

Abstract
Labeling approaches using isobaric chemical tags (e.g., isobaric tagging for relative and absolute quantification, iTRAQ and tandem mass tag, TMT) have been widely applied for the quantification of peptides and proteins in bottom-up MS. However, until recently, successful applications of these approaches to top-down proteomics have been limited because proteins tend to precipitate and “crash” out of solution during TMT labeling of complex samples making the quantification of such samples difficult. In this study, we report a top-down TMT MS platform for confidently identifying and quantifying low molecular weight intact proteoforms in complex biological samples. To reduce the sample complexity and remove large proteins from complex samples, we developed a filter-SEC technique that combines a molecular weight cutoff filtration step with high-performance size exclusion chromatography (SEC) separation. No protein precipitation was observed in filtered samples under the intact protein-level TMT labeling conditions. The proposed top-down TMT MS platform enables high-throughput analysis of intact proteoforms, allowing for the identification and quantification of hundreds of intact proteoforms from Escherichia coli cell lysates. To our knowledge, this represents the first high-throughput TMT labeling-based, quantitative, top-down MS analysis suitable for complex biological samples.
Keywords: top-down proteomics, quantitative proteomics, mass spectrometry, liquid chromatography, isobaric labeling, tandem mass tags (TMT)
Graphical Abstract
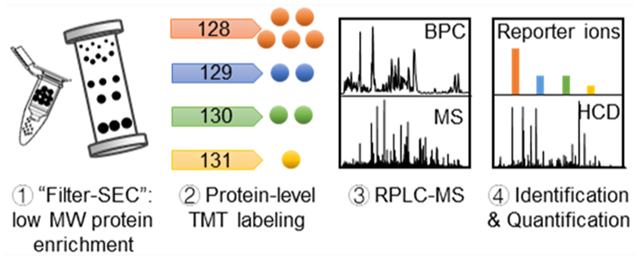
INTRODUCTION
Quantitative top-down MS analysis of intact proteoforms with different post-translational modifications (PTMs) that arise from one gene is important because PTMs often cause the proteoforms to exhibit differing functions in biological processes.1–3 Currently, peak intensity-based, label-free quantitation is the most commonly used quantitation approach in top-down proteomics. It has been successfully applied to the quantitation of proteoforms in biological samples such as tumor samples,4 animal tissues,5,6 human saliva,7 murine mitochondria,8 and the yeast proteome.9 Moreover, label-free, top-down MS quantitation has been successfully used to quantify 1000+ human proteoforms below 30 kDa based on a pregenerated protein identification database.10 However, one of the major challenges of label-free quantitation is run-to-run LC-MS instrument variation, which may result in inconsistent analysis among multiple runs. In addition, multidimensional separation techniques, which have been applied for increasing the throughput of top-down MS, may split a single proteoform into different fractions and make accurate, label-free quantitation difficult.
Various isotope-labeling techniques, both in vivo and in vitro, have also been applied to quantitative, top-down proteomics. Stable isotope labeling by amino acids in cell culture (SILAC) is an in vivo metabolic labeling technology that relies on the incorporation of isotopically labeled amino acids in the growth medium during cell growth.11,12 In top-down SILAC MS, peak detection and spectral deconvolution are challenging because multiple isotopic distributions in parent scans are often partially overlapped.13 The spectral complexity can be further increased by even a small amount of incomplete labeling. More recently, a neutron coding (NeuCode) SILAC method has been introduced to incorporate isotope-labeled amino acids with subtle mass differences. High-resolution MS instruments (i.e., mass resolution >120 K) are coupled with NeuCode SILAC and can alleviate some of the drawbacks of the traditional SILAC approach for the quantitation of intact proteoforms.14 However, like other in vivo isotope-labeling approaches, the application of NeuCode SILAC is largely limited to lab-cultured samples. Alternatively, in vitro isotope-labeling approaches can be applied to lab-cultured or clinical samples. For example, the pseudo isobaric dimethyl labeling (pIDL) approach utilizes isotopically labeled formaldehyde to label lysine residues, and protein quantification is based on the fragment ion pairs.15,16
Isobaric labeling, such as isobaric tagging for relative and absolute quantification (iTRAQ),17 tandem mass tags (TMT),18 and N,N-dimethyl leucine (DiLeu),19,20 has become one of the most applied quantitative approaches in bottom-up proteomics.21,22 Currently, isobaric labeling approaches can simultaneously quantify up to 21 biological samples in a single run, which largely increases the throughput and reduces the need to control run-to-run instrument variation.20 With isobaric chemical tags, signals across different fractions from multidimensional separation can be normalized, which increases the accuracy of quantification and enables deeper identification and quantification than can be achieved using one-dimensional separation. Coupling isobaric labeling and multidimensional separation could improve protein quantification in complex biological samples such as cell lysates; however, under typical labeling conditions, intact proteins are challenging to label using isobaric chemical tags. In fact, isobaric chemical tag labeling of intact proteins has only been achieved using pure proteins or simple protein mixtures.23–26 Previously, TMT label-based protein quantification of complex protein mixtures such as cell lysates has not been achieved, and one major challenge with intact complex sample labeling is that proteins tend to precipitate and “crash” out of solution due to the organic solvents required for chemical labeling.
We applied a protein-level TMT labeling platform to quantify intact proteins and proteoforms in complex protein samples, such as Escherichia coli cell lysates, using top-down proteomics. To reduce sample complexity and to remove large proteins in the complex sample, we developed a filter-SEC approach that combines a molecular weight cutoff (MWCO) step with high-performance size exclusion chromatography (SEC) separation. Our results indicated that the filter-SEC approach efficiently enriched proteins with molecular weight (MW) less than 30 kDa and, most importantly, avoided protein precipitation for efficient labeling. We further combined filter-SEC enrichment, intact protein-level TMT labeling, and top-down MS to identify and quantify low MW proteins and proteoforms (i.e., < 30 kDa) in an E. coli cell lysate. A total of 411 intact proteoforms of 95 proteins were identified and quantified from two LC-MS/MS runs after manual evaluation. Our results demonstrate that the proposed protein-level TMT labeling approach can efficiently label and quantify proteoforms in complex protein samples.
EXPERIMENTAL SECTION
Chemicals and Materials.
Acetonitrile (ACN), trifluoroacetic acid (TFA), formic acid, water, triethylammonium bicarbonate (TEAB), 50% (w/w) hydroxylamine, iodoacetamide (IAA), phenylmethanesulfonylfluoride (PMSF), and ammonium bicarbonate were purchased from Sigma-Aldrich (St. Louis, MO, USA). ACN, TFA, and water used for LC mobile phases were LC-MS grade. Tris (2-carboxyethyl) phosphine hydrochloride (TCEP), BCA assay, Bradford assay, and the TMT 6-plex isobaric label reagent set were obtained from Thermo Fisher (Waltham, MA, USA). Molecular weight cutoff spin filters were purchased from Sartorius (Gottingen, Germany).
E. coli Cell Lysate Preparation.
E. coli K12 cells were incubated in LB medium at 37 °C for 12 h. The cells were collected and pelleted through centrifugation (10000g) at 4 °C for 20 min. Cell lysates were prepared using the bead-beating approach as previously described.27 Cell lysis buffer (1 mM PMSF and 25 mM NH4HCO3), cell pellets, and 0.1 mm zirconia/silica beads were mixed at a ratio of 2:1:1 (v/v/v), and the cells were lysed in a bullet blender (Next Advance, Atkinson, NH) at 4 °C for 3 min. After bead-beating, the cell debris was removed by centrifugation at 20000g and 4 °C for 40 min. Protein concentration of collected cell lysates was measured by a BCA assay before storage at −80 °C.
Filter-SEC Separation.
Large proteins in cell lysates were removed using a 100 kDa MWCO spin filter at 4 °C. After filtration, the low MW protein fraction was concentrated to 8 μg/μL using a 3 kDa MWCO filter (low MWCO sample) or a 10 kDa MWCO filter (high MWCO sample). 100 μL of each sample was injected onto a BioSuite UHR column for high-pressure SEC separation (UHR SEC 250, 4.6 mm i.d., 30 cm length, 4 μm particle size). The mobile phase was 150 mM ammonium acetate (pH 7.4), and the flow rate was 200 μL/min. Thirty (30) 1 min fractions were collected. These fractions were analyzed using SDS-PAGE before TMT labeling and top-down MS analysis.
Protein-level TMT Labeling.
SEC fractions were combined into two samples based on the molecular weight range determined by SDS-PAGE analysis. For the protein-level labeling, the protein concentration of samples was adjusted to 1 μg/μL. This is comparable to previous literature which reports protein concentrations between 1–5 μg/μL before reduction and alkylation.24–26 300 μg of the proteins were diluted to 300 μL using 150 mM ammonium acetate, reduced with 10 μL of 200 mM TCEP for 1 h at 55 °C, and alkylated with 20 μL of 375 mM iodoacetamide (IAA) at room temperature in the dark for 30 min. The samples were then buffer exchanged with 100 mM TEAB (pH 8.5). A total of 200 μg of each protein sample was diluted to 300 μL (protein concentration: 0.67 μg/μL) using 100 mM TEAB (pH 8.5) and separated into four aliquots with a volume ratio of 5:2:2:1. A total of 41 μL of ACN was added to each TMT reagent vial. Each aliquot of protein sample was mixed at room temperature with the corresponding TMT reagent at a protein-to-TMT reagent ratio of 1:4 (w/w). The reaction was quenched after 1 h by incubation with 8 μL of 5% hydroxylamine at room temperature for 15 min.
Top-Down LC-MS.
Labeled proteins were injected onto a home-packed C5 RPLC capillary column (75 μm i.d., 70 cm length, Jupiter particles, 5 μm diameter, 300 Å pore size) using a Thermo Scientific (Waltham, MA, USA.) Accela LC System, as previously described.28,29 The packing materials for the C5 column (Jupiter particles, 5 μm diameter, 300 Å pore size) were purchased from Phenomenex (Torrance, CA). Mobile phase A was 0.01% TFA, 0.585% acetic acid, 2.5% 2-propanol, and 5% acetonitrile in water. Mobile phase B was 0.01% TFA, 0.585% acetic acid, 45% 2-propanol, and 45% ACN in water. A 200 min gradient from 10% to 65% mobile phase B was applied at a flow rate of 400 nL/min. The LC was coupled online to an LTQ Orbitrap Velos Pro mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) through a customized nano-ESI interface. The temperature of the inlet capillary was 300 °C, and the electrospray voltage was 2.6 kV. Full MS scans used a resolving power setting of 100 000 (at m/z 400) with two micro scans. Higher energy collisional induced dissociation (HCD) with a normalized collision energy of 30% was applied. The MS/MS data were collected using data-dependent acquisition and the top six most abundant precursor ions were selected. The MS/MS data were obtained at a resolving power setting of 60 000 (at m/z 400) with two micro scans and an isolation window of 3.0 m/z and 60 s dynamic exclusion. Electron transfer dissociation (ETD) was applied for the MS/MS acquisition as well. The activation time used for ETD was 20 ms. The ETD MS/MS data were obtained at a resolving power setting of 100 000 (at m/z 400). All other parameters were the same as used for HCD. All the data were collected with the Xcalibur 3.0 software (Thermo Fisher Scientific, Bremen, Germany).
Data Analysis.
The top-down proteomics raw data were converted to mzXML files using MSConvert,30 deconvoluted using Biopharma Finder (Thermo Fisher Scientific), and searched against the annotated E. coli protein database (UniProt 2017-03-18) using TopPIC Suite.31 TMT on lysine residues and the N-terminus were set as fixed PTMs, as well as carbamidomethylation of cysteine residues. The error tolerance was set to 15 ppm, the maximum number of unexpected mass shifts in a proteoform spectrum-match was 1, and the maximum mass shift was 500 Da. Detailed parameters for TopPIC can be found in Supplementary Table S1A. MASH Suite32 and ProSight Lite33 were used for manual interpretation and spectrum presentation. A python program was developed in-house to extract the ion intensities of individual TMT reporter ions from the corresponding MS/MS spectrum in the converted mzML files.34,35
The labeling efficiency was calculated from the deconvolution and identification results in top-down analysis using an in-house python package. Proteoforms with mass shifts of ±229.163 Da (TMT6-plex monoisotopic mass) were grouped together as a “protein group” if the mass shift was less than 200 ppm and the retention time shift was less than 6 min. Here, a protein group indicates the same proteoform with different numbers of TMT labels. The identified proteoforms from TopPIC Suite were utilized to assign the TMT labeling status of each proteoform in a protein group. The labeling efficiency was calculated using the following equation (Ic: intensity of completely labeled proteoform; Io: intensity of overlabeled proteoforms; Iu: intensity of underlabeled proteoforms):
RESULTS AND DISCUSSION
Low Molecular Weight Protein Enrichment Using the Filter-SEC.
To enrich low molecular weight proteins, a 100 kDa MWCO spin filter was used to remove high MW proteins, and the remaining low MW fraction was concentrated using a 3 kDa MWCO spin filter. A sample of low molecular weight enriched proteins prepared with the MWCO filters and an analogous unfiltered sample was injected individually onto the SEC column to compare the separation efficiency (Supplementary Figure S1A,B). The MWCO filtering step significantly improved the SEC separation of low MW proteins in this complex sample and decreased the background and peak broadening caused by the high abundance of large MW proteins in the unfiltered samples. In addition, the filter-SEC allowed for the enrichment of low MW proteins resulting in the injection of more low MW proteins in a single LC-MS run. After the filter-SEC step, low MW fractions (fraction 19–26) were combined into two samples based on the molecular weight as determined by SDS-PAGE analysis. Sample 1 contained fractions 19–21 (<66 kDa), and Sample 2 contained fractions 22–26 (<35 kDa). These two samples were used for the subsequent intact protein TMT labeling. In addition, several replicate experiments were performed to demonstrate the reproducibility of the filter-SEC (Supplementary Figure S1C).
Intact Protein-level TMT Labeling for the Low MW Proteins Enriched by Filter-SEC.
A common problem for intact protein TMT labeling is the reduction in solubility of the intact proteins after reduction and alkylation of the disulfide bonds. However, the cleavage of disulfide bonds is important for efficient protein-level TMT labeling, so we examined the effect of the cleavage of disulfide bonds on protein solubility under the labeling conditions (Figure 1A). The protein recovery after reduction and alkylation of disulfide bonds for a nonfiltered sample of E. coli cell lysate was approximately 60%, but there was no significant loss of protein for the filter-SEC prepared low MW samples. There was also visible protein precipitation in the nonfiltered sample after the reduction and alkylation, while no visible precipitation formed in the filter-SEC prepared low MW samples (fractions 19–26). These results indicate that the filter-SEC enrichment of low MW proteins prevents protein precipitation under labeling conditions.
Figure 1.
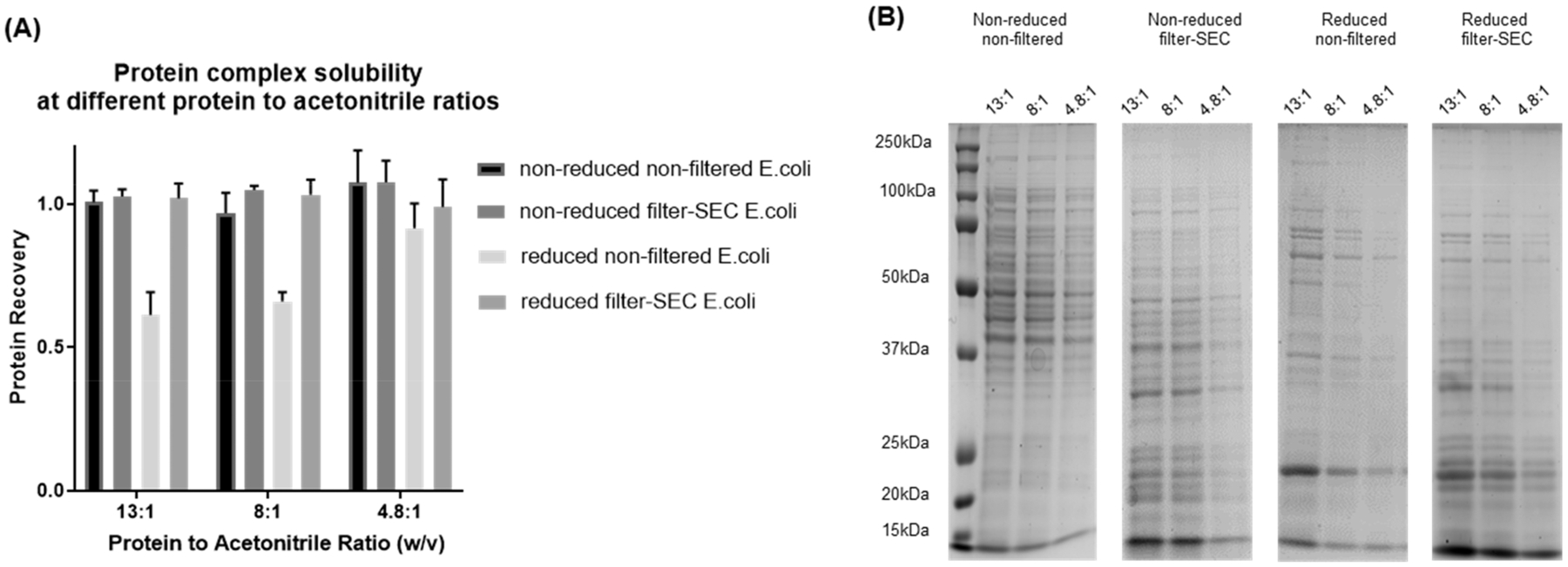
Evaluation of protein solubility under different TMT labeling conditions. Reduced (reduced by TCEP and alkylated by iodoacetamide) and nonreduced E. coli proteins were mixed with acetonitrile at different ratios. (A) Protein concentrations of the supernatants were measured using BCA assay. (B) The protein content of each supernatant was evaluated using SDS-PAGE.
We then evaluated the ratio of the labeling reagent to protein for TMT labeling. Since the organic solvent (ACN) in the reaction solution may cause the proteins to precipitate, we evaluated the protein solubility in ACN with no TMT reagent added at varying protein concentrations. According to the manufacturer instructions, TMT reagent should be dissolved in ACN at a ratio of 800 μg of TMT to 41 μL of ACN. A sample-to-reagent ratio between 1:1 and 1:8 (w/w) is suggested for peptide labeling, which corresponds to a peptide-to-ACN ratio from 19.2:1 to 2.4:1 (w/v).36 We analyzed the percent recovery of samples with protein-to-ACN ratios of 13:1, 8:1, and 4.8:1 (w/v), which is in the middle of the range of recommended ratios for peptide labeling. In the filter-SEC low MW samples, an average protein recovery of 98.9% was observed with no visible precipitation.
The protein patterns of the soluble proteins were analyzed by SDS-PAGE before and after reduction and alkylation for the filter-SEC and nonfiltered samples, and no significant changes were observed for the filter-SEC samples under all three protein concentration conditions (Figure 1B). The filter-SEC sample also maintained similar protein patterns before and after the cleavage of disulfide bonds as observed using SDS-PAGE; a significant loss of large MW proteins was observed on the SDS-PAGE after the reduction and alkylation of the nonfiltered samples. Our results indicate that the cleavage of disulfide bonds can significantly reduce the solubility of large MW proteins due to the exposure of hydrophobic groups as suggested in previous studies.37 The filter-SEC procedure removed most of the large MW proteins, and the remaining low MW proteins were soluble before and after the reduction and alkylation of cysteine residues. Moreover, these filter-SEC enriched low MW proteins remained soluble under various protein-to-ACN ratio conditions. In summary, our filter-SEC prevented protein precipitation and preserved similar protein patterns under denaturing TMT labeling conditions.
Intact Protein and Proteoform Identification after TMT Labeling.
Top-down LC-MS/MS was performed for the identification of labeled proteoforms in the filter-SEC low MW samples (e.g., Sample 1 and Sample 2, Figure 2A). There are 411 proteoforms of 95 proteins identified from these 2 samples: 309 proteoforms from 72 proteins identified in Sample 1 and 222 proteoforms from 60 proteins identified in sample 2 after TMT labeling (Supplementary Table S1B,C). The labeling efficiency for identified proteoforms was listed in a heatmap (Figure 2B). Each row in the heatmap represents a “protein group”, which contains all TMT labeled products from 1 proteoform (i.e., completely/under-/overlabeled products), and the relative intensity of each labeled product is indicated in greyscale. In sample 1, 175 out of 211 protein groups display only the completely labeled products with no detection of improperly labeled side products (Figure 2B). 17.1% (36 of 211) of the protein groups demonstrated overlabeled products, and 10.4% (22 of 211) of the protein groups demonstrated underlabeled products. In addition, 88.6% (187 of 211) of the protein groups show the completely labeled product with >80% intensity. In sample 2, 114 of 183 protein groups showed only the completely labeled species, 31.1% (57 of 183) of the protein groups included overlabeled products, and 20.8% (38 of 183) of the protein groups included underlabeled products. 74.9% (137 of 183) of the protein groups show a completely labeled product with >80% intensity. The average labeling efficiency is 87.7% ± 5.8% and 81.3% ± 7.6% in sample 1 and sample 2, respectively, where the average labeling efficiency is the mean of the labeling efficiency of all the protein groups.
Figure 2.
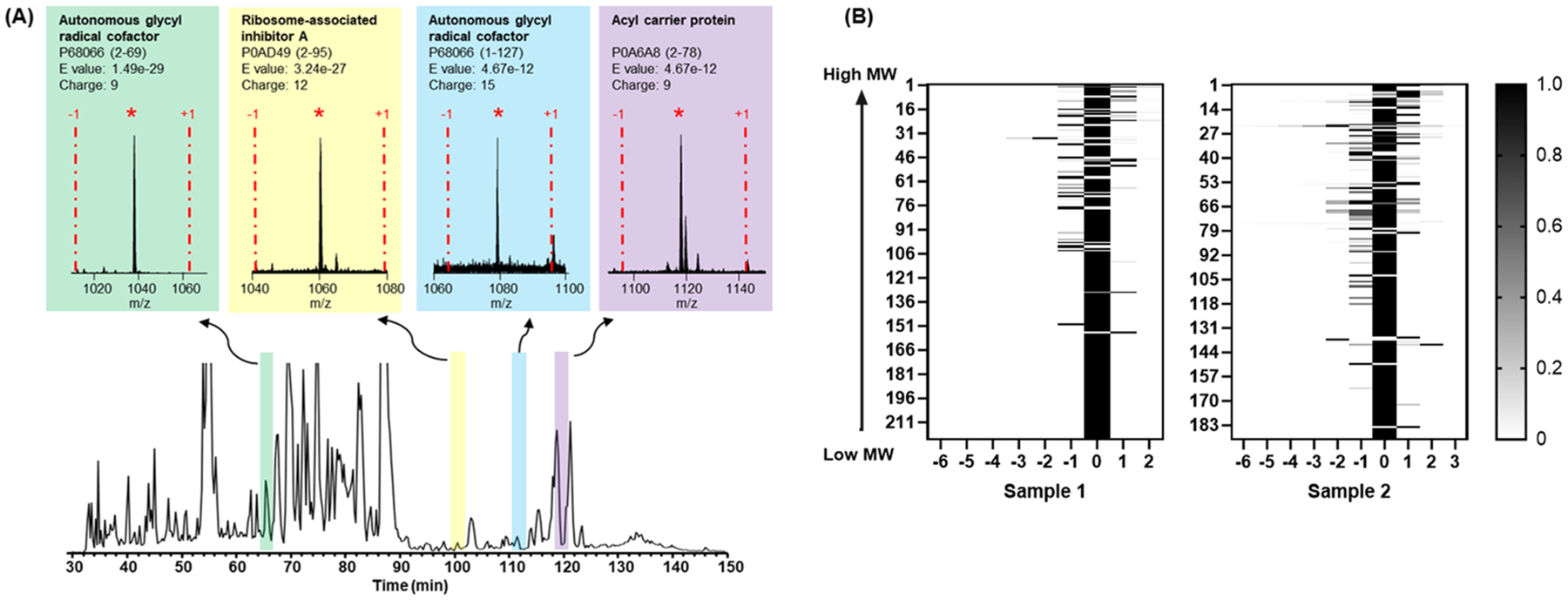
Intact protein labeling and identification. (A) Examples of labeled proteoforms identified in one LC-MS run. The completely labeled forms are indicated by an asterisk and the −1 and +1 represent proteoforms that are underlabeled or overlabeled by one tag. (B) The identification heatmap of samples 1 and 2. The labeling status is indicated for each intact proteoform after labeling.
Overlabeling was observed in these two samples when the NHS-ester reactive group on the TMT labels reacted with amino acid residues other than K. For example, the pKa of Y (10.3 ± 1.2) is similar to K (10.5 ± 1.1),38 which suggests that Y may react with TMT under similar pH conditions.36 The pKa of H (6.6 ± 1.0) is lower than the N-terminus (7.7 ± 0.5)38 and, accordingly, the N-imidazole of H is more deprotonated than N-terminus, which promotes TMT labeling on H. Moreover, the hydrogen bonds within the secondary structure increase the nucleophilicity of hydroxyl-group, leading to the O-acylation of S, T, and Y residues.39,40 As suggested in the previous bottom-up study, the optimization of the quenching solution may help to reduce the overlabeled products.36
We also found that a significantly lower number of proteins were identified with MW larger than 15 kDa when compared with the number of proteins identified smaller than 15 kDa (Supplementary Figure S2A). To evaluate the ability of TMT to efficiently label proteins >15 kDa, we compared the filter-SEC samples before and after TMT labeling (high MWCO sample) using SDS-PAGE (Supplementary Figure S2B). The SDS results suggested that there was no significant loss of larger proteins (i.e., between 20 kDa and 50 kDa) before and after labeling. However, it is still possible that large proteins precipitated after labeling in organic condition and were reconstituted in when SDS was added prior to SDS-PAGE analysis. To remove any proteins that may have precipitated due to addition of organic solvents, the samples were centrifuged at 13 000 rpm for 30 min before SDS-PAGE or LC-MS. Removal of the large proteins was not observed in the centrifuged samples, and, furthermore, continuous injection of these samples did not clog the LC column. This suggests that proteins were not precipitated under labeling conditions or LC buffer conditions. The low identification number of larger proteins may be related to the MS instrument limitations (e.g., LTQ-Orbitrap). Because of the limited charge capacity of MS instruments, it is a challenge to identify proteoforms >30 kDa as has been discussed previously.41 Kelleher and co-workers also discussed that protein identifications are dominated by species under 30 kDa due to both inefficient front-end separations and increased difficulty in detection of high mass proteins.42 Yang et al. reported identified proteoforms that were smaller than 35 kDa from both E. coli and HepG2 cells using an CZE separation coupled to Orbitrap QE-HF.43 Another possible reason for these issues may be the heterogeneity caused by unwanted species such as underlabeled or overlabeled products. These species are often coeluted with each other and/or with other protein species. In ion trap instruments with limited charge capacity, these complications can significantly reduce the signal-to-noise ratio (S/N). With improved MS instrumentation (e.g., ion–ion proton transfer and parallel ion parking44) and efficient enrichment/separation approaches for large proteins, we expect that the proposed TMT labeling approach may be applied to proteins with relatively high MW (up to 50 kDa).
Additionally, we noticed that a significant number of proteoform features cannot be identified, even when good quality MS/MS spectra were collected. This may be due to limitations associated with the identification software (TopPIC) which limits the number of dynamic modifications (up to two) without significantly increasing the FDR. The current software considers any N-terminal protein processing (e.g., N-terminal methionine cleavage, signal peptide removal, and N-terminal acetylation) as well as other known or unknown modifications as dynamic modifications. In addition, because we defined TMT as a fixed modification on the lysine residues and N-terminus, any underlabeling or overlabeling events are considered as dynamic modifications. If more than two underlabeling and/or PTM events are present, our customized TopPIC software cannot automatically identify the detected proteoforms. Therefore, we need to improve the current bioinformatic tools for TMT top-down MS to allow the characterization of unexpected modifications as well as underlabeled or overlabeled proteoforms.
Quantification of Intact Proteins and Proteoforms Using TMT Labeling.
To evaluate the quality of protein-level TMT labeling and its quantification accuracy and reproducibility, the filter-SEC sample was separated to four aliquots with a 5:2:2:1 volume ratio, and the aliquots were labeled independently with TMT reagent 128, 129, 130, and 131. The intensity of reporter ions was distributed from 1e2 to 1e6, which suggests a potentially large dynamic range of relative quantification with top-down proteomics (Supplementary Table S2). To verify the labeling accuracy, the intensity ratios among each pair of samples were compared (128:131, 129:131, and 130:131) for all the scans in which all reporter ions were detected (Figure 3A). The average reporter ion ratios were 6.22 ± 2.17, 2.42 ± 0.97, and 2.60 ± 0.99 for Sample 1 and 5.12 ± 3.33, 2.20 ± 0.88, and 2.40 ± 0.90 for Sample 2. To evaluate the reproducibility of the method, we tested two replicate samples that were individually labeled with TMT 129 and 130. The reporter ion intensities in all the identified MS/MS scans were plotted between these two samples (Figure 3B,C), and good linear correlations were observed for both Sample 1 and Sample 2 (R2 = 0.9922 and 0.9844, respectively). We also examined the quantification results of underlabeled and overlabeled proteoforms and found that the results were similar to completely labeled proteoforms. However, the over/underlabeled species tended to have lower intensities, which may result from lower parent ion intensities of the side products (Supplementary Figure S3). These results indicate that it is possible to quantify complex protein mixtures with intact protein labeling and top-down proteomics.
Figure 3.
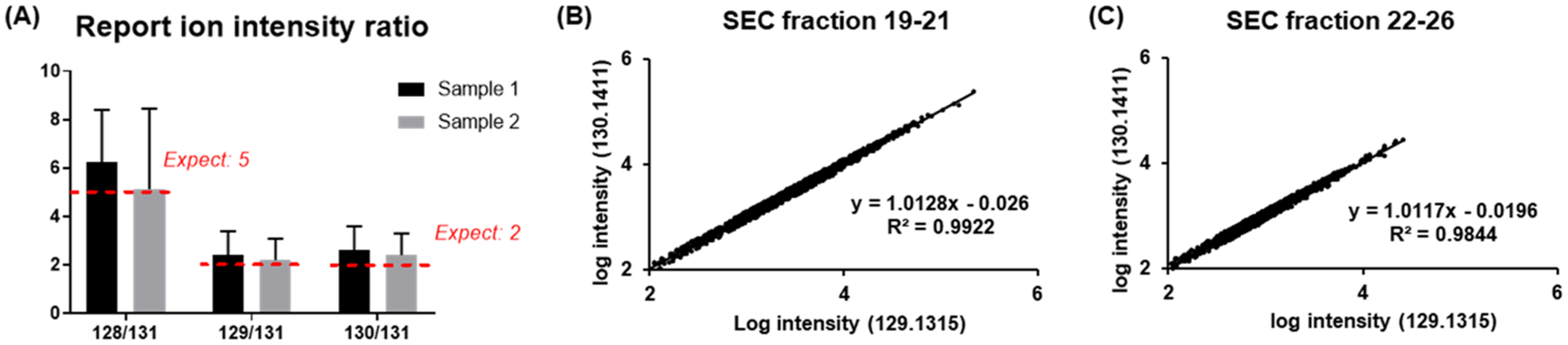
Evaluation of protein-level TMT quantification. (A) The normalized intensity ratio (128/131, 129/131, and 130/131) of the two samples. The error-bars represent the calculated standard deviation of reporter ion ratios from all MS/MS scans. The intensity of reporter ions 129 and 130 are correlated to evaluate the reproducibility of the quantification: (B) Sample 1 (fractions 19–21) and (C) Sample 2 (fractions 22–26).
Examples of Protein-level TMT Labeling.
Two 50S ribosomal protein L6 (P0AG55) proteoforms were observed (Figure 4). 50S ribosomal protein L6 contains six lysine residues and an amine group on the N-terminus. As such, the completely labeled proteoform should contain 7 TMT tags. In the labeled sample, one peak that represented the completely labeled proteoform and a small shoulder peak that represented the underlabeled proteoform containing six labels were observed. The HCD MS/MS spectrum confirmed that the proteoform with 7 TMT tags was the completely labeled ribosomal L6 protein. The reporter ions were confidently detected in the HCD spectrum, and the ratio correlated well with the theoretical ratio (5:2:2:1). Another interesting observation is that the charge state distributions were similar between the unlabeled proteoforms and the labeled proteoforms. This observation indicates that the proton affinity on the labeled group is similar to that of the lysine amino group, which is consistent with previous reports.45,46 We also identified the proteoform with 6 TMT tags and confirmed that the TMT underlabeling was either on the N-terminus or on Lys-5 (additional example in Supplementary Figure S4 suggested that the missing label was on the N-terminus for the ribosome-associated inhibitor A protein). Interestingly, reporter ion intensity ratios among different samples correlated well with theoretical ratios suggesting that underlabeling may not affect the quantification results. It has been reported that ETD can also be used to identify and quantify TMT labeled peptides,47 so we performed an ETD analysis on the same sample. However, the overall fragmentation efficiency was low, and no observable reporter ions were detected in the ETD spectra (Figure 4D, Supplementary Figure S4D). Additional optimization or evaluation should be performed for ETD-based top-down TMT experiments.
Figure 4.
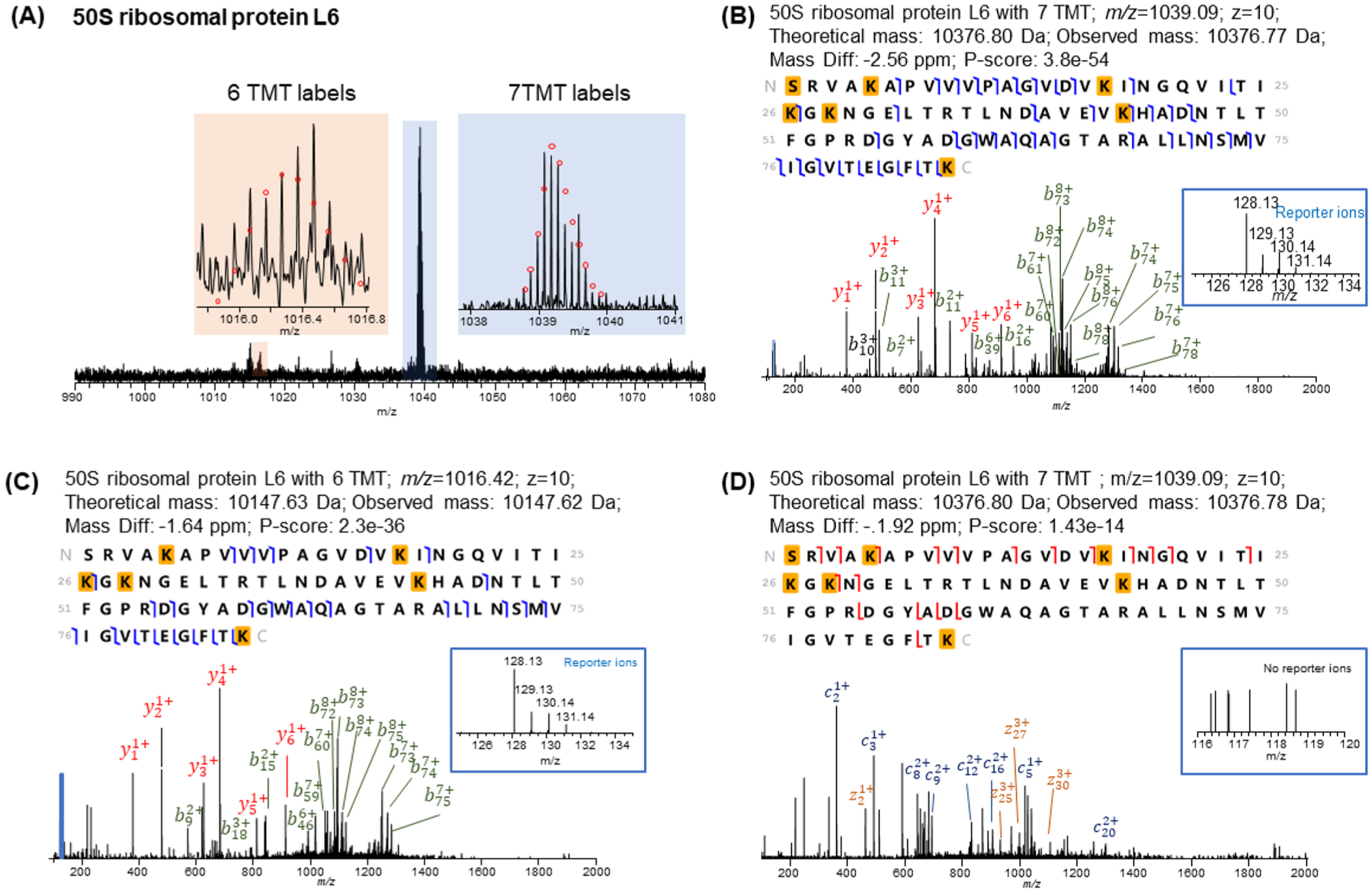
Identification of 50S ribosomal protein L6 with 6 and 7 TMT labels. (A) Full MS spectrum of the protein with 6 and 7 TMT labels. The HCD MS/MS spectra and identification of (B) protein with 7 TMT labels and (C) protein with 6 TMT labels. (D) ETD MS/MS spectrum of protein with 7 TMT labels.
The thiol peroxidase (P0A862) proteoforms were studied to evaluate the quantitative accuracy of overlabeled species (Supplementary Figure S5A). Thiol peroxidase contains seven lysine residues and an amine group on the N-terminus. In the labeled sample, three peaks indicating an underlabeled proteoform (7 TMT), completely labeled proteoform (8 TMT; Supplementary Figure S5B), and overlabeled proteoform (9 TMT; Supplementary Figure S5C) were observed. The completely labeled and overlabeled proteoforms were confirmed with HCD MS/MS spectrum. In the overlabeled proteoform, the excess label was located between Ser-1 and His-5 as determined using continuous b ions from MS/MS fragmentation. It has been reported that overlabeling may occur on S/T/Y residues separated by 1 residue from H.36,40,48 Therefore, it is possible that the overlabeled residue is Thr-3. The reporter ions were confidently detected in the HCD spectrum for the overlabeled species and the experimenal ratio (5.28:2.73:2.79:1) of the overlabeled species correlated well with the experimental ratio of the completely labeled species (5.04:2.43:2.88:1) and the theoretical ratio (5:2:2:1). It is apparent that over/underlabeled side products provide accurate quantitation results and may be used as replicates for quantification.
We also evaluated the labeling efficiency on large proteins. The completely labeled ribose import binding protein (RbsB) was detected with all 24 lysine residues and the N-terminus labeled in the MS spectrum (Figure 5A) and was confirmed by the MS/MS spectrum (Figure 5B). The reporter ions were also detected in the HCD spectrum with good intensities, and the ratio correlated well with the theoretical ratio. However, the underlabeled proteoform that lacked TMT labeling on the N-terminus had relatively high abundance. Underlabeling of the proteoform indicates the ratio between labeling reagent and protein needs to be optimized for larger proteins (i.e., larger than 20 kDa).
Figure 5.
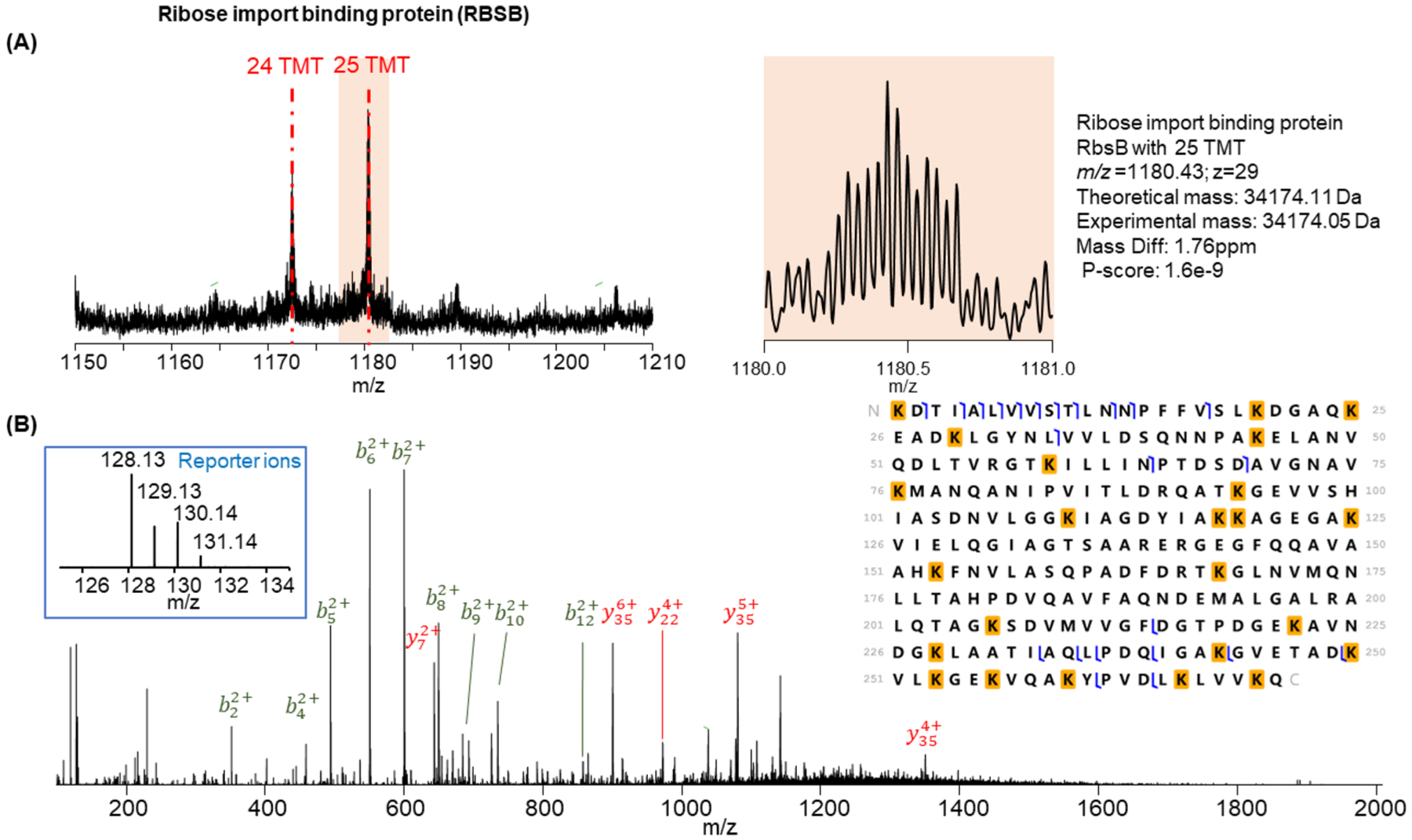
Identification of completely labeled ribose import binding protein RbsB. (A) MS spectrum of the completely labeled proteoform (25 TMT labels) and one underlabeled proteoform (24 TMT labels). (B) HCD MS/MS spectrum and identification of the completely labeled RbsB. The lysine on the N-terminus is labeled on both its side chain and N-terminus primary amine.
CONCLUSION
We have developed a method to identify and quantify intact TMT-labeled proteins using top-down proteomics. Our work provides a tool to enrich proteins smaller than 30 kDa, a workflow for intact protein-level TMT labeling, and a quantitative top-down analysis of TMT-labeled proteins with high-resolution FTMS instruments (e.g., orbitrap or FTICR). We have demonstrated that most identified proteins were efficiently labeled (e.g., >80% labeling efficiency). Additionally, our quantification results with proteins smaller than 30 kDa were highly replicable, even for proteins that were not completely labeled. Further studies will be focused on the optimization of the protein-level TMT labeling conditions to improve the intact protein labeling efficiency such as TMT reagent to protein ratio, TMT reagent and protein concentrations, quenching solution concentration and pH, labeling time, and reaction buffers. In addition, other separation methods that select for different molecule sizes may be used, such as GELFrEE49 or serial size exclusion chromatography50 to enrich low molecular weight proteins. Our filter-SEC method enriches proteins smaller than 30 kDa from the E. coli lysate, which excludes some larger proteins in the E. coli proteome, so complete proteome analysis cannot be achieved. For larger proteins, different organic solvents can be evaluated to improve sample solubility. In summary, the intact TMT labeling technique extends isobaric labeling quantification to a wide variety of applications in top-down proteomics and could be easily incorporated into many areas of quantitative proteomics research including the discovery of therapeutic proteins and disease biomarkers.
Supplementary Material
Figure S1. Filter-SEC separation performance evaluation.
Figure S2. Evaluation of detected protein MW ranges after TMT labeling.
Figure S3. Correlation of the intensity of reporter ions for under-labeled and overlabeled proteoforms.
Figure S4. Identification of ribosome-associated inhibitor A with 9 and 8 TMT labels. Figure S5. Identification of thiol peroxidase with 9, 8, and 7 TMT labels (PDF)
Table S2. Report ion intensities in TMT-labeled samples (XLSX)
Table S1. Intact proteoform identification after labeling (XLSX)
ACKNOWLEDGMENTS
This work was partly supported by grants from NIH NIAID R01AI141625, NIAID CSGADP Pilot project (NIH 5U01AI101990-04, BRI no. FY15109843), NIH NIGMS R01GM118470, OCAST HR16-125, and OU FIP program. We thank Mulin Fang for helping with the data analysis. D.H.Y. also wants to thank Hongyan Ma and Toni Woodard for the initial wet lab training.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jasms.0c00464.
Complete contact information is available at: https://pubs.acs.org/10.1021/jasms.0c00464
Contributor Information
Qiang Kou, School of Informatics and Computing, Indiana University-Purdue University Indianapolis, Indianapolis, Indiana 46202, United States;.
Kenneth Smith, Department of Arthritis and Clinical Immunology, Oklahoma Medical Research Foundation, Oklahoma City, Oklahoma 73104, United States.
Xiaowen Liu, School of Informatics and Computing, Indiana University-Purdue University Indianapolis, Indianapolis, Indiana 46202, United States.
Si Wu, Department of Chemistry and Biochemistry, University of Oklahoma, Norman, Oklahoma 73019, United States;.
REFERENCES
- (1).Ansong C; Wu S; Meng D; Liu X; Brewer HM; Deatherage Kaiser BL; Nakayasu ES; Cort JR; Pevzner P; Smith RD; Heffron F; Adkins JN; Pasa-Tolic L Top-down proteomics reveals a unique protein S-thiolation switch in Salmonella Typhimurium in response to infection-like conditions. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 10153–10158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Zhang J; Guy MJ; Norman HS; Chen Y-C; Xu Q; Dong X; Guner H; Wang S; Kohmoto T; Young KH; Moss RL; Ge Y Top-Down Quantitative Proteomics Identified Phosphorylation of Cardiac Troponin I as a Candidate Biomarker for Chronic Heart Failure. J. Proteome Res 2011, 10, 4054–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Chamot-Rooke J; Mikaty G; Malosse C; Soyer M; Dumont A; Gault J; Imhaus A-F; Martin P; Trellet M; Clary G; Chafey P; Camoin L; Nilges M; Nassif X; Dumenil G Posttranslational modification of pili upon cell contact triggers N. meningitidis dissemination. Science 2011, 331, 778–782. [DOI] [PubMed] [Google Scholar]
- (4).Ntai I; LeDuc RD; Fellers RT; Erdmann-Gilmore P; Davies SR; Rumsey J; Early BP; Thomas PM; Li S; Compton PD; Ellis MJC; Ruggles KV; Fenyo D; Boja ES; Rodriguez H; Townsend RR; Kelleher NL Integrated bottom-up and top-down proteomics of patient-derived breast tumor xenografts. Molecular & Cellular Proteomics 2016, 15, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Zhang J; Guy MJ; Norman HS; Chen Y-C; Xu Q; Dong X; Guner H; Wang S; Kohmoto T; Young KH; Moss RL; Ge Y Top-down quantitative proteomics identified phosphorylation of cardiac troponin I as a candidate biomarker for chronic heart failure. J. Proteome Res 2011, 10, 4054–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wei L; Gregorich ZR; Lin Z; Cai W; Jin Y; McKiernan SH; McIlwain S; Aiken JM; Moss RL; Diffee GM; Ge Y Novel Sarcopenia-related Alterations in Sarcomeric Protein Post-translational Modifications (PTMs) in Skeletal Muscles Identified by Top-down Proteomics. Molecular & Cellular Proteomics 2018, 17, 134–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wu S; Brown JN; Tolic N; Meng D; Liu X; Zhang H; Zhao R; Moore RJ; Pevzner P; Smith RD; Pasa-Tolic L Quantitative analysis of human salivary gland-derived intact proteome using top-down mass spectrometry. Proteomics 2014, 14, 1211–1222. [DOI] [PubMed] [Google Scholar]
- (8).Schaffer LV; Rensvold JW; Shortreed MR; Cesnik AJ; Jochem A; Scalf M; Frey BL; Pagliarini DJ; Smith LM Identification and Quantification of Murine Mitochondrial Proteoforms Using an Integrated Top-Down and Intact-Mass Strategy. J. Proteome Res 2018, 17, 3526–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Du Y; Parks BA; Sohn S; Kwast KE; Kelleher NL Top-down approaches for measuring expression ratios of intact yeast proteins using Fourier transform mass spectrometry. Anal. Chem 2006, 78, 686–694. [DOI] [PubMed] [Google Scholar]
- (10).Fornelli L; Durbin KR; Fellers RT; Early BP; Greer JB; LeDuc RD; Compton PD; Kelleher NL Advancing top-down analysis of the human proteome using a benchtop quadrupole-Orbitrap mass spectrometer. J. Proteome Res 2017, 16, 609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ong S-E; Blagoev B; Kratchmarova I; Kristensen DB; Steen H; Pandey A; Mann M Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Molecular & cellular proteomics 2002, 1, 376–386. [DOI] [PubMed] [Google Scholar]
- (12).Collier TS; Hawkridge AM; Georgianna DR; Payne GA; Muddiman DC Top-down identification and quantification of stable isotope labeled proteins from Aspergillus flavus using online nano-flow reversed-phase liquid chromatography coupled to a LTQ-FTICR mass spectrometer. Anal. Chem 2008, 80, 4994–5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Hebert AS; Merrill AE; Bailey DJ; Still AJ; Westphall MS; Strieter ER; Pagliarini DJ; Coon JJ Neutron-encoded mass signatures for multiplexed proteome quantification. Nat. Methods 2013, 10, 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Rhoads TW; Rose CM; Bailey DJ; Riley NM; Molden RC; Nestler AJ; Merrill AE; Smith LM; Hebert AS; Westphall MS; Pagliarini DJ; Garcia BA; Coon JJ Neutron-encoded mass signatures for quantitative top-down proteomics. Anal. Chem 2014, 86, 2314–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Fang H; Xiao K; Li Y; Yu F; Liu Y; Xue B; Tian Z Intact Protein Quantitation Using Pseudoisobaric Dimethyl Labeling. Anal. Chem 2016, 88, 7198–7205. [DOI] [PubMed] [Google Scholar]
- (16).Liu Z; Wang R; Liu J; Sun R; Wang F Global Quantification of Intact Proteins via Chemical Isotope Labeling and Mass Spectrometry. J. Proteome Res 2019, 18, 2185. [DOI] [PubMed] [Google Scholar]
- (17).Ross PL; Huang YN; Marchese JN; Williamson B; Parker K; Hattan S; Khainovski N; Pillai S; Dey S; Daniels S; Purkayastha S; Juhasz P; Martin S; Bartlet-Jones M; He F; Jacobson A; Pappin DJ Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Molecular & cellular proteomics 2004, 3, 1154–1169. [DOI] [PubMed] [Google Scholar]
- (18).Thompson A; Schäfer J; Kuhn K; Kienle S; Schwarz J; Schmidt G; Neumann T; Hamon C Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem 2003, 75, 1895–1904. [DOI] [PubMed] [Google Scholar]
- (19).Xiang F; Ye H; Chen R; Fu Q; Li LN, N-dimethyl leucines as novel isobaric tandem mass tags for quantitative proteomics and peptidomics. Anal. Chem 2010, 82, 2817–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Frost DC; Feng Y; Li L 21-plex DiLeu isobaric tags for high-throughput quantitative proteomics. Anal. Chem 2020, 92, 8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Cox J; Mann M Quantitative, high-resolution proteomics for data-driven systems biology. Annu. Rev. Biochem 2011, 80, 273–299. [DOI] [PubMed] [Google Scholar]
- (22).Bantscheff M; Schirle M; Sweetman G; Rick J; Kuster B Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanal. Chem 2007, 389, 1017–1031. [DOI] [PubMed] [Google Scholar]
- (23).Sinclair J; Timms JF Quantitative profiling of serum samples using TMT protein labelling, fractionation and LC–MS/MS. Methods 2011, 54, 361–369. [DOI] [PubMed] [Google Scholar]
- (24).Wiese S; Reidegeld KA; Meyer HE; Warscheid B Protein labeling by iTRAQ: a new tool for quantitative mass spectrometry in proteome research. Proteomics 2007, 7, 340–350. [DOI] [PubMed] [Google Scholar]
- (25).Prudova A; auf dem Keller U; Butler GS; Overall CM Multiplex N-terminome analysis of MMP-2 and MMP-9 substrate degradomes by iTRAQ-TAILS quantitative proteomics. Mol. & Cell. Proteomics 2010, 9, P894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Hung C-W; Tholey A Tandem mass tag protein labeling for top-down identification and quantification. Anal. Chem 2012, 84, 161–170. [DOI] [PubMed] [Google Scholar]
- (27).Chowdhury SM; Du X; Tolic N; Wu S; Moore RJ; Mayer MU; Smith RD; Adkins JN Identification of cross-linked peptides after click-based enrichment using sequential collision-induced dissociation and electron transfer dissociation tandem mass spectrometry. Anal. Chem 2009, 81, 5524–5532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wang Z; Ma H; Smith K; Wu S Two-dimensional separation using high-pH and low-pH reversed phase liquid chromatography for top-down proteomics. Int. J. Mass Spectrom 2018, 427, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Wang Z; Liu X; Muther J; James JA; Smith K; Wu S Top-down Mass Spectrometry Analysis of Human Serum Autoantibody Antigen-Binding Fragments. Sci. Rep 2019, 9, 2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Chambers MC; Maclean B; Burke R; Amodei D; Ruderman DL; Neumann S; Gatto L; Fischer B; Pratt B; Egertson J A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol 2012, 30, 918–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kou Q; Xun L; Liu X TopPIC: a software tool for top-down mass spectrometry-based proteoform identification and characterization. Bioinformatics 2016, 32, 3495–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Guner H; Close PL; Cai W; Zhang H; Peng Y; Gregorich ZR; Ge Y MASH Suite: a user-friendly and versatile software interface for high-resolution mass spectrometry data interpretation and visualization. J. Am. Soc. Mass Spectrom 2014, 25, 464–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Fellers RT; Greer JB; Early BP; Yu X; LeDuc RD; Kelleher NL; Thomas PM ProSight Lite: graphical software to analyze top-down mass spectrometry data. Proteomics 2015, 15, 1235–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Levitsky LI; Klein JA; Ivanov MV; Gorshkov MV Pyteomics 4.0: five years of development of a Python proteomics framework. J. Proteome Res 2019, 18, 709–714. [DOI] [PubMed] [Google Scholar]
- (35).Goloborodko AA; Levitsky LI; Ivanov MV; Gorshkov MV Pyteomics—a Python framework for exploratory data analysis and rapid software prototyping in proteomics. J. Am. Soc. Mass Spectrom 2013, 24, 301–304. [DOI] [PubMed] [Google Scholar]
- (36).Zecha J; Satpathy S; Kanashova T; Avanessian SC; Kane MH; Clauser KR; Mertins P; Carr SA; Kuster B TMT labeling for the masses: a robust and cost-efficient, in-solution labeling approach. Molecular & Cellular Proteomics 2019, 18, 1468–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Rossky PJ Protein denaturation by urea: slash and bond. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 16825–16826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Grimsley GR; Scholtz JM; Pace CN A summary of the measured pK values of the ionizable groups in folded proteins. Protein Sci. 2009, 18, 247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Uchida K; Kawakishi S Sequence-dependent reactivity of histidine-containing peptides with copper (II)/ascorbate. J. Agric. Food Chem 1992, 40, 13–16. [Google Scholar]
- (40).Miller BT; Kurosky A Elevated intrinsic reactivity of seryl hydroxyl groups within the linear peptide triads His-Xaa-Ser or Ser-Xaa-His. Biochem. Biophys. Res. Commun 1993, 196, 461–467. [DOI] [PubMed] [Google Scholar]
- (41).Schaffer LV; Millikin RJ; Miller RM; Anderson LC; Fellers RT; Ge Y; Kelleher NL; LeDuc RD; Liu X; Payne SH; Sun L; Thomas PM; Tucholski T; Wang Z; Wu S; Wu Z; Yu D; Shortreed MR; Smith LM Identification and quantification of proteoforms by mass spectrometry. Proteomics 2019, 19, 1800361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Li Y; Compton PD; Tran JC; Ntai I; Kelleher NL Optimizing capillary electrophoresis for top-down proteomics of 30–80 kDa proteins. Proteomics 2014, 14, 1158–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Yang Z; Shen X; Chen D; Sun L Towards a universal sample preparation method for denaturing top-down proteomics of complex proteomes. J. Proteome Res 2020, 19, 3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Ugrin SA; English AM; Syka JE; Bai DL; Anderson LC; Shabanowitz J; Hunt DF Ion-Ion Proton Transfer and Parallel Ion Parking for the Analysis of Mixtures of Intact Proteins on a Modified Orbitrap Mass Analyzer. J. Am. Soc. Mass Spectrom 2019, 30, 2163–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Bush MF; Oomens J; Williams ER Proton affinity and zwitterion stability: new results from infrared spectroscopy and theory of cationized lysine and analogues in the gas phase. J. Phys. Chem. A 2009, 113, 431–438. [DOI] [PubMed] [Google Scholar]
- (46).Thingholm TE; Palmisano G; Kjeldsen F; Larsen MR Undesirable charge-enhancement of isobaric tagged phosphopeptides leads to reduced identification efficiency. J. Proteome Res 2010, 9, 4045–4052. [DOI] [PubMed] [Google Scholar]
- (47).Phanstiel D; Unwin R; McAlister GC; Coon JJ Peptide quantification using 8-plex isobaric tags and electron transfer dissociation tandem mass spectrometry. Anal. Chem 2009, 81, 1693–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Wiktorowicz JE; English RD; Wu Z; Kurosky A Model studies on iTRAQ modification of peptides: sequence-dependent reaction specificity. J. Proteome Res 2012, 11, 1512–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Tran JC; Zamdborg L; Ahlf DR; Lee JE; Catherman AD; Durbin KR; Tipton JD; Vellaichamy A; Kellie JF; Li M; Wu C; Sweet SMM; Early BP; Siuti N; LeDuc RD; Compton PD; Thomas PM; Kelleher NL Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature 2011, 480, 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Cai W; Tucholski T; Chen B; Alpert AJ; McIlwain S; Kohmoto T; Jin S; Ge Y Top-down proteomics of large proteins up to 223 kDa enabled by serial size exclusion chromatography strategy. Anal. Chem 2017, 89, 5467–5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Filter-SEC separation performance evaluation.
Figure S2. Evaluation of detected protein MW ranges after TMT labeling.
Figure S3. Correlation of the intensity of reporter ions for under-labeled and overlabeled proteoforms.
Figure S4. Identification of ribosome-associated inhibitor A with 9 and 8 TMT labels. Figure S5. Identification of thiol peroxidase with 9, 8, and 7 TMT labels (PDF)
Table S2. Report ion intensities in TMT-labeled samples (XLSX)
Table S1. Intact proteoform identification after labeling (XLSX)