

Abstract

The light-promoted conversion of extensively used cyanine dyes to blue-shifted emissive products has been observed in various contexts. However, both the underlying mechanism and the species involved in this photoconversion reaction have remained elusive. Here we report that irradiation of heptamethine cyanines provides pentamethine cyanines, which, in turn, are photoconverted to trimethine cyanines. We detail an examination of the mechanism and substrate scope of this remarkable two-carbon phototruncation reaction. Supported by computational analysis, we propose that this reaction involves a singlet oxygen-initiated multistep sequence involving a key hydroperoxycyclobutanol intermediate. Building on this mechanistic framework, we identify conditions to improve the yield of photoconversion by over an order of magnitude. We then demonstrate that cyanine phototruncation can be applied to super-resolution single-molecule localization microscopy, leading to improved spatial resolution with shorter imaging times. We anticipate these insights will help transform a common, but previously mechanistically ill-defined, chemical transformation into a valuable optical tool.
Short abstract
Broadly used cyanine dyes undergo photoconversion to two-carbon truncated products. These studies detail the scope, mechanistic basis, and application of this novel reaction.
Introduction
Photochemical reactions involving organic fluorophores occur during nearly every microscopy experiment. Most common are irreversible “photobleaching” reactions that lead to nonemissive products. Also encountered are photoconversion reactions in which irradiation leads to the formation of new molecules with altered optical properties. Such photochemical transformations have been employed to create valuable tools. For example, photoconvertible green fluorescent proteins (e.g., mEos) undergo an oxidative reaction that leads to red-shifted variants (Figure 1a).1 This transformation enabled seminal single-molecule localization microscopy (SMLM) experiments, and various photoconversion reactions are central elements of these methods.2−4 In other cases, these processes can be problematic artifacts that alter the optical properties of the probe molecule. For example, photooxidative dealkylation and consequent hypsochromic shift of rhodamines and related structures are a component of the photodegradation chemistry of these important chromophores.5−7 A detailed understanding of the chemistry underlying these transformation is critical to efforts seeking to avoid, or to productively harness, these processes.
Figure 1.

Key background and current studies. (a) Photoconversion reactions in common fluorophores. (b) Cyanine phototruncation reaction reported here. Wavelengths in nanometers.
Cyanine fluorophores are broadly used in applications that span super-resolution microscopy to clinical imaging.4,8,9 The hypsochromic photoconversion of these broadly used dyes, and its potential role as a “photoblueing” artifact in multicolor experiments, has been described in several reports.10−13 Collectively, these microscopy-centered efforts illuminate several features of the cyanine photoconversion reaction. These include the requirement of oxygenated aqueous conditions and that it can be avoided by the addition of thiols and triplet quenchers, a common component of SMLM buffers.13 Furthermore, the yield of this reaction had been estimated to be approximately 2% or less under all conditions examined previously.13,14 However, despite the widespread use of the cyanine scaffold, the chemistry underpinning this “photoblueing” artifact had remained opaque. We recently found that the irradiation of the simplest pentamethine cyanine, 2, leads to formation of the corresponding trimethine cyanine, 3 (Figure 1b).14 This reaction, involving the formal excision of the ethene diradical from a polymethine chain, has no precedent in the photochemical literature.
Here we report the first study examining the scope and mechanistic basis of the cyanine phototruncation reaction. These efforts enable its deliberate use as a new photoconversion reaction for SMLM. We show the reaction occurs via an intramolecular rearrangement and is mediated by singlet oxygen (1O2). Unmodified heptamethine cyanines, such as 1, convert to the corresponding pentamethine cyanine, 2 (Figure 1b), but commonly used ring-substituted cyanines do not photoconvert. We propose that phototruncation is a multistep process initiated by attack of the C1′ methine of the cyanine on 1O2, leading to a zwitterionic peroxy intermediate. Subsequent water addition and ring closure form a cyclobutane intermediate, which then eliminates hydroperoxyethenol (HOOCH=CHOH) to form the truncated cyanine. Taking note of the critical role of the electrophilic attack by an exogenous nucleophile on the 1O2-modified polymethine, screening efforts identified nucleophilic additives that improve the yield of the reaction by over an order of magnitude. Using these optimized conditions, we then demonstrate photoconversion can be employed for SMLM experiments with Cy7-labeled secondary antibodies supported by an artificial neural network.15 Finally, we find that DNA-based point accumulation for imaging nanoscale topography (DNA-PAINT) experiments can be carried out with up to three orders of magnitude higher concentration of the imager strand at sub-20 nm spatial resolution.
Results
Mechanistic Studies
We first addressed a series of fundamental mechanistic questions. In our initial analysis, we were uncertain if the reaction involved derivatization of products derived from the well-characterized cyanine photooxidative fragmentation process or, alternatively, some unprecedented intramolecular rearrangement.16−18 To test this, we prepared deuterated variants of cyanine 2, 2-d3, and 2-d6 (see Supporting Information for synthesis and NMR analysis). Compound 2-d3 was irradiated with a 630 nm LED (0.2 mW cm–2), and the product mixture was analyzed by LC/MS and high-resolution electrospray ionization mass spectrometry (HRESIMS). We observed only 3-d3 (>98%) but not 3 and 3-d6, as well as the expected photooxidative cleavage products, which include 4 and 5 (Figure 2a and Figure S1). Furthermore, irradiation and analysis of an equimolar ratio of 2 and 2-d6 led to only 3 and 3-d6 and not 3-d3 (Figure 2a and Figure S2). These studies conclusively demonstrate that the reaction occurs through an intramolecular process and not from fragmentation and intermolecular derivatization.
Figure 2.
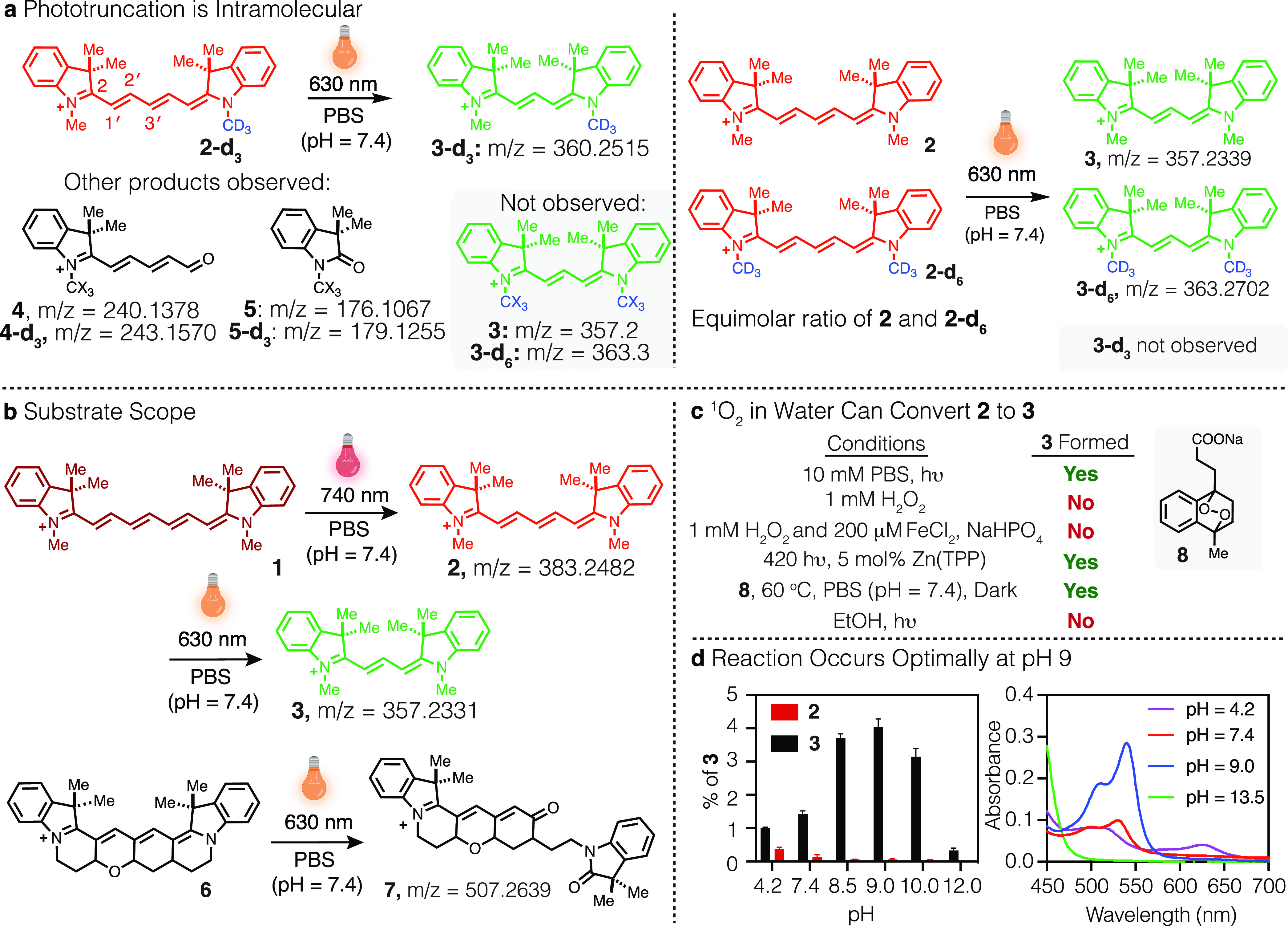
Mechanistic studies of cyanine phototruncation. (a) Products observed following irradiation of 2-d3 or an equimolar ratio of 2 and 2-d6 in PBS (100 μM, 1 mL, with 0.01% DMSO, pH = 7.4) with 630 nm LED at 0.2 W cm–2 for 60 min and monitored by HRESIMS. (b) Evaluating the scope of cyanine phototruncation in PBS (100 μM, 1 mL, with 0.01% DMSO, pH = 7.4) upon irradiation of a heptamethine probe (1) with 740 nm LED (at 0.5 mW cm–2, 30 min) and pentamethine probes (2 and 6) with 630 nm LED (at 0.2 mW cm–2, 60 min). (c) Evaluating the truncation of 2 to 3 with independently generated ROS (see Methods for detailed experimental procedures). (d) Effect of pH on conversion of 2 (50 μM) to 3 in Britton–Robinson buffer (0.04 M boric acid, 0.04 M phosphoric acid, and 0.04 M acetic acid that has been titrated to the desired pH with 1 M sodium hydroxide) upon irradiation with 630 nm LED at 0.2 W cm–2 for 60 min and monitored by UV–vis. Yields were calculated based on the absorption coefficients (ε) of 2 (230 400 M–1 cm–1) and 3 (150 000 M–1 cm–1) in water. Experiments were conducted in triplicate with the error expressed as the standard deviation of the mean.
We then examined the substrate scope of the cyanine phototruncation reaction. Irradiation of the simplest heptamethine cyanine, 1, at 740 nm with an LED (0.5 mW cm–2) led to formation of 2 (in PBS, pH = 7.4, see Table S1), which could be truncated to 3 upon irradiation with a 630 nm LED (Figure 2b). We also examined Cy5, Cy7, Alexa Fluor (AF) 647, AF 750, and indocyanine green (ICG), which are commercial sulfonated variants of 1 and 2 and the most commonly used penta- and heptamethine cyanines (Figure S3). All five undergo phototruncation to their corresponding tri- and pentamethine analogues (see Figure S4). We then examined the conversion of our recently disclosed conformationally constrained pentamethine cyanine 6.19 Compound 6 does not form a blue-shifted cyanine-like product upon 630 nm irradiation, and the only product we could identify is the result of photooxidation (+ O2), which we hypothesize to be compound 7 based on analogy to the previously reported cyanine photochemistry (Figure 2b).16 We also evaluated FNIR-Tag and IR-800CW, heptamethine cyanines with a six-membered ring fused to the polymethine chain (Figure S4).9,20 In both cases, irradiation (740 nm, 0.5 mW cm–2) did not result in any new products with absorbance maxima in the 650–700 nm range, and LC-MS analysis only revealed aldehyde products resulting from photooxidative cleavage (Figure S4). The latter two results suggest that the incorporation of a ring or rings onto polymethine chromophore inhibits the phototruncation reaction.
The next question we sought to address is the responsible reactive oxygen species (ROS). We exposed 2 to a series of independently generated ROS species (Figure 2c, Table S2). Hydrogen peroxide, superoxide, and the hydroxy radical, generated through a Fenton process (200 μM FeCl2, 1 mM H2O2, pH = 6), did not induce the phototransformation, even at concentrations exceeding those attainable by photolysis.21 We also ensured that the observed conversion was not thermally driven. By contrast, 1O2 generated either from endoperoxide 8 (at 60 °C in pH = 7.4 PBS for 4 h, dark) or through photosensitization (with a 2 mW cm–2 420 nm LED at r.t. in 1:2 MeCN/water for 2 h w/5 mol % tetraphenylporphyrin zinc (Zn(TPP)) induced the conversion of 2 to 3 (Table S2).22,23 We validated this observation by heating 1 and 8 and measuring the yield of the formation of 2 (3.8%) using HPLC (Figures S7 and S8). Notably, this reaction is highly solvent dependent and only proceeds in aqueous conditions. For example, using EtOH, DMF, and ethylene glycol led to only trace amounts of 3 (<0.1%) and predominantly induced the photooxidative cleavage reaction to form 4 and 5.
Finally, we examined the role of water in the reaction. We first established that, as seen with the independent generation of 1O2, photoconversion does not occur in any nonaqueous solvent we have examined to date. We probed the impact of pH using Britton-Robinson universal buffers (boric acid, phosphoric acid, and acetic acid). The yield of 2 and 3 was calculated using their absorption coefficients (ε) at their absorption maxima in water following irradiation with a 630 nm LED at 0.2 W cm–1 (Figure 2d). Strikingly, the reaction is highly pH-dependent. At pH 7.4, the yield is 1.4 ± 0.1%, but at pH 9 it is 4.1 ± 0.2%. This observation suggests a probable role for hydroxide in promoting the intramolecular rearrangement of a photooxidized intermediate. At elevated pH (13.5), the reaction is nearly completely suppressed, which may suggest the potential for additional hydrolytic reactions to occur on intermediates in this process. We also examined the role of irradiation power density. Consistent with a single photon process, phototruncation occurs with a linear dependence on irradiation power (Tables S3 and S4).
Building on these experimental results, we developed the mechanistic proposal shown in Figure 3a. For the following reasons, we propose the first step is an asynchronous attack of 1O2 on cyanine 2 at C1′ to provide peroxy intermediate 9 (see Supporting Information for further discussion). First, our prior studies demonstrated that dioxetane formation and subsequent fragmentation to carbonyls 4 and 5 occurs only at the C2/C1′ position.16 Additionally, the computational results, at the M06-2X/6-31G(d,p) level, show that the peroxy intermediate 9 exists as a minimum. By contrast, no perepoxide intermediates were found to be a minimum on the potential energy surface. Furthermore, in examining the regioselectivity of 1O2 addition, only peroxyl formation at C1′ provides an energy minima, whereas addition of 1O2 to the C2, C2′, or C3′ sites leads to O2 dissociation. This is because peroxy intermediate 9 is unique in that the positive charge is delocalized along the polymethine, which stabilizes this intermediate. Additionally, the resonance-delocalized positive charge renders the molecule susceptible to hydration to form hydroperoxyethanol 11. This step is reminiscent of the reaction of 2,4-hexadienes with 1O2, wherein alcoholysis of the zwitterionic peroxy intermediate provides hydroperoxyalkoxyethenols.24−26 On forming the hydroperoxyethanol 11, facile rotation about the C2′–C1′ sets up an intramolecular attack by the vinylogous enamine to provide the hydroperoxycyclobutanol 12. Intermediate 12 subsequently releases hydroperoxyethenol 13 to afford cyanine 3.a Both the four-membered ring, 12, and five-membered ring, 14, intermediates are feasible from 11. However, the formation of the hydroperoxycyclobutanol intermediate 12 is likely due to the enhanced nucleophilicity of the incoming carbon nucleophile, as well as an energetic advantage to forming the enamine-stabilized cation found in 12 relative to the secondary cation 14. Additionally, density functional theory (DFT) calculations show the unsymmetrical product, 15, that results from 14, to be distorted and thus energetically disfavored by 24.5 kcal/mol relative to 3.
Figure 3.
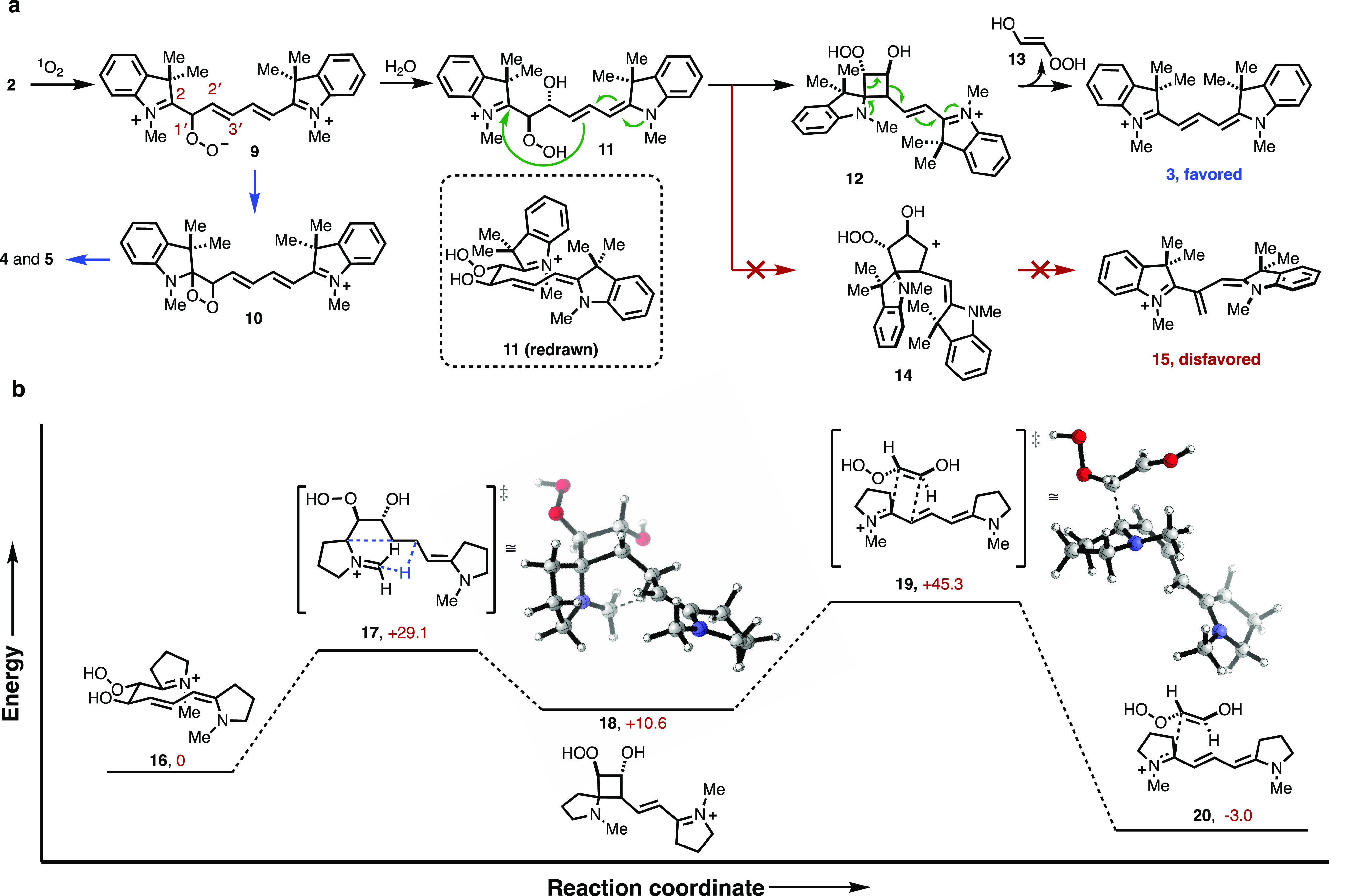
Mechanistic proposal and computational analysis. (a) Proposed mechanism of cyanine phototruncation with the representative conversion of 2 to 3. (b) DFT-computed potential energy surface for conversion of 16 to 20. Computed surfaces showing the results of ground state DFT calculations using M062-X/6-31G(d,p). Relative enthalpies (ΔH) are given in kcal/mol.
A study of the reaction mechanism was conducted using the M06-2X method. We set out to model reaction progress, for which we used a pared version of the cyanine framework. Our starting point was compound 16, which results from hydration of the peroxy intermediate according to the mechanism described above. We then examined the conversion of 16 to cyclobutane intermediate 18 through transition state 17 (+29.1 kcal/mol). There are two features of this computational model that justify the preferential formation of the cyclobutane intermediate 12. The first is a chairlike syn-rotamer conformation in the ground state of 16 that orients the reactive centers. The second is a proton on the methyl group favorably assisting in the formation of the four-membered product 18 by stabilizing the generated partial positive charge. Final elimination of an hydroperoxyethenol unit proceeds via a transition state 19, with an energy of 34.7 kcal/mol relative to 18 from an exothermic product 20. We hypothesize that solvation, which may promote a hydrogen-bond assisted elimination reaction, may significantly lower the energy of this transition state.
Optimization of Phototruncation Reaction
With these mechanistic insights in hand, we then set out to optimize the photoconversion reaction. Guiding this effort was the hypothesis that a water-solvated nucleophile other than hydroxide might improve the yield. Motivated by the potential utility of a hepta- to pentamethine conversion reaction for the SMLM applications described below, we chose to focus our efforts on this reaction. We screened the conversion of 1 to 2 in over 300 conditions using a range of additives and buffers at varying pH (Figure 4a). This was done using a sealed 384-well plate (Figure 4b), which was irradiated with a 0.5 mW cm–2 740 nm LED for 40 min (see Methods for further information), and the approximate conversion to 2 was measured directly by using the absorbance of the product. At pH 7.4, the yield of 2 is ∼1%. As shown in Figure 4a,b, the reaction is highly sensitive to additives and pH. Any conditions below pH 3 and above 11 provided only trace conversion to 2, whereas the yield at pH 9 was improved to ∼2%. A screen of amino acids found that arginine (pH = 9.0 in water), histidine, and lysine (both pH = 10 in water) improved the apparent phototruncation yield. Notably, histidine (10 mM) at pH = 7.4 in PBS also provided considerable photoconversion. By contrast, various nucleophiles including azide, imidazole, ethanolamine, mercaptopropionic acid, and tetrabutylammonium iodide (TBAI) led to only a modest improvement in the yields. We also screened additives with multiple nucleophilic heteroatoms (Figure S9) and found that 1,3-diaminopropan-2-ol (DAPOL) (100 mM) at pH = 9.0 (in water) significantly improves the phototruncation. Inspired by these results, we conducted a screen of Good’s buffers and found 1 M N-cyclohexyl-3-aminopropanesulfonic acid (CAPSO) pH = 9.5 resulted in a dramatic order of magnitude improvement in yield.
Figure 4.

Optimization of phototruncation reaction. (a) Screening phototruncation conditions with varying buffers, additives, and pH. A 50 μM solution of 1 in a 384-well plate was irradiated with 740 nm LED at 0.5 mW cm–2 for 40 min. Every data point represents the buffer (square) and pH (circle). (b) A representative image of the 384-well plate before/after irradiation. (c) HPLC yields of representative conditions. (d) Ensemble fluorescence experiments following irradiating 1 (50 μM) for the indicated time with a 630 nm LED in 10 mM PBS (pH = 7.4), 1 and 0.5 M CAPSO (pH = 9.5) buffer. Fluorescence traces measured following excitation using a 630 nm LED (0.2 W cm–2) for up to 20 min (see Figures S13 and S14 for longer absorption and emission experiments in CAPSO).
While the 384-well approach enabled this initial screening, the absorbance-based readout introduces some uncertainty regarding the yield of the conversion. To address this, we then obtained highly accurate photoconversion yields for several conditions (Figure 4c, Figures S10–12 and Table S5). Specifically, reactions were carried out in sealed glass HPLC vials (in triplicate) in larger volumes, and the yield was measured using the UV–vis absorbance (Table S5) and validated using HPLC (Figure 4c and Figure S12). Under our optimal conditions (1 M CAPSO, pH 9.5), we measured a HPLC yield of 17.2 ± 0.36% (s.d.), whereas neutral conditions (pH = 7.4, PBS) resulted in an overall yield of only 1.31 ± 0.12% (s.d.). Notably, our best neutral conditions (10 mM histidine, pH = 7.4) also resulted in a meaningful improvement in yield to 4.00 ± 0.23% (s.d.).
Application of Phototruncation to SMLM
With these optimization studies complete, we sought to examine the application of phototruncation to SMLM. SMLM methods such as PALM and (d)STORM enable fluorescence imaging beyond Abbe’s diffraction limit with virtually molecular-level resolution in cells.27 SMLM depends on the availability of bright photoactivatable or photoswitchable probes.2,3,28−32 The core element of this approach is the stochastic generation of only a sparse subset of single-molecule emitters per frame at any time during the experiment. We hypothesized that the phototruncation of hepta- to pentamethine cyanines could be carried out using the common 640 nm laser line, which could then concurrently be used to image individual photoconverted pentamethine cyanine molecules. This was first tested in ensemble fluorescence experiments using a 630 nm LED at 0.2 mW cm–2 in 10 mM PBS (pH = 7.4) and 1 M CAPSO (pH = 9.5) buffers (Figure 4d and Figure S13). In 10 mM PBS, short irradiation times resulted in the transient appearance of a Cy5 fluorescence; however, this signal quickly disappeared due to photobleaching. By contrast, when the same experiment was carried out in 1 M CAPSO buffer, 70-fold and 60-fold higher amounts of 1 (remaining) and 2 (photoconverted), respectively, were observed after 10 min (Figure 4d and Figure S14).
We then carried out initial wide-field fluorescence-imaging experiments of microtubules in COS7 cells. Cells were immunostained with secondary IgG-goat-anti-rabbit antibodies labeled with Cy7 at a degree of labeling (DOL) of ∼4.0. We used total internal reflection fluorescence (TIRF) imaging and irradiated the cells solely at 640 nm with a low irradiation intensity of 0.1 kW cm–2 to minimize photoconversion of the heptamethine cyanine dye, which starts immediately at 640 nm irradiation (Figure 5a). Upon irradiation, the overall fluorescence intensity in the Cy7 channel increased over time (Figure 5a,b), with subsequent increases in the Cy5 channel prior to bleaching of the fluorescence signal in both channels. Due to the high labeling density, these Cy7-antibodies are initially largely aggregated with a characteristic H-aggregate shoulder in the absorption spectrum (Figure S15). The fluorescence intensity recorded in the Cy7 channel increases during the early part of irradiation as the Cy7 aggregates are disrupted, presumably through a mixture of photoconversion and photobleaching. The delayed rise in the Cy5 signal suggests that only the fraction of heptamethine cyanines in the nonquenched monomeric state undergoes phototruncation. Similar TIRF-imaging experiments performed in PBS, pH 7.4 showed identical fluorescence time traces albeit at lower photoconversion efficiency (Figure S16).
Figure 5.
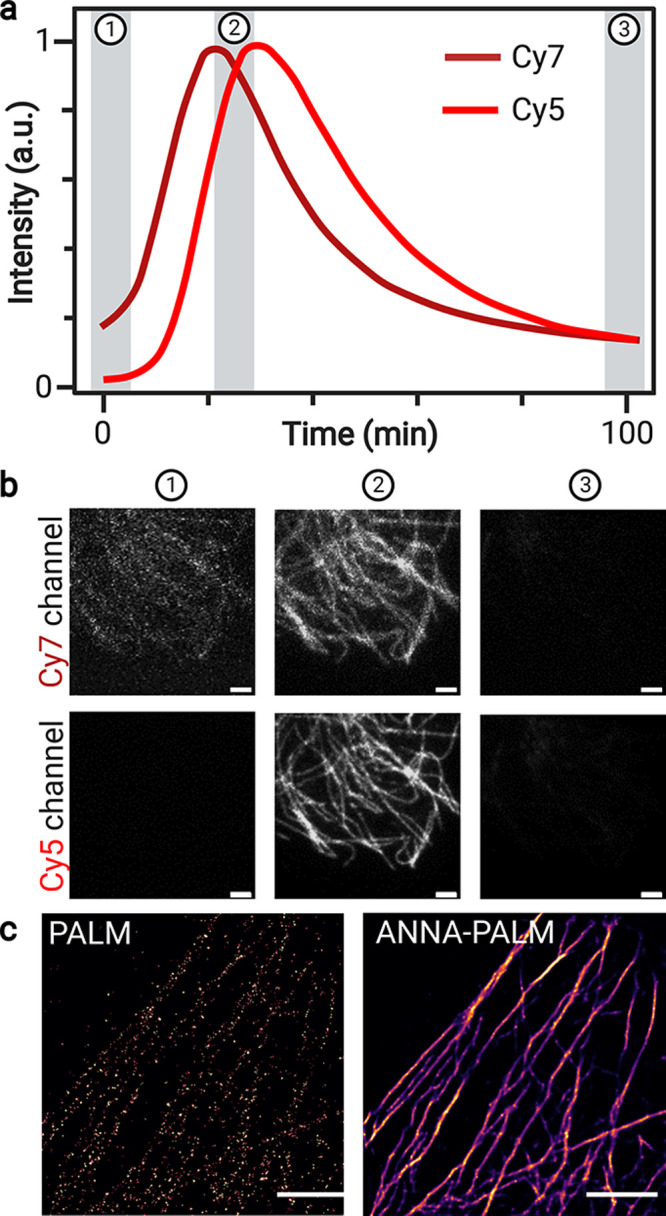
Cyanine phototruncation enables SMLM. TIRF imaging of IgG-gar-Cy7 (DOL ≈ 4) immunolabeled microtubules in COS7 cells using solely 640 nm irradiation. (a) Time course of fluorescence signals recorded on a longer wavelength Cy7 channel and a shorter wavelength Cy5 channel. (b) Fluorescence imaging was performed in 1 M CAPSO, pH 9.5 at the designated time points (1–3) using an irradiation intensity of 0.1 kW cm–2. Band-pass filters used: Cy5 red channel (679/41 nm) and Cy7 far-red channel (835/35 nm). (c) SMLM image of microtubules reconstructed after an acquisition time of 83.3 min at 100 ms exposure time using solely 640 nm irradiation at an intensity of 0.5 kW cm–2 in 1 M CAPSO, pH 9.5. The left image shows the raw reconstructed PALM-like; i.e., Cy5 is photoactivated via phototruncation Cy7 → Cy5 followed by detection, localization, and photobleaching or further photoconversion. SMLM image obtained after applying a trained artificial neural network (ANNA-PALM) algorithm15 to the reconstructed raw PALM image. Scale bars: 2 μm (b, c).
Next, we applied immunolabeling of microtubules with IgG-gar-Cy7 antibodies to SMLM imaging in 1 M CAPSO, pH 9.5 using 640 nm laser irradiation at intensities of 0.5 and 1.0 kW cm–2 in TIRF-mode (Figure 5c and Figure S17). Here, imaging with the lowest irradiation intensity (0.5 kW cm–2) delivered the highest fluorescence intensities recorded for photoconverted Cy5 fluorophores of 945 ± 487 (s.d.) photons corresponding to an average localization precision of 6.9 ± 3.0 nm (s.d.) before photobleaching or conversion to a nonfluorescent form. With a maximum photoconversion yield of ∼17% in 1 M CAPSO, pH 9.5 (Figure 4c), as expected we obtained only relatively sparse reconstructed SMLM images (Figure 5c and Figure S18). However, the somewhat sparse localization images can be improved using a deep learning technique that employs artificial neural networks (ANNs) to learn complex nonlinear mappings.15 This method, known as ANNA-PALM, exploits the structural redundancy of the captured image to reconstruct a high-quality image (Figure 5c and Figure S18).
These results show that phototruncation-SMLM with heptamethine cyanines is possible, although the imaging quality is limited by the photoconversion yield when using the current constructs. We hypothesized that phototruncation-SMLM in combination with DNA-PAINT33−35 should enable high-density super-resolution imaging. In DNA-PAINT, the molecule of interest is labeled (directly or via standard immunolabeling approaches) with a short DNA sequence termed docking strand, and the transient binding of a complementary short imager strand carrying a fluorescent probe is used for high precision localization and image reconstruction (Figure 6a). Despite its now established utility, the fluorescence of the unbound imager strands can make implementing these experiments a significant challenge. Hence, it is usually performed at picomolar concentrations of imager strands (typically 100 pM), requiring TIRF imaging and long acquisition times for high-resolution image reconstruction.33−35 If, however, imager strands are nonfluorescent initially and only are fluorescent, or become fluorescent, when hybridized transiently and irradiated in the evanescent field, higher imager strand concentrations can be used and faster acquisition times can be attained. Such approaches have been realized using dual-labeling FRET approaches,36,37 and we hypothesized might also be possible using a cyanine photoconversion strategy using only singly labeled Cy7 imager strands. Once photoconverted (e.g., Cy7 → Cy5), the pentamethine cyanine is rapidly photobleached or further converted. Hence, under ideal conditions, the background fluorescence does not increase during the experiment independent of the imager strand concentration (Figure 6a). In practice, we have found that TIRF illumination at 640 nm and detection behind a far-red Cy5 band-pass filter (679 ± 21 nm) allowed us to increase the concentration of the imager strand by three orders of magnitude (100 nM of Cy7-imager strand) to provide SMLM images of immunolabeled microtubules in COS7 cells (Figure 6c). To optimize the photoconversion yield, we performed Cy7-photoconversion DNA-PAINT experiments in PBS, containing 500 mM NaCl and 10 mM histidine at pH 7.4 (Figure 4c). Interestingly, we found that an irradiation intensity of 0.5 kW cm–2 provides better image qualities than with higher intensities (Figure 6 and Figure S17).
Figure 6.

Cy7-Phototruncation DNA-PAINT. (a) Schematic to apply cyanine phototruncation for conventional and phototruncation DNA-PAINT imaging. (b, c) DNA-PAINT images of immunolabeled microtubules in COS7 cells using conventional Cy5-imager strands (100 pM) and phototruncation Cy7-imager strands (100 nM) and corresponding Fourier ring correlation (FRC) analysis38 estimated spatial resolutions. Measurements were performed in PBS, pH 7.4 containing 500 mM NaCl in the absence (conventional DNA-PAINT) and presence of 10 mM histidine (phototruncation DNA-PAINT) at an irradiation of 0.5 kW cm–2 at 640 nm (τb = bright time, τd = dark time). Scale bars: 5 μm (b, c).
For comparison, we performed DNA-PAINT imaging experiments in PBS containing 500 mM NaCl at pH 7.4 and identical irradiation intensity (0.5 kW cm–2) using 100 pM of Cy5-imager strands (Figure 6b). Under the applied experimental conditions (0.5 kW cm–2 at 640 nm, 100 ms frame–1, 50 000 frames) Cy7-photoconversion DNA-PAINT measured in the presence of 10 mM histidine outperformed Cy5 DNA-PAINT with a localization precision of 8.3 ± 3.2 nm (s.d.), and 10.2 ± 2.3 nm (s.d.), respectively. This finding was corroborated by Fourier ring correlation analysis (Figure 6b,c).38 Despite the 1000-fold higher concentration, background signals originating from nonspecifically bound and photoconverted Cy7-imager strands only modestly increase with increasing measurement time, with results similar to the conventional DNA-PAINT protocol.
To investigate the temporal behavior of Cy7-photoconversion in more detail, we performed DNA-PAINT experiments with 100 nM Cy7-imager and 100 pM Cy5-imager strands in PBS, containing 500 mM NaCl and 10 mM histidine at pH 7.4 using an integration time of 20 ms per frame. For Cy7-phototruncation DNA-PAINT, we determined shorter average off-state durations compared to standard DNA-PAINT using Cy5-imager strands at three orders of magnitude lower concentration (Figure S19). Overall, DNA-PAINT using 100 nM imager strands is ∼2-fold faster than conventional DNA-PAINT using 100 pM Cy5 imager strands (Figure S19).39 Additional optimization of imaging conditions to improve the photoconversion process and enable the use of higher Cy7-imager strand concentration will likely enhance the utility of Cy7-photoconversion DNA-PAINT.
Conclusions
Here we described efforts to define the basis of the cyanine phototruncation reaction and apply it to SMLM. Mechanistic studies implicate roles for both 1O2 and hydroxide in a process that proceeds via an intramolecular rearrangement. We propose the reaction involves stepwise intermediacy of peroxy alcohol and cyclobutane species, which then undergo an elimination reaction to provide the two-carbon truncated product. Computational analysis has identified plausible transition states in route to the observed product and help to define the basis for the regioselective formation of the initial peroxy intermediate. Motivated by a key role for electrophilic attack of the peroxy intermediate, we carried out a screening effort to identify optimal conditions for truncation of hepta- to pentamethine cyanines. These efforts identify conditions that dramatically improve the yield of the photoconversion, though the exact role of the additives remains to be fully defined. We then demonstrate that this reaction can be used for SMLM, including for DNA-PAINT experiments that benefit from increased imager concentration and, consequently, reduced acquisition time.
Several features of cyanine phototrunction make it distinct from other commonly used photoconversion reactions. First, the reaction involves the interconversion of some of the most broadly used fluorescent probes, which means that emission and excitation optics are in place on numerous instruments. Second, as the 640 nm laser line can be used to excite both pentamethine and heptamethine cyanine probes, we anticipate that the Cy7 channel could be used to determine the initial field of view prior to photoconversion and SMLM. This is an advantage relative to conventional caging strategies where the probe starts in a dark state.40 Additionally, it is quite feasible that green laser lines can promote the conversion of pentamethine cyanines to trimethine cyanines. Going forward, chemical approaches to optimize the cyanine phototruncation reaction will likely be able to enhance its utility. The remarkable impact of nucleophilic additives on photoconversion yield suggests that efforts to incorporate the nucleophile into either the probe itself or a self-labeling protein may have a beneficial impact on photoconversion. Additionally, these studies have also shown that the quenched form of the Cy7 probe can act as a convenient reservoir for dark-state unconverted dye. We are currently investigating strategies to deliberately take advantage of this feature. More broadly, as this reaction leads to bond cleavage, cyanine phototruncation has the potential to act as a new approach to NIR photocaging.41 Overall, we believe the chemical insights provided here open the door to future efforts to use cyanine phototruncation in the development of new optical methods.
Methods
Detailed procedures for the synthesis of all compounds, their characterization, and imaging experiments are given in the Supporting Information.
Photoconversion Experiments
A 50 or 100 μM solution of 1 or 2 in the buffer/solution of interest was generated from a 5 mM DMSO stock solution. The sample was irradiated at 22 °C with 0.5 W cm–2 (740 nm ± 20 nm) or 0.2 W cm–2 (630 nm ± 20 nm) LED source (Marubeni America Co.) in a sealed HPLC vial (containing 240 μL of sample in a borosilicate glass insert) for the designated time. Depending on the study, the sample was then analyzed by UV–vis, fluorimeter, HPLC, or HRESIMS, according to the method outlined in the general Methods section. Yields were calculated based on the absorption coefficients (ε) of 1 (107 000 M–1 cm–1), 2 (230 400 M–1 cm–1), and 3 (150 000 M–1 cm–1) in water. Experiments were conducted in triplicate with the error expressed in the standard deviation of the mean. In prior work, we established that the in-house synthesized samples of 2 do not contain detectable 1, and we have repeated the same analysis to show that 3 does not contain 2 or 1.14
420 nm Irradiation with ZnTPP
A 20 μM solution of 2 in 50% MeCN/H2O was generated from a 1 mM DMSO stock solution. A portion of a 1 mM THF stock solution of 5,10,15,20-tetraphenyl-21H,23Hporphine zinc (ZnTPP) was added to achieve 5 mol % concentration of ZnTPP. The sample was irradiated at 22 °C with 2 mW cm–2 420 nm LED in an HPLC vial for 240 min.
Hydrogen Peroxide
A 100 μM solution of 2 in 10 mM PBS (pH = 7.4) was generated from a 5 mM DMSO stock solution. A solution of hydrogen peroxide (100 mM in H2O stock) was added such that the final concentration of H2O2 was 100 μM, and the sample was incubated for 120 min.
Fenton Conditions
A 20 μM solution of 2 in 50 mM NaHPO4 (pH = 6) was generated from a 5 mM DMSO stock solution. Solutions of FeCl2 tetrahydrate (50 mM H2O stock) and hydrogen peroxide (100 mM H2O stock) were added in succession such that the final concentration of each was 500 μM, and the sample was incubated for 5 min.
Thermal 1O2 Generation
Endoperoxide 8 was synthesized according to a known procedure.42 A 20 mM solution of 8 in H2O was prepared. A solution of 100 μM 2 and 4 mM 8 in 10 mM PBS was generated from their respective stock solutions. The sample was heated to 60 °C in a HPLC vial for 4 h in the dark.
Photoconversion Screening in a 384-Well Plate
A 50 μM solution of 1 in the buffer/solution of interest was generated from a 5 mM DMSO stock solution. The solutions were first prepared in an Eppendorf tubes and added to the designated vials in a 384-well plate. The plate was then sealed with ThermalSeal RT film. The sample was irradiated such that 16-wells were illuminated in a single run using the LED. Irradiations were conducted at 22 °C using 630 nm LED (Marubeni America Co.) set to 0.5 W cm–2 for the designated time. The sample was then analyzed by UV–vis absorbance.
Preparation of 1 M CAPSO (pH = 9.5) buffer
CAPSO commercial source: Obtained as a 99% pure crystalline solid from Millipore Sigma. A total pf 3.69 g of CAPSO was suspended in 10.0 mL of water (pH 7.4), resulting in a suspension of pH < 5.0. Following which, the pH of the solution was adjusted to 9.5 using ∼2 mL of 5 M NaOH (and 10 M HCl, if required in the end). The final volume of the mixture was adjusted to 15.5 mL using ∼3.5 mL of water and stored at r.t. The obtained 1 M solution was then used for analysis. In case of precipitation upon storage, it was reconstituted using vortex/sonication.
Computational Chemistry
Optimizations, frequency calculations, and the intrinsic reaction coordinate calculations were carried out with Gaussian1643 and visualized with Gaussview 5.0.44 M062-X calculations were conducted with the 6-31G(d,p) basis set. Thermal corrections to 298 K were used in the reported energies. Structures were optimized to minima or maxima. Transition structures were verified as transition states by frequency calculations.45 In some cases, calculations were carried out by scanning of bond rotations and bond dissociations by constraining compound geometries The energetics are reported as the thermal enthalpies. For the potential energy surface (PES) in Figure 3, frequency calculations established the nature of the stationary point obtained. Vibrational analyses showed that 17 and 19 species were transition structures, while 16, 18, and 20 were the minima. The intrinsic reaction coordinate calculations were used to verify the transition state structures 17 and 19.
DNA Labeling with Cy7
Imager strands were custom modified with Cy7-NHS monosuccinimidyl ester (Biomol, 239054). Imager strand labeling was performed at 20 °C for 4 h in labeling buffer (100 mM sodium tetraborate (Fulka, 71999), pH 9.5) following the manufacturers standard protocol using a concentration of 10 μM imager strand (AmC6-5′-GTAATGAAGA-3′)33 and a 25× excess of Cy7-NHS. Cy7-imager strands were purified by HPLC on a Kinetex biphenyl column (150 × 4.6 mm) consisting of 2.6-μm particles at 100-Å pore size (Phenomenex, 00F-4622-E0) with a flow rate of 1 mL/min. Solvent A consisted of 0.1 M triethylammonium acetate (TEAA) (AppliChem, A3846, 100); solvent B was 75% acetonitrile (Sigma-Aldrich, 34998-2.5L) in 0.1 M aqueous TEAA (AppliChem, A3846, 100). Purification was done using a linear gradient of 0% B to 65% B over 25 min. The resulting elution peak was collected and dried with a speed vac, consisting of a centrifuge (ThermoFisher, SPD111V), a refrigerated vapor trap (ThermoFisher, RVT400), and a vacuum pump (ThermoFisher, VLP80). The pellet was resuspended in double-distilled water, and the final concentration was determined by UV–vis absorption spectrometry (Jasco V-650).
Antibody Labeling with Cy7
For direct antibody labeling with Cy7-NHS (Biomol, 239054) a goat anti-rabbit IgG (Invitrogen, 31212) was used as a secondary antibody. Antibody labeling was performed at 20 °C for 4 h in labeling buffer (100 mM sodium tetraborate (Fulka, 71999), pH 9.5) following the manufacturers standard protocol. Briefly, 100 μg of antibody was reconstituted in labeling buffer using 0.5 mL spin-desalting columns (40K MWCO, ThermoFisher, 87766). A 35× excess of Cy7-NHS was used to achieve a DOL ≈ 4. Antibody conjugates were purified and washed up to three times using spin-desalting columns (40K MWCO) in PBS (Sigma-Aldrich, D8537-500 ML) to remove excess dyes. Finally, antibody concentration and DOL were determined by UV–vis absorption spectrometry (Jasco V-650).
Antibody Modification for DNA-PAINT
For labeling of goat anti-rabbit IgG (H+L) secondary antibody (Invitrogen, 31212) with PEG4-TCO, an excess of TCO-PEG4-NHS was used. Antibody labeling was performed at 20 °C for 4 h in labeling buffer (100 mM sodium tetraborate (Fulka, 71999), pH 9.5) following the manufacturer’s standard protocol. Goat anti-rabbit IgG (H+L) secondary antibody was reconstituted in labeling buffer using 0.5 mL spin-desalting columns (40K MWCO, ThermoFisher, 87766). The conjugated antibody was purified and washed up to three times using spin-desalting columns (40K MWCO) in PBS (Sigma-Aldrich, D8537-500 ML). Finally, the antibody concentration was determined by UV–vis absorption spectrometry (Jasco V-650).
Cell Culture
African green monkey kidney fibroblast-like cells (COS7, Cell Lines Service GmbH, Eppelheim, #605470) were cultured in DMEM (Sigma, #D8062) containing 10% FCS (Sigma-Aldrich, #F7524), 100 U/mL penicillin and 0.1 mg/mL streptomycin (Sigma-Aldrich, #P4333) at 37 °C and 5% CO2. Cells were grown in standard T25-culture flasks (Greiner Bio-One).
Immunofluorescence Labeling for SMLM Measurements
For immunostaining, cells were seeded at a concentration of 2.5 × 104 cells/well into eight-chambered cover glass systems with a high-performance cover glass (Cellvis, C8-1.5H-N) and stained after 3 h of incubation at 37 °C and 5% CO2. Cells were washed with prewarmed (37 °C) PBS (Sigma-Aldrich, D8537-500 ML) and permeabilized for 2 min with 0.3% glutaraldehyde (GA) + 0.25% Triton X-100 (EMS, 16220 and ThermoFisher, 28314) in prewarmed (37 °C) cytoskeleton buffer (CB), consisting of 10 mM MES ((Sigma-Aldrich, M8250), pH 6.1), 150 mM NaCl (Sigma-Aldrich, 55886), 5 mM EGTA (Sigma-Aldrich, 03777), 5 mM glucose (Sigma-Aldrich, G7021), and 5 mM MgCl2 (Sigma-Aldrich, M9272). After permeabilization, cells were fixed with a prewarmed (37 °C) solution of 2% GA for 10 min. After fixation, cells were washed twice with PBS (Sigma-Aldrich, D8537-500 ML). After fixation, samples were reduced with 0.1% sodium borohydride (Sigma-Aldrich, 71320) in PBS for 7 min. Cells were washed three times with PBS (Sigma-Aldrich, D8537-500 ML) before blocking with 5% BSA (Roth, #3737.3) for 30 min. Subsequently, microtubule samples were incubated with 2 ng/μL rabbit anti-α-tubulin primary antibody (Abcam, #ab18251) in blocking buffer for 1 h. After primary antibody incubation, cells were rinsed with PBS (Sigma-Aldrich, D8537-500 ML) and washed twice with 0.1% Tween20 (ThermoFisher, 28320) in PBS (Sigma-Aldrich, D8537-500 ML) for 5 min. After washing, cells were incubated in blocking buffer with 4 ng/μL of Cy7-labeled goat anti-rabbit IgG secondary antibodies (Invitrogen, 31212) for 45 min. After secondary antibody incubation, cells were rinsed with PBS (Sigma-Aldrich, D8537-500 ML) and washed twice with 0.1% Tween20 (ThermoFisher, 28320) in PBS (Sigma-Aldrich, D8537-500 ML) for 5 min. After washing, cells were fixed with 4% formaldehyde (Sigma-Aldrich, F8775) for 10 min and washed three times in PBS (Sigma-Aldrich, D8537-500 ML) prior to imaging.
Immunofluorescence Labeling for DNA-PAINT Measurements
For immunostaining, cells were seeded at a concentration of 2.5 × 104 cells/well into eight-chambered cover glass systems with a high-performance cover glass (Cellvis, C8-1.5H-N) and grown until proper adhesion at 37 °C and 5% CO2. For microtubule immunostaining, cells were washed with prewarmed (37 °C) PBS (Sigma-Aldrich, D8537-500 ML) and permeabilized for 2 min with 0.3% glutaraldehyde (GA) + 0.25% Triton X-100 (EMS, 16220 and ThermoFisher, 28314) in prewarmed (37 °C) cytoskeleton buffer (CB), consisting of 10 mM MES ((Sigma-Aldrich, M8250), pH 6.1), 150 mM NaCl (Sigma-Aldrich, 55886), 5 mM EGTA (Sigma-Aldrich, 03777), 5 mM glucose (Sigma-Aldrich, G7021), and 5 mM MgCl2 (Sigma-Aldrich, M9272). After permeabilization, cells were fixed with a prewarmed (37 °C) solution of 2% GA for 10 min. Afterward, cells were incubated for 5 min with 100 mM glycine (Ajinomoto, G5417) to stop fixation and then washed twice with PBS (Sigma-Aldrich, D8537-500 ML). Samples were reduced with 0.1% sodium borohydride (Sigma-Aldrich, 71320) in PBS for 7 min and again washed three times with PBS (Sigma-Aldrich, D8537-500 ML) before blocking with 5% BSA (Roth, #3737.3) for 30 min. After washing with PBS (Sigma-Aldrich, D8537-500 ML) cells were incubated with primary antibody (rabbit anti-alpha tubulin antibody, abcam, ab18251, 5 μg/mL) for 1 h at 20 °C followed by three washing steps with PBS (Sigma-Aldrich, D8537-500 ML). Next, cells were incubated with secondary goat anti-rabbit IgG labeled with PEG4-TCO (10 μg/mL) for 1 h at 20 °C and again washed three times with PBS (Sigma-Aldrich, D8537-500 ML). After washing, cells were incubated with methyl-tetrazine-modified DNA strands (biomers.net GmbH, custom-made, 10 μg/mL) for 15 min at RT. The docking strand was attached via a click chemical reaction using a tetrazine-functionalized oligonucleotide. The oligonucleotide sequence 5′-TTTCTTCATTA-3′ was 5′-modified with a methyl-tetrazine (biomers.net GmbH, Ulm, Germany).33 Finally, cells were washed three times with PBS (Sigma-Aldrich, D8537-500 ML) and stored at 4 °C until imaging.
Ensemble Absorption and Fluorescence Emission Measurements
Steady-state absorption spectra were recorded on a V-650 spectrophotometer (Jasco). Samples were measured in a 0.3 mm path-length fluorescence cuvette (Hellma, 105.251-QS) in PBS (Sigma-Aldrich, D8537-500 ML) or 1 M CAPSO (CarlRoth, 5584.2). The temperature was controlled using a Peltier thermocouple set to 25 °C. Concentrations were kept at 1 μM. Dimerization and aggregation of dyes, respectively, were analyzed in a 0.3 mm path-length fluorescence cuvette (Hellma, 105.251-QS) using 1 μM concentrated dye-labeled secondary antibody solutions in PBS (Sigma, D8537-500 ML), pH 7.4, or 1 M CAPSO (CarlRoth, 5584.2), pH 10.
Ensemble Irradiation Experiments
Irradiation experiments were carried out in a custom-made cuvette holder using laser irradiation at 647 nm provided by an argon–krypton laser (Toptica, iBEAM smart_640S_11598) and fluorescence detection by a fiber optic spectrometer (Ocean Optics, USB2000). For the experiments, a 0.3 mm path-length fluorescence cuvette (Hellma, 105.251-QS) containing 50 μL of a 1 μM dye-labeled antibody solution was used to ensure that the whole volume was irradiated. The emission was recorded behind a band-pass filter (FF01-835/70, Semrock).
Wide-Field Photoconversion Experiments
The immunostained cells were measured on a custom-built wide-field setup equipped with an inverted wide-field fluorescence microscope (IX-71, Olympus) containing an oil-immersion objective (Olympus, APON 60XOTIRF, NA 1.49) and a nosepiece stage (IX2-NPS, Olympus) and two electron-multiplying CCD cameras (both iXon Ultra 897). The samples were excited with the appropriate laser systems (Coherent, Genesis MX 639 and MX 561) in TIRF illumination mode. A dichromatic mirror (FF650-Di01-25×36, Semrock) was used to separate excitation and emitted light. The fluorescence emission was collected by the same objective and transmitted by the dichroic beam splitter and several detection filters (FF01-679/41-25 and FF01-835/70, Semrock), before being projected onto two electron-multiplying CCD cameras (both iXon Ultra 897, Andor).
SMLM Imaging
Super-resolution imaging was performed using an inverted wide-field fluorescence microscope (IX-71; Olympus). A 641 nm diode laser (Cube 640-100C, Coherent), in combination with a clean-up filter (laser clean-up filter 640/10, Chroma) was used. The laser beam was focused onto the back focal plane of the oil-immersion objective (60×, NA 1.45; Olympus). Emission light was separated from the illumination light using a dichroic mirror (HC 560/659; Semrock) and spectrally filtered by a band-pass filter (FF01-679/41-25, Semrock). Images were recorded with an electron-multiplying CCD camera chip (iXon DU-897; Andor). The pixel size for data analysis was measured to 128 nm. Microtubules were imaged by total internal reflection illumination. Experiments were performed in 1 M CAPSO buffer (CarlRoth, 5584.2) adjusted to pH 9.5. All SMLM data were analyzed with rapidSTORM3.3.46 The localization precision was calculated according to Mortensen et al.47
DNA-PAINT Imaging
Super-resolution imaging via DNA-PAINT was performed using an inverted wide-field fluorescence microscope (IX-71; Olympus). For excitation of Cy5 and Cy7, a 641 nm diode laser (Cube 640-100C, Coherent) in combination with a clean-up filter (laser clean-up filter 640/10, Chroma) was used. The laser beam was focused onto the back focal plane of the oil-immersion objective (60×, NA 1.45; Olympus). Emission light was separated from the illumination light using a dichroic mirror (HC 560/659; Semrock) and spectrally filtered by a band-pass filter (FF01-679/41-25, Semrock). Images were recorded with an electron-multiplying CCD camera chip (iXon DU-897; Andor). Pixel size for data analysis was measured to 128 nm. For DNA-PAINT measurement (Figure 6), 50 000 images with an exposure time of 100 ms (frame rate 10 Hz) and irradiation intensity of 0.5 kW cm–2 were recorded. For DNA-PAINT measurement (Figure S19), 24 000 images with an exposure time of 20 ms (frame rate 50 Hz) and irradiation intensity of 1.5 kW cm–2 were recorded. Microtubules were imaged by TIRF microscopy. Experiments were performed in imaging buffer (Figure 6b and Figure S19b) or imaging buffer supplemented with 10 mM histidine (Figure 6c and Figure S19a,c) at pH 7.4. All DNA-PAINT results were analyzed with rapidSTORM3.3.46 The resolution was estimated via the Fourier ring correlation (FRC)28 plug-in in Fiji with the threshold set to 1/7.48
Improvement of Sparse Localization Images Using Deep Learning
After fitting, localization images with a pixel size (binning) of 20 nm were reconstructed. Subsequently, the reconstructed images were sent through an artificial neural network (ANN) trained to reconstruct dense images of microtubules from sparse localization data.15 Prior to ANN prediction, images were padded to a size of 512 × 512 pixels to match the size of the network input layer.
Safety Hazards
No unexpected or unusually high safety hazards were encountered.
Acknowledgments
S.S.M. and M.J.S. were supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute (NCI), Center for Cancer Research. We thank the Biophysics Resource in the Structural Biophysics Laboratory, Center for Cancer Research, NCI at Frederick for assistance with liquid chromatography–mass spectrometry analysis. S.S.M thanks Ms. Monika Arumalla for assistance with Tableau. M.M. and M.S. acknowledge financial support by the Deutsche Forschungsgemeinschaft (SFB/TR 240, project number 374031971, A07). D.A.H., G.B., and M.S. acknowledge financial support by the European Regional Development Fund (EFRE project: “Center for Personalized Molecular Immunotherapy”). A.G. acknowledges the National Science Foundation (CHE-1856765). Computations were carried out using Comet, the Extreme Science and Engineering Discovery Environment (XSEDE) cluster at the San Diego Supercomputer Center, which is supported by the NSF (ACI-1548562) through allocation CHE-200050. The figures were in part created with BioRender.com and CYLview.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.1c00483.
Supplementary figures and tables, general experimental details, synthetic procedures, including NMR spectra, for the synthesis of 2-d3 and 2-d6, computational methods, including coordinates (PDF)
Author Contributions
# S.S.M. and D.A.H. contributed equally. S.S.M. carried out the mechanistic experiments. S.S.M., M.J.S., and A.G. developed the mechanistic model. A.G. carried out the DFT calculations. D.A.H., M.M., G.B, and P.K. carried out the microscopy experiments. M.J.S. and M.S. initiated the project. S.S.M., A.G., M.J.S., and M.S. wrote the manuscript. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Footnotes
The identification of 13, or byproducts derived from it, has not been possible due to the low concentrations (low micromolar) employed for these reactions.
Supplementary Material
References
- Wiedenmann J.; Ivanchenko S.; Oswald F.; Schmitt F.; Rocker C.; Salih A.; Spindler K. D.; Nienhaus G. U. EosFP, a fluorescent marker protein with UV-inducible green-to-red fluorescence conversion. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 15905. 10.1073/pnas.0403668101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzig E.; Patterson G. H.; Sougrat R.; Lindwasser O. W.; Olenych S.; Bonifacino J. S.; Davidson M. W.; Lippincott-Schwartz J.; Hess H. F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642. 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Jradi F. M.; Lavis L. D. Chemistry of photosensitive fluorophores for single-molecule localization microscopy. ACS Chem. Biol. 2019, 14, 1077. 10.1021/acschembio.9b00197. [DOI] [PubMed] [Google Scholar]
- Gorka A. P.; Nani R. R.; Schnermann M. J. Cyanine polyene reactivity: scope and biomedical applications. Org. Biomol. Chem. 2015, 13, 7584. 10.1039/C5OB00788G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butkevich A. N.; Bossi M. L.; Lukinavičius G. v.; Hell S. W. Triarylmethane fluorophores resistant to oxidative photobluing. J. Am. Chem. Soc. 2019, 141, 981. 10.1021/jacs.8b11036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans N. Photofading of rhodamine dyes: I—Identification of some rhodamine B photoproducts. J. Soc. Dyers Colour. 1970, 86, 174. 10.1111/j.1478-4408.1970.tb02946.x. [DOI] [Google Scholar]
- Robinson-Duggon J.; Marino-Ocampo N.; Barrias P.; Zuniga-Nunez D.; Gunther G.; Edwards A. M.; Greer A.; Fuentealba D. Mechanism of visible-light photooxidative demethylation of toluidine blue O. J. Phys. Chem. A 2019, 123, 4863. 10.1021/acs.jpca.9b03588. [DOI] [PubMed] [Google Scholar]
- Gorka A. P.; Nani R. R.; Schnermann M. J. Harnessing cyanine reactivity for optical imaging and drug delivery. Acc. Chem. Res. 2018, 51, 3226. 10.1021/acs.accounts.8b00384. [DOI] [PubMed] [Google Scholar]
- Sato K.; Nagaya T.; Nakamura Y.; Harada T.; Nani R. R.; Shaum J. B.; Gorka A. P.; Kim I.; Paik C. H.; Choyke P. L.; Schnermann M. J.; Kobayashi H. Impact of C4′-O-alkyl linker on in vivo pharmacokinetics of near-infrared cyanine/monoclonal antibody conjugates. Mol. Pharmaceutics 2015, 12, 3303. 10.1021/acs.molpharmaceut.5b00472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone M. B.; Veatch S. L. Far-red organic fluorophores contain a fluorescent impurity. ChemPhysChem 2014, 15, 2240. 10.1002/cphc.201402002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok S. J.; Choi M.; Bhayana B.; Zhang X.; Ran C.; Yun S.-H. Two-photon excited photoconversion of cyanine-based dyes. Sci. Rep. 2016, 6, 23866. 10.1038/srep23866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson A. L.; Fujisaki J.; Wu J.; Runnels J. M.; Turcotte R.; Celso C. L.; Scadden D. T.; Strom T. B.; Lin C. P. Tracking single cells in live animals using a photoconvertible near-infrared cell membrane label. PLoS One 2013, 8, e69257. 10.1371/journal.pone.0069257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirix L.; Kennes K.; Fron E.; Debyser Z.; Van der Auweraer M.; Hofkens J.; Rocha S. Photoconversion of far-red organic dyes: Implications for multicolor super-resolution imaging. ChemPhotoChem. 2018, 2, 433. 10.1002/cptc.201700216. [DOI] [Google Scholar]
- Helmerich D. A.; Beliu G.; Matikonda S. S.; Schnermann M. J.; Sauer M. Photoblueing of organic dyes can cause artifacts in super-resolution microscopy. Nat. Methods 2021, 18, 253–257. 10.1038/s41592-021-01061-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W.; Aristov A.; Lelek M.; Hao X.; Zimmer C. Deep learning massively accelerates super-resolution localization microscopy. Nat. Biotechnol. 2018, 36, 460. 10.1038/nbt.4106. [DOI] [PubMed] [Google Scholar]
- Nani R. R.; Kelley J. A.; Ivanic J.; Schnermann M. J. Reactive species involved in the regioselective photooxidation of heptamethine cyanines. Chemical Science 2015, 6, 6556. 10.1039/C5SC02396C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers G. W.; Gross S.; Henrichs P. M. Direct and sensitized photooxidation of cyanine dyes. Photochem. Photobiol. 1976, 23, 37. 10.1111/j.1751-1097.1976.tb06768.x. [DOI] [PubMed] [Google Scholar]
- Gorka A. P.; Schnermann M. J. Harnessing cyanine photooxidation: from slowing photobleaching to near-IR uncaging. Curr. Opin. Chem. Biol. 2016, 33, 117. 10.1016/j.cbpa.2016.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michie M. S.; Götz R.; Franke C.; Bowler M.; Kumari N.; Magidson V.; Levitus M.; Loncarek J.; Sauer M.; Schnermann M. J. Cyanine conformational restraint in the far-red range. J. Am. Chem. Soc. 2017, 139, 12406. 10.1021/jacs.7b07272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciano M. P.; Crooke S. N.; Nourian S.; Dingle I.; Nani R. R.; Kline G.; Patel N. L.; Robinson C. M.; Difilippantonio S.; Kalen J. D.; Finn M. G.; Schnermann M. J. A nonaggregating heptamethine cyanine for building brighter labeled biomolecules. ACS Chem. Biol. 2019, 14, 934. 10.1021/acschembio.9b00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan M.; Xu D.; Guo L.; Tam D.; Liu L.; Chen Y.; Wong M.; Lo P. Cyanine fluorophores for cellular protection against ROS in stimulated macrophages and two-photon ROS detection. Org. Biomol. Chem. 2015, 13, 7307. 10.1039/C5OB00643K. [DOI] [PubMed] [Google Scholar]
- Redmond R. W.; Gamlin J. N. A compilation of singlet oxygen yields from biologically relevant molecules. Photochem. Photobiol. 1999, 70, 391. 10.1111/j.1751-1097.1999.tb08240.x. [DOI] [PubMed] [Google Scholar]
- Turro N. J.; Chow M. F. Mechanism of thermolysis of endoperoxides of aromatic compounds. Activation parameters, magnetic field, and magnetic isotope effects. J. Am. Chem. Soc. 1981, 103, 7218. 10.1021/ja00414a029. [DOI] [Google Scholar]
- O’Shea K. E.; Foote C. S. Chemistry of singlet oxygen. 51. Zwitterionic intermediates from 2,4-hexadienes. J. Am. Chem. Soc. 1988, 110, 7167. 10.1021/ja00229a035. [DOI] [Google Scholar]
- Clennan E. L.; Lewis K. K. Nucleophilic trapping of intermediates in the singlet oxygenations of isomeric 1,4-Di-tert-butoxy-1,3-butadienes. J. Org. Chem. 1986, 51, 3721. 10.1021/jo00369a037. [DOI] [Google Scholar]
- Turque O.; Greer A.; Wauchope O. R. Synthetic feasibility of oxygen-driven photoisomerizations of alkenes and polyenes. Org. Biomol. Chem. 2020, 18, 9181. 10.1039/D0OB01993C. [DOI] [PubMed] [Google Scholar]
- Sauer M.; Heilemann M. Single-molecule localization microscopy in eukaryotes. Chem. Rev. 2017, 117, 7478. 10.1021/acs.chemrev.6b00667. [DOI] [PubMed] [Google Scholar]
- Chozinski T. J.; Gagnon L. A.; Vaughan J. C. Twinkle, twinkle little star: Photoswitchable fluorophores for super-resolution imaging. FEBS Lett. 2014, 588, 3603. 10.1016/j.febslet.2014.06.043. [DOI] [PubMed] [Google Scholar]
- Sengupta P.; van Engelenburg S. B.; Lippincott-Schwartz J. Superresolution imaging of biological systems using photoactivated localization microscopy. Chem. Rev. 2014, 114, 3189. 10.1021/cr400614m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozma E.; Kele P. Fluorogenic probes for super-resolution microscopy. Org. Biomol. Chem. 2019, 17, 215. 10.1039/C8OB02711K. [DOI] [PubMed] [Google Scholar]
- Lukinavicius G.; Umezawa K.; Olivier N.; Honigmann A.; Yang G.; Plass T.; Mueller V.; Reymond L.; Correa Jr I. R.; Luo Z.-G.; Schultz C.; Lemke E. A.; Heppenstall P.; Eggeling C.; Manley S.; Johnsson K. A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins. Nat. Chem. 2013, 5, 132. 10.1038/nchem.1546. [DOI] [PubMed] [Google Scholar]
- Uno S.-n.; Kamiya M.; Yoshihara T.; Sugawara K.; Okabe K.; Tarhan M. C.; Fujita H.; Funatsu T.; Okada Y.; Tobita S.; Urano Y. A spontaneously blinking fluorophore based on intramolecular spirocyclization for live-cell super-resolution imaging. Nat. Chem. 2014, 6, 681. 10.1038/nchem.2002. [DOI] [PubMed] [Google Scholar]
- Schnitzbauer J.; Strauss M. T.; Schlichthaerle T.; Schueder F.; Jungmann R. Super-resolution microscopy with DNA-PAINT. Nat. Protoc. 2017, 12, 1198. 10.1038/nprot.2017.024. [DOI] [PubMed] [Google Scholar]
- Jungmann R.; Avendaño M. S.; Woehrstein J. B.; Dai M.; Shih W. M.; Yin P. Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and Exchange-PAINT. Nat. Methods 2014, 11, 313. 10.1038/nmeth.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schueder F.; Lara-Gutiérrez J.; Beliveau B. J.; Saka S. K.; Sasaki H. M.; Woehrstein J. B.; Strauss M. T.; Grabmayr H.; Yin P.; Jungmann R. Multiplexed 3D super-resolution imaging of whole cells using spinning disk confocal microscopy and DNA-PAINT. Nat. Commun. 2017, 8, 2090. 10.1038/s41467-017-02028-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auer A.; Strauss M. T.; Schlichthaerle T.; Jungmann R. Fast, background-free DNA-PAINT imaging using FRET-based probes. Nano Lett. 2017, 17, 6428. 10.1021/acs.nanolett.7b03425. [DOI] [PubMed] [Google Scholar]
- Lee J.; Park S.; Hohng S. Accelerated FRET-PAINT microscopy. Mol. Brain 2018, 11, 70. 10.1186/s13041-018-0414-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwenhuizen R. P.; Lidke K. A.; Bates M.; Puig D. L.; Grünwald D.; Stallinga S.; Rieger B. Measuring image resolution in optical nanoscopy. Nat. Methods 2013, 10, 557. 10.1038/nmeth.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schueder F.; Stein J.; Stehr F.; Auer A.; Sperl B.; Strauss M. T.; Schwille P.; Jungmann R. An order of magnitude faster DNA-PAINT imaging by optimized sequence design and buffer conditions. Nat. Methods 2019, 16, 1101. 10.1038/s41592-019-0584-7. [DOI] [PubMed] [Google Scholar]
- Grimm J. B.; Klein T.; Kopek B. G.; Shtengel G.; Hess H. F.; Sauer M.; Lavis L. D. Synthesis of a far-red photoactivatable silicon-containing rhodamine for super-resolution microscopy. Angew. Chem., Int. Ed. 2016, 55, 1723. 10.1002/anie.201509649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nani R. R.; Gorka A. P.; Nagaya T.; Yamamoto T.; Ivanic J.; Kobayashi H.; Schnermann M. J. In vivo activation of duocarmycin-antibody conjugates by near-infrared light. ACS Cent. Sci. 2017, 3, 329. 10.1021/acscentsci.7b00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubry J.-M.; Cazin B.; Duprat F. Chemical sources of singlet oxygen. 3. Peroxidation of water-soluble singlet oxygen carriers with the hydrogen peroxide-molybdate system. J. Org. Chem. 1989, 54, 726. 10.1021/jo00264a046. [DOI] [Google Scholar]
- Frisch M.; Trucks G.; Schlegel H.; Scuseria G.; Robb M.; Cheeseman J.; Scalmani G.; Barone V.; Petersson G.; Nakatsuji H.. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, 2016.
- Dennington R.; Keith T.; Millam J.. GaussView 5.0; Semichem Inc.: Shawnee Mission, KS, 2009.
- Jensen F.Introduction to Computational Chemistry, 2nd ed.; Wiley: Chichester, U.K., 2007. [Google Scholar]
- Wolter S.; Löschberger A.; Holm T.; Aufmkolk S.; Dabauvalle M.-C.; Van De Linde S.; Sauer M. rapid STORM: accurate, fast open-source software for localization microscopy. Nat. Methods 2012, 9, 1040. 10.1038/nmeth.2224. [DOI] [PubMed] [Google Scholar]
- Mortensen K. I.; Churchman L. S.; Spudich J. A.; Flyvbjerg H. Optimized localization analysis for single-molecule tracking and super-resolution microscopy. Nat. Methods 2010, 7, 377. 10.1038/nmeth.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J.; Arganda-Carreras I.; Frise E.; Kaynig V.; Longair M.; Pietzsch T.; Preibisch S.; Rueden C.; Saalfeld S.; Schmid B.; Tinevez J.-Y.; White D. J.; Hartenstein V.; Eliceiri K.; Tomancak P.; Cardona A. Fiji: an open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.