a spot morning sample; haematuria or red blood cell casts: >5 red blood cells/HPF or red blood cells casts in the urinary sediment or ≥2+ on dipstick |
The presence of at least two criteria yields a sensitivity of 87.1% and a specificity of 87.7%.
The presence of purpura with lower limb predominance and at least one of the other four criteria yields a sensitivity of 100% and a specificity of 87%.
For purpura with atypical distribution a demonstration of an IgA deposit in a biopsy is required.
IgAV is characterized by the tetrad of palpable purpura, arthralgia/arthritis, gastrointestinal involvement and glomerulonephritis, but not all features are necessarily present and other organ systems can be involved. Except for life-threatening gastrointestinal and pulmonary involvement at initial presentation, the morbidity and mortality of IgAV are mainly determined by the severity of renal involvement. Palpable purpura is the most prevalent symptom, reported in up to 100% of adults and children. Joint involvement is present in 51–65% of adults and 67–75% of children, while gastrointestinal involvement is reported in 57–84% of adults and 67–71% of children [6, 7]. Glomerulonephritis is more frequent in adults (58–84% versus 27–56% of children), more severe (14% of adults versus 5% of children have nephrotic syndrome, 11% versus 4% nephritic syndrome and 13–46% versus 3–7% renal insufficiency) [6–8] and has worse long-term renal outcome (Table 2).
Table 2.
Long-term renal outcome in adult IgAV
| References | Patients | Outcome | FU |
|---|---|---|---|
|
Garcia-Porrúa et al. [6] |
31 adults 73 children |
Renal insufficiencya: 10% of adults, 0% of children Persistent haematuria and/or proteinuria without renal insufficiency: 29% of adults, 11% of children |
5–6 years median |
| Blanco et al. [9] |
38 adults 114 children |
Mild renal impairmentb: 11% of adults, 0% of children Microscopic haematuria: 0% of adults, 5% of children |
12–15 months median |
| Coppo et al. [10] |
83 adults 136 children biopsy-proven nephritis |
Dialysis-dependency: 13% of adults, 7% of children Severe renal impairmentc: 10% of adults, 5% of children Moderate renal impairmentd: 15% of adults, 12% of children Moderate proteinuria with normal renal function: 15% of adults, 12% of children Proteinuria ≤1 g/24 h with normal renal function: 40% of adults, 45% of children Nephrotic-range proteinuria: 2% of adults, 4% of children |
4.5 years median |
| Pillebout et al. [11] |
250 adults biopsy-proven nephritis |
ESRD: 11% Severe renal insufficiencye: 13% Moderate renal insufficiencyf: 14% Renal remissiong: 20% Nephrotic-range proteinuria: 8% Non-nephrotic proteinuria: 47% Haematuria: 50% |
14.8 years median |
| Shrestha et al. [12] |
37 adults biopsy-proven nephritis |
Clinical remissionh: 43% ESRD: 27% Renal insufficiencyi: 8% Proteinuria with normal renal function: 19% |
9.4 ± 7.1 years mean ± SD |
| Rauta et al. [13] |
38 adults biopsy-proven nephritis |
Normal serum creatinine + absence of haematuria + absence of proteinuria: 12% ESRD: 11% Proteinuria: 45% Nephrotic-range proteinuria: 3% |
6.1 ± 4.3 years mean ± SD |
Plasma creatinine >125% upper limit of normal.
Plasma creatinine 1.6–2.4 mg/dL.
Serum creatinine >3 mg/dL in adults and CrCl <60 mL/min/1.73 m2 (Schwartz formula) in children.
Serum ceatinine >1.5 mg/dL and ≤3 mg/dL in adults, and CrCl <90 mL/min/1.73 m2 and ≥60 mL/min/1.73 m2 (Schwartz formula) in children.
CrCl <30 mL/min (Cockroft and Gault formula).
CrCl <50 mL/min and CrCl ≥30 mL/min (Cockroft and Gault formula).
CrCl ≥50 mL/min + absence of proteinuria + absence of haematuria.
Absence of extra-renal involvement + creatinine <120 μmol/L in females, <130 μmol/L in males + absence of significant haematuria (≤1 × 106/24 h in males, ≤2 × 106/24 h in females) and/or proteinuria (<0.3 g/24 h).
Creatinine ≥120 μmol/L in females, ≥130 μmol/L in males. ESRD, end-stage renal disease; CrCl, creatinine clearance.
Pillebout et al. [11] reported renal insufficiency and proteinuria >1 g/24 h at baseline, the proportion of interstitial fibrosis, fibrinoid necrosis and glomerular sclerosis to be associated with worse renal prognosis, while Coppo et al. [10] found proteinuria during follow-up to be the main risk factor for the progression of IgAV nephritis. Data regarding the disease course and extra-renal outcome in the adult population are scarce. Relapse rates of 14–43% have been described in children, while in adults 26–48% of patients experience relapses [6, 11, 14]. The goal of this retrospective study was to investigate the disease course and outcome of all involved organ systems in the adult IgAV population.
MATERIALS AND METHODS
Patients
We retrospectively analysed the medical records of all IgAV patients aged ≥16 years, attending a vasculitis clinic at a University Hospital, between 1 March 2004 and 31 August 2015. The diagnosis of IgAV was made by an experienced nephrologist or rheumatologist based on clinical judgement with or without histological confirmation. We excluded patients with thrombocytopaenia (defined as a platelet count <150 000 × 109/L), those with isolated IgA nephropathy (IgAN) and patients suffering from other autoimmune diseases explaining the clinical presentation. Ethical approval was not required for this audit review under National Health Service guidelines.
Evaluation and clinical definitions
The initial clinical and laboratory presentation and the presence of predisposing factors were evaluated. The initial presentation was assessed for the presence of palpable purpura, joint involvement (polyarthralgia and/or joint effusion), gastrointestinal involvement (nausea, vomiting, abdominal pain, diarrhoea and/or rectal bleeding), hypertension, renal involvement, constitutional symptoms and other organ involvement. Hypertension was defined as a systolic blood pressure (BP) ≥140 mmHg and/or diastolic BP ≥90 mmHg and/or treatment with antihypertensive medication prior to IgAV diagnosis. Renal involvement was the presence of proteinuria (>30 mg/mmol albumin to creatinine ratio or ≥1+ on urine dipstick) and/or haematuria [>5 red cells per high-power field (HPF) or ≥2+ on dipstick] and/or new estimated glomerular filtration rate (eGFR) [Modification of Diet in Renal Disease (MDRD) equation] <60 mL/min/1.73 m2, further classified as acute kidney injury (AKI) staged according to the Kidney Disease: Improving Global Outcomes (KDIGO) criteria (Stage 1: increase in serum creatinine to 1.5–1.9 times baseline or increase in serum creatinine by ≥26.5 μmol/L, Stage 2: increase in serum creatinine to 2.0–2.9 times baseline, Stage 3: increase in serum creatinine to 3.0 times baseline or increase in serum creatinine to ≥353.6 μmol/L or the initiation of renal replacement therapy). Nephrotic-range proteinuria was defined as proteinuria >300 mg/mmol albumin to creatinine ratio. The levels of C-reactive protein (CRP), erythrocyte sedimentation rate (ESR) and IgA were recorded.
In patients with at least 6 months of follow-up, organ involvement and disease status (categorized as complete remission, partial remission, progressive disease or relapsing disease) were evaluated at different time points (6, 12, 24, 36 and 48 months of follow-up) and relapses were recorded during follow-up. Time to remission and time to relapse was calculated for every patient reaching complete remission and experiencing relapsing disease, respectively. For extra-renal disease, complete remission was defined as the absence of extra-renal symptoms with a prednisolone dose <10 mg/day, and partial remission was defined as marked improvement without complete disappearance of extra-renal symptoms with a prednisolone dose <10 mg/day or the absence of extra-renal symptoms with a prednisolone dose ≥10 mg/day. For renal disease, complete remission was defined as an eGFR ≥60 mL/min/1.73 m2 (MDRD), proteinuria ≤30 mg/mmol albumin to creatinine ratio or <1+ on dipstick and haematuria ≤5 red cells per HPF or <2+ on dipstick with a prednisolone dose <10 mg/day. Partial renal remission was defined as >50% eGFR improvement if previous eGFR was <60 mL/min/1.73 m2 (MDRD) and proteinuria ≤70 mg/mmol albumin to creatinine ratio or <2+ on dipstick with or without microscopic haematuria with a prednisolone dose <10 mg/day, or as the presence of complete renal remission with a prednisolone dose of ≥10 mg/day. Progressive extra-renal disease was defined as the lack of improvement of respective extra-renal symptoms and progressive renal disease as the failure to achieve partial renal remission. Relapsing disease was defined as the recurrence of disease activity after a 1-month period of complete or partial remission since the last evaluation time point. Disease status was labelled as progressive, relapsing or partial remission if a minimum of one organ system presented with progressive disease, relapsing disease or partial remission respectively, respecting the following order of rank: progressive disease > relapsing disease > partial remission > complete remission. Chronic kidney disease (CKD) Stage G3a was defined as eGFR 45–59 mL/min/1.73 m2 for ≥3 months, Stage G3b eGFR 30–44 mL/min/1.73 m2 for ≥3 months, Stage G4 eGFR 15–29 mL/min/1.73 m2 for ≥3 months and Stage G5 eGFR < 15 mL/min/1.73 m2 for ≥3 months or requiring renal replacement therapy.
Statistical analysis
Results are expressed as numbers (percentages) for categorical variables and as median (range) for continuous variables. Kaplan–Meier functions were used for survival and relapse curve calculations.
RESULTS
Patient demographics, initial presentation and follow-up
Eighty-five patients were assessed at the time of diagnosis (Table 3). Fifteen of 85 (17.6%) had histological evidence of IgAV on skin biopsy (leukocytoclastic vasculitis with IgA deposition), and 32 (37.6%) had a renal biopsy with characteristic features (proliferative glomerulonephritis with predominant mesangial IgA deposition). Possible predisposing drugs were ibuprofen, adalimumab, infliximab, oral contraception (unspecified) and sertraline in one patient each. In two, malignancy was related to the onset of vasculitis (prostate adenocarcinoma and metastatic sigmoid carcinoma). Median time between disease onset and diagnosis was 1.3 months (range 0–60.8 months). Eighty-four patients (98.8%) fulfilled EULAR/PRINTO/PRES classification criteria and 67 (78.8%) fulfilled ACR classification criteria at diagnosis. Median follow-up of all patients was 27.5 months (range 0–204.8 months), while median follow-up for 67 patients with at least 6 months of follow-up was 42.7 months (range 6.0–204.8 months). Sixty patients were followed for a minimum of 12 months, 46 for 24 months, 39 for 36 months and 29 for 48 months.
Table 3.
Patient demographics, predisposing factors and initial presentation (n = 85)
| Age at disease onset [median (range)], years | 41 (16–75) |
| Sex (male), n (%) | 46 (54.1) |
| Ethnicity, n (%) | |
| White | 84 (98.8) |
| Black | 0 (0.0) |
| Asian | 1 (1.2) |
| Season at disease onset, n (%) | |
| Summer | 13 (15.3) |
| Autumn | 19 (22.4) |
| Winter | 21 (24.7) |
| Spring | 27 (31.8) |
| Not recorded | 5 (5.9) |
| Predisposing factor, n (%) | |
| Upper respiratory tract infection | 39 (45.9) |
| Alcohol | 9 (10.6) |
| Drug | 5 (5.9) |
| Physical stress | 5 (5.9) |
| Immunization | 2 (2.4) |
| Psychological stress | 2 (2.4) |
| Malignancy | 2 (2.4) |
| No predisposing factor identified | 33 (38.8) |
| Clinical presentation, n (%) | |
| Palpable purpura | 85 (100) |
| Ulcerative rash | 10 (11.8) |
| Joint involvement | 59 (69.4) |
| Gastrointestinal involvement | 40 (47.1) |
| Renal involvement | 66 (77.6) |
| eGFR <60 mL/min/1.73 m2 | 11 (12.9) |
| AKI Stage 1 | 7 (8.2) |
| AKI Stage 2 | 1 (1.2) |
| AKI Stage 3 | 3 (3.5) |
| Proteinuria | 51 (60.0) |
| Nephrotic-range proteinuria | 3 (3.5) |
| Haematuria | 57 (67.1) |
| Macroscopic haematuria | 8 (9.4) |
| Hypertension | 35 (41.2) |
| Not recorded | 9 (10.6) |
| Ear, nose and throat involvement | 7 (8.2) |
| Pulmonary involvement | 6 (7.1) |
| Oral mucosal ulceration | 2 (2.4) |
| Testicular involvement | 1 (1.2) |
| Ocular involvement | 1 (1.2) |
| Constitutional symptoms | 42 (49.4) |
| Laboratory examinations, n (%) | |
| IgA >4 g/L | 27 (31.8) |
| Not recorded | 7 (9.4) |
| CRP >5 mg/L | 43 (50.6) |
| Not recorded | 6 (7.1) |
| ESR >20 mm/h | 27 (31.8) |
| Not recorded | 9 (10.6) |
General disease status and outcome
Of 67 patients with at least 6 months of follow-up, 33 (49.3%) reached complete remission, a median of 7.6 months after diagnosis (range 0.3–145.0 months) (Figure 1A), while 27 (40.3%) reached partial remission, a median of 7.1 months after diagnosis (range 0.2–81.7 months). At 6 months of follow-up, 58 (86.6%) were not in complete remission, while at 12 months of follow-up this was 49 (81.7%), at 24 months 35 (76.1%), at 36 months 25 (64.1%) and finally 19 (65.5%) at 48 months of follow-up (Figure 2). For those not achieving complete remission, extra-renal disease was present in 37 (63.8%) at 6 months, 32 (65.3%) at 12 months, 18 (51.4%) at 24 months, 13 (52.0%) at 36 months and 10 (52.6%) at 48 months of follow-up (Table 4). Twenty-nine of 67 patients (43.3%) had at least one relapse during follow-up (Figure 1B) and in 18/67 (26.9%) there was more than one relapse. Characteristics of relapse were skin involvement in 18 (62.1%) of 29 relapsing patients, nephritis in 12 (41.4%), joint involvement in seven (24.1%), gastrointestinal involvement in five (17.2%), constitutional symptoms in three (10.3%) and episcleritis in one (3.4%). One patient died during follow-up, due to acute myocardial infarction and sepsis, at the age of 51 years.
FIGURE 1:

Kaplan–Meier curves for (A) the proportion of patients achieving complete remission and (B) the proportion of patients experiencing a relapse, in patients with at least 6 months of follow-up.
FIGURE 2:
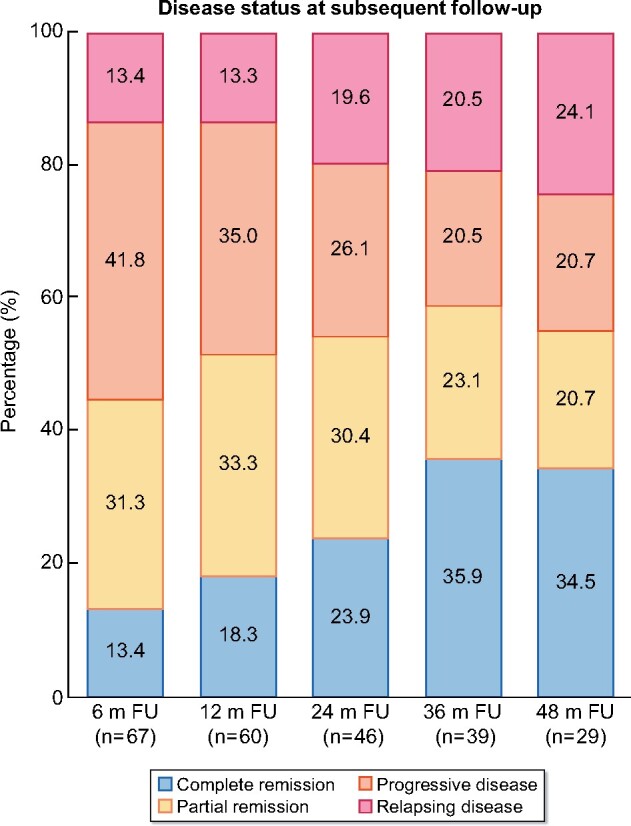
Disease status at subsequent follow-up. m FU, months of follow-up.
Table 4.
Organ involvement at subsequent follow-up in patients not achieving complete remission
| Organ involvement | 6 m FU (n = 58) | 12 m FU (n = 49) | 24 m FU (n = 35) | 36 m FU (n = 25) | 48 m FU (n = 19) |
|---|---|---|---|---|---|
| Renal | 51 (87.9) | 41 (83.7) | 28 (80.0) | 20 (80.0) | 17 (89.5) |
| Extra-renal | 37 (63.8) | 32 (65.3) | 18 (51.4) | 13 (52.0) | 10 (52.6) |
| Skin | 31 (53.5) | 24 (49.0) | 14 (40.0) | 8 (32.0) | 7 (36.8) |
| Joint | 23 (39.7) | 20 (40.8) | 15 (42.9) | 12 (48.0) | 10 (52.6) |
| Gastrointestinal | 6 (10.3) | 7 (14.3) | 7 (20.0) | 5 (20.0) | 7 (36.8) |
| Ocular | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (4.0) | 1 (5.3) |
| Constitutional | 8 (13.8) | 8 (16.3) | 5 (14.3) | 4 (16.0) | 4 (21.1) |
Values expressed as n (%). m FU, months of follow-up.
Renal involvement and outcome
Fifty-four of 67 (80.6%) patients with at least 6 months of follow-up had renal involvement at diagnosis, while five (7.5%) developed nephritis during follow-up, a median of 1.5 months after diagnosis (range 0.9–5.1 months). New renal involvement was characterized by proteinuria and haematuria in three, by isolated haematuria in one and by AKI and proteinuria in another. Eight (11.9%) had no renal involvement during follow-up.
Of patients with at least 6 months of follow-up, 20 (29.9%) developed AKI, of whom 10 (14.9%) had AKI at presentation (Stage 1 in six, Stage 2 in one and Stage 3 in three patients). Only one required renal replacement therapy. Three (4.5%) showed complete resolution of their kidney function at 6 months of follow-up, while seven (10.4%) developed CKD. Ten (14.9%) developed AKI during follow-up (Stage 1 in eight and Stage 2 in two patients), at a median of 9.3 months after diagnosis (range 0.9–62.1 months). Seven (10.4%) had complete recovery of their kidney function by the subsequent follow-up time point, while three (4.5%) developed CKD.
At a median final follow-up of 42.7 months (range 6.0–204.8 months) five patients (7.5%) had CKD Stage G3a, four (6.0%) Stage G3b and one (1.5%) Stage G4, 18 (26.9%) had haematuria and 13 (19.4%) had proteinuria.
The characterization of renal involvement at subsequent follow-up is summarized in Table 5.
Table 5.
Renal involvement at subsequent follow-up
| Renal involvement | 6 m FU (n = 67) | 12 m FU (n = 60) | 24 m FU (n = 46) | 36 m FU (n = 39) | 48 m FU (n = 29) |
|---|---|---|---|---|---|
| Nephritis | 50 (74.6) | 41 (68.3) | 27 (58.7) | 20 (51.3) | 16 (55.2) |
| CKD Stage ≥G3a | 7 (10.4) | 6 (10.0) | 3 (6.5) | 3 (7.7) | 5 (17.2) |
| CKD Stage G3a | 4 (6.0) | 4 (6.7) | 2 (4.4) | 2 (5.1) | 4 (13.8) |
| CKD Stage G3b | 2 (3.0) | 1 (1.7) | 1 (2.2) | 1 (2.6) | 1 (3.5) |
| CKD Stage G4 | 1 (1.5) | 1 (1.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| CKD Stage G5 | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Proteinuria | 35 (52.2) | 24 (40.0) | 19 (41.3) | 14 (35.9) | 8 (27.6) |
| Haematuria | 48 (71.6) | 39 (65.0) | 23 (50.0) | 17 (43.6) | 12 (41.4) |
Values expressed as n (%). m FU, months of follow-up.
Treatment
Of 67 patients with at least 6 months of follow-up, 51 (76.1%) received oral glucocorticoids, 18 (26.9%) intravenous glucocorticoids and 29 (43.3%) additional immunosuppressive or immunomodulatory therapy (Table 6). Mycophenolate mofetil (MMF) or mycophenolate sodium was administered in 19 (28.4%), of whom 17 received the therapy for at least 1 month (Figure 3). In patients in whom MMF was started during the first 6 months of follow-up, only six of 13 demonstrated a response at 6 months and only five of 11 at 12 months. None of the patients in whom MMF was started after 6 months of follow-up showed a response at 12 months.
Table 6.
Immunosuppressive and immunomodulatory therapy (n = 67)
| Oral glucocorticoids | 51 (76.1) |
| Intravenous glucocorticoids | 18 (26.9) |
| MMF/mycophenolate sodium | 19 (28.4) |
| Cyclophosphamide | 9 (13.4) |
| Azathioprine | 9 (13.4) |
| Plasma exchange | 6 (9.0) |
| Hydroxychloroquine | 6 (9.0) |
| Methotrexate | 5 (7.5) |
| Dapsone | 4 (6.0) |
| Tonsillectomy | 4 (6.0) |
| Intravenous Ig | 4 (6.0) |
| Rituximab | 4 (6.0) |
| Alemtuzumab | 3 (4.5) |
| Colchicine | 3 (4.5) |
| Leflunomide | 2 (3.0) |
| Bortezomib | 1 (1.5) |
| Infliximab | 1 (1.5) |
| Thalidomide | 1 (1.5) |
Values expressed as n (%).
FIGURE 3:

Disease status at 6 and 12 months of follow-up in patients treated with MMF.
Two patients experienced relapses during treatment with hydroxychloroquine, one relapsed during treatment with azathioprine and one during treatment with colchicine. Two relapsed after stopping or reducing steroid dose and one relapsed after stopping MMF.
DISCUSSION
We aimed to describe the disease course of an unselected cohort of adult IgAV patients in order to highlight the frequency of remission failure, relapse and difficulties of treatment. Renal involvement was present in 88%. Complete remission was only achieved in 35% by 4 years, lower than in previous reports [6, 9, 12] but consistent with the remission rates seen in IgAN. Although renal involvement was the most common reason for remission failure, extra-renal features were also present in >50% of patients at every follow-up time point. This is a higher rate than described in a previous report [12] where the cohort was defined by the presence of nephritis. Relapse rate was high at 43%, 62% of whom suffered multiple relapses, similar to previous reports [6, 11, 12, 14].
Current classification criteria, ACR and EULAR/PRINTO/PRES, performed reasonably with 79% and 99% of patients meeting the criteria, respectively, contributing to the validation of the EULAR/PRINTO/PRES paediatric criteria in adults. An ongoing global study, Diagnostic and Classification Criteria for Vasculitis [15], aims to update, improve and validate primary vasculitis classification criteria. The pattern of clinical features of our cohort was compatible with previous publications [6, 7, 16, 17]. Delayed development of nephritis was observed in a minority, as previously described by Pillebout et al. [11] in adults and by Narchi [18] in children, but not after 6 months of follow-up. This highlights the need for ongoing clinical review in those without nephritis at diagnosis.
Patient survival was good, with only one death during follow-up. Long-term renal involvement was common with long-term haematuria in 40% and proteinuria in 30%. Proteinuria has previously been shown to be a predictor of worse renal outcome [10, 11]. Fewer patients (17%) had CKD at long-term follow-up, with no patients having Stage G5, results that are better than described in earlier reports (Table 2) and that might indicate that although complete remission rates are low, the use of glucocorticoids and additional immunosuppressive or immunomodulatory therapy may have impacted on renal progression.
Due to the highly variable clinical outcome and lack of correlation between initial presentation and long-term outcome with a substantial proportion of patients exhibiting a self-limiting and benign disease course, prospective, randomized or even controlled studies regarding the treatment of IgAV are scarce. The treatment of both childhood and adult IgAV remains controversial, and consensus-based recommendations have only been formulated in the childhood population [19]. Treatment of adult IgAV is limited to symptom control with the addition of oral corticosteroids in persistent and moderate organ involvement and other immunosuppressive agents in severe gastro-intestinal or renal involvement. We used oral immunosuppressives such as MMF to permit steroid sparing and consolidate treatment response, but this did not appear to effectively allow complete remission and prevent subsequent relapse in our study. Earlier, Ren et al. [20] described the effectiveness of MMF as a steroid-sparing agent in a retrospective analysis of 53 adults with biopsy-proven IgAV with nephritis. Overall, the efficacy of treatment of adult IgAV with corticosteroids and other immunosuppression still remains to be demonstrated [21]. The Randomized Therapeutic Study of Steroid vs. Steroid Plus Cyclosphosphamide for Severe Visceral Henoch-Schönlein Purpura (CESAR) trial [22] in severe IgAV and IgAN found no benefit of cyclophosphamide, but was underpowered. An observational study has reported responses to rituximab [23] and experience with other agents in adult IgAV is too small to draw firm conclusions. The decision to start immunosuppression in IgAN, a disease that is pathophysiologically indistinguishable from IgAV, is currently controversial, as the Supportive Versus Immunosuppressive Therapy for the Treatment Of Progressive IgAN (STOP-IgAN) trial [24] found no effect of immunosuppression on the outcome in IgAN and as the Therapeutic Evaluation of Steroids in IgA Nephropathy Global (TESTING) trial [25], comparing corticosteroids with placebo, was terminated early due to unacceptably high serious adverse event rates in the group randomized to corticosteroids. Novel targeted therapies, including a targeted-release formulation of budesonide [26], the spleen tyrosine kinase inhibitor fostamatinib (NCT02112838) and complement-directed therapies, for example, the complement factor B inhibitor LNP023 (NCT03373461), are being investigated in clinical trials for IgAN and may extend to IgAV in the foreseeable future.
The limitations of our study are the retrospective nature, potential selection bias for more refractory cases with greater potential for patients in stable remission being lost to follow-up or discharged early. Another limitation is the variable length of follow-up. Nonetheless, our findings highlight the refractory and relapsing disease course of adult IgAV, with a minority of patients reaching complete remission and up to 45% of patients experiencing relapses. Adult IgAV patients suffer a high rate of nephritis, which can develop after diagnosis within the first 6 months. Although renal involvement is the main determinant of progressive or relapsing disease, the persistence of extra-renal manifestations is encountered in 50% of patients, sometimes causing severe morbidity and mortality. Our data also highlight the need for better treatments for IgAV.
CONFLICT OF INTEREST STATEMENT
None declared.
REFERENCES
- 1. Gardner-Medwin JM, Dolezalova P, Cummins C. et al. Incidence of Henoch-Schonlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet 2002; 360: 1197–1202 [DOI] [PubMed] [Google Scholar]
- 2. Piram M, Mahr A.. Epidemiology of immunoglobulin A vasculitis (Henoch-Schonlein): current state of knowledge. Curr Opin Rheumatol 2013; 25: 171–178 [DOI] [PubMed] [Google Scholar]
- 3. Hočevar A, Rotar Z, Ostrovrsnik J. et al. Incidence of IgA vasculitis in the adult Slovenian population. Br J Dermatol 2014; 171: 524–527 [DOI] [PubMed] [Google Scholar]
- 4. Mills JA, Michel BA, Bloch DA. et al. The American College of Rheumatology 1990 criteria for the classification of Henoch-Schönlein purpura. Arthritis Rheum 2010; 33: 1114–1121 [DOI] [PubMed] [Google Scholar]
- 5. Ozen S, Pistorio A, Iusan SM. et al. ; for the Paediatric Rheumatology International Trials Organisation (PRINTO). EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: final classification criteria. Ann Rheum Dis 2010; 69: 798–806 [DOI] [PubMed] [Google Scholar]
- 6. Garcia-Porrúa C, Calvino MC, Llorca J. et al. Henoch-Schonlein purpura in children and adults: clinical differences in a defined population. Semin Arthritis Rheum 2002; 32: 149–156 [DOI] [PubMed] [Google Scholar]
- 7. Calvo-Rio V, Loricera J, Mata C. et al. Henoch-Schonlein purpura in northern Spain: clinical spectrum of the disease in 417 patients from a single center. Medicine (Baltimore )2014; 93: 106–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Selewski DT, Ambruzs JM, Appel GB. et al. Clinical characteristics and treatment patterns of children and adults with IgA nephropathy or IgA vasculitis: findings from the CureGN study. Kidney Int Rep 2018; 3: 1373–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blanco R, Martínez-Taboada V, Rodríguez-Valverde V. et al. Henoch-Schönlein purpura in adulthood and childhood: two different expressions of the same syndrome. Arthritis Rheum 1997; 40: 859–864 [DOI] [PubMed] [Google Scholar]
- 10. Coppo R, Andrulli S, Amore A. et al. Predictors of outcome in Henoch-Schonlein nephritis in children and adults. Am J Kidney Dis 2006; 47: 993–1003 [DOI] [PubMed] [Google Scholar]
- 11. Pillebout E, Thervet E, Hill G. et al. Henoch-Schonlein purpura in adults: outcome and prognostic factors. J Am Soc Nephrol 2002; 13: 1271–1278 [DOI] [PubMed] [Google Scholar]
- 12. Shrestha S, Sumingan N, Tan J. et al. Henoch Schonlein purpura with nephritis in adults: adverse prognostic indicators in a UK population. QJM 2006; 99: 253–265 [DOI] [PubMed] [Google Scholar]
- 13. Rauta V, Törnroth T, Grönhagen-Riska C.. Henoch-Schoenlein nephritis in adults - clinical features and outcome in Finnish patients. Clin Nephrol 2002; 58: 1–8 [DOI] [PubMed] [Google Scholar]
- 14. Calvo-Rio V, Hernandez JL, Ortiz-Sanjuan F. et al. Relapses in patients with Henoch-Schonlein purpura: analysis of 417 patients from a single center. Medicine (Baltimore) 2016; 95: e4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Craven A, Robson J, Ponte C. et al. ACR/EULAR-endorsed study to develop diagnostic and classification criteria for vasculitis (DCVAS). Clin Exp Nephrol 2013; 17: 619–621 [DOI] [PubMed] [Google Scholar]
- 16. Hocevar A, Rotar Z, Jurcic V. et al. Patient age, gender and extent of purpura may suggest short-term outcomes in adults with IgA vasculitis. Rheumatology (Oxford )2015; 54: 1330–1332 [DOI] [PubMed] [Google Scholar]
- 17. Audemard-Verger A, Terrier B, Dechartres A. et al. ; on behalf of the French Vasculitis Study Group. Characteristics and management of IgA vasculitis (Henoch-Schonlein) in adults: data from 260 patients included in a French multicenter retrospective survey. Arthritis Rheumatol 2017; 69: 1862–1870 [DOI] [PubMed] [Google Scholar]
- 18. Narchi H. Risk of long term renal impairment and duration of follow up recommended for Henoch-Schönlein purpura with normal or minimal urinary findings: a systematic review. Arch Dis Child 2005; 90: 916–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ozen S, Marks SD, Brogan P. et al. European consensus-based recommendations for diagnosis and treatment of immunoglobulin A vasculitis-the SHARE initiative. Rheumatology (Oxford )2019; 58: 1607–1616 [DOI] [PubMed] [Google Scholar]
- 20. Ren P, Han F, Chen L. et al. The combination of mycophenolate mofetil with corticosteroids induces remission of Henoch-Schönlein purpura nephritis. Am J Nephrol 2012; 36: 271–277 [DOI] [PubMed] [Google Scholar]
- 21. Audemard-Verger A, Pillebout E, Guillevin L. et al. IgA vasculitis (Henoch-Schönlein purpura) in adults: diagnostic and therapeutic aspects. Autoimmun Rev 2015; 14: 579–585 [DOI] [PubMed] [Google Scholar]
- 22. Pillebout E, Alberti C, Guillevin L. et al. Addition of cyclophosphamide to steroids provides no benefit compared with steroids alone in treating adult patients with severe Henoch Schonlein purpura. Kidney Int 2010; 78: 495–502 [DOI] [PubMed] [Google Scholar]
- 23. Maritati F, Fenoglio R, Pillebout E. et al. Brief report: rituximab for the treatment of adult-onset IgA vasculitis (Henoch-Schonlein). Arthritis Rheumatol 2018; 70: 109–114 [DOI] [PubMed] [Google Scholar]
- 24. Rauen T, Eitner F, Fitzner C. et al. Intensive supportive care plus immunosuppression in IgA nephropathy. N Engl J Med 2015; 373: 2225–2236 [DOI] [PubMed] [Google Scholar]
- 25. Lv J, Zhang H, Wong MG. et al. ; for the TESTING Study Group. Effect of oral methylprednisolone on clinical outcomes in patients with IgA nephropathy: the TESTING randomized clinical trial. JAMA 2017; 318: 432–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fellstrom BC, Barratt J, Cook H. et al. Targeted-release budesonide versus placebo in patients with IgA nephropathy (NEFIGAN): a double-blind, randomised, placebo-controlled phase 2b trial. Lancet 2017; 389: 2117–2127 [DOI] [PubMed] [Google Scholar]