

Abstract
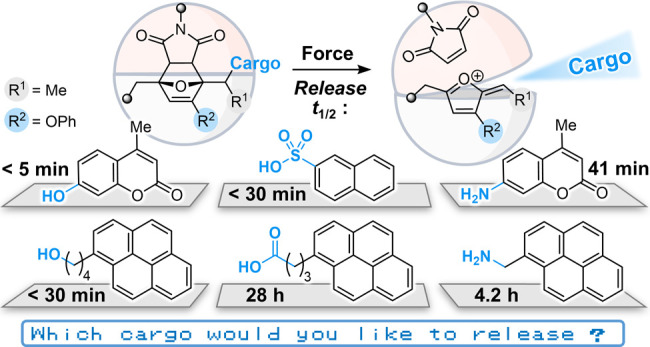
Polymers that release functional small molecules in response to mechanical force are appealing targets for drug delivery, sensing, catalysis, and many other applications. Mechanically sensitive molecules called mechanophores are uniquely suited to enable molecular release with excellent selectivity and control, but mechanophore designs capable of releasing cargo with diverse chemical functionality are limited. Here, we describe a general and highly modular mechanophore platform based on masked 2-furylcarbinol derivatives that spontaneously decompose under mild conditions upon liberation via a mechanically triggered reaction, resulting in the release of a covalently installed molecular payload. We identify key structure–property relationships for the reactivity of 2-furylcarbinol derivatives that enable the mechanically triggered release of functionally diverse molecular cargo with release kinetics being tunable over several orders of magnitude. In particular, the incorporation of an electron-donating phenoxy group on the furan ring in combination with an α-methyl substituent dramatically lowers the activation barrier for fragmentation, providing a highly active substrate for molecular release. Moreover, we find that phenoxy substitution enhances the thermal stability of the mechanophore without adversely affecting its mechanochemical reactivity. The generality and efficacy of this molecular design platform are demonstrated using ultrasound-induced mechanical force to trigger the efficient release of a broad scope of cargo molecules, including those bearing alcohol, phenol, alkylamine, arylamine, carboxylic acid, and sulfonic acid functional groups.
Short abstract
Polymers that release small molecules under mechanical force are desirable targets for a wide range of applications. We report a highly effective platform for mechanically triggered molecular release.
Introduction
Polymers that release functional molecules in response to a specific stimulus are desirable for a variety of applications including sensing, catalysis, self-healing, and targeted drug delivery.1−3 Mechanically triggered release is a particularly appealing target. To this end, several different approaches have been demonstrated, including physically entrapped payloads within a polymeric matrix,4 dissociation of supramolecular assemblies,5,6 and the use of fluid-filled microcapsules7 or vascular networks8 embedded within a material that release their payload after being ruptured. Recently, the use of mechanical force as an external stimulus to drive covalent chemical transformations has emerged as an attractive strategy.9 Force is typically transduced via polymer chains to mechanically sensitive molecules known as mechanophores that respond in a chemoselective fashion, resulting in a productive chemical reaction.10,11 In the context of targeted drug delivery, for example, ultrasound is capable of penetrating deep within biological tissues to stimulate mechanochemical transformations noninvasively with spatial and temporal precision.12 In light of these advantages, the field of polymer mechanochemistry has attracted significant interest for the design of autonomous materials that respond innately to mechanically dynamic environments,3 as well as abundant opportunities to advance the fundamental understanding of mechanochemical reactivity, which is underdeveloped in comparison to other areas of organic chemistry.13
Several mechanophores have been designed to achieve the mechanically triggered release of functional organic molecules, although the scope of molecules that can be released is still relatively limited. Moore and Craig have designed mechanophores based on gem-dichlorocyclopropane motifs that undergo mechanochemical rearrangement reactions with subsequent release of HCl.14,15 Boydston has developed mechanophores based on a flex-activation manifold demonstrating release of a benzyl furfuryl ether molecule via a mechanically induced cycloelimination reaction16,17 and the release of N-heterocyclic carbenes.18 Notably, each approach uses a judiciously designed mechanophore to release a specific compound upon mechanical activation, which consequently limits the scope of molecules that can be released. Small-molecule release has also been achieved through the mechanically triggered heterolytic scission and subsequent depolymerization of poly(o-phthalaldehyde) to regenerate its constituent monomers.19,20 Finally, Herrmann and Göstl have introduced an elegant mechanophore design that relies on the mechanically activated reduction21,22 of a chain-centered disulfide unit and ensuing 5-exo-trig cyclization to release an alcohol attached via a β-carbonate linker.5,23 While the release of several different alcohols has been successfully demonstrated using this disulfide mechanophore platform, it is susceptible to nonspecific activation via chemical reduction or thiol exchange and the cargo scope appears to be somewhat limited, as indicated by the low release efficiency observed for amine payloads.24
In 2019, our group reported a strategy for molecular release via a mechanically triggered cascade reaction in which a mechanochemical retro-Diels–Alder reaction unveils an unstable furfuryl carbonate motif that subsequently decomposes under mild conditions to release its molecular payload. The mechanically triggered release of the fluorescent probe 7-hydroxy-4-methylcoumarin was demonstrated to occur efficiently at room temperature with a half-life (t1/2) on the order of 1 h following mechanical activation (Scheme 1a).25 The α-methyl group on the furfuryl carbonate was a key structural feature that enabled relatively fast release of hydroxycoumarin, presumably due to stabilization of the developing positive charge in the transition state leading to the secondary furfuryl cation intermediate.26,27 This general strategy relies on the concept of mechanically gated reactivity,28 in which the mechanochemical reaction is decoupled from the ultimate functional response, and follows our earlier conceptualization of mechanically gated photoswitching.29 This paradigm offers a powerful approach for the design of highly modular systems, as the mechanochemical behavior of the mechanophore and the functional properties of the masked intermediate can be controlled independently. While the molecular design strategy is promising, our first-generation mechanophore is nevertheless limited to the release of phenols and a more general platform capable of releasing functionally diverse molecular cargo on reasonable time scales is desired.
Scheme 1. Mechanically Triggered Molecular Release via a Retro-Diels–Alder/Fragmentation Cascade.

Here we investigate the effect of substitution on the reactivity of 2-furylcarbinol derivatives, identifying structure–activity relationships (SAR) that enable the mechanically triggered release of functionally diverse molecular payloads from a second-generation mechanophore platform (Scheme 1b). Using density functional theory (DFT) calculations to guide molecular design, we demonstrate that the rate of molecular release is significantly modulated by varying the substitution of the 2-furylcarbinol scaffold. In particular, substitution at the 3-position of the furan ring with an electron-donating phenoxy group, combined with an α-methyl substituent, renders a highly reactive substrate for molecular release. Masked 2-furylcarbinol derivatives are incorporated into polymers and activated using ultrasound to achieve the mechanically triggered release of alkyl/aryl alcohols and amines and further extended to cargo molecules containing carboxylic acid and sulfonic acid functional groups conjugated to the mechanophore via carboxylate and sulfonate linkages.
Results and Discussion
Furfuryl carbonates decompose in polar protic media by the mechanism depicted in Scheme 1a via a putative furfuryl cation intermediate.30 Primary furfuryl carbonates possessing only an alkyl group at the 5-position of the furan ring are relatively unreactive and decompose slowly at room temperature; however, installation of an additional α-methyl group significantly reduces the activation barrier for carbonate fragmentation.25 While this substitution pattern is sufficient to enable the release of a phenolic cargo molecule with a half-life of approximately 1 h, the rate of fragmentation is still prohibitively slow for alcohol- and amine-derived furfuryl carbonates and carbamates, respectively. For example, preliminary kinetic studies performed on a small-molecule model compound reveal that the release of a primary alcohol from our earlier furfuryl carbonate substrate occurs with a half-life of approximately 4 days, or nearly 100× slower than the release of hydroxycoumarin (Figure S1). We reasoned that the addition of an electron-donating substituent31 at the 3-position of the furan would further suppress the activation barrier for fragmentation since this substituent is in resonance with the furfuryl carbocation, potentially enabling the efficient release of even more challenging payloads such as amines32 under mild conditions.
To test this hypothesis, we first computed the activation energies for fragmentation of a series of model primary and secondary furfuryl carbonate (FC1(O)–FC4(O)) and furfuryl carbamate (FC1(NH)–FC4(NH)) substrates with varying substitution at the 3-position of the furan ring (Figure 1). Activation energies were calculated using DFT at the M06-2X/6-311+G** level of theory using a polarizable continuum model to simulate a polar solvent environment (see the Supporting Information for details). For the furfuryl carbonate series, α-methyl substitution reduces the activation energy for fragmentation by 3.2–3.8 kcal/mol relative to the primary furfuryl carbonate substrates. Similarly, the addition of an electron-donating phenoxy substituent at the 3-position of the furan ring lowers the computed activation energies by 3.7–4.3 kcal/mol. Significantly, the combination of α-methyl and 3-phenoxy substitution on FC1(O) results in a computed activation energy of 18.3 kcal/mol, suggesting a nearly instantaneous reaction at room temperature. These values indicate that the half-life for reaction of FC1(O) is nearly 5 orders of magnitude shorter than that of the unsubstituted primary furfuryl carbonate represented by FC4(O) and approximately 500× shorter than that of our originally reported secondary furfuryl carbonate substrate depicted by FC3(O). A similar trend in reactivity is observed for the furfuryl carbamate model series; however, calculated activation energies are 5.5–7.6 kcal/mol higher for the furfuryl carbamates in comparison to the analogous furfuryl carbonate substrates in all cases. The higher activation energies calculated for the furfuryl carbamate series are consistent with experimentally determined reaction kinetics for different self-immolative spacers,33 which suggest that the more electron withdrawing carbonate leaving group is able to better stabilize the partial negative charge on the oxygen atom of the fragmenting C–O bond in the transition state relative to a carbamate leaving group. The activation energies calculated for furfuryl carbamates FC2(NH)–FC4(NH) suggest that decomposition of similarly substituted substrates occurs on time scales that are impractical for triggered release. For furfuryl carbamate model FC1(NH), however, the combination of α-methyl and 3-phenoxy substitution significantly reduces the activation barrier to 23.8 kcal/mol, which approaches the activation energy calculated for our first-generation furfuryl carbonate and suggests that the release of challenging amine-based molecular payloads may be accessible on reasonable time scales.
Figure 1.
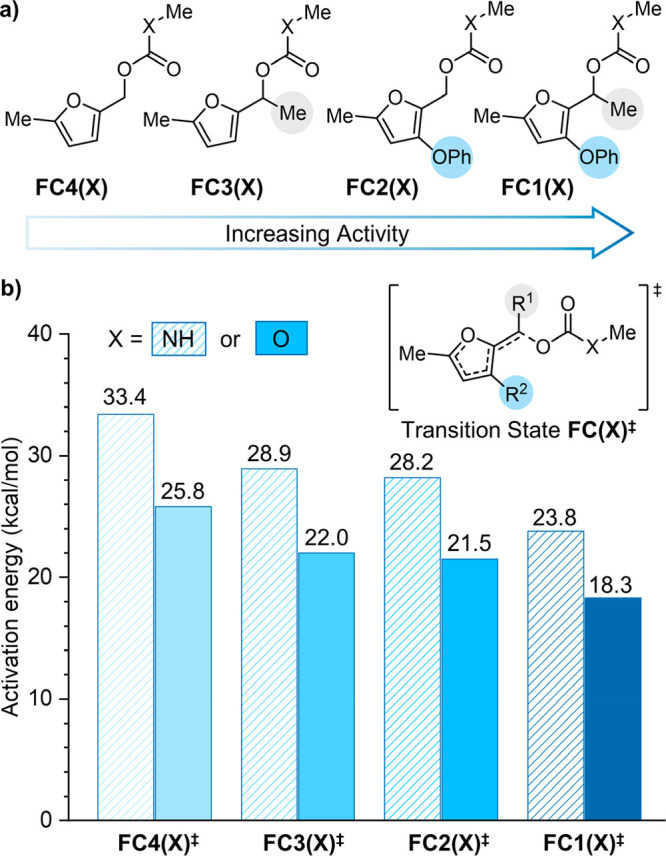
Substituent effects on the reactivity of 2-furylcarbinol derivatives. (a) Structures of model furfuryl carbonates (X = O) and furfuryl carbamates (X = NH) with varying substitution patterns, and (b) corresponding activation energies for fragmentation of the α-C–O bond calculated at the M06-2X/6-311+G** level of density functional theory.
To validate the computational predictions, we synthesized fluorogenic furfuryl carbamate model compound 1 containing α-methyl and 3-phenoxy substituents and investigated its reactivity experimentally (Figure 2a). The coumarin payload exhibits a fluorescence turn-on after release, allowing the reaction to be conveniently monitored using photoluminescence (PL), in addition to NMR spectroscopy. The addition of methanol to a room-temperature solution of 1 in acetonitrile-d3 (19 μM, 3:1 MeCN/MeOH) triggers decomposition and results in clean conversion to aminocoumarin 3 and furfuryl methyl ether 4, as evidenced by NMR spectroscopy (Figure 2b). The formation of furfuryl methyl ether 4 is consistent with the transient formation of a furfuryl cation intermediate that is intercepted by methanol. Interestingly, when the reaction is performed at significantly higher concentrations, another set of peaks was observed in the 1H NMR spectra corresponding to the formation of a side product that was identified to be the furfuryl amine derived from nucleophilic attack of the furfuryl cation intermediate by liberated aminocoumarin 3 (Figures S2 and S3). A similar reaction was not observed for the furfuryl carbonate studied previously, highlighting the increased nucleophilicity of the amine cargo. Importantly, however, this furfuryl amine side product is formed in <2% yield in reactions with a substrate concentration of 19 μM, which is similar to the concentration of mechanophores in typical ultrasonication experiments (vide infra). These results confirm that at these relatively low substrate concentrations, the reaction depicted in Figure 2a is sufficiently descriptive. The kinetics of furfuryl carbamate decomposition were further studied by monitoring the conversion of starting material and the generation of aminocoumarin 3 as a function of time using NMR and PL spectroscopy, respectively (Figure 2c). Furfuryl carbamate 1 is fully converted to products in approximately 5 h, with a concomitant increase in fluorescence corresponding to the generation of aminocoumarin 3 (Figure S4). The data from both time course experiments were fitted to first-order rate expressions, providing half-lives of t1/2 = 34 and 45 min from NMR and PL measurements, respectively. In direct contrast, secondary furfuryl carbamate model compound 2, which does not contain a 3-phenoxy substituent but is otherwise identical to our previously reported furfuryl carbonate that is active toward phenol release,25 is completely unreactive under the same conditions (Figure 2c and Figure S5). The striking difference in decomposition behavior between model compounds 1 and 2 highlights the effect of an electron-donating phenoxy substituent on the furan ring and supports the molecular design for a second-generation mechanophore platform enabling the molecular release of previously inaccessible payloads.
Figure 2.
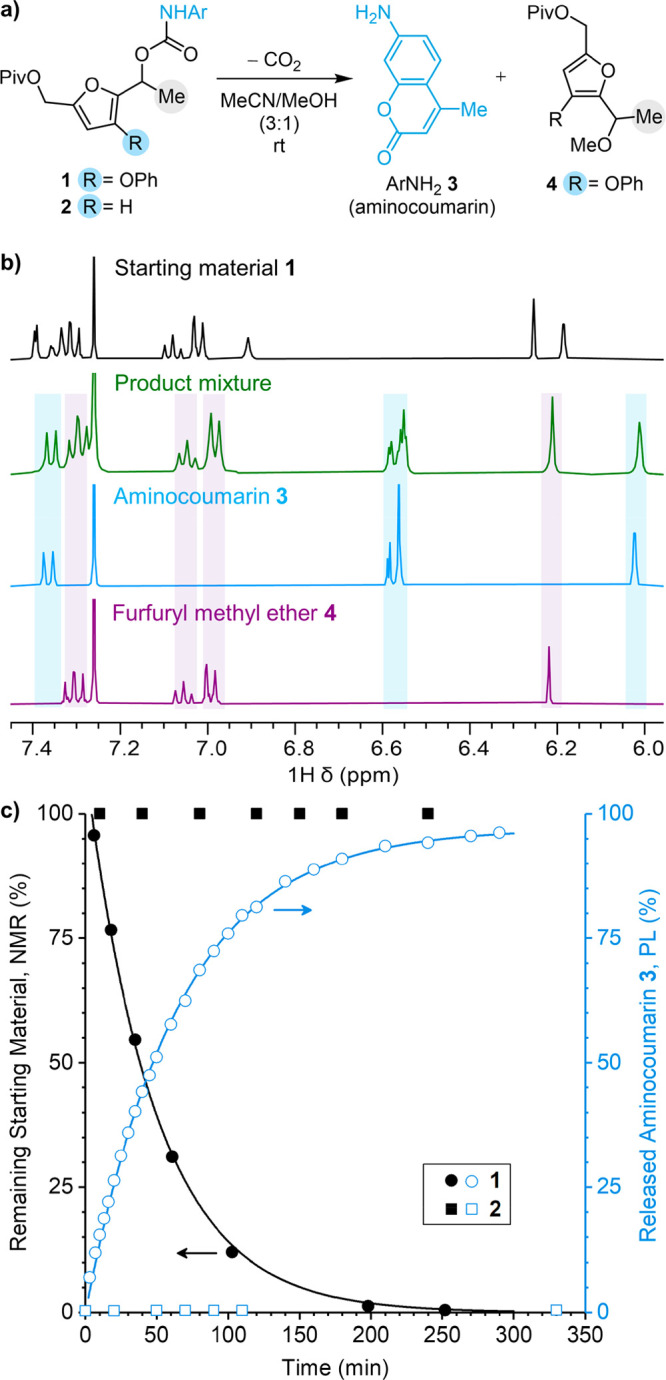
Characterization of the decomposition reactions of model furfuryl carbamates 1 and 2. (a) Decomposition of 1 in MeCN/MeOH (3:1) at room temperature generates fluorescent aminocoumarin 3 and furfuryl methyl ether 4 via a putative furfuryl cation intermediate; (b) partial 1H NMR spectra (400 MHz, CDCl3) demonstrating the clean conversion of 1 to products ([1]0 = 19 μM); (c) time course experiments following the conversion of furfuryl carbamates 1 and 2 by NMR spectroscopy (in 3:1 MeCN-d3/MeOH; [1]0 = 14 mM; [2]0 = 14 mM) and the generation of aminocoumarin 3 by photoluminescence spectroscopy (3:1 MeCN/MeOH; λex = 365 nm; λem = 424 nm; [1]0, [2]0 = 7.6 μM).
We next synthesized a series of furan–maleimide Diels–Alder adducts serving as masked furfuryl carbonates/carbamates with varying substitution and incorporated them into polymers to study their mechanochemical behavior. Polymers containing a chain-centered mechanophore are mechanically activated in solution using ultrasonication, which produces elongational forces that are maximized near the chain midpoint.34 The synthesis of polymers containing a masked phenoxy-substituted secondary furfuryl carbonate/carbamate is illustrated in Scheme 2, while details for the synthesis of polymers containing a masked phenoxy-substituted primary furfuryl carbonate/carbamate (1°, 3-OPh) as well as a secondary furfuryl carbonate/carbamate without a phenoxy substituent (2°, 3H) matching our first-generation molecular design are provided in the Supporting Information. Starting from 3-bromofurfural, a phenoxy group was installed via a nucleophilic substitution reaction with phenol, followed by Grignard addition and protection to yield furfuryl silyl ether 6. Next, a formylation reaction and subsequent desilylation with TBAF yielded 2,3,5-trisubstituted furfuryl alcohol 7. Reduction of the aldehyde with sodium borohydride and a [4 + 2] cycloaddition reaction with a prefunctionalized maleimide dienophile in a two-step sequence furnished an isomeric mixture of Diels–Alder adducts, from which endo diastereomer (±)-7 was isolated by silica gel chromatography. Esterification of the primary alcohol proceeded with reasonable selectivity using α-bromoisobutyryl bromide to give the modular bis-initiator (±)-9 containing a secondary alcohol for cargo attachment. The precursor bis-initiator (±)-9 was then conveniently elaborated to carbonate (±)-10(O) and carbamate (±)-10(NH) containing fluorogenic coumarin payloads via a reaction with the corresponding chloroformate or isocyanate, respectively. After cargo installation, the bis-initiators were employed in the controlled radical polymerization of methyl acrylate with Cu wire/Me6TREN in DMSO35 to afford poly(methyl acrylate) (PMA) polymers PMA-1(O) and PMA-1(NH) containing a chain-centered mechanophore. An analogous synthetic approach enabled the preparation of chain-centered polymers PMA-2(X) (1°, 3-OPh) and PMA-3(X) (2°, 3-H) with differing mechanophore substitution. The structure of each polymer is illustrated in Scheme 3 along with the number-average molecular weight (Mn), which was determined to be in the range 94.7–102 kDa with Đ ≤ 1.06 by gel permeation chromatography (GPC) monitored with refractive index and multiangle light scattering detectors. In addition, chain-end functional control polymers were synthesized similarly by starting from the masked furfuryl carbonates/carbamates containing a single α-bromo ester initiating group (see the Supporting Information for details).
Scheme 2. Synthesis of Poly(methyl acrylate) (PMA) Polymers Containing a Chain-Centered Mechanophore with α-Methyl/Phenoxy Substitution and a Fluorogenic Coumarin Payload.
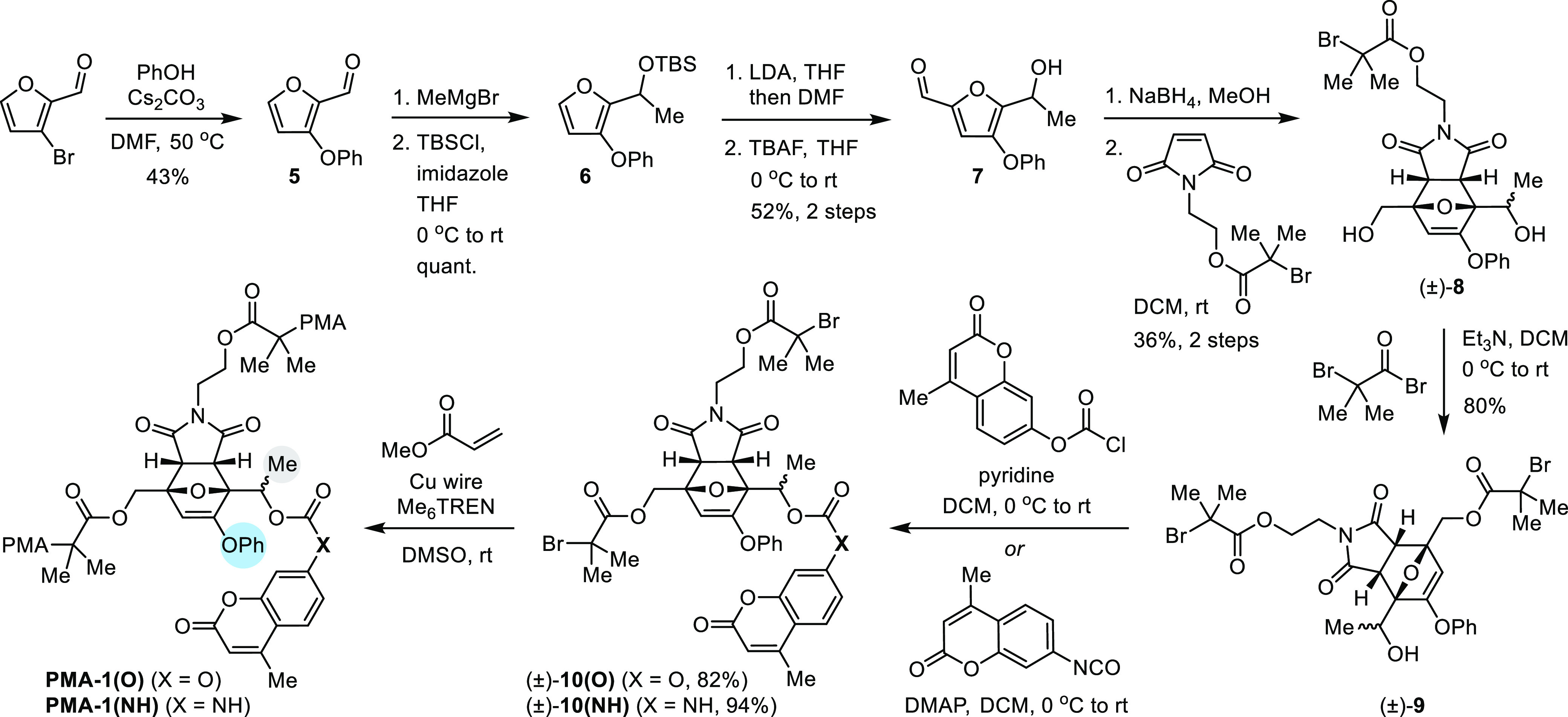
Scheme 3. Ultrasound-Induced Mechanical Activation of Substituted Mechanophores and Release of Fluorescent Hydroxycoumarin or Aminocoumarin Cargo.

Đ = 1.03–1.06 for all polymers.
Prior to evaluating their molecular release behavior, we sought to investigate how substitution of the 2-furylcarbinol derivatives affects the mechanochemical and thermal properties of the Diels–Alder adducts. First, DFT calculations were performed on truncated models of each carbamate-functionalized mechanophore using the constrained geometries simulate external force (CoGEF) method,36 which is a simple and reliable computational technique for predicting mechanochemical reactivity.37 The mechanical elongation of each furan–maleimide adduct results in a predicted retro-[4 + 2] cycloaddition reaction to produce the expected furfuryl carbamate and maleimide fragments with nearly identical energy–displacement profiles (Figure S6). The calculated rupture force (Fmax) is essentially the same regardless of substitution, which occurs at 4.0–4.1 nN and suggests similar mechanochemical activity of each mechanophore that is primarily dictated by pulling geometry.38−40 In contrast to the predicted mechanochemical invariability, however, the addition of a phenoxy group leads to a pronounced increase in the thermal stability of the Diels–Alder adduct, which has been observed previously for cycloadducts derived from 3-alkoxyfurans and ascribed to a reduction in the energetic penalty for loss of furan aromaticity.41,42 Heating a solution of (±)-9 in toluene-d8 at 70 °C results in <2% cycloelimination after 5 h, while the same conditions lead to approximately 46% reversion of the analogous Diels–Alder adduct without the phenoxy substituent (Figure S7). At room temperature, phenoxy-substituted mechanophore (±)-9 is stable indefinitely in toluene-d8 in comparison to the slow, but detectable reversion of the compound with no phenoxy group, as monitored over several months using 1H NMR spectroscopy (Figure S8). The improved thermal stability of the phenoxy-substituted mechanophores in combination with the predicted mechanochemical activity being similar to that of our originally reported masked furfuryl carbonate mechanophore renders this second-generation molecular design an attractive platform for mechanically triggered release.
The mechanically triggered release of hydroxycoumarin or aminocoumarin from PMA-1(X)–PMA-3(X) in 3:1 MeCN/MeOH was evaluated using pulsed ultrasonication (1 s on/1 s off, 8–10 °C, 20 kHz, 8.2 W/cm2), and the effect of substitution on the rate of coumarin release from the mechanically liberated 2-furylcarbinol derivatives was measured using photoluminescence spectroscopy (Scheme 3). Each polymer solution was subjected to 60 min of ultrasonication (“on” time), warmed to room temperature, and then fluorescence was monitored over time. Kinetic data for the release of hydroxycoumarin from PMA-1(O)–PMA-3(O) are illustrated in Figure 3a, while the data for release of aminocoumarin from PMA-1(NH)–PMA-3(NH) are illustrated in Figure 3b. For clarity and to account for slight differences in average polymer molecular weight and dispersity that influence the extent of mechanophore conversion during ultrasonication,43−45 the initial fluorescence intensity (t = 0) is subtracted from each measurement and the data are normalized to emphasize the relative rates of molecular release (see the Supporting Information for additional details). The fluorescence emission from solutions of PMA-1(O) and PMA-2(O) reached a maximum prior to the first measurement and remained essentially constant over time, indicating that the release of hydroxycoumarin from both primary and secondary furfuryl carbonates containing a 3-phenoxy substituent completed nearly instantaneously upon formation (t1/2 < 5 min). These results are contrasted by the release of hydroxycoumarin from mechanically activated PMA-3(O) containing our first-generation mechanophore, which occurs steadily and predictably over several hours postactivation. Fitting the time-dependent photoluminescence data for release of hydroxycoumarin from PMA-3(O) to a first-order rate expression gives an estimated half-life for decomposition of the 3H secondary furfuryl carbonate of 46 min (average of two trials).
Figure 3.
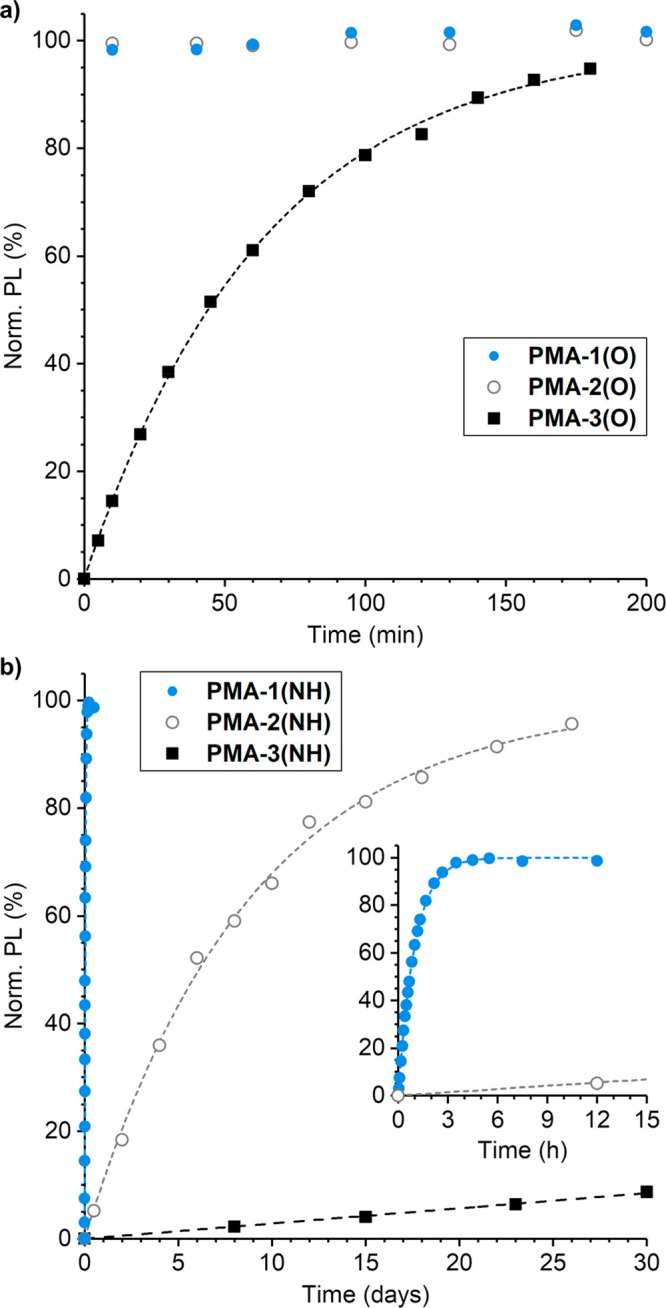
Mechanically triggered release of (a) hydroxycoumarin and (b) aminocoumarin from polymers as a function of mechanophore substitution. Polymer solutions (2 mg/mL in 3:1 MeCN/MeOH) were sonicated for 60 min (“on” time), warmed to room temperature, and the release of coumarin cargo from the mechanically liberated 2-furylcarbinol derivatives was monitored by photoluminescence spectroscopy. PL parameters: λex = 330 nm, λem = 378 nm (hydroxycoumarin); λex = 365 nm, λem = 424 nm (aminocoumarin). The initial PL intensity was subtracted from each measurement and the data were normalized to the plateau value. For PMA-3(NH), the data were normalized assuming 36% mechanophore activation; the black dashed line represents a first-order reaction with a half-life of 240 days.
The release of aminocoumarin from mechanically activated PMA-1(NH)–PMA-3(NH) provides an even clearer demonstration of the effect of substitution on the kinetics of furfuryl carbamate decomposition, with reaction half-lives spanning 4 orders of magnitude (Figure 3b). The time-dependent photoluminescence of the sonicated solution of PMA-1(NH) is described by a first-order rate expression and reaches a maximum intensity after approximately 4 h postactivation, corresponding to the release of aminocoumarin with an average half-life of 41 min from two replicate experiments. This remarkably fast release from the furfuryl carbamate containing both α-methyl and 3-phenoxy substituents is consistent with the substantially diminished activation energy calculated by DFT for corresponding model substrate FC1(NH). In comparison, the release of aminocoumarin from mechanically activated PMA-2(NH) containing a masked primary furfuryl carbamate with a 3-phenoxy substituent is over 200× slower with an average half-life of 6.5 days, again highlighting the stabilizing effect of the α-methyl substituent identified previously. For PMA-3(NH) containing a chain-centered mechanophore analogous to our first-generation molecular design with an α-methyl substituent and no phenoxy group, only 8% release of aminocoumarin was observed after 30 days postactivation. This calculation assumes a mechanophore conversion of 36% as determined previously for our original masked furfuryl carbonate under nearly identical conditions.25 The time-dependent photoluminescence data for release of aminocoumarin from PMA-3(NH) fall on the line for a first-order reaction with a half-life of approximately 240 days. It is worth noting that the yield of hydroxycoumarin or aminocoumarin released from each polymer studied, with the exception of PMA-3(NH), was 34–39% relative to the mechanophore concentration in each experiment, which is consistent with the anticipated mechanophore conversion after a relatively short exposure to ultrasound (Table S1).
The results above illustrate the ability to control the rate of mechanically triggered release by fine-tuning the molecular structure of the furan–maleimide mechanophore, and in particular, highlight the potential for releasing diverse chemical payloads from masked secondary 2-furylcarbinol derivatives containing a 3-phenoxy group. Therefore, we sought to further investigate the scope of molecular cargo that can be effectively released upon mechanical activation of the second-generation mechanophore (Scheme 4). Starting again from modular bis-initiator precursor (±)-9 containing a secondary alcohol, a variety of molecular cargo were installed via different functional group connectivity and the mechanophores were incorporated into polymers following the same protocol as before (Table S2; see the Supporting Information for additional details). In addition to the hydroxycoumarin (phenol) and aminocoumarin (arylamine) payloads attached via carbonate and carbamate groups, respectively, four other cargo molecules were installed, including those bearing alcohol, alkylamine, carboxylic acid, and sulfonic acid functional groups. Conjugation of the alcohol and alkylamine functional cargo molecules was achieved using carbonate and carbamate spacers, respectively, where a decarboxylation step is required for molecular release similar to the coumarin-based phenol and arylamine payloads. On the other hand, cargo molecules bearing carboxylic acid and sulfonic acid functional groups were conjugated directly to the mechanophore substrate through carboxylate and sulfonate linkages. Each payload molecule was chosen to be strongly absorbing in the UV region to facilitate the characterization of their release using high-performance liquid chromatography (HPLC) equipped with a UV detector. For each derivative, a corresponding chain-end functional control polymer was also synthesized and evaluated under the same conditions to confirm the mechanical origin of molecular release (Table S3).9
Scheme 4. Scope of Mechanically Triggered Cargo Release from the Second-Generation Mechanophore with α-Methyl/Phenoxy Substitution.

Values of percent release and half-lives are averages from two replicate experiments. Percent release is reported relative to the initial mechanophore concentration and does not account for incomplete mechanophore conversion after 60 min of ultrasonication. The values of percent release were determined by HPLC.
Half-life for cargo release was measured by photoluminescence spectroscopy.
Half-life for cargo release was measured by HPLC.
Similar to the kinetic studies performed above and following the same ultrasonication procedures, solutions of each polymer (2.0 mg/mL in 3:1 MeCN/MeOH) were subjected to pulsed ultrasonication (60 min “on” time) and then payload release from the mechanically liberated 2-furylcarbinol derivative was monitored at room temperature by HPLC and quantified using an internal standard (see the Supporting Information for details). The identity of each cargo molecule, the average half-life and yield of payload release measured from two replicate experiments, and the values of Mn and Đ of the parent chain-centered polymers are summarized in Scheme 4. Similar to hydroxycoumarin, the mechanically triggered release of 1-pyrenebutanol was sufficiently rapid such that it was complete prior to the first HPLC measurement (t1/2 < 30 min). Release of 1-pyrenemethylamine from the corresponding furfuryl carbamate occurred with a moderate half-life of 4.2 h, albeit approximately 6× slower than the release of aminocoumarin. The difference in release kinetics between the alkyl- and arylamines is ascribed to the difference in pKa values of the conjugate acids,33 with the aniline derivative being a better leaving group. The second-generation mechanophore design not only enables the successful release of alcohols and amines via carbonate and carbamate linkages but is also capable of effecting the release of payloads incorporating carboxylic acid and sulfonic acid functional groups conjugated to the mechanophore via carboxylate and sulfonate linkages. The mechanically triggered release of 1-pyrenebutanoic acid proceeded with a half-life of approximately 28 h, while the release of 2-naphthalenesulfonic acid was complete before the first HPLC measurement (t1/2 < 30 min). Again, this trend is consistent with the significantly lower pKa value of the sulfonic acid in comparison to the carboxylic acid, reflecting the relative stabilities of sulfonate and carbonate leaving groups. The mechanically triggered release of organic acids enabled by this second-generation platform significantly expands upon the limited repertoire of mechanophores that generate HCl14,15 and is also highly modular in nature owing to the generality of the mechanophore design.
The percent release for each cargo molecule determined by HPLC is reported in Scheme 4 relative to the initial concentration of the mechanophore. It is important to note, however, that only a fraction of mechanophores is converted after 60 min of ultrasonication, which again is expected to be ∼36% based on the experimental conditions and the average molecular weight of the polymers (Mn ≈ 100 kDa).25 As demonstrated previously, increasing the sonication time to 150 min results in 64% release of hydroxycoumarin from PMA-3(O).25 Among other factors,46 the rate of mechanophore activation is particularly sensitive to the length of the attached polymer chains, with longer chains producing faster mechanochemical reactions.44,45 Here the duration of ultrasonication was selected on the basis of experimental expediency. With the exception of the alkylamine cargo, the yields for payload release after 60 min of ultrasonication are within the range of 33–41%. These results suggest that payload release from the mechanochemically generated 2-furylcarbinol derivative in each case is highly efficient. Release of the alkylamine plateaus at approximately 8%, and we tentatively attribute the reduced yield to a reaction between the amine and polymer-bound furfuryl cation intermediate, similar to the side reaction observed in the decomposition of model compound 1 at relatively high concentrations. In this case, the enhanced nucleophilicity of the alkylamine is anticipated to promote this reaction pathway to a greater extent in comparison to aminocoumarin. On the other hand, the higher yield of 41% measured for the release of both organic acid payloads is consistent with the slightly higher average molecular weight of those polymers, which results in increased mechanophore conversion during the same period of ultrasonication. Finally, we note that payload release was not observed from any of the chain-end functional control polymers under identical experimental conditions (see the Supporting Information for details), confirming that molecular release from polymers bearing a chain-centered mechanophore was indeed triggered by mechanical force.9
Conclusions
We have demonstrated the mechanically triggered release of functionally diverse small molecules with tunable release kinetics from a second-generation mechanophore platform. The mechanophore design leverages a mechanically triggered cascade reaction in which mechanochemical activation of a furan–maleimide Diels–Alder adduct reveals an unstable 2-furylcarbinol derivative that spontaneously decomposes under mild conditions to release a covalently bound payload molecule. Using DFT calculations to guide the molecular design, we have identified key structure–property relationships for the reactivity of 2-furylcarbinol derivatives that enable significant advancements over an earlier mechanophore design, which was limited to the release of phenolic cargo molecules with relatively slow release kinetics. The incorporation of an electron-donating 3-phenoxy substituent on the furan heterocycle reduces the activation barrier for fragmentation of the furfuryl C–O bond, through the putative resonance stabilization of the developing positive charge in the transition state leading to the furfuryl cation intermediate. Moreover, phenoxy substitution was found to enhance the thermal stability of the Diels–Alder mechanophore without adversely affecting its mechanochemical reactivity. Changing the substitution on the masked 2-furylcarbinol derivatives allows the rates of molecular release to be varied by several orders of magnitude, while the combination of α-methyl and 3-phenoxy substitution results in a highly active substrate for the triggered release of functionally diverse molecular payloads. Starting from a modular mechanophore initiator, a variety of molecular cargoes were conveniently installed and then incorporated into polymers by controlled radical polymerization. The mechanically triggered release of functionally diverse cargo molecules bearing alkyl/aryl alcohols and amines as well as carboxylic acid and sulfonic acid functional groups was demonstrated using ultrasonication, exhibiting fast rates of release and high reaction efficiencies. The generality and efficacy of the mechanophore design make it a promising platform for the mechanically triggered release of a wide variety of functional molecules to address applications in catalysis, sensing, drug delivery, and other areas.
Acknowledgments
This research was supported by Caltech and the Arnold and Mabel Beckman Foundation through a Beckman Young Investigator Award. We thank the Center for Catalysis and Chemical Synthesis of the Beckman Institute at Caltech and the CCE Multiuser Mass Spectrometry Laboratory for access to equipment.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.1c00460.
Experimental details, synthetic procedures, DFT calculations, fluorescence and HPLC data, and NMR spectra (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Swager T. M. Sensor Technologies Empowered by Materials and Molecular Innovations. Angew. Chem., Int. Ed. 2018, 57, 4248–4257. 10.1002/anie.201711611. [DOI] [PubMed] [Google Scholar]
- Roth M. E.; Green O.; Gnaim S.; Shabat D. Dendritic, Oligomeric, and Polymeric Self-Immolative Molecular Amplification. Chem. Rev. 2016, 116, 1309–1352. 10.1021/acs.chemrev.5b00372. [DOI] [PubMed] [Google Scholar]
- Patrick J. F.; Robb M. J.; Sottos N. R.; Moore J. S.; White S. R. Polymers with autonomous life-cycle control. Nature 2016, 540, 363–370. 10.1038/nature21002. [DOI] [PubMed] [Google Scholar]
- Lee K. Y.; Peters M. C.; Mooney D. J. Controlled Drug Delivery from Polymers by Mechanical Signals. Adv. Mater. 2001, 13, 837–839. 10.1002/1521-4095(200106)13:11<837::AID-ADMA837>3.0.CO;2-D. [DOI] [Google Scholar]
- Huo S.; Zhao P.; Shi Z.; Zou M.; Yang X.; Warszawik E.; Loznik M.; Göstl R.; Herrmann A. Mechanochemical bond scission for the activation of drugs. Nat. Chem. 2021, 13, 131–139. 10.1038/s41557-020-00624-8. [DOI] [PubMed] [Google Scholar]
- Küng R.; Pausch T.; Rasch D.; Göstl R.; Schmidt B. M. Mechanochemical Release of Non-Covalently Bound Guests from a Polymer-Decorated Supramolecular Cage. Angew. Chem., Int. Ed. 2021, 60, 13626–13630. 10.1002/anie.202102383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White S. R.; Sottos N. R.; Geubelle P. H.; Moore J. S.; Kessler M. R.; Sriram S. R.; Brown E. N.; Viswanathan S. Autonomic healing of polymer composites. Nature 2001, 409, 794–797. 10.1038/35057232. [DOI] [PubMed] [Google Scholar]
- Toohey K. S.; Sottos N. R.; Lewis J. A.; Moore J. S.; White S. R. Self-healing materials with microvascular networks. Nat. Mater. 2007, 6, 581–585. 10.1038/nmat1934. [DOI] [PubMed] [Google Scholar]
- Li J.; Nagamani C.; Moore J. S. Polymer Mechanochemistry: From Destructive to Productive. Acc. Chem. Res. 2015, 48, 2181–2190. 10.1021/acs.accounts.5b00184. [DOI] [PubMed] [Google Scholar]
- Beyer M. K.; Clausen-Schaumann H. Mechanochemistry: The Mechanical Activation of Covalent Bonds. Chem. Rev. 2005, 105, 2921–2948. 10.1021/cr030697h. [DOI] [PubMed] [Google Scholar]
- Caruso M. M.; Davis D. A.; Shen Q.; Odom S. A.; Sottos N. R.; White S. R.; Moore J. S. Mechanically-Induced Chemical Changes in Polymeric Materials. Chem. Rev. 2009, 109, 5755–5798. 10.1021/cr9001353. [DOI] [PubMed] [Google Scholar]
- Kim G.; Lau V. M.; Halmes A. J.; Oelze M. L.; Moore J. S.; Li K. C. High-intensity focused ultrasound-induced mechanochemical transduction in synthetic elastomers. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 10214–10222. 10.1073/pnas.1901047116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbulatov S.; Boulatov R. Experimental polymer mechanochemistry and its interpretational frameworks. ChemPhysChem 2017, 18, 1422–1450. 10.1002/cphc.201601354. [DOI] [PubMed] [Google Scholar]
- Diesendruck C. E.; Steinberg B. D.; Sugai N.; Silberstein M. N.; Sottos N. R.; White S. R.; Braun P. V.; Moore J. S. Proton-Coupled Mechanochemical Transduction: A Mechanogenerated Acid. J. Am. Chem. Soc. 2012, 134, 12446–12449. 10.1021/ja305645x. [DOI] [PubMed] [Google Scholar]
- Lin Y.; Kouznetsova T. B.; Craig S. L. A Latent Mechanoacid for Time-Stamped Mechanochromism and Chemical Signaling in Polymeric Materials. J. Am. Chem. Soc. 2020, 142, 99–103. 10.1021/jacs.9b12861. [DOI] [PubMed] [Google Scholar]
- Larsen M. B.; Boydston A. J. Flex-Activated” Mechanophores: Using Polymer Mechanochemistry To Direct Bond Bending Activation. J. Am. Chem. Soc. 2013, 135, 8189–8192. 10.1021/ja403757p. [DOI] [PubMed] [Google Scholar]
- Larsen M. B.; Boydston A. J. Successive Mechanochemical Activation and Small Molecule Release in an Elastomeric Material. J. Am. Chem. Soc. 2014, 136, 1276–1279. 10.1021/ja411891x. [DOI] [PubMed] [Google Scholar]
- Shen H.; Larsen M. B.; Roessler A.; Zimmerman P.; Boydston A. J. Mechanochemical Release of N-heterocyclic Carbenes from Flex-Activated Mechanophores. Angew. Chem., Int. Ed. 2021, 60, 13559. 10.1002/anie.202100576. [DOI] [PubMed] [Google Scholar]
- Diesendruck C. E.; Peterson G. I.; Kulik H. J.; Kaitz J. A.; Mar B. D.; May P. A.; White S. R.; Martínez T. J.; Boydston A. J.; Moore J. S. Mechanically triggered heterolytic unzipping of a low-ceiling-temperature polymer. Nat. Chem. 2014, 6, 623–628. 10.1038/nchem.1938. [DOI] [PubMed] [Google Scholar]
- Peterson G. I.; Boydston A. J. Kinetic Analysis of Mechanochemical Chain Scission of Linear Poly(phthalaldehyde). Macromol. Rapid Commun. 2014, 35, 1611–1614. 10.1002/marc.201400271. [DOI] [PubMed] [Google Scholar]
- Wiita A. P.; Ainavarapu S. R. K.; Huang H. H.; Fernandez J. M. Force-dependent chemical kinetics of disulfide bond reduction observed with single-molecule techniques. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 7222–7227. 10.1073/pnas.0511035103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dopieralski P.; Ribas-Arino J.; Anjukandi P.; Krupicka M.; Marx D. Unexpected mechanochemical complexity in the mechanistic scenarios of disulfide bond reduction in alkaline solution. Nat. Chem. 2017, 9, 164–170. 10.1038/nchem.2632. [DOI] [PubMed] [Google Scholar]
- Shi Z.; Song Q.; Göstl R.; Herrmann A. Mechanochemical activation of disulfide-based multifunctional polymers for theranostic drug release. Chem. Sci. 2021, 12, 1668. 10.1039/D0SC06054B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z.Ultrasound-mediated activation of drugs. Ph.D. Dissertation, RWTH Aachen University, Aachen, Germany, 2021. https://publications.rwth-aachen.de/record/814605 (accessed 2021-05-07). [Google Scholar]
- Hu X.; Zeng T.; Husic C. C.; Robb M. J. Mechanically Triggered Small Molecule Release from a Masked Furfuryl Carbonate. J. Am. Chem. Soc. 2019, 141, 15018–15023. 10.1021/jacs.9b08663. [DOI] [PubMed] [Google Scholar]
- Hay M. P.; Sykes B. M.; Denny W. A.; O’Connor C. J. Substituent effects on the kinetics of reductively-initiated fragmentation of nitrobenzyl carbamates designed as triggers for bioreductive prodrugs. J. Chem. Soc., Perkin Trans. 1 1999, (19), 2759–2770. 10.1039/a904067f. [DOI] [Google Scholar]
- Mosey R. A.; Floreancig P. E. Versatile approach to α-alkoxy carbamate synthesis and stimulus-responsive alcohol release. Org. Biomol. Chem. 2012, 10, 7980–7985. 10.1039/c2ob26571k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Kouznetsova T. B.; Boulatov R.; Craig S. L. Mechanical gating of a mechanochemical reaction cascade. Nat. Commun. 2016, 7, 13433. 10.1038/ncomms13433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X.; McFadden M. E.; Barber R. W.; Robb M. J. Mechanochemical Regulation of a Photochemical Reaction. J. Am. Chem. Soc. 2018, 140, 14073–14077. 10.1021/jacs.8b09628. [DOI] [PubMed] [Google Scholar]
- Fan B.; Trant J. F.; Hemery G.; Sandre O.; Gillies E. R. Thermo-responsive self-immolative nanoassemblies: direct and indirect triggering. Chem. Commun. 2017, 53, 12068–12071. 10.1039/C7CC06410A. [DOI] [PubMed] [Google Scholar]
- Schmid K. M.; Jensen L.; Phillips S. T. A Self-Immolative Spacer That Enables Tunable Controlled Release of Phenols under Neutral Conditions. J. Org. Chem. 2012, 77, 4363–4374. 10.1021/jo300400q. [DOI] [PubMed] [Google Scholar]
- Nichol M. F.; Clark K. D.; Dolinski N. D.; Read de Alaniz J. Multi-stimuli responsive trigger for temporally controlled depolymerization of self-immolative polymers. Polym. Chem. 2019, 10, 4914–4919. 10.1039/C9PY00301K. [DOI] [Google Scholar]
- Alouane A.; Labruère R.; Le Saux T.; Schmidt F.; Jullien L. Self-Immolative Spacers: Kinetic Aspects, Structure-Property Relationships, and Applications. Angew. Chem., Int. Ed. 2015, 54, 7492–7509. 10.1002/anie.201500088. [DOI] [PubMed] [Google Scholar]
- Berkowski K. L.; Potisek S. L.; Hickenboth C. R.; Moore J. S. Ultrasound-Induced Site-Specific Cleavage of Azo-Functionalized Poly(ethylene glycol). Macromolecules 2005, 38, 8975–8978. 10.1021/ma051394n. [DOI] [Google Scholar]
- Nguyen N. H.; Rosen B. M.; Lligadas G.; Percec V. Surface-Dependent Kinetics of Cu(0)-Wire-Catalyzed Single-Electron Transfer Living Radical Polymerization of Methyl Acrylate in DMSO at 25 °C. Macromolecules 2009, 42, 2379–2386. 10.1021/ma8028562. [DOI] [Google Scholar]
- Beyer M. K. The mechanical strength of a covalent bond calculated by density functional theory. J. Chem. Phys. 2000, 112, 7307–7312. 10.1063/1.481330. [DOI] [Google Scholar]
- Klein I. M.; Husic C. C.; Kovács D. P.; Choquette N. J.; Robb M. J. Validation of the CoGEF Method as a Predictive Tool for Polymer Mechanochemistry. J. Am. Chem. Soc. 2020, 142, 16364–16381. 10.1021/jacs.0c06868. [DOI] [PubMed] [Google Scholar]
- Konda S. S. M.; Brantley J. N.; Varghese B. T.; Wiggins K. M.; Bielawski C. W.; Makarov D. E. Molecular Catch Bonds and the Anti-Hammond Effect in Polymer Mechanochemistry. J. Am. Chem. Soc. 2013, 135, 12722–12729. 10.1021/ja4051108. [DOI] [PubMed] [Google Scholar]
- Robb M. J.; Kim T. A.; Halmes A. J.; White S. R.; Sottos N. R.; Moore J. S. Regioisomer-Specific Mechanochromism of Naphthopyran in Polymeric Materials. J. Am. Chem. Soc. 2016, 138, 12328–12331. 10.1021/jacs.6b07610. [DOI] [PubMed] [Google Scholar]
- Stevenson R.; De Bo G. Controlling Reactivity by Geometry in Retro-Diels-Alder Reactions under Tension. J. Am. Chem. Soc. 2017, 139, 16768–16771. 10.1021/jacs.7b08895. [DOI] [PubMed] [Google Scholar]
- Boutelle R. C.; Northrop B. H. Substituent Effects on the Reversibility of Furan-Maleimide Cycloadditions. J. Org. Chem. 2011, 76, 7994–8002. 10.1021/jo201606z. [DOI] [PubMed] [Google Scholar]
- Foster R. W.; Benhamou L.; Porter M. J.; Bučar D.-K.; Hailes H. C.; Tame C. J.; Sheppard T. D. Irreversible endo-Selective Diels-Alder Reactions of Substituted Alkoxyfurans: A General Synthesis of endo-Cantharimides. Chem. - Eur. J. 2015, 21, 6107–6114. 10.1002/chem.201406286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryger M. J.; Munaretto A. M.; Moore J. S. Structure-Mechanochemical Activity Relationships for Cyclobutane Mechanophores. J. Am. Chem. Soc. 2011, 133, 18992–18998. 10.1021/ja2086728. [DOI] [PubMed] [Google Scholar]
- May P. A.; Munaretto N. F.; Hamoy M. B.; Robb M. J.; Moore J. S. Is Molecular Weight or Degree of Polymerization a Better Descriptor of Ultrasound-Induced Mechanochemical Transduction?. ACS Macro Lett. 2016, 5, 177–180. 10.1021/acsmacrolett.5b00855. [DOI] [PubMed] [Google Scholar]
- Schaefer M.; Icli B.; Weder C.; Lattuada M.; Kilbinger A. F. M.; Simon Y. C. The Role of Mass and Length in the Sonochemistry of Polymers. Macromolecules 2016, 49, 1630–1636. 10.1021/acs.macromol.5b02362. [DOI] [Google Scholar]
- Lenhardt J. M.; Black Ramirez A. L.; Lee B.; Kouznetsova T. B.; Craig S. L. Mechanistic Insights into the Sonochemical Activation of Multimechanophore Cyclopropanated Polybutadiene Polymers. Macromolecules 2015, 48, 6396–6403. 10.1021/acs.macromol.5b01677. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.