

Abstract
Despite its central role in tumor progression and treatment resistance, poor vascularization that necessitates penetration of therapeutics through tumor extracellular matrix (ECM) constitutes a significant challenge to managing tumor hypoxia via conventional systemic treatment regimens. In addition, methods to target hypoxic tumor cells are lacking. Here, we discovered that human ferritin nanocages (FTn) possess an intrinsic ability to preferentially engage with hypoxic tumor tissues, in addition to normoxic tumor areas. We also developed a simple method of endowing FTn with spatially controlled “mosaic” surface polyethylene glycol (PEG) coatings that facilitate deep penetration of FTn through ECM to reach hypoxic tumor tissues while retaining its inherent hypoxia-tropic property. Hypoxia-inhibiting agents systemically delivered via this surface-PEGylated FTn were readily accumulated in hypoxic tumor tissues, thereby providing significantly enhanced therapeutic benefits compared to the identical agents delivered in solution as a stand-alone therapy or an adjuvant to restore efficacy of conventional systemic chemotherapy.
Keywords: ferritin nanocage, PEGylation, hypoxia, tumor penetration, HIF-1α, drug resistance
Graphical Abstract
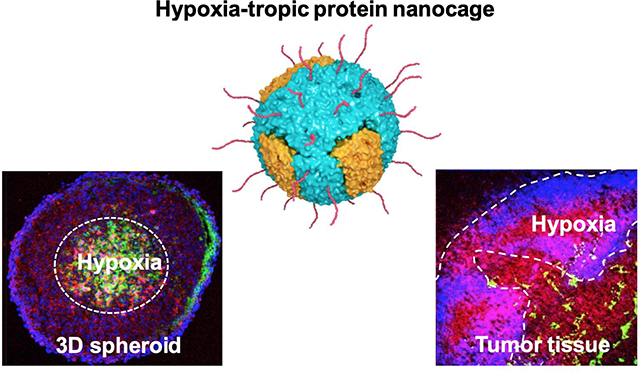
Hypoxia, attributed to spatially disorganized tumor vasculature and unstable blood perfusion, is a common feature shared by most solid tumors in humans.1 Tumor hypoxia is associated with increased invasiveness and metastasis of tumors, development of immunosuppressive environment, resistance to chemo- and radiotherapy and high mortality rates.2–5 Tumor hypoxia mediates upregulation and/or activation of hypoxia-inducible factor-1α (HIF-1α), a key transcription factor that triggers expression of a panel of genes essential for tumor adaption to an oxygen-deficient hypoxic environment.6 For example, HIF-1α upregulates multidrug resistance protein 1 (MDR1), which leads to treatment resistance to standard-of-care chemotherapy.7 Thus, pharmacological HIF-1α inhibition can potentially restore tumor susceptibility to chemotherapy.
Despite the clear rationale for combination therapies with HIF-1α inhibitors and chemotherapy, efficient delivery of HIF-1α inhibitors to hypoxic regions within tumor is challenging due to the microenvironment that brings about multiple biological barriers.8, 9 Hypoxic regions of tumors lack blood vessels and, thus, are not readily accessible by systemically administered therapeutic agents.10 Once beyond the systemic circulation, therapeutic agents must percolate an adhesive and dense extracellular matrix (ECM) to reach hypoxic tumor tissues,11 followed by specific tumor cell uptake. Nanoparticle-based drug delivery platforms have been widely explored as a potential means to overcome biological barriers.12 However, nano-systems capable of simultaneously tackling multiple barriers, thereby efficiently delivering therapeutic payloads to the hypoxic region of tumors, have been rarely reported.
A ferritin nanocage (FTn), composed of 24 human ferritin heavy chain subunits,13 is an attractive drug delivery platform for cancer therapy due to its intrinsic ability to extravasate tumor vasculature,14–18 to penetrate tumor tissue19 and to mediate uptake by cancer cells via a receptor-mediated endocytosis (i.e., transferrin receptor 1 or TfR1 for human; T cell immunoglobulin mucin receptor 2 or TIM-2 for mouse).20–24 Recently, we reported that surface modification of FTn with polyethylene glycol (PEG) improved their colloidal stability and reduced non-specific adhesive interactions in the physiological lung environment following inhaled administration,19 reflecting well-established roles of PEGylation on in vivo behaviors of drug delivery systems.25–27 Interestingly, TfR1 expression has been shown to increase under various hypoxic conditions,28–31 which has led to our hypothesis that FTn, in conjuction with PEGylation, may be utilized to enhance therapeutic delivery to the hypoxic regions within tumor tissues, in addition to the tumor as a whole. However, “PEG delimma” is a primary limiting factor that precludes full exploitation of benefits potentially provided by conventional PEGylation strategies regarding nanoparticle-based drug delivery.25, 32 To this end, we engineered PEGylated hybrid FTn (hybrid PEG-FTn) using a uniquely controllable disassembly/reassembly method to precisely modulate the surface density and distribution pattern of the PEG coatings. We then tested the ability of hybrid PEG-FTn to overcome a series of biological barriers and deliver HIF-1α inhibitor payloads to lung cancer, including the hypoxic tumor regions, when administered systemically.
RESULTS AND DISCUSSION
TfR1 and TIM-2 upregulation in hypoxia
To determine the relevance of using FTn for tumor hypoxia targeting, we first confirmed the prior findings that TfR1 expression was elevated in hypoxic conditions.28–31 We found that the level of TfR1 expression significantly increased in vitro in A549 human lung carcinoma cells that were maintained at hypoxia (i.e., 1% O2) compared to those grown at normoxia (i.e., 21% O2) (Figure S1a). Further, enhanced TfR1 expression was observed regardless of oxygen level when A549 cells were transfected with a plasmid vector encoding HIF-1α (Figure S1b), in agreement with the previously established HIF-1α-mediated TfR1 upregulation.28–30 Of note, molecular linkage between HIF-1α and TfR1 has been mechanistically determined by the identification of a binding site for HIF-1α located within the the human TfR1 promoter region.28, 33
TIM-2 has been previously uncovered as a receptor for human ferritin heavy chain protein in mouse,22, 23 but impact of hypoxia on TIM-2 expression has not been reported. We thus analyzed for co-regulating transcript factors using INSECT 2.0 (IN-silico SEarch for Co-occurring Transcription factors)34 where the mouse TIM-2 promotor was found to possess five potential transcription factor binding sites (−1139 to −1116) for the transcriptionally active HIF-1α/ HIF-1β heterodimer.35 To this end, we hypotheszed that hypoxia would upregulate TIM-2 in mice in a similar manner as TfR1 in humans. We then confirmed using western blot analysis that TIM-2 was upregulated when 3LL mouse Lewis lung carcinoma cells were incubated in a hypoxic condition or induced to overexpress HIF-1α (Figures S1a, b). We next established a 3LL-based flank tumor model in mice to validate our in vitro observation. Immunostaining revealed abundant stroma as well as limited blood vessel distribution in hypoxic regions within the tumor with vessel density less than one third of that in normoxic tumor regions of the same tumor (Figure S2). Consistent with the in vitro observation, the TIM-2 level was significantly elevated in hypoxic over normoxic regions, as evidenced by the significantly greater co-localization of TIM-2 and hypoxia stainings (Figure S1c). We further observed preferential accumulation of FTn within the hypoxic area developed in the core of 3LL multicellular spheroids (Figure S1d), providing a rationale for the use of FTn or perhaps hybrid PEG-FTn as a delivery system for targeting hypoxic tumors.
Tumor penetration of hybrid PEG-FTn
We recently introduced a simple method to endow FTn with controlled surface functionality by blending 12 of each non-PEGylated and PEGylated ferritin subunits to produce hybrid PEG-FTn for inhaled delivery of chemotherapeutic agents for the local treatment of lung cancer.19 Here, we further optimized the hybrid PEG-FTn specifically for efficient delivery of payloads to solid tumors, particularly hypoxic areas, after systemic administration. Hybrid PEG-FTn candidates were engineered at varying ratios of non-PEGylated and PEGylated ferritin subunits to determine a formulation providing excellent serum stability and blood circulation time, thus potentially maximizing the benefit of its ability to penetrate tumor tissue and target cancer cells, including those in hypoxic tumor areas. Using a pH-based disassembly/reassembly method,36 hybrid PEG-FTn candidates were formulated with 25%, 37.5%, 50%, 75% or 87.5% inclusion of PEGylated ferritin subunits (i.e., 6, 9, 12, 18 and 21 PEGylated subunits, respectively, out of the total of 24 subunits required to form a single hybrid PEG-FTn) and the respective rest of non-PEGylated subunits (Figure 1a).
Figure 1. In vitro characterization of hybrid PEG-FTn: identification of an optimal hybrid PEG-FTn formulation.
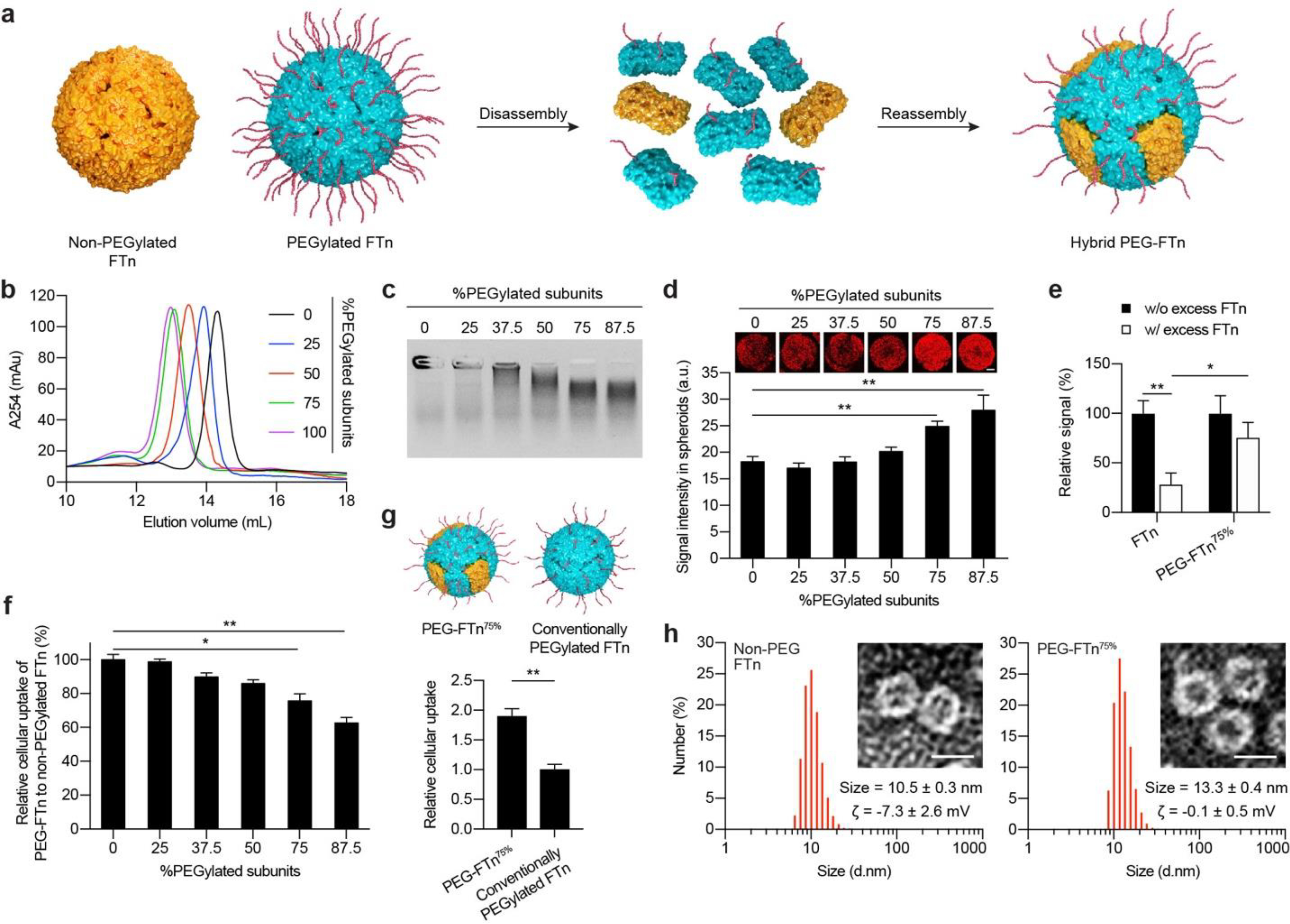
(a) Schematic illustration of hybrid PEG-FTn preparation via a sequential disassembly and reassembly procedure. (b) Size exclusion chromatography of hybrid PEG-FTn engineered with varying factions of PEGylated ferritin subunits. (c) Colloidal stability of hybrid PEG-FTn candidates in 10% human plasma. (d) Penetration of hybrid PEG-FTn candidates through 3LL multicellular spheroids. Representative 3D-reconstructed images of the spheroids treated with Cy5-labeled PEG-FTn are shown above the graph. Scale bar = 100 pm. (e) Relative penetration of FTn or PEG-FTn75% through 3LL-based spheroids in the absence or presence of an excess of unlabeled FTn. (f) Cellular uptake of hybrid PEG-FTn candidates by 3LL cells. The FTn and hybrid PEG-FTn tested in (d-g) include three Cy5-labeled ferritin subunits out of 24 subunits (12.5%) for their fluorometric and microscopic detection. (g) Comparative cellular uptake of PEG-FTn75% and conventionally PEGylated FTn by 3LL cells. (Upper) Schematic illustration of PEG-FTn75% and conventionally PEGylated FTn. Both formulations possess comparable amounts of overall surface PEG content while the spatial distributions are different. (Lower) Fluorometric analysis of cellular uptake. (h) Hydrodynamic diameters and morphology of (Left) non-PEGylated FTn and (Right) PEG-FTn75% determined by dynamic light scattering and TEM, respectively. Scale bar = 10 nm. The average hydrodynamic diameters and the ζ-potentials (ζ) of each formulation are presented. *P < 0.05, **P < 0.01.
Size exclusion chromatography analysis showed that the molecular size of hybrid PEG-FTn became larger, as indicated by the shorter elution time, when the incorporated portion of PEGylated ferritin subunit increased (Figure 1b). Importantly, a clear single peak observed for each hybrid PEG-FTn confirms successful formation of uniform hybrid structures. We then evaluated colloidal stability of hybrid PEG-FTn candidates with a varying degree of PEGylation in an isotonic plasma solution. An agarose gel electrophoresis analysis revealed that particle migration enhanced as the ratio of incorporated PEGylated ferritin subunits increased, indicating that the greater the PEGylation, the less the particle aggregation and/or protein adsorption (Figure 1c).
We next assessed the ability of hybrid PEG-FTn to penetrate multicellular tumor spheroids established with 3LL cells. Multicellular spheroids have been considered as an excellent in vitro model for evaluating the penetration of hybrid PEG-FTn through tumor tissue, since ECM is formed between the tumor cells within spheroids, thereby mimicking the tumor found in vivo.37, 38 While non-PEGylated FTn and hybrid PEG-FTn engineered with ≤ 50% of PEGylated subunits exhibited comparable distribution within the spheroids, those composed of ≥ 75% of PEGylated subunits provided significantly enhanced tumor penetration in vitro (P < 0.01) (Figure 1d). This was unexpected given our prior observation that hybrid PEG-FTn composed of ≤ 50% PEGylated subunits appeared to penetrate the spheroid in a receptor-dependent manner,19 and thus, further PEGylation was expected to reduce hybrid PEG-FTn engagement with the receptor. To confirm this observation, we conducted a competition assay where in vitro tumor penetration of fluorescently labeled nanocages, including non-PEGylated FTn and hybrid PEG-FTn composed of 75% PEGylated subunits (i.e., 25% non-PEGylated subunits; PEG-FTn75% hereafter), was evaluated with or without blocking the receptors with an excess of unlabeled FTn. While the penetration of both formulations decreased upon receptor blockade by unlabeled FTn, the level of reduction was not statistically significant for the PEG-FTn75% (P ~ 0.06), in contrast to FTn that exhibited a significant dicrease (P < 0.01; Figure 1e). This finding suggests that an alternative mechanism of tumor penetration may exist for PEG-FTn75%.
To evaluate whether the PEGylation compromised the ability of FTn to interact with the receptor, we compared in vitro cellular uptake of non-PEGylated FTn and hybrid PEG-FTn candidates in 3LL cells. We found that the uptake by hybrid PEG-FTn with ≥ 75% of PEGylated subunits was significantly reduced unlike those with lower PEGylation degrees; however, the PEG-FTn75% retained ~80% of the uptake observed with non-PEGylated FTn (Figure 1f). We hypothesized that this relatively small decrease in the uptake would be attributed to the unique spatial distribution of surface PEG on the PEG-FTn75% as 25% of its surface is contributed by non-PEGylated subunits and thus allows TIM-2-mediated cancer cell uptake. To test this, we engineered a conventionally PEGylated FTn possessing a total amount of surface PEG identical to the PEG-FTn75% but evenly distributed throughout the surface (schematics in Figure 1g), and compared in vitro cellular uptake. The PEG-FTn75% exhibited double the uptake achieved by the conventionally PEGylated FTn (Figure 1g), suggesting that the hybrid strategy, providing a “mosaic” surface PEGylation pattern, was essential for retaining the intrinsic ability of FTn to bind and subsequently internalize cancer cells. Based on the panel of careful in vitro characterization thus far, we have determined PEG-FTn75% as a lead hybid system to be evaluated in subsequent studies.
Prior to in vivo assessment, we further compared physicochemical properties of non-PEGylated FTn and PEG-FTn75% where transmission electron microscopy (TEM) revealed the same cage-like structures for these two formulations. This finding underscore that our PEGylation strategy did not interfere with the reassembly cage-formation process. The incorporation of PEG on the surface of PEGF-Tn75% was confirmed by the increase in hydrodynamic diameters from 10.5 ± 0.3 to 13.3 ± 0.3 nm as well as the decrease in ζ-potentials from −7.3 ± 2.6 to −0.1 ± 0.5 mV compared to non-PEGylated FTn (Figure 1h). The more neutral surface charge of PEG-FTn75% would reduce potential electrostatic interactions of FTn surfaces with ECM components. We next confirmed excellent colloidal stability of PEG-FTn75% in human serum (i.e. 25% in PBS) using size exclusion chromatography. The PEG-FTn75% did not exhibit any measurable level of aggregation, whereas non-PEGylated FTn progressively aggregated in serum over time (Figure S3a). The stablilty of PEG-FTn75% was also retained at acidic endolysosomal pH of 4.5 (Figure S3b).
Collectively, our PEGylation approach appeared to facilitate deep penetration of FTn through the tumor tissue via two distinct mechanisms, including receptor-mediated transcytosis and passive diffusion through tumor interstitium. As expected from the relatively small but significant decrease in their receptor binding capacity by the inclusion of PEG (Figure 1f), the tumor penetration of PEG-FTn75% via the former mechanism is likely compromised to a certain degree. However, enhanced tumor penetration mediated by the latter scenario is seemingly more than enough to offset the opposite effect incurred by the reduced receptor binding capacity. To this end, the improved interstitial diffusion of PEG-FTn75% may be attributed to the reduction in interactions with ECM components within the tumor tissue39, 40 as well as the attenuation of the binding site barrier effect,41 which is enabled by partial loss of their cancer cell-binding ability. We hypothesized that the ability of PEG-FTn75% to efficiently penetrate tumor tissues, regardless of the mechanism, would be a key to achieving preferential accumulation in vessel-lacking hypoxic tumor areas following systemic administration. We thus sought to investigate whether this specific hybrid formulation, PEG-FTn75%, provided enhanced hypoxia accumulation compared to the non-PEGylated FTn which should theoretically possess the greatest intrinsic ability to accumulate in TfR1- or TIM-2-upregulated hypoxic tumors in absence of other delivery barriers found in vivo.
Hypoxia-tropic translocation of systemically administered PEG-FTn75%
We then compared in vivo behaviors of non-PEGylated FTn and PEG-FTn75%. Following systemic administration, PEG-FTn75% provided significantly improved blood pharmacokinetics compared to non-PEGylated FTn, exhibiting 5.8- and 2.9-fold greater area under a curve (AUC) and plasma half-life, respectively (Figure 2a and Table S1). We also found significantly improved accumulation of PEG-FTn75% in a 3LL-based flank tumor compared to non-PEGylated FTn, 18 h after systemic administration (P < 0.05) (Figure 2b), which correlated with a significantly enhanced accumulation of PEG-FTn75% in hypoxic tumor areas lacking blood vessels (P < 0.01) (Figure 2c). To test whether the preferential accumulation of PEG-FTn75% in hypoxic tumor was attributed not only to the prolonged blood circulation by PEGylation, but also to its superior ability to penetrate the tumor tissue, we compared in vivo distribution of non-PEGylated FTn and PEG-FTn75% following intratumoral injection. We found that PEG-FTn75% provided 3.5-fold greater distribution compared to the non-PEGylated formulation in a 3LL-based flank tumor (P < 0.05) (Figure 2d), in good agreement with our in vitro observation with 3D spheroids (Figure 1d).
Figure 2. In vivo behaviors of non-PEGylated FTn and PEG-FTn75%: superior ability of PEG-FTn75% to overcome multiple biological barriers.

(a) Plasma concentration profiles of systemically administered nanocages. (b) Accumulation of systemically administered nanocages in 3LL-based flank tumors. (Left) Representative fluorescence images and (Right) image-based quantification of nanocage accumulation in the flank tumors. (c) Accumulation of systemically administered nanocages (red) within hypoxic areas within the 3LL-based flank tumor (blue). Green color represents blood vessels. (Left) Representative confocal images and (Right) image-based quantification of nanocage accumulation within hypoxic tumor areas (n = 4). Scale bar = 100 pm. (d) Distribution of intratumorally infused nanocages in the 3LL-based flank tumor. (Left) Representative 3D-reconstructed images and (Right) image-based quantification of volumetric distribution of nanocages within the flank tumor. Scale bar = 4 mm. *P < 0.05, **P < 0.01.
To further evaluate the intratumoral behaviors of the systemically administered PEG-FTn75%, we assessed the translocation of PEG-FTn75% from vessel lumen to extravascular space within the 3LL flank tumor over time. The PEG-FTn75% were primarily found within blood vessels at 0.5 h post-administration, but gradually diffused out to the extravascular spaces (Figure S4a). Quantitative analysis showed an enhanced penetration of PEG-FTn75% away from the blood vessels through the flank tumor tissue over time; the maximum distances reached by PEG-FTn75% were approximately 20 and 100 μm at 0.5 and 3 h post-administration, respectively (Figure S4b). TEM revealed translocation of iron oxide particle-loaded PEG-FTn75% (PEG-FTn75%/IO) (Figure 3a) from tumor endothelial linings into subendothelial tumor tissues 0.5 h after systemic administration (Figure 3b). PEG-FTn75%/IO appeared to extravasate tumor vasculature via vesicle-, vesiculo-vacuolar organelle- or fenestrae-mediated pathways (Figure 3c–g), similar to a prior observation with non-PEGylated FTn,16, 18 suggesting that the PEGylation here did not undermine the intrinsic extravasating ability of FTn. In parallel, we evaluated time course distribution of systemically administered PEG-FTn75% into hypoxic regions of the flank tumor after extravasation (Figure 4a). The PEG-FTn75% were primarily found in normoxic over hypoxic tumor at the initial time point of 0.5 h post-administration most likely due to the abundance of blood vessels in the normoxic area. However, PEG-FTn75% gradually accumulated in the hypoxic area over time and roughly double the amount of PEG-FTn75% were found in hypoxic over normoxic areas 18 h after the administration (P < 0.05) (Figure 4b). The finding was further confirmed by flow cytometry analysis of cells isolated from the tumor (Figure 4c). While preferential accumulation of both non-PEGylated FTn and PEG-FTn75% in hypoxic over normoxic cells was apparent at 18 h post-administration, the amount of PEG-FTn75% found in hypoxic cells was significantly greater than that of non-PEGylated FTn (P < 0.01) (Figure 4c), in good accordance with the microscopic observations (Figure 2c).
Figure 3. Mechanisms of PEG-FTn75% extravasation across tumor blood vessels.
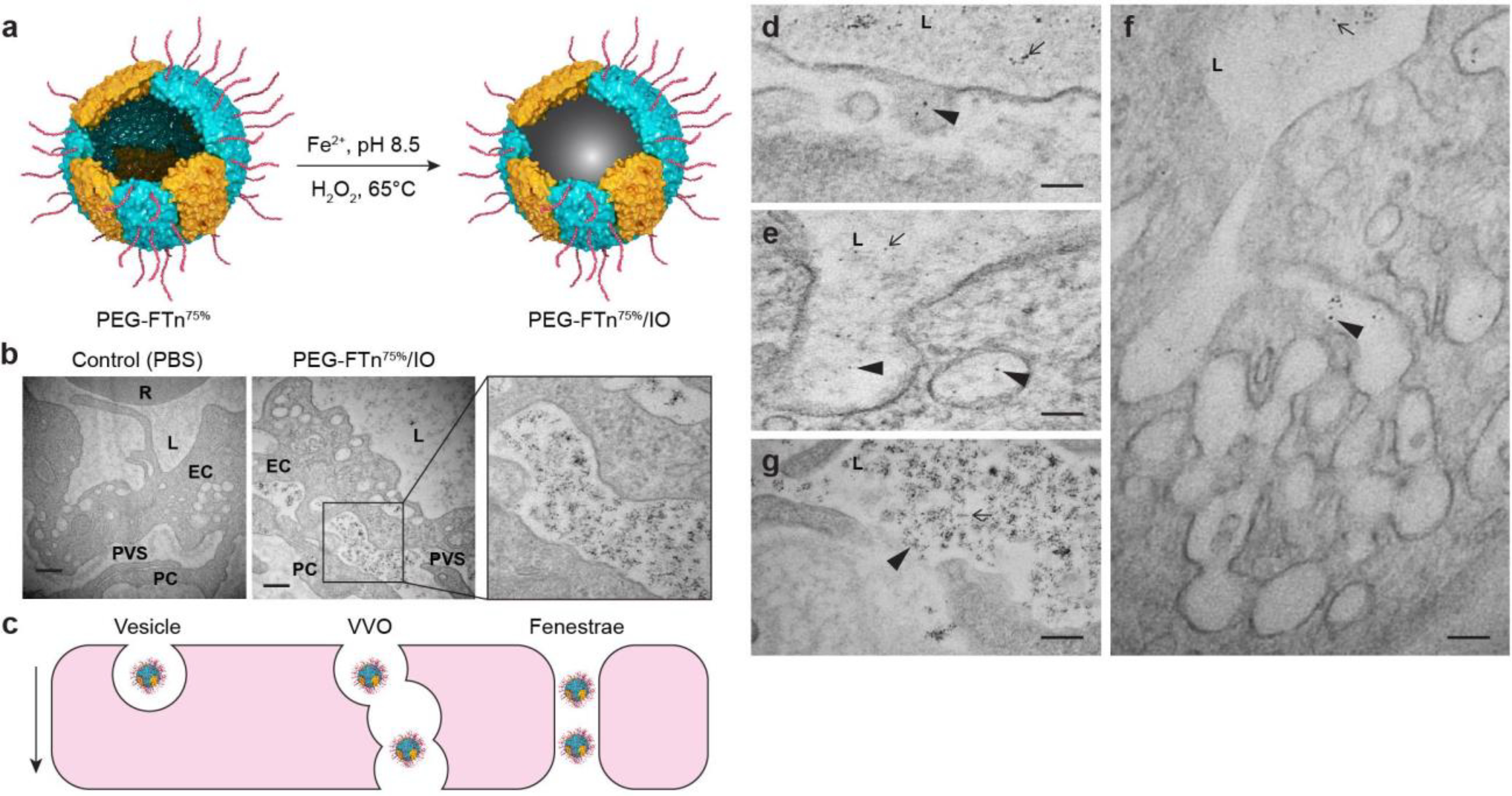
(a) Schematic illustration of iron oxide-loaded PEG-FTn75% (i.e. PEG-FTn75%/IO) preparation. (b) Representative TEM images of PEG-FTn75%/IO localization in the perivascular space within a 3LL-based flank tumor 30 minutes after the systemic administration. R: red blood cells; L: lumen; EC: endothelial cells; PC: pericytes; PVS: perivascular space. Scale bar = 500 nm. (c) Schematic illustration of various extravasation pathways exploited by systemically administered PEG-FTn75%/IO. Representative TEM images showing extravasation of systemically administered PEG-FTn75%/IO via (d, e) vesicles, (f) VVO, and (g) fenestraes. L: lumen; arrows: PEG-FTn75%/IO in the vessel lumen; arrow head: extravasation of PEG-FTn75%/IO. Scale bar = 100 nm.
Figure 4. Accumulation of systemically administered PEG-FTn75% in hypoxic areas within 3LL-based flank tumors over time.
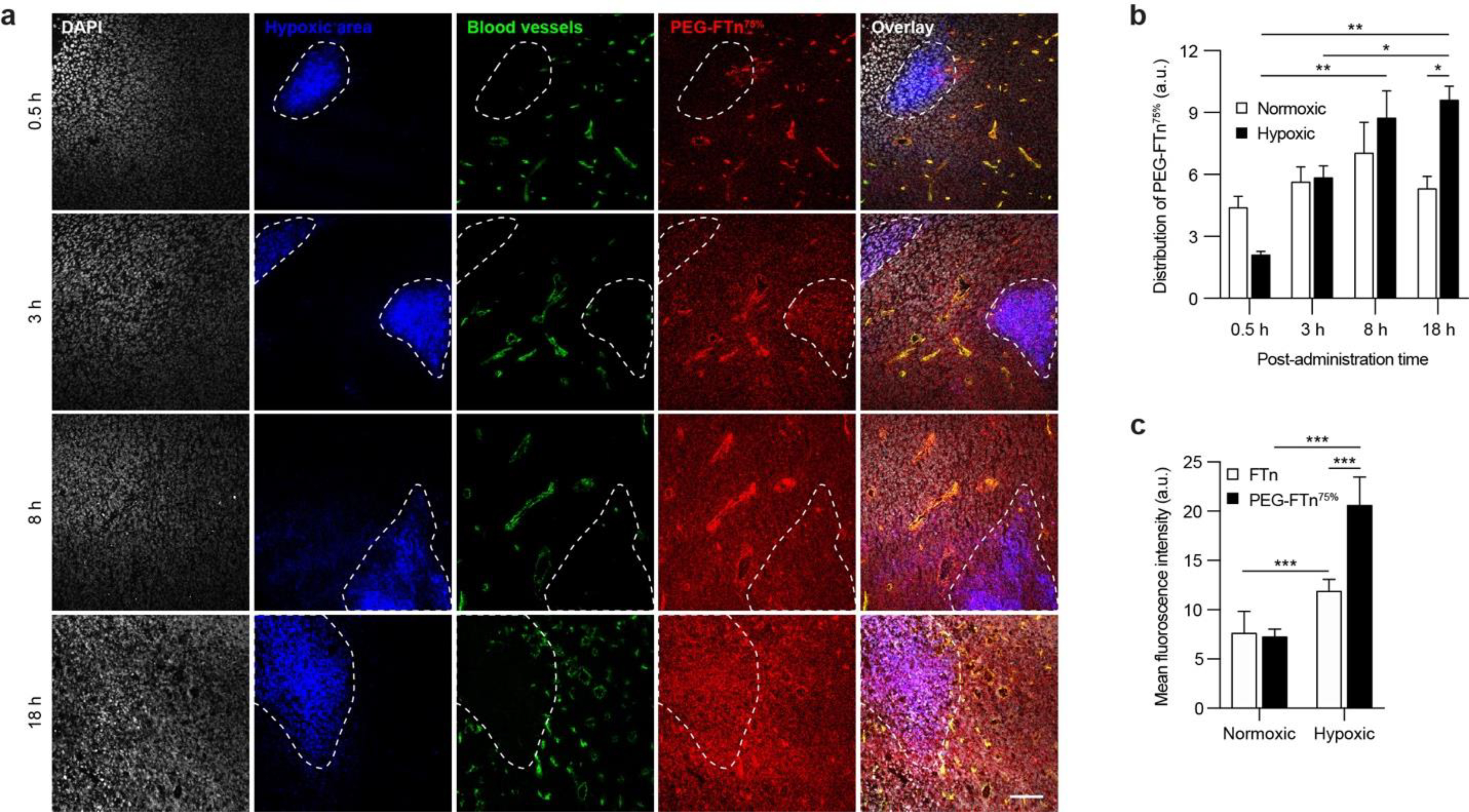
(a) Representative confocal images showing time-course accumulation of systemically administered PEG-FTn75% (red) in hypoxic tumor areas (blue). White and green colors represent cell nuclei and blood vessels, respectively. Scale bar = 100 pm. (b) Image-based quantification of PEG-FTn75% localization in normoxic and hypoxic tumor areas at different time points after the administration (n = 4). (c) Flow cytometry analysis of accumulation of PEG-FTn75%, in comparison to non-PEGylated FTn, in normoxic and hypoxic tumor areas 18 h after the administration.
Enhanced HIF-1 inhibition in tumor by acriflavine delivered by PEG-FTn75%
Based on the observed hypoxia-tropic accumulation of PEG-FTn75%, we loaded a HIF-1α inhibitor, acriflavine (AF), into the PEG-FTn75% (Figure 5a) for its delivery to hypoxic tumor regions following systemic administration. Of note, AF has been shown to restore chemotherapeutic efficacy by suppressing the HIF-1-mediated upregulation of MDR1.7, 42 We first found that the pH adjustment method used36 for the preparation and mechanistic evaluation of the PEG-FTn75% was not optimal for producing AF-loaded PEG-FTn75% (PEG-FTn75%/AF) due to the relatively low AF loading efficiency (i.e. 18 ± 6 AF molecules of per PEG-FTn75%) (Table S2). This is likely attributed to the highly acidic disassembly pH (~2) of this method where drug encapsulation takes place; acidic pH may neutralize negative charges in the inner cavity of FTn, thereby hampering electrostatic interactions between the cavity and positively charged AF molecules (pKa > 12).43 We thus employed an alternative, but equally applicable, method of using gradient concentrations of urea solution at neutral pH,13 yielding a high drug loading efficiency of 62 ± 4 AF molecules of per PEG-FTn75%. We confirmed that the alternative loading strategy did not alter the key properties of PEG-FTn75%, including its physicochemical characteristics and abilities to interact with TIM-2-positive cancer cells as well as to penetrate tumor tissues (Figure 5b and Figure S5). We then evaluated the release kinetics of AF from the PEG-FTn75%/AF where we found that the AF release was pH-dependent; less than 30% of AF were released at pH 7.4 over 48 h, whereas nearly 80% of AF were released at pH 5.0 during the same time period (Figure 5c). This finding and the observed nanocage stability in an acidic environment (i.e. pH 4.5; Figure S3b) suggest that AF will likely be released from the PEG-FTn75% primarily in intracellular acidic vesicles without nanocage disassembly following the uptake by cells.
Figure 5. In vitro characterization of, and in vitro/vivo HIF-1α inhibition by, PEG-FTn75%/AF.

(a) Schematic illustration of AF loading into the internal cavity of PEG-FTn75%. (b) Representative TEM image of PEG-FTn75%/AF. Scale bar = 100 nm. (c) Drug release kinetics of PEG-FTn75%/AF incubated at pH 5.0 and 7.4. (d) Western blot analysis of the dose- and time-dependent HIF-1a inhibition by AF and PEG-FTn75%/AF in 3LL cells in vitro. (e) Plasma concentration profiles of AF after intravenous administration of AF or PEG-FTn75%/AF. (f) The amount of AF in different organs and tumor tissue at 24 h post-administration of AF or PEG-FTn75%/AF. (g) Real-time PCR and (h) western blot analysis of in vivo expression of HIF-1a and its associated genes in 3LL-based flank tumors following systemic treatment with saline, AF or PEG-FTn75%/AF (n = 4). *P < 0.05, **P < 0.01.
To determine the ability of PEG-FTn75%/AF to inhibit HIF-1α in vitro, we first assessed the levels of HIF-1α expression in 3LL cells grown at a hypoxic condition following the treatment with AF or PEG-FTn75%/AF at different exposure times and concentrations (Figure 5d). Although the change in the HIF-1α levels were marginal when the cells were exposed to AF or PEG-FTn75%/AF at an identical dose of 2.5 μM up to 24 h, the protein expression were markedly reduced to similar levels at 48 h post-exposure. This is in good agreement with recent studies that demonstrated significant decrease in HIF-1α level by prolonged exposure to AF (> 24 h),2, 44 in contrast to an earlier observation that AF blocked the dimerization of HIF-1α rather than affecting the protein level.45 Both AF and PEG-FTn75%/AF similarly reduced HIF-1α protein levels in a dose-dependent manner after 48 h of the drug exposure, underscoring that PEG-FTn75% did not compromise the ability of AF payloads to inhibit HIF-1α.
We next explored the in vivo behaviors of PEG-FTn75%/AF in mice bearing highly aggressive 3LL-based flank tumors following systemic administration. We found that AF delivered via PEG-FTn75% exhibited significantly improved plasma profiles with a 72-fold greater AUC and significantly prolonged circulation time compared to dose-matched AF administered as free drugs (Figure 5e and Table S3). While the amounts of AF found in major organs were comparable between AF- and PEG-FTn75%/AF-treated animals at 24 h post-administration, the nanocage provided a significant 7-fold greater tumor accumulation of AF at the same time (P < 0.01; Figure 5f). This finding is most likely attributed to the ability of PEG-FTn75% to overcome multiple delivery barriers and to accumulate in tumor hypoxia as demonstrated above in a series of mechanistical studies. We subsequently evaluated in vivo HIF-1α inhibition by AF delivered via PEG-FTn75% using the identical tumor model. Each mouse bearing the flank tumor received 5 doses of either AF or PEG-FTn75%/AF (at the AF dose of 2 mg/kg) every other day. The effects of the treatments on expression of HIF-1α and HIF-1α-driven downstream genes, including vascular endothelial growth factor (VEGF), lysyl oxidase (LOX), glucose transporters 1 (GLUT1), stromal-derived factor 1 (SDF1) and stem cell factor (SCF), were assessed using real-time PCR and/or western blot analysis. We found that PEG-FTn75%/AF significantly decreased the levels of mRNA transcripts for all target genes tested; in contrast, AF was unable to do so except for the GLUT1 (Figure 5g). Similarly, the levels of HIF-1α, LOX, and VEGF proteins in the flank tumor appeared to decrease to greater extents when mice were treated with PEG-FTn75%/AF than with AF; while the levels of all monitored proteins were significantly decreased by PEG-FTn75%/AF, VEGF level alone was significantly reduced by AF (Figure 5h). We also evaluated whether systemically administered AF and/or PEG-FTn75%/AF delayed the growth of the flank tumor. Despite not being generally recognized as a strong stand-alone cytotoxic drug, AF significantly attenuated the tumor growth (P < 0.05) and the effect was further enhanced when delivered by PEG-FTn75% (P < 0.01 and 0.05 compare to the control and the AF-treated group, respectively) (Figure S6a). The observed anti-cancer effect may be due to the downregulation of tumor-promoting genes by AF (Figure 5g, h). Importantly, body weight and serum biochemistry parameters remained unperturbed when mice received repeated or high-dose administration of PEG-FTn75%/AF (Figure S6b and Table S4), suggesting no apparent systemic toxicity of our formulation.
Combinatory anti-cancer effect of cisplatin and AF-loaded PEG-FTn75%
We sought to investigate a potential role of HIF-1α in developing drug resistance in cancer cells in vitro. We first confirmed that HIF-1α and MDR1 protein levels were elevated at hypoxia but suppressed by AF treatments (Figure 6a). Based on previously observation that 3LL cells are inherently resistant to cisplatin,46 we then investigated whether mechanism of the drug resistance involved amplification of HIF-1α activity. We found that two different doses of cisplatin increased both HIF-1α and MDR1 protein levels in 3LL cells grown at hypoxia, which were downregulated by either AF or PEG-FTn75%/AF (Figure 6b), suggesting a potential restoration of chemo-sensitivity of cisplatin by AF.
Figure 6. In vivo anti-efficacy of combined treatment with PEG-FTn75%/AF and cisplatin.
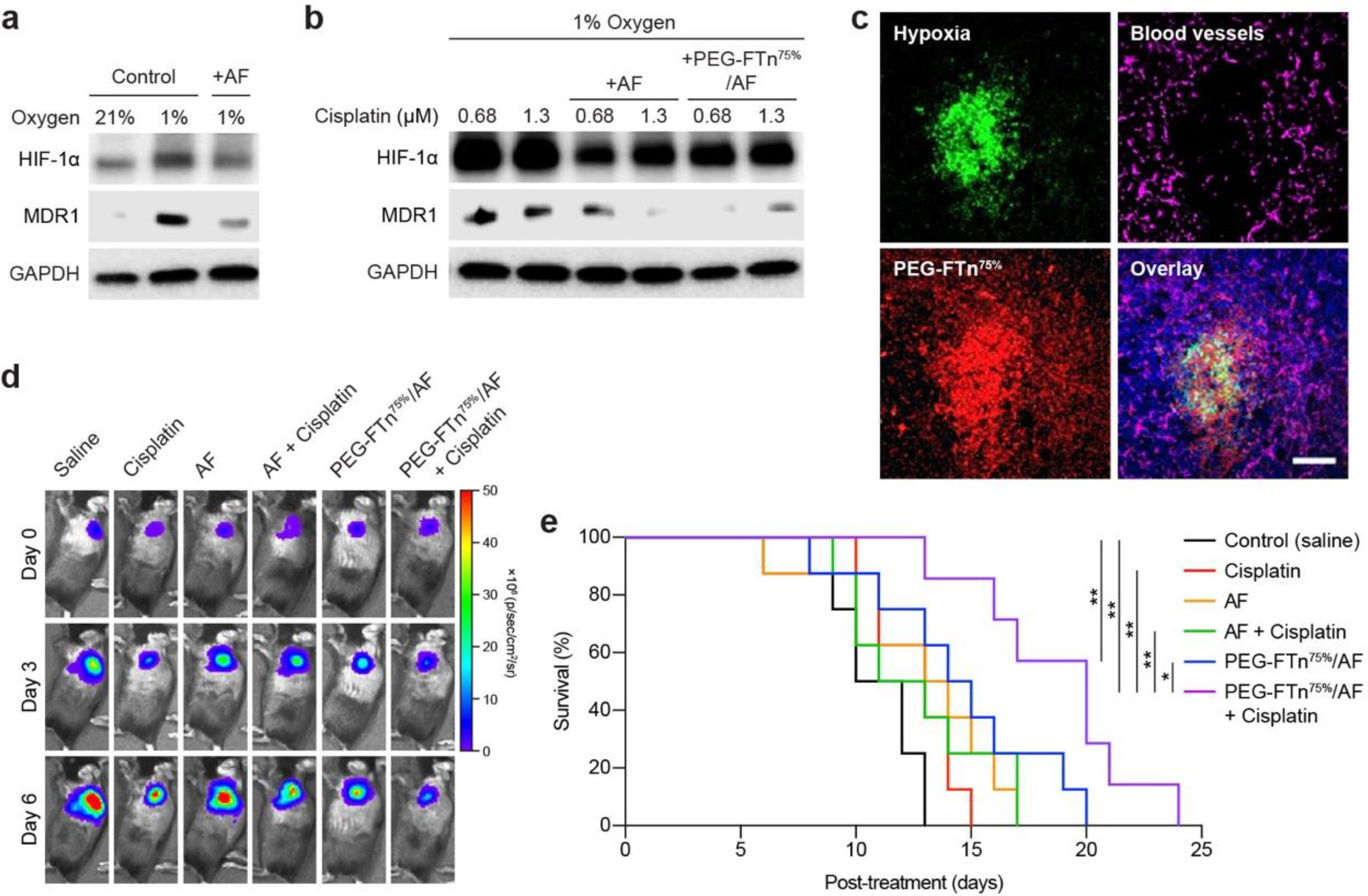
Western blot analysis of in vitro HIF-1a and MDR1 expression in 3LL cells incubated in normoxic and/or hypoxic environment (a) with or without AF treatment and (b) with cisplatin alone or in combination with AF or PEG-FTn75%/AF. (c) Representative confocal images showing accumulation of systemically administered PEG-FTn75% (red) in hypoxic areas (green) within a 3LL-based orthotopic lung tumor tissue. Magenta color represents blood vessels. Scale bar = 100 pm. In vivo anti-cancer efficacy assessed with the orthotopic tumor-bearing mice under various systemic treatment regimens (n = 7–8), in terms of (d) time-course tumor bioluminescence and (e) Kaplan-Meier survival curves. *P < 0.05, **P < 0.01.
To determine whether PEG-FTn75%/AF restores susceptibility of 3LL cells to cisplatin in vivo, we established a highly aggressive orthotopic lung cancer model by a direct intrathoracic injection of 3LL cells constitutively expressing luciferase (3LL-Luc) into the mouse lung. Bioluminescence imaging (i.e. IVIS) revealed a formation of significant tumor mass within the lung five days after the inoculation (Figure S7a). We also confirmed using western blot analysis an induction of HIF-1α and MDR1 protein expression within the tumor, which was amplified when mice were treated with cisplatin at a systemic dose of 2 mg/kg (Figure S7b). We next tested whether systemically administered PEG-FTn75% were capable of reaching the hypoxic lung tumor tissues of this orthotopic model. We found that PEG-FTn75% were highly accumulated in the hypoxic tumor area that lacked vascularization at 18 h post-administration (Figure 6c). We then went on to evaluate in vivo anti-cancer efficacy of a combined therapy of PEG-FTn75%/AF and cisplatin, compared to appropriate control groups, as depicted in Figure S8a. All treatment groups showed delayed tumor progression 6 days after the initial treatment, but the combination of PEG-FTn75%/AF and cisplatin exhibited the greatest suppressive effect on the tumor growth, as confirmed by bioluminescence signal intensity (Figure 6d and Figure S8b). We found that systemically administered cisplatin was unable to provide a survival benefit presumably due to the development of drug resistance (Figure 6e), which is supported by our observation that MDR1 levels were elevated by cisplatin in cell cultures (Figure 6b) and in the orthotopic tumor model (Figure S7b). Likewise, the survival of mice treated with either AF alone or the combination of AF and cisplatin did not improve (Figure 6e). However, PEG-FTn75%/AF alone was able to significantly enhance the survival, underscoring that the improved delivery of AF to the tumor by PEG-FTn75% was critical to achieving its therapeutic benefit in this in vivo study. Importantly, PEG-FTn75%/AF deployed with cisplatin further enhanced the survival compared to PEG-FTn75%/AF alone group with a statistically significant difference (P < 0.05). This is likely due to the ability of PEG-FTn75%/AF to downregulate MDR1 and thus to restore chemo-sensitivity of cisplatin, consistent with our in vitro observation (Figure 6b). Of note, the median survival time was doubled by this combined therapy compared to the untreated control (P < 0.01) (Figure 6e).
CONCLUSION
Here, we introduced a human protein nanocage-based drug carrier capable of overcoming multiple physiological barriers and delivering therapeutic payloads to solid tumors, including the poorly vascularized hypoxic areas, following systemic administration. We newly identified a hypoxia-tropic property of FTn, which is associated with its intrinsic ability to target TfR1 or TIM-2 of which expression is particularly elevated in hypoxic tumor tissues. Further, we developed an effective approach of endowing FTn with spatially controlled surface PEG engraftment that facilitates the penetration of FTn through the highly dense tumor ECM to reach hypoxic tumor areas, while minimally affecting their natural ability to target cancer cells via the receptors. Given the significance of hypoxia in promoting tumor progression, the concept of effective drug delivery to hypoxic tumor areas, in addition to normoxic areas, would pose a critical impact on the treatment of advanced solid tumors.
Methods
Western blot analysis.
3LL and A549 cells were incubated at 21% or 1% oxygenated environment for 48 h. 3LL and A549 cells transfected with empty or HIF-1α-expressing plasmid vectors were also incubated at same conditions. The tumor samples were prepared by establishing 3LL tumor in female C57BL/6 mice (6 – 8 weeks) by subcutaneous (1 × 106 cells) or direct thoracic (5 × 105 cells) inoculation and harvesting 10 or 5 days after the inoculation, respectively. The healthy proximal and distal lung tissues were also collected from the animals with orthotopic lung tumor. All animals were handled in accordance with the policies and guidelines of the Johns Hopkins University Animal Care and Use Committee. The cell and tumor tissue samples were lysed and subjected to western blot according to the standard procedure. The membranes were probed using the primary antibodies against TfR1 (BD Pharmingen, cat# 555534), TIM-2 (Abcam, cat# ab86480) or HIF-1α (Cayman, cat# 10006421).
Preparation and characterization of nanocages.
FTn, composed of 24 human ferritin heavy chain protein subunits, was prepared as per our recently published protocol.19 Briefly, the plasmid vector encoding the human ferritin heavy chain protein was transformed into chemically competent E.coli BL21 (DE3) (Thermo Fisher Scientific). Isopropyl b-D-1-thiogalactopyranoside (IPTG; Sigma-Aldrich) was used to induce the protein production. Cell lysate, prepared by probe sonication of the bacterial culture, was centrifuged to collect supernatant, which was then heated at 70°C to remove heat-labile proteins that appeared as a precipitate. For further purification, the supernatant was subjected to a size exclusion chromatography using Superose 6 increase 10/300 GL column (GE Healthcare). The concentration of FTn was determined by the absorbance at 280 nm using a NanoDrop Spectrophotometer (Thermo Fisher Scientific).
PEGylated FTn was prepared by reacting 120 molar equivalents of methoxyl-PEG-succinimidyl ester (mPEG-NHS, PEG MW 2000; Nanocs) to FTn in PBS (pH 7.4). Following a 4 h reaction under gentle swirling at room temperature, unreacted mPEG-NHS was removed by an extensive membrane dialysis (MWCO 10 kDa; Thermo Fisher Scientific). Subsequently, the number of PEG molecules on the surface of each FTn was determined to be 24 – 48 using SDS-PAGE and MALDI-TOF MS (Voyager-DE STR Biospectrometry Workstation, Applied Biosystems) analyses (Figure S9). For the fluorescent labeling of FTn, 50 molar equivalents of amine-reactive dyes, including Cy5-NHS (GE Healthcare) or Alexa Fluor 488-NHS (Life Technologies), were reacted to FTn in PBS (pH 7.4) and subsequently the product was purified using a PD-10 column (GE Healthcare). The number of conjugated Cy5 or Alexa Fluor 488 per FTn was fluorometrically determined to be 24 or 28, respectively.
The hybrid PEG-FTn was prepared by two different disassembly/reassembly methods, including pH adjustment and gradient concentrations of urea. For mechanistic studies, the more conventional and simple pH adjustment method was employed. Briefly, non-PEGylated FTn, including fluorescently labeled FTn if needed, and PEGylated FTn were mixed at a range of molar ratios in PBS (pH 7.4) at an overall protein concentration of 0.4 μM. The nanocages were disassembled by adjusting pH to 2 for a 20 minute incubation, the pH was brought back to 7.0–7.4 for a 6 h incubation to yield hybrid PEG-FTn formulations with varying surface PEG contents and distribution patterns. For example, the lead formulation, PEG-FTn75% that consists of 6 and 18 non-PEGylated and PEGylated ferritin subunits, respectively, was engineered by mixing FTn and PEGylated FTn at a 1:3 molar ratio. Fluorescently labeled nanocages were constructed to include 3 (out of 24) subunits of fluorescently labeled, non-PEGylated ferritin protein. The urea gradient method was used to prepare AF-loaded PEG-FTn75% (i.e. PEG-FTn75%/AF) in order to achieve a high AF loading efficiency as described earlier.13 Briefly, non-PEGylated and PEGylated FTn were mixed at a 1:3 molar ratio in an 8 M urea solution (Sigma-Aldrich) and incubated at room temperature for 10 minutes to trigger nanocage disassembly. Subsequently, 2 mM acriflavine (AF; Sigma-Aldrich) was added to the solution, which was transferred to a dialysis membrane bag (MWCO 10 kDa, Thermo Scientific) for a series of dialysis in solutions (pH 7.4) with decreasing urea concentrations (7, 5, 3, 2, 1 and 0 M; each for at least 2 h) at 4°C to slowly reassemble the FTn. The product was further purified using PD-10 column and the number of AF molecules loaded per each FTn was determined by measuring the FTn and AF concentrations using Bradford protein assay (Bio-Rad) and absorbance measurement at 595 nm, respectively.
The molecular sizes of different hybrid PEG-FTn were determined by size exclusion chromatography as described above. The morphology of non-PEGylated FTn and PEG-FTn75% was characterized by TEM (H7600, Hitachi) following the negative staining of the specimen with 1% uranyl acetate. The hydrodynamic diameters and ζ-potentials of non-PEGylated FTn and PEG-FTn75% were measured by dynamic light scattering and laser Doppler anemometry, respectively, using a Zetasizer Nano ZE (Malvern Instruments).
Stability tests.
The plasma stability of non-PEGylated and hybrid PEG-FTn was characterized by gel electrophoresis. Briefly, Alexa Fluor 488-labeled nanocages composed of varying fractions of PEGylated ferritin subunits were incubated in 10% human plasma at 37°C for 30 minutes and subjected to electrophoresis in a 1% agarose gel (Thermo Fisher Scientific). The bands showing migrated nanocages within the gel were visualized by UV light excitation. The stability of Cy5-labeled FTn and/or PEG-FTn75% prepared as described above was further evaluated using size exclusion chromatography. Briefly, nanocages were incubated in 25% human serum in PBS, 100 mM sodium acetate buffer (pH 4.5) or in PBS (pH 7.2) at a final concentration of 5 mg/ml for specified times at 37 °C. The samples were then subjected to size exclusion chromatography using Superose 6 increase 10/300 GL column (GE Healthcare) and the peaks were monitored under a UV detector (280 nm).
Penetration through multicellular tumor spheroids.
The 3LL-based multicellular spheroids were prepared as described elsewhere with some modifications.47 Briefly, 50 μl of DMEM (Gibco) containing 1% (w/v) agarose was added to, and solidified in, each well of a 96-well microplate in which 3LL cells were seeded at 1 × 104 cell/well and cultured for 4 – 5 days to obtain 3D multicellular tumor spheroids with 400 – 500 μm in diameters. The spheroids were carefully transferred to glass bottom dishes where Cy5-labeled nanocages (40 nM) were added and incubated for 2 h. The competition assay was performed by incubating the spheroids with the Cy5-labeled nanocages, either non-PEGylated or PEGylated FTn, in the absence or presence of 15-fold molar excess of unlabeled FTn for 1 h. After washing with PBS, Z-stack images of the spheroids were acquired at 10 μm intervals using a confocal microscope (Carl Zeiss) and subsequently 3D-rendered images were constructed using a ZEN microscopy software (Carl Zeiss). All image-based quantitative analyses were performed using ImageJ (NIH). For the microscopic observation of hypoxia, pimonidazole HCl (200 μM; Hypoxyprobe) was additionally added to the spheroids for a 2 h incubation and cryosectioned to be immunostained using the Hypoxyprobe-1 kit (Hypoxyprobe) following the manufacturer’s instruction.
In vitro cellular uptake.
To compare the cellular uptake of non-PEGylated and hybrid PEG-FTn, Cy5-labeled nanocages (40 nM) composed of varying fractions of PEGylated ferritin subunits were incubated with 3LL cells for 2 h. Nanocages that were not associated with the cells were then washed and subsequently, cellular uptake was analyzed using an AccuriC6 flow cytometer (BD Biosciences).
To test our hypothesis that the spatial distribution pattern of PEG polymers on the surface of PEG-FTn75% is critical for retaining its ability to interact with the receptor, we engineered conventionally (i.e. uniformly) PEGylated FTn, possessing the same overall surface PEG content as the PEG-FTn75% (see schematics in Figure 1g), to be evaluated for receptor-mediated cellular uptake. We first PEGylated FTn using 120 molar equivalents of FITC-conjugated PEG-succinimidyl ester (FITC-PEG-NHS, PEG MW 2000; Nanocs) to yield FITC-PEG-FTn PEG. Subsequently, we formulated FITC-labeled PEG-FTn75% using the pH adjustment method at the non-PEGylated FTn to FITC-PEG-FTn molar ratio of 1:3 and flourometrically determined the surface PEG content. We next formulated uniformly PEGylated FTn exhibiting the identical FITC-associated fluorescence as the FITC-labeled PEG-FTn75%, by gradually increasing the amount of FITC-PEG-NHS conjugated to the FTn surface. The cellular uptake of these two formulations was evaluated by incubating 3LL cells with nanocages at 40 nM for 2 h, followed by subsequent washing and analysis using flow cytometery.
Extravasation of circulating nanocages.
For intratumoral tracking of PEG-FTn75% after systemic administration, iron oxide nanoparticle-loaded PEG-FTn75% (PEG-FTn75%/IO) was prepared using a previously reported method.48 A single 250 mg/kg dose of PEG-FTn75%/IO was injected via tail vein to 3LL tumor-bearing C57BL/6 mice and the tumor tissues were harvested 5 or 30 minutes after the administration. The tumor tissues were fixed and processed according to the standard procedure for preparing TEM samples of biological specimens.
Plasma pharmacokinetics and tumor accumulation of nanocages.
Cy5-labeled nanocages (60 mg/kg), either non-PEGylated FTn or PEG-FTn75%, were injected to the C57BL/6 mice (6 – 8 weeks) via tail vein. Plasma samples containing systemically administered nanocages were isolated from 50 μL of whole blood drained from individual mice via tail vein at the specific time points. The amounts of the non-PEGylated FTn and PEG-FTn75% in the plasma were fluorometrically determined using a standard curve generated by measuring Cy5-labeled nanocages in plasma isolated from mice without nanocage administration. The pharmacokinetic parameters were calculated using WinNonlin 6.0 (Pharsight). To visualize the tumor accumulation, 3LL tumor-bearing C57BL/6 mice received PBS or Cy5-labeled nanocages (60 mg/kg), either non-PEGylated FTn or PEG-FTn75%, via tail vein. Animals were sacrificed 18 h after the administration and tumor tissues were harvested to be imaged by the IVIS Spectrum In Vivo Imaging System (Perkin Elmer).
In vivo nanocage distribution within tumor.
Intratumoral nanocage infusion and 3D image reconstruction were performed following our previously reported procedures49. The C57BL/6 mice bearing 3LL-based flank tumor were anesthetized and Cy5-labeled nanocages, either non-PEGylated FTn or PEG-FTn75%, were intratumorally administered into a depth of 4.0 mm underneath the mouse dermis over 30 minutes at an infusion rate of 0.66 μL/min using a Chemyx Nanojet Injector Module (Chemyx, Stafford, TX). Animals were sacrificed immediately after the administration and the tumor tissues were harvested and frozen. The whole tumor tissues were then cryosectioned at a thickness of 50 μm (Leica Biosystems) and a series of sections were imaged using confocal microscopy. The fluorescence background was determined with tissue section devoid of nanocages. Three-dimensional images depicting volumetric distribution of nanocages were reconstructed by stacking and aligning individual images using Metamorph Microscopy Automation and Image Analysis Software (Molecular Devices), Image J and Imaris Software (Bitplane). The volume of nanocage distribution was quantified using a custom-designed MATLAB code.49
In vivo hypoxia accumulation of nanocages.
Fluorescently labeled nanocages (60 mg/kg) were administered to the tumor-bearing mice at specified time points before the animals were sacrificed. The hypoxic area within the tumor was immunostained using Hypoxyprobe-1 kit. Briefly, pimonidazole HCl (60 mg/kg) was given to the animals 1.5 h prior to the euthanization, and tumor tissues were harvested and prepared as frozen sections. The hypoxic regions of the tumor were stained using the FITC-conjugated antibody provided in the kit. Additionally, the blood vessels were immunostained using PE-conjugated anti-CD31 antibody (1:200; Biolegend, cat# 102407). The immunostaining was performed according to the standard procedure. The immunofluorescent images were acquired using LSM 510 Meta confocal microscope (Carl Zeiss). Quantitative analysis of the acquired images was performed using ImageJ. The extent of nanocage penetration was determined by measuring the radial distances of the nanocage-associated fluorescence signal from the blood vessels. For flow cytometry analysis, isolated tumors were processed as a single cell suspension by mincing the tumor, followed by a 40 minute incubation in PBS containing 0.08% collagenase (Sigma-Aldrich). Subsequently, 0.06% DNase (Sigma-Aldrich) was added to the suspension, vortexed and passed through a 40 μm cell strainer. The cells were fixed with 70% ethanol and permeabilized using a permeabilization buffer (Thermo Scientific), followed by an addition of FITC-conjugated antibody (1:500) included in the HypoxyProbe-1 kit. After 2.5 h of incubation at 37°C, the cells were washed and resuspended in PBS containing 1% BSA and 0.1% sodium azide for flow cytometry analysis. The cells were subjected to SH800 Cell Sorter (SONY Biotechnology) and the results were further analyzed using FlowJo software (FlowJo LLC).
In vitro drug release kinetics.
To investigate pH-dependent AF release, PEG-FTn75%/AF were placed in a dialysis bag (10 kDa MWCO) and dialyzed in either PBS (pH 7.4) or acetate buffer (pH 5.0) at 37°C. At designated time points, 150 μl of the buffer outside the dialysis bag was collected and the same amount of fresh buffer was added back. The concentrations of AF released from the nanocages at varying time points were determined using an analytical HPLC (Prominence-i LC-2030C, Shimadzu) equipped with C18 5 μm analytical column (150 mm × 4.6 mm; Phenomenex) and monitored using a fluorescent detector (450/490 nm).
Plasma pharmacokinetics and quantitative biodistribution of AF.
After tail vein injection of AF (2 mg/kg) or PEG-FTn75%/AF (2 mg/kg equivalent dose to AF) into female C57BL/6 mice (6 – 8 weeks, n = 3 per time point), the blood samples (200 μl) were collected from the retro-orbital plexus and stabilized using EDTA followed by separation of plasma using a refrigerated centrifuge (1500 × g, 10 minutes). A single blood sample was collected at a given time point and subsequently major organs and tumor tissues were harvested as well from each mouse. The AF concentrations in plasma and tissues were determined using a previously described method with some modifications50. Tissue homogenates were prepared by adding three volumes (v/w) of chilled 50 mM Tris-HCl buffer (pH 7.4) containing 0.25 M sucrose to the tissue followed by bead homogenization. The plasma and tissue homogenates were then spiked by 9-aminoacridine (2 μg/ml; Sigma-Aldrich) as an internal standard and 10 volumes of an acidified methanol (1% acetic acid) were subsequently added to each sample. The mixture was vigorously vortexed for 30 minutes and then sonicated for 10 minutes. After centrifugation (10,000 rpm, 15 minutes), supernatant was collected, filtered and subjected to HPLC analysis as described above. The standards were prepared in the plasma or tissue homogenates from untreated mice.
In vivo HIF-1α inhibition.
Mice bearing 3LL-based flank tumor received 5 doses of saline or AF-loaded nanocages, either AF in solution or PEG-FTn75%/AF, at the AF dose of 2 mg/kg via tail vein every other day, and tumor volume and body weight were monitored over time. Ten days after the initial treatment, tumor tissues were harvested. Total RNA was extracted and purified from the tumor tissues using TRIzol (Invitrogen) and DNase (Ambion) treatment. A 1 μg aliquot of total RNA was reverse-transcribed using iScript cDNA Synthesis system and then qRT-PCR was performed using iQ SYBR Green Supermix and iCycler Real-time PCR Detection System (Bio-Rad). Primers for VEGF, LOX, GLUT1, SCF and SDF1 mRNA and 18S rRNA were designed using Beacon Designer software (Bio-Rad) and determined to be specific by BLAST and dissociation curve analysis. The level of each mRNA was normalized to that of 18S rRNA in the same sample. The sequences of primers are as follows: mouse VEGF (F-GTGACAAGCCAAGGCGGTGAG; R-GGCGAATCCAGTCCCACGAGG), mouse LOX (F-TGCCAGTGGATTGATATTACAGATGT; R-AGCGAATGTCACAGCGTACAA), mouse GLUT1 (F-CGGGCCAAGAGTGTGCTAAA; R-TGACGATACCGGAGCCAATG), mouse SCF (F-ATGGTTTCCACCACCTCTAAC; R-CCAACTACACCTAACTGCCTAC), mouse SDF1 (F-GCTAAGGTTTGCCAGCATAAAG; R-CGAGGGCAGGGATGAATATAAG). In parallel, protein in the tumor tissue was extracted and purified for western blot analysis. The membranes were probed using the primary antibodies against HIF-1α (1:200; Cayman, cat# 10006421), LOX (1:500; Millipore, cat# ABT112), VEGF (1:500; Santa Cruz, cat# sc-152), and MDR1 (1:200; Novus Biologicals, cat# 80870).
Serum biochemistry analysis.
For serum biochemical evaluation, saline, PEG-FTn75% (320 mg/kg), AF (10 mg/kg) or PEG-FTn75%/AF (10 mg/kg AF equivalents) was injected into C57BL/6 healthy mice via tail vein (n = 4). Whole blood sample were collected 14 days after the administraiton from which serum samples were prepared and evaluated for systemic toxicity by standard biochemistry analysis.
In vitro analysis of hypoxia and cisplatin-mediated MDR1 upregulation.
To evaluate the hypoxia-mediated MDR1 upregulation, 3LL cells were incubated at 21% or 1% oxygenated environment for 48 h with or without AF (1.25 μM). For assessing the cisplatin-mediated MDR1 upregulation, the cells were incubated at 1% oxygenated environment in the presence of cisplatin (0.675 or 1.3 μM; Sigma-Aldrich); cells were subjected to an addition co-treatment with AF or PEG-FTn75%/AF (1.25 μM AF equivalent) to investigate whether AF prevented cisplatin-mediated MDR1 elevation. The cells were then lysed and subjected to western blot analysis according to the standard procedure. The membranes were probed using the primary antibodies against HIF-1α and MDR1 as described above.
In vivo anti-cancer efficacy.
An orthotopic lung cancer model was established by direct intrathoracic injection of the 3LL cells into the left lungs of C57BL/6 mice as described above, but cells constitutively expressing luciferase (i.e. 3LL-Luc) were used for monitoring the tumor growth in live animals.
For the anti-cancer efficacy study, animals were divided into 6 groups and subjected to treatments with multiple doses of saline, cisplatin (2 mg/kg), AF (2 mg/kg), AF + cisplatin, PEG-FTn75%/AF (2 mg/kg AF equivalent) or PEG-FTn75%/AF + cisplatin, following the schedule depicted in Figure S7. The tumor growth was evaluated by quantifying the bioluminescence signals in the lungs using the IVIS Spectrum In Vivo Imaging System. In parallel, survival of animals was monitored in a daily basis.
Statistical analysis.
All data are presented as means ± s.e.m. The statistical differences between two groups and among multiple groups were determined using Student’s t-test and one-way analysis of variance (ANOVA), respectively. The differences in the survival were analyzed using the log-rank test. All statistical analysis was two-sided and P values less than 0.05 were considered statistically significant. The GraphPad Prism version 7.0a (GraphPad Software, San Diego, CA) software was used for the statistical calculations.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (R01HL125169, R01HL127413, R01HL136617 and P30EY001765), the Cystic Fibrosis Foundation (HANES16XX0 and SUK18I0), the National Natural Science Foundation of China (31870999), the Natural Science Foundation of Tianjin (18JCYBJC40900) and the Foundation of the Thousand Talents Plan for Young Professionals.
References
- 1.Wilson WR; Hay MP, Targeting hypoxia in cancer therapy. Nature reviews. Cancer 2011, 11 (6), 393–410. [DOI] [PubMed] [Google Scholar]
- 2.Samanta D; Gilkes DM; Chaturvedi P; Xiang L; Semenza GL, Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc Natl Acad Sci U S A 2014, 111 (50), E5429–38. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 3.Khan KA; Kerbel RS, Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat Rev Clin Oncol 2018, 15 (5), 310–324. [DOI] [PubMed] [Google Scholar]
- 4.Harris AL, Hypoxia--a key regulatory factor in tumour growth. Nature reviews. Cancer 2002, 2 (1), 38–47. [DOI] [PubMed] [Google Scholar]
- 5.Dewhirst MW; Cao Y; Moeller B, Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nature reviews. Cancer 2008, 8 (6), 425–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Semenza GL, Targeting HIF-1 for cancer therapy. Nature reviews. Cancer 2003, 3 (10), 721–32. [DOI] [PubMed] [Google Scholar]
- 7.Comerford KM; Wallace TJ; Karhausen J; Louis NA; Montalto MC; Colgan SP, Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res 2002, 62 (12), 3387–94. [PubMed] [Google Scholar]
- 8.Shi J; Kantoff PW; Wooster R; Farokhzad OC, Cancer nanomedicine: progress, challenges and opportunities. Nature reviews. Cancer 2017, 17 (1), 20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Minchinton AI; Tannock IF, Drug penetration in solid tumours. Nature reviews. Cancer 2006, 6 (8), 583–92. [DOI] [PubMed] [Google Scholar]
- 10.Tredan O; Galmarini CM; Patel K; Tannock IF, Drug resistance and the solid tumor microenvironment. Journal of the National Cancer Institute 2007, 99 (19), 1441–54. [DOI] [PubMed] [Google Scholar]
- 11.Jain RK; Stylianopoulos T, Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol 2010, 7 (11), 653–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blanco E; Shen H; Ferrari M, Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nature biotechnology 2015, 33 (9), 941–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liang M; Fan K; Zhou M; Duan D; Zheng J; Yang D; Feng J; Yan X, H-ferritin-nanocaged doxorubicin nanoparticles specifically target and kill tumors with a single-dose injection. Proc Natl Acad Sci U S A 2014, 111 (41), 14900–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nagy JA; Feng D; Vasile E; Wong WH; Shih SC; Dvorak AM; Dvorak HF, Permeability properties of tumor surrogate blood vessels induced by VEGF-A. Lab Invest 2006, 86 (8), 767–80. [DOI] [PubMed] [Google Scholar]
- 15.Nagy JA; Chang SH; Shih SC; Dvorak AM; Dvorak HF, Heterogeneity of the tumor vasculature. Semin Thromb Hemost 2010, 36 (3), 321–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng D; Nagy JA; Dvorak AM; Dvorak HF, Different pathways of macromolecule extravasation from hyperpermeable tumor vessels. Microvasc Res 2000, 59 (1), 24–37. [DOI] [PubMed] [Google Scholar]
- 17.Zhen Z; Tang W; Guo C; Chen H; Lin X; Liu G; Fei B; Chen X; Xu B; Xie J, Ferritin nanocages to encapsulate and deliver photosensitizers for efficient photodynamic therapy against cancer. ACS nano 2013, 7 (8), 6988–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khoshnejad M; Parhiz H; Shuvaev VV; Dmochowski IJ; Muzykantov VR, Ferritin-based drug delivery systems: Hybrid nanocarriers for vascular immunotargeting. J Control Release 2018, 282, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang X; Chisholm J; Zhuang J; Xiao Y; Duncan G; Chen X; Suk JS; Hanes J, Protein nanocages that penetrate airway mucus and tumor tissue. Proc Natl Acad Sci U S A 2017, 114 (32), E6595–E6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li L; Fang CJ; Ryan JC; Niemi EC; Lebron JA; Bjorkman PJ; Arase H; Torti FM; Torti SV; Nakamura MC; Seaman WE, Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. Proc Natl Acad Sci U S A 2010, 107 (8), 3505–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fan K; Cao C; Pan Y; Lu D; Yang D; Feng J; Song L; Liang M; Yan X, Magnetoferritin nanoparticles for targeting and visualizing tumour tissues. Nature nanotechnology 2012, 7 (7), 459–64. [DOI] [PubMed] [Google Scholar]
- 22.Chen TT; Li L; Chung DH; Allen CD; Torti SV; Torti FM; Cyster JG; Chen CY; Brodsky FM; Niemi EC; Nakamura MC; Seaman WE; Daws MR, TIM-2 is expressed on B cells and in liver and kidney and is a receptor for H-ferritin endocytosis. J Exp Med 2005, 202 (7), 955–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han J; Seaman WE; Di X; Wang W; Willingham M; Torti FM; Torti SV, Iron uptake mediated by binding of H-ferritin to the TIM-2 receptor in mouse cells. PLoS One 2011, 6 (8), e23800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fan K; Jia X; Zhou M; Wang K; Conde J; He J; Tian J; Yan X, Ferritin Nanocarrier Traverses the Blood Brain Barrier and Kills Glioma. ACS nano 2018, 12 (5), 4105–4115. [DOI] [PubMed] [Google Scholar]
- 25.Suk JS; Xu QG; Kim N; Hanes J; Ensign LM, PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliver Rev 2016, 99, 28–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mastorakos P; da Silva AL; Chisholm J; Song E; Choi WK; Boyle MP; Morales MM; Hanes J; Suk JS, Highly compacted biodegradable DNA nanoparticles capable of overcoming the mucus barrier for inhaled lung gene therapy. Proc Natl Acad Sci U S A 2015, 112 (28), 8720–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schneider CS; Xu Q; Boylan NJ; Chisholm J; Tang BC; Schuster BS; Henning A; Ensign LM; Lee E; Adstamongkonkul P; Simons BW; Wang SS; Gong X; Yu T; Boyle MP; Suk JS; Hanes J, Nanoparticles that do not adhere to mucus provide uniform and long-lasting drug delivery to airways following inhalation. Sci Adv 2017, 3 (4), e1601556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lok CN; Ponka P, Identification of a hypoxia response element in the transferrin receptor gene. The Journal of biological chemistry 1999, 274 (34), 24147–52. [DOI] [PubMed] [Google Scholar]
- 29.Tacchini L; Bianchi L; Bernelli-Zazzera A; Cairo G, Transferrin receptor induction by hypoxia. HIF-1-mediated transcriptional activation and cell-specific post-transcriptional regulation. The Journal of biological chemistry 1999, 274 (34), 24142–6. [DOI] [PubMed] [Google Scholar]
- 30.Tacchini L; Gammella E; De Ponti C; Recalcati S; Cairo G, Role of HIF-1 and NF-kappaB transcription factors in the modulation of transferrin receptor by inflammatory and anti-inflammatory signals. The Journal of biological chemistry 2008, 283 (30), 20674–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herbison CE; Thorstensen K; Chua AC; Graham RM; Leedman P; Olynyk JK; Trinder D, The role of transferrin receptor 1 and 2 in transferrin-bound iron uptake in human hepatoma cells. Am J Physiol Cell Physiol 2009, 297 (6), C1567–75. [DOI] [PubMed] [Google Scholar]
- 32.Fang Y; Xue J; Gao S; Lu A; Yang D; Jiang H; He Y; Shi K, Cleavable PEGylation: a strategy for overcoming the “PEG dilemma” in efficient drug delivery. Drug Deliv 2017, 24 (sup1), 22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bianchi L; Tacchini L; Cairo G, HIF-1-mediated activation of transferrin receptor gene transcription by iron chelation. Nucleic Acids Res 1999, 27 (21), 4223–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parra RG; Rohr CO; Koile D; Perez-Castro C; Yankilevich P, INSECT 2.0: a web-server for genome-wide cis-regulatory modules prediction. Bioinformatics 2016, 32 (8), 1229–31. [DOI] [PubMed] [Google Scholar]
- 35.Jiang BH; Rue E; Wang GL; Roe R; Semenza GL, Dimerization DNA binding, and transactivation properties of hypoxia-inducible factor 1. The Journal of biological chemistry 1996, 271 (30), 17771–8. [DOI] [PubMed] [Google Scholar]
- 36.Lin X; Xie J; Niu G; Zhang F; Gao H; Yang M; Quan Q; Aronova MA; Zhang G; Lee S; Leapman R; Chen X, Chimeric ferritin nanocages for multiple function loading and multimodal imaging. Nano Lett 2011, 11 (2), 814–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nederman T; Norling B; Glimelius B; Carlsson J; Brunk U, Demonstration of an extracellular matrix in multicellular tumor spheroids. Cancer Res 1984, 44 (7), 3090–7. [PubMed] [Google Scholar]
- 38.Herrmann D; Conway JR; Vennin C; Magenau A; Hughes WE; Morton JP; Timpson P, Three-dimensional cancer models mimic cell-matrix interactions in the tumour microenvironment. Carcinogenesis 2014, 35 (8), 1671–9. [DOI] [PubMed] [Google Scholar]
- 39.Nance EA; Woodworth GF; Sailor KA; Shih TY; Xu Q; Swaminathan G; Xiang D; Eberhart C; Hanes J, A dense poly(ethylene glycol) coating improves penetration of large polymeric nanoparticles within brain tissue. Sci Transl Med 2012, 4 (149), 149ra119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mastorakos P; Zhang C; Berry S; Oh Y; Lee S; Eberhart CG; Woodworth GF; Suk JS; Hanes J, Highly PEGylated DNA Nanoparticles Provide Uniform and Widespread Gene Transfer in the Brain. Adv Healthc Mater 2015, 4 (7), 1023–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miao L; Newby JM; Lin CM; Zhang L; Xu F; Kim WY; Forest MG; Lai SK; Milowsky MI; Wobker SE; Huang L, The Binding Site Barrier Elicited by Tumor-Associated Fibroblasts Interferes Disposition of Nanoparticles in Stroma-Vessel Type Tumors. ACS nano 2016, 9243–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen J; Ding Z; Peng Y; Pan F; Li J; Zou L; Zhang Y; Liang H, HIF-1alpha inhibition reverses multidrug resistance in colon cancer cells via downregulation of MDR1/P-glycoprotein. PLoS One 2014, 9 (6), e98882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ladoulis CT; Gill TJ, Physical chemical studies on the specific interaction of an acriflavine-phosphotungstic Acid complex with double-stranded nucleic acids. The Journal of cell biology 1970, 47 (2), 500–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang H; Lu H; Xiang L; Bullen JW; Zhang C; Samanta D; Gilkes DM; He J; Semenza GL, HIF-1 regulates CD47 expression in breast cancer cells to promote evasion of phagocytosis and maintenance of cancer stem cells. Proc Natl Acad Sci U S A 2015, 112 (45), E6215–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee K; Zhang H; Qian DZ; Rey S; Liu JO; Semenza GL, Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc Natl Acad Sci U S A 2009, 106 (42), 17910–5. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 46.van Moorsel CJ; Pinedo HM; Veerman G; Vermorken JB; Postmus PE; Peters GJ, Scheduling of gemcitabine and cisplatin in Lewis lung tumour bearing mice. Eur J Cancer 1999, 35 (5), 808–14. [DOI] [PubMed] [Google Scholar]
- 47.Friedrich J; Seidel C; Ebner R; Kunz-Schughart LA, Spheroid-based drug screen: considerations and practical approach. Nat Protoc 2009, 4 (3), 309–24. [DOI] [PubMed] [Google Scholar]
- 48.Uchida M; Flenniken ML; Allen M; Willits DA; Crowley BE; Brumfield S; Willis AF; Jackiw L; Jutila M; Young MJ; Douglas T, Targeting of cancer cells with ferrimagnetic ferritin cage nanoparticles. Journal of the American Chemical Society 2006, 128 (51), 16626–33. [DOI] [PubMed] [Google Scholar]
- 49.Mastorakos P; Zhang C; Song E; Kim YE; Park HW; Berry S; Choi WK; Hanes J; Suk JS, Biodegradable brain-penetrating DNA nanocomplexes and their use to treat malignant brain tumors. Journal of Controlled Release 2017, 262, 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Song S; Kwon OS; Chung YB, Pharmacokinetics and metabolism of acriflavine in rats following intravenous or intramuscular administration of AG60, a mixture of acriflavine and guanosine, a potential antitumour agent. Xenobiotica 2005, 35 (7), 755–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.