


Keywords: brain, central nervous system, factor, inflammation, injury, pathways, repair, stroke
Abstract
Oxidative stress is a crucial pathological process that contributes to secondary injury following intracerebral hemorrhage. P2X7 receptor (P2X7R), which is activated by the abnormal accumulation of extracellular ATP, plays an important role in the regulation of oxidative stress in the central nervous system, although the effects of activated P2X7R-associated oxidative stress after intracerebral hemorrhage remain unclear. Mouse models of intracerebral hemorrhage were established through the stereotactic injection of 0.075 U VII collagenase into the right basal ganglia. The results revealed that P2X7R expression peaked 24 hours after intracerebral hemorrhage, and P2X7R expressed primarily in neurons. The inhibition of P2X7R, using A438079 (100 mg/kg, intraperitoneal), reduced nicotinamide adenine dinucleotide phosphate oxidase 2 (NOX2) expression and malondialdehyde generation, increased superoxide dismutase and glutathione/oxidized glutathione levels, and alleviated neurological damage, brain edema, and apoptosis after intracellular hemorrhage. The P2X7R inhibitor A438079 (100 mg/kg, intraperitoneal injection) inhibited the activation of extracellular signal-regulated kinase 1/2 (ERK1/2) and nuclear factor kappa-B (NF-κB) after intracerebral hemorrhage. Blocking ERK1/2 activation, using the ERK1/2 inhibitor U0126 (2 µg, intraventricular injection), reduced the level of NOX2-mediated oxidative stress induced by P2X7R activation after intracellular hemorrhage. Similarly, the inhibition of NF-κB, using the NF-κB inhibitor JSH-23 (3.5 µg, intraventricular), reduced the level of NOX2-mediated oxidative stress induced by P2X7R activation. Finally, GSK2795039 (100 mg/kg, intraperitoneal), a NOX2 antagonist, attenuated P2X7R-mediated oxidative stress, neurological damage, and brain edema after intracerebral hemorrhage. The results indicated that P2X7R activation aggravated NOX2-induced oxidative stress through the activation of the ERK1/2 and NF-κB pathways following intracerebral hemorrhage in mice. The present study was approved by the Ethics Committee of Huazhong University of Science and Technology, China (approval No. TJ-A20160805) on August 26, 2016.
Chinese Library Classification No. R453; R743.34; Q554+.5
Introduction
Intracerebral hemorrhage (ICH) is a destructive stroke subtype that comprises 6.5–19.6% of all stroke events (Feigin et al., 2003; O’Donnell et al., 2016) and is associated with high mortality (approximately 60% after 1 year and 76% after 10 years) and a low rate of functional independence (Sacco et al., 2009). The treatment of ICH should consist of a composite strategy that both reduces primary injury and alleviates secondary injury (Keep et al., 2012). The initial bleeding that occurs after the rupture of small arteries causes the physical destruction of the brain’s cellular architecture, and resulting hematoma increases intracranial pressure, leading to primary injury (Xi et al., 2006; Gao et al., 2020). Although many clinical trials have been conducted to explore strategies for reducing primary injury after ICH, such as surgical clot removal and the prevention of hematoma expansion, the effects on the recovery of neurological damage has been minimal (Gross et al., 2019). Only a few clinical trials have explored intervention strategies for the prevention of secondary injury, caused by the release of clot components following ICH. However, additional preclinical evidence examining the alleviation of secondary injury after ICH remains necessary to support clinical trials (Wang, 2010).
Oxidative stress describes the grievous imbalance between antioxidant resistance and oxidation, which leads to the formation of large quantities of oxidized intermediates (Sies et al., 2017; Cheng et al., 2019; Shefa et al., 2019). Numerous studies have illustrated that oxidative stress is a likely culprit for the mediation of secondary injury after ICH (Duan et al., 2016; Hu et al., 2016). Mitochondria and other enzymatic pathways are the primary sources of reactive oxygen species (ROS) (Sies et al., 2017). Nicotinamide adenine dinucleotide phosphate oxidase 2 (NOX2) is the primary enzyme that mediates ROS production (Drummond et al., 2011). Previous studies have found that NOX2 expression is upregulated after ICH and subarachnoid hemorrhage (Yang et al., 2017; Zhang et al., 2017). However, the mechanism underlying NOX2 upregulation after ICH is still poorly understood.
P2X7 receptor (P2X7R), a member of the P2X receptor family, is an ATP-gated, non-selective, ionotropic channel (Di Virgilio et al., 2017). P2X7R is activated by the abnormal accumulation of extracellular ATP and plays a vital function in the regulation of oxidative stress in the central nervous system (Miras-Portugal et al., 2019). The pharmacological blockade or genetic knockout of P2X7R has been shown to alleviate oxidative stress in a variety of neurodegenerative disease models (Apolloni et al., 2013; Butterfield and Halliwell, 2019). Several studies have explored the pathophysiological role played by P2X7R following ICH, focusing on the P2X7R-mediated inflammatory response, through the promotion of the NOD-, LRR- and pyrin domain-containing protein 3 inflammasome (Feng et al., 2015; Zhao et al., 2017). However, the role played by P2X7R activation in the regulation of oxidative stress after ICH remains still poorly understood. Mitogen-activated protein kinases (MAPKs) and nuclear factor kappa-B (NF-κB) are important pathways involved in ICH-induced secondary injury (Zhu et al., 2019). Several studies have shown that P2X7R activation mediates NOX2-dependent ROS production by activating extracellular signal-regulated kinase 1/2 (ERK1/2) (Martel-Gallegos et al., 2013; Apolloni et al., 2016). Additional research has demonstrated that NF-κB activation is involved in NOX2 expression (Anrather et al., 2006).
Therefore, this study aimed to explore whether P2X7R activation aggravates NOX2-induced oxidative stress following ICH and explored the underlying mechanism.
Materials and Methods
Animals
Male C57BL/6 mice (specific-pathogen-free, 8- to 10-week-old, 20–23 g, Beijing HFK Bioscience Co. Ltd. China, license No. SCXK [Jing] 2014-0004) were used for this experiment. Mice were housed in a comfortable environment (humidity: 55% ± 5%, temperature: 22 ± 2°C, 12-hour dark/light cycle) and were randomly assigned to five separate experiments. Except for 8 hours of fasting before model induction, mice had free access to food and water. The experimental protocol was approved by the Ethics Committee of Huazhong University of Science and Technology, China (approval No. TJ-A20160805) on August 26, 2016. The number of mice used for each experiment is shown in Additional Table 1. To generate the ICH model, mice were anesthetized with 1% sodium pentobarbital (70 mg/kg; intraperitoneal injection; Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) and fixed in a prone position, on a stereotaxic instrument (Model 500; Kopf Instruments, Beijing, China). To perform sampling, mice were first euthanized by deep anesthesia, using 1% sodium pentobarbital (100 mg/kg; intraperitoneal injection), and then transcardially perfused with saline (Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) and 4% paraformaldehyde (Sinopharm Chemical Reagent Co., Ltd.).
Additional Table 1.
Experimental grouping and animal mortality in this study
| Experimental groups | Inclusion | Mortality [n(%)] | Subtotal |
|---|---|---|---|
| Experiment I | |||
| Sham | 6 | 0 | 6 |
| ICH (3, 6, 12, 24, 48, 72 h, 5 d) | 48 | 6(12) | 54 |
| Experiment II | |||
| Sham | 28 | 0 | 28 |
| ICH | 28 | 2(7) | 30 |
| ICH + vehicle | 28 | 3(11) | 31 |
| ICH + BzATP | 28 | 3(11) | 31 |
| ICH + A438079 | 28 | 2(7) | 30 |
| Experiment III | |||
| Sham | 6 | 0 | 6 |
| ICH + vehicle | 6 | 0 | 6 |
| ICH + U0126 | 12 | 1(8) | 13 |
| ICH + BzATP | 6 | 0 | 6 |
| ICH + U0126+ BzATP | 12 | 2(17) | 14 |
| Experiment IV | |||
| Sham | 6 | 0 | 6 |
| ICH + vehicle | 6 | 0 | 6 |
| ICH + JSH-23 | 12 | 2(17) | 14 |
| ICH + BzATP | 6 | 1 (17) | 7 |
| ICH + JSH-23 + BzATP | 12 | 1(8) | 13 |
| Experiment V | |||
| Sham | 22 | 0 | 22 |
| ICH+ vehicle | 22 | 1(5) | 23 |
| ICH + GSK2795039 | 22 | 2(9) | 24 |
| ICH + BzATP | 22 | 3(14) | 25 |
| ICH + BzATP + GSK2795039 | 22 | 1(5) | 23 |
| Total | 388 | 30(7) | 418 |
A438079: P2X7 receptor inhibitor; BzATP: P2X7 receptor agonist; GSK2795039: selective NAPDH oxidase 2 inhibitor; ICH: intracerebral hemorrhage; JSH-23: nuclear factor kappa-B inhibitor; U0126: extracellular signal-regulated kinase 1/2 inhibitor.
Design
Experiment I
To investigate the P2X7R expression pattern after ICH, the experimental plan included the following groups (n = 6): sham, 3, 6, 12, 24, 48, and 72 hours, and 5 days after ICH, and then all mice were processed for western blot detection.
Experiment II
To evaluate the role played by P2X7R in the activation of secondary injury following ICH, the experimental plan included the following groups: sham, ICH, ICH + vehicle (0.15 mL dimethyl sulfoxide), ICH + BzATP (P2X7R agonist), and ICH + A438079 (P2X7R inhibitor). The mice in each group were then subdivided for oxidative stress assessment (n = 6), immunofluorescence analysis (n = 6), brain edema analysis (n = 12), neurobehavioral evaluation (n = 10), and western blot detection (n = 6), and samples were processed accordingly.
Experiment III
To further explore whether P2X7R activation aggravates NOX2-mediated oxidative stress through the activation of ERK1/2 after ICH, the experimental plan included the following groups: Sham, ICH + vehicle, ICH + U0126 (ERK1/2 inhibitor), ICH + BzATP, and ICH + U0126 + BzATP. The mice in each group were then subdivided for immunofluorescence analysis (n = 6), oxidative stress assessment (n = 6), and western blot detection (n = 6), and samples were processed accordingly.
Experiment IV
To examine whether P2X7R activation aggravates NOX2-mediated oxidative stress through the activation of NF-κB after ICH, the experimental plan included the following groups: sham, ICH + vehicle, ICH + JSH-23 (NF-κB inhibitor), ICH + BzATP, and ICH + JSH-23 + BzATP. The mice in each group were then subdivided for immunofluorescence analysis (n = 6), oxidative stress assessment (n = 6), and western blot detection (n = 6), and samples were processed accordingly.
Experiment V
To confirm whether P2X7R-induced oxidative stress is mediated by the promotion of NOX2 expression after ICH, the experimental plan included the following groups: sham, ICH + vehicle, ICH + GSK2795039 (selective NOX2 inhibitor), ICH + BzATP, and ICH + BzATP + GSK2795039. The mice in each group were then subdivided for oxidative stress assessment (n = 6), brain edema analysis (n = 12), and neurobehavioral evaluation (n = 10), and samples were processed accordingly.
Drug administration
The protocol followed for the intraventricular administration of the various treatments was similar to that described in our previous study (Pan et al., 2018). Briefly, a microsyringe was slowly implanted into the lateral ventricle (0.7 mm lateral, 0.5 mm posterior to Bregma, and 2.3 mm below the dura). The rate of ventricular administration was 0.2 µL/min for all treatments.
The drug doses used in this study were based on those reported in previous studies (Koo et al., 2010; Hirano et al., 2015; Dang et al., 2016; Munoz et al., 2017). The P2X7R agonist BzATP (30 nmol in 1 µL saline; Sigma-Aldrich, St. Louis, MO, USA) was administered by intraventricular injection 1 hour before ICH model induction (Munoz et al., 2017). The P2X7R inhibitor A438079 (100 mg/kg) and the NOX2 inhibitor GSK2795039 (100 mg/kg) were each administered by intraperitoneal injection 30 minutes after ICH (Hirano et al., 2015; Munoz et al., 2017). The ERK1/2 activation inhibitor U0126 (2 µg in 1 µL 10% dimethyl sulfoxide, intraventricular injection) and the NF-κB transcriptional activity inhibitor JSH-23 (3.5 µg in 1 µL 10% dimethyl sulfoxide, intraventricular injection) were each administered 30 minutes before ICH (Koo et al., 2010; Dang et al., 2016). A438079, GSK2795039, U0126, and JSH-23 were purchased from MedChemExpress (Princeton, NJ, USA). For the experiments lasting 72 hours, GSK2795039 (100 mg/kg) and A438079 (100 mg/kg) were each administered again at 24 and 48 hours after ICH.
ICH model
The collagenase-induced ICH model was similar to the described in our previous research (Zhang et al., 2019). Briefly, after the mice were anesthetized (1% sodium pentobarbital; 70 mg/kg), the dura mater was exposed by drilling a hole in the skull. A microsyringe was gently and slowly implanted into the right basal ganglia (0.5 mm posterior and 2.0 mm lateral to Bregma, and 3.5 mm below the dura). Then, 0.075 U type VII collagenase (Sigma-Aldrich), diluted in 1 µL phosphate-buffered saline (PBS), was injected into the brain using a micro-infusion pump (0.2 µL/min). The needle was left in situ for 10 minutes and was then withdrawn slowly (over more than three minutes). The scalp was sutured, and the mice were placed in an incubator (Reward Life Technology Co., Ltd., Shenzhen, China), at 37°C, until they woke up from anesthesia. In the sham group, the same procedure was performed but with needle insertion only.
Western blot analysis
To study P2X7R activation-induced changes in NOX2 expression after ICH, mouse brain tissues were probed by western blot analysis. After the mice were anesthetized (1% sodium pentobarbital; 100 mg/kg), brain tissues were separated from the perihematomal region (within 1 mm from the edge of the hematoma) for use in western blot analysis. Tissues were homogenized in RIPA lysis buffer (Beyotime Biotechnology Co., Ltd., Shanghai, China) and centrifuged at 4°C for 10 minutes at 10,000 × g. Nuclear proteins were extracted by NE-PER nuclear and cytoplasmic extraction reagents (Thermo Fisher Scientific, Waltham, MA, USA). Western blot analysis was performed as previously described (Pei et al., 2015). Nitrocellulose membranes were incubated at 4°C, overnight, with the following primary antibodies: mouse anti-P2X7R (1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit anti-NOX2/gp91phox (1:1000; Abcam, Cambridge, UK), rabbit anti-p38/MAPK (1:1000; CST, Danvers, MA, USA), rabbit anti-phospho-p38/MAPK (1:1000; CST), rabbit anti-ERK1/2 (1:1000; CST), rabbit anti-phospho-ERK1/2 (1:1000; CST), rabbit anti-phospho-c-Jun N-terminal kinase (JNK) 1/2 (1:1000; CST), rabbit anti-JNK1/2 (1:1000; CST), rabbit anti-NF-κB p65 (1:1000; CST), rabbit anti-phospho-NF-κB p65 (1:1000; CST), rabbit anti-histone3 (1:1000; CST), and rabbit anti-β-actin (1:1000; CST). The membranes were then incubated with Dylight™ 800 4XPEG conjugated goat anti-mouse IgG or goat anti-rabbit IgG (1:12,000; CST), for 1 hour at 25°C. The bands were visualized using the Odyssey Infrared Imaging System (Licor Biosciences, Lincoln, NE, USA) and analyzed with ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Immunofluorescence and terminal deoxynucleotidyl transferase dUTP nick-end labeling staining
After mice were anesthetized (1% sodium pentobarbital; 100 mg/kg), brain tissues were extracted and dehydrated in 15% sucrose for 24 hours, followed by 30% sucrose for 24 hours. Brains were cut into 10-µm-thick sections, using a cryostat. Brain sections were incubated with the following primary antibodies, at 4°C for over 12 hours: mouse anti-P2X7 (1:50; Santa Cruz Biotechnology), rabbit anti-NeuN, rabbit anti-glial fibrillary acidic protein, mouse anti-NeuN, rabbit anti-phospho-ERK1/2 (all antibodies at 1:200; CST), and rabbit anti-ionized calcium-binding adaptor molecule 1 (1:200; Wako, Osaka, Japan). The next day, the brain slices were incubated with corresponding secondary antibodies, including Alexa Fluor 488-conjugated goat anti-rabbit IgG and goat anti-mouse IgG (1:200; Thermo Fisher Scientific) and Cy3-conjugated goat anti-mouse IgG and goat anti-rabbit IgG (1:200; Jackson Immuno-Research), for 1 hour at 25°C. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed using an in situ apoptosis detection kit (Roche, Basel, Switzerland), according to the manufacturer’s instructions. Brain slices were observed under an Olympus confocal laser scanning microscope (Olympus, Tokyo, Japan). For statistical analysis, three brain slices were selected for each mouse. For each brain slice, three visual fields near the hematoma were selected. Image analysis was performed with Image J software.
Oxidative stress assessment
After the mice were anesthetized (1% sodium pentobarbital; 100 mg/kg), the brain tissues around the hematoma (within 1 mm from the edge of the hematoma) were immediately separated for further testing. Malondialdehyde (MDA), superoxide dismutase (SOD), glutathione (GSH), and oxidized glutathione (GSSG) were assessed as representative biomarkers, to measure oxidative stress (Seminotti et al., 2013). The detection of MDA, SOD, GSH, and GSSG was performed using a kit developed by Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
Brain water content
The mice were euthanized by deep anesthesia (1% sodium pentobarbital; 100 mg/kg), and the brains, excluding the olfactory bulbs, were each divided into three pieces: the contralateral hemisphere, the hemorrhage hemisphere, and the cerebellum. Each piece was weighed directly to obtain the wet weight and then incubated at 100°C, for 24 hours, after which each piece was weighed again to obtain the dry weight. The brain water content was calculated as follows: water content (%) = [(wet weight – dry weight)/wet weight] × 100.
Motor function and neurobehavioral evaluation
All neurobehavioral evaluations were performed by a laboratory technician who was blinded to the groupings. The forelimb placement test was performed to detect the integrity of motor and sensory pathways (Hua et al., 2002). The testing was induced by brushing the respective vibrissae on the corner edge of a countertop. When the vibrissae were stimulated, animals without neurological dysfunction quickly place the ipsilateral forelimb on the countertop. According to the degree of neurological deficit, the placing of the forelimb may be impaired. After testing each mouse 10 times, the proportion of tests during which each mouse touched the countertop with the left forelimb was recorded. Mice were placed in a 30° corner, composed of two pieces of cardboard, 30 and 20 cm in length, to perform the corner turn test (Hua et al., 2002). To leave the corner, the mouse could turn to the left or right. When one side of the somatic nerve function is damaged, the mouse is more likely to turn to the opposite side of the damage. After testing each mouse 10 times, the proportion of right turns was recorded for each mouse. The modified Garcia score consists of seven independent subtests: free movement, tentacle proprioception, axial sensation, limb motor symmetry, lateral turning, climbing, and forelimb walking (Chen et al., 2018). Each item was graded from 0 (severe deficit) to 3 (no deficit), with a maximum overall score of 21 (indicating no neurological impairment).
Statistical analysis
The results are presented as the mean ± standard error of the mean (SEM) and were statistically analyzed using GraphPad Prism 7.0 (GraphPad Software Inc., San Diego, CA, USA). The Shapiro-Wilk test was used to examine the normal distribution of the data. A two-way repeated-measures analysis of variance, followed by Bonferroni’s post hoc comparison, was employed to analyze the neurobehavioral data. A one-way analysis of variance, followed by Tukey’s post hoc comparison, was employed to perform all other data analysis among more than two groups. P < 0.05 was considered significant.
Results
Spatiotemporal expression of P2X7R after ICH in mice
To explore the dynamic changes in the P2X7R expression pattern after ICH, the brain tissues surrounding the hematoma (Figure 1C) were removed at different time points following ICH model induction, and western blot analysis was performed on the tissue lysates. The results indicated that P2X7R expression began to increase 6 hours after ICH and peaked 24 hours after ICH (P < 0.001; Figure 1A and B). Immunofluorescence staining was used to detect the localization of P2X7R expression in the brain. P2X7R appeared to be expressed on neurons, microglia, and astrocytes, 24 hours after collagenase injection (Figure 1D). Additionally, the expression levels of P2X7R in neurons was remarkably higher than those in astrocytes or microglia (P < 0.001; Figure 1E). These results showed that P2X7R expression increased and was primarily expressed in neurons following ICH.
Figure 1.

Spatiotemporal expression of P2X7R in brain tissues near the hematoma after ICH.
(A and B) Quantitative determination of P2X7R expression levels by western blot assay. The relative expression of P2X7R is expressed as the optical density ratio of P2X7R relative to that of β-actin. (C) Mouse brain slice showing the areas of brain tissues near the hematoma (indicated by black squares). These areas were examined by immunofluorescence and sampled for western blotting analysis. (D) P2X7R localization (green, stained by Alexa Fluor 488) was determined by immunofluorescence staining, 24 hours after ICH. The neuronal expression level of P2X7R was significantly higher than those observed for astrocytes and microglia. Iba-1, NeuN, and GFAP immunopositivity was detected using Cy3 (red). Scale bars: 50 µm. (E) Quantitative analysis of P2X7R expression in microglia (Iba-1+), neurons (NeuN+), and astrocytes (GFAP+). Data are presented as the mean ± SEM (n = 6). ###P < 0.001, vs. sham group; †††P < 0.001, vs. Iba-1+ or GFAP+ (one-way analysis of variance followed by Tukey’s post hoc test). GFAP: Glial fibrillary acidic protein; Iba-1: ionized calcium-binding adaptor molecule 1; ICH: intracerebral hemorrhage; P2X7R: P2X7 receptor.
P2X7R activation aggravates apoptosis, brain edema, and neurological impairments after ICH
We next examined the role played by P2X7R following ICH model induction by using a P2X7R agonist, BzATP, and antagonist, A438079. Based on the dynamic expression of P2X7R observed following ICH, the experimental time point used in this analysis was 24 hours after ICH. TUNEL staining showed that a significant reduction in the proportion of apoptotic cells in the ICH + A438079 treatment group compared with the ICH and ICH + vehicle groups, whereas the ICH + BzATP treatment group presented with an increased proportion of apoptotic cells in the tissues near the hematoma compared with the control groups (P < 0.05; Figure 2A and B). BzATP treatment aggravated neurological deficits 24 hours after collagenase injection, compared with the ICH alone and ICH + treatment. In contrast, mice in the ICH + A438079 group presented fewer neurological deficits (forelimb placement test, P < 0.01; corner turn test, P < 0.05; modified Garcia score, P < 0.01; Figure 2C–E) than mice in the ICH or ICH + vehicle groups. The brain water content in the hemorrhage hemisphere was markedly attenuated in mice treated with A438079 compared with the contents measured in the ICH and ICH + vehicle groups, 24 and 72 hours after collagenase injection (P < 0.001). In contrast, treatment with BzATP further aggravated brain edema after ICH (P < 0.01; Figure 2F). These results suggested that P2X7R activation aggravated apoptosis, brain edema, and neurological impairments after ICH.
Figure 2.
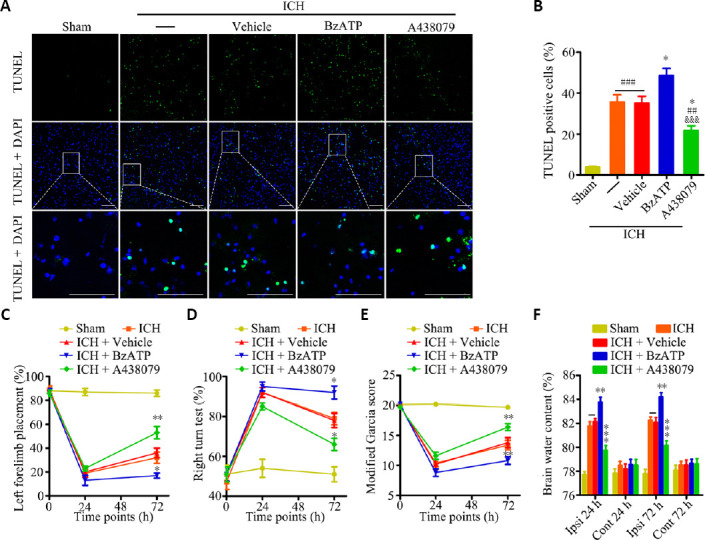
P2X7R activation aggravates apoptosis, brain edema, and neurological impairments after ICH.
(A) Apoptotic cells were detected by TUNEL staining, 24 hours after ICH. Low magnification, full-field and partial, high-magnification visual fields (indicated by the white frame) are displayed. The A438079 treatment group displayed a significantly reduced proportion of apoptotic cells compared with the ICH and ICH + vehicle groups, whereas the BzATP treatment group displayed an increased proportion of apoptotic cells. Scale bars: 100 µm. (B) Quantitative analysis showing the percentage of apoptotic cells (n = 6). (C) Forelimb placement test (n = 10). (D) Corner turn test (n = 10). (E) Modified Garcia score (n = 10). (F) Brain water contents (n = 6). Data are expressed as the mean ± SEM (n = 6). ##P < 0.01, ###P < 0.001, vs. sham group; *P < 0.05, **P < 0.01, ***P < 0.001, vs. ICH or ICH + vehicle group; &&&P < 0.001, vs. ICH + BzATP group (apoptotic cells and brain water contents: one-way analysis of variance, followed by Tukey’s post hoc test; neurobehavioral data: two-way repeated-measures analysis of variance, followed by Bonferroni’s post hoc test). Cont: Contralateral hemisphere; DAPI: 4′,6-diamidino-2-phenylindole; GFAP: glial fibrillary acidic protein; Iba-1: ionized calcium-binding adaptor molecule 1; ICH: intracerebral hemorrhage; Ipsi: ipsilateral hemisphere; P2X7R: P2X7 receptor; TUNEL: terminal deoxynucleotidyl transferase dUTP nick-end labeling.
P2X7R activation promotes oxidative stress by upregulating NOX2 after ICH
The inhibition of P2X7R, using A438079, significantly reduced P2X7R expression levels, compared with those in the ICH and ICH + vehicle groups, 24 hours after collagenase injection (P < 0.05). P2X7R expression was further increased in the ICH + BzATP group (P < 0.05; Figure 3A and C). Consistently, the immunofluorescence staining results showed that treatment with A438079 downregulated P2X7R expression in neurons 24 hours after ICH compared with the expression levels in the control groups (Figure 3E). We found BzATP treatment resulted in increased NOX2 expression compared with the expression levels observed in the ICH and ICH + vehicle groups (P < 0.01). In contrast, treatment with A438079 significantly decreased NOX2 expression compared with the levels observed in the control groups (P < 0.05; Figure 3B and D). When we measured the levels of SOD, MDA, GSH, and GSH/GSSG in the tissues near the hematoma, 24 hours after ICH, the inhibition of P2X7R was found to significantly reduce the MDA concentration (P < 0.05), and increase SOD (P < 0.05), GSH (P < 0.01), and GSH/GSSG levels (P < 0.05; Figure 3F–I) compared with the levels in the control groups. Although the MDA, SOD, GSH, and GSH/GSSG levels were also altered in the BzATP + ICH group compared with the control groups, only the changes in the SOD and MDA levels were significant (P < 0.05; Figure 3F–I). These results suggested that P2X7R aggravated oxidative stress following ICH, which might be mediated by the promotion of NOX2 expression.
Figure 3.

P2X7R activation increases NOX2 expression and oxidative stress at 24 hours after ICH.
(A–D) Quantitative determination of P2X7R (A and C) and NOX2 (B and D) expression levels by western blot assay. The relative expression levels of P2X7R or NOX2 was expressed as the optical density relative to the optical density of β-actin. (E) Immunofluorescence staining showing P2X7R immunopositivity (green, stained by Alexa Fluor 488) in neurons (NeuN+cells, red, stained by Cy3). Compared with the ICH or ICH + vehicle groups, the ICH + A438079 treatment group displayed downregulated P2X7R expression in neurons, whereas the ICH + BzATP treatment group presented the further upregulation of P2X7R expression. Scale bars: 50 µm. (F–I) Quantitative analysis of MDA, GSH, SOD, and GSH/GSSG levels. Data are expressed as the mean ± SEM (n = 6). #P < 0.05, ###P < 0.001, vs. sham group; *P < 0.05, **P < 0.01, vs. ICH or ICH + vehicle group; &&P < 0.01, &&&P < 0.001, vs. ICH + BzATP group (one-way analysis of variance followed by Tukey’s post hoc test). A438079: P2X7 receptor inhibitor; BzATP: P2X7 receptor agonist; GSH: glutathione; GSSG: oxidized glutathione; ICH: intracerebral hemorrhage; MDA: malondialdehyde; NOX2: nicotinamide adenine dinucleotide phosphate oxidase 2; P2X7R: P2X7 receptor; SOD: superoxide dismutase.
Effects of P2X7R activation on MAPK and NF-κB pathways
The western blot results showed that phosphorylated ERK1/2 (P < 0.001), phosphorylated P38 (P < 0.01), phosphorylated JNK1/2 (P < 0.01), and phosphorylated NF-κB (P < 0.001) levels significantly increased 24 hours after ICH (ICH group vs. sham group; Figures 4A–D and 5). The western blot results also showed that treatment with BzATP further promoted the phosphorylation of ERK1/2 (vs. ICH + vehicle group; P < 0.001) and the nuclear translocation of NF-κB (vs. ICH + vehicle group; P < 0.05), whereas the opposite results were observed for the administration of the P2X7R inhibitor A438079 (vs. ICH + vehicle group; P < 0.05; Figures 4A and B and 5). Similarly, the immunofluorescence results showed that A438079 treatment significantly reduced the phospho-ERK1/2 levels in neurons 24 hours after ICH, whereas BzATP treatment further increased phospho-ERK1/2 levels (Figure 4E). However, no significant changes in P38 and JNK1/2 phosphorylation levels were observed in the ICH + BzATP and ICH + A438079 groups compared with the control groups (vs. ICH + vehicle group; P > 0.05; Figure 4A, C, and D). These results demonstrated that P2X7R promoted the activation of NF-κB and ERK1/2 after ICH.
Figure 4.

The effects of P2X7R activation on the MAPK signaling pathway at 24 hours after ICH.
(A–D) Quantitative evaluation of p-ERK1/2, p-p38, and p-JNK1/2 levels compared to total protein levels by western blot assay. Data are expressed as the mean ± SEM (n = 6). ##P < 0.01, ###P < 0.001, vs. sham group; *P < 0.05, ***P < 0.001, vs. ICH or ICH + vehicle group; &&&P < 0.001, vs. ICH + BzATP group (one-way analysis of variance followed by Tukey’s post hoc test). (E) Immunofluorescence staining showing p-ERK1/2 immunopositivity (green, stained by Alexa Fluor 488) in neurons (NeuN+ cells, red, stained by Cy3). Compared with the ICH or ICH + vehicle groups, the ICH + A438079 treatment group showed reduced phospho-ERK1/2 levels in neurons, whereas the ICH + BzATP treatment group showed increased phospho-ERK1/2 levels. Scale bars: 50 µm. A438079: P2X7 receptor inhibitor; BzATP: P2X7 receptor agonist; ICH: intracerebral hemorrhage; MAPK: mitogen-activated protein kinases; (p-)ERK1/2: (phospho-)extracellular signal-regulated kinase 1/2; (p-)JNK1/2: phospho-c-Jun N-terminal kinase; p-p38: phospho-p38; P2X7R: P2X7 receptor.
Figure 5.
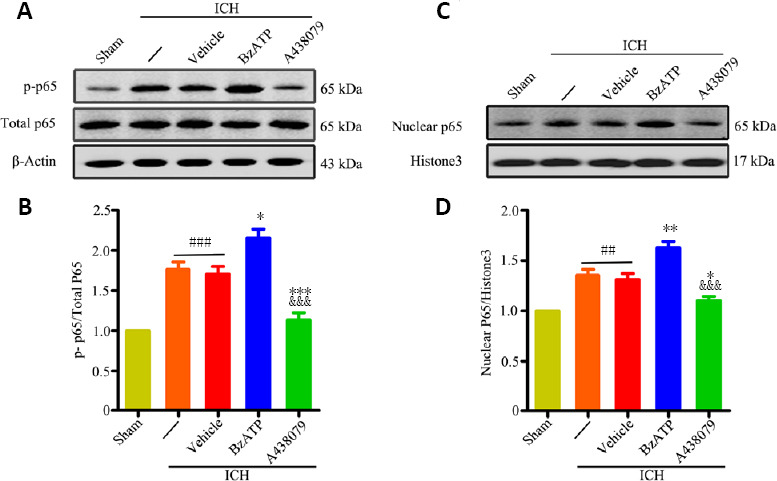
P2X7R activation promotes NF-κB activation at 24 hours after ICH.
(A–D) Quantitative evaluation of p-NF-κB p65 (A and B) and nuclear NF-κB p65 (C and D) levels, by western blot assay. Data are expressed as the mean ± SEM (n = 6). ##P < 0.01, ###P < 0.001, vs. sham group; *P < 0.05, **P < 0.01, ***P < 0.001, vs. ICH or ICH + vehicle group; &&&P < 0.001, vs. ICH + BzATP group (one-way analysis of variance followed by Tukey’s post hoc test). A438079: P2X7 receptor inhibitor; BzATP: P2X7 receptor agonist; ICH: intracerebral hemorrhage; p-NF-κB: (phospho-)nuclear factor kappa-B.
Inhibition of ERK1/2 activation ameliorates P2X7R-mediated oxidative stress following ICH
The western blot results showed that the administration of U0126 was able to effectively inhibit ERK1/2 phosphorylation after ICH (vs. ICH + vehicle group; P < 0.001), even in the presence of BzATP (vs. ICH + vehicle group; P < 0.01; Figure 6A and C). Moreover, U0126 treatment reversed the NOX2 upregulation induced by BzATP treatment (P < 0.01; Figure 6B and D). Consistently, the inhibition of ERK1/2 phosphorylation was associated with a significant decrease in the MDA concentration (ICH + U0126 group vs. ICH + vehicle group, P < 0.001; ICH + BzATP +U0126 group vs. ICH + vehicle group, P < 0.05) and significant increases in the SOD (ICH + U0126 group vs. ICH + vehicle group, P < 0.01; ICH + BzATP +U0126 group vs. ICH + vehicle group, P < 0.05), GSH (ICH + U0126 group vs. ICH + vehicle group, P < 0.01; ICH + BzATP +U0126 group vs. ICH + vehicle group, P < 0.05), and GSH/GSSG levels (ICH + U0126 group vs. ICH + vehicle group, P < 0.01; ICH + BzATP +U0126 group vs. ICH + vehicle group, P < 0.05) in the ICH + U0126 and ICH + BzATP + U0126 groups, compared with the control groups, 24 hours after ICH (Figure 6E–H). These data suggested that the inhibition of ERK1/2 phosphorylation attenuated P2X7R-induced NOX2 expression and oxidative stress after ICH. TUNEL staining also showed that U0126 treatment protected cells from apoptosis after ICH (vs. ICH + vehicle group; P < 0.01), even when BzATP was administered (vs. ICH + vehicle group; P < 0.01; Figure 6I and J). These data demonstrated that the inhibition of ERK1/2 phosphorylation alleviated NOX2-mediated oxidative stress caused by P2X7R activation.
Figure 6.

Inhibition of ERK1/2 activation reduces P2X7R-mediated upregulation of NOX2 expression, oxidative stress, and apoptosis at 24 hours after ICH.
(A–D) Quantitative assessment of p-ERK1/2 (A and C) and NOX2 (B and D) levels by western blot assay. (E-H) Quantitative assessment of MDA, SOD, GSH, and GSH/GSSG levels. (I) Apoptotic cells were detected by TUNEL staining. Low-magnification, full-field and high-magnification, partial visual fields (indicated by the white frame) are displayed. The ICH + U0126 treatment group displayed a significantly reduced proportion of apoptotic cells compared with the ICH + vehicle or ICH + BzATP groups. Scale bars: 100 µm. (J) Quantitative analysis of the percentage of apoptotic cells. Data are expressed as the mean ± SEM (n = 6). #P < 0.05, ##P < 0.01, ###P < 0.001, vs. sham group; *P < 0.05, **P < 0.01, ***P < 0.001, vs. ICH + vehicle group; &&P < 0.01, &&&P < 0.001, vs. ICH + BzATP group (one-way analysis of variance followed by Tukey’s post hoc test). BzATP: P2X7 receptor agonist; GSH: glutathione; DAPI: 4′,6-diamidino-2-phenylindole; GSSG: oxidized glutathione; ICH: intracerebral hemorrhage; MDA: malondialdehyde; NOX2: NAPDH oxidase 2; P2X7R: P2X7 receptor; (p-)ERK1/2: (phospho-)extracellular signal-regulated kinase 1/2; SOD: superoxide dismutase; TUNEL: terminal deoxynucleotidyl transferase dUTP nick end labeling; U0126: extracellular signal-regulated kinase 1/2 inhibitor.
Inhibition of the NF-κB ameliorates P2X7R-mediated oxidative stress following ICH
JSH-23 is a selective inhibitor of NF-κB transcriptional activity that specifically inhibits the nuclear translocation of NF-κB. The western blot results showed that nuclear NF-κB p65 levels were significantly reduced in the ICH + JSH-23 (vs. ICH + vehicle, P < 0.001) and ICH + BzATP + JSH-23 groups (vs. ICH + vehicle, P < 0.01; vs. ICH + BzATP groups, P < 0.001) compared with those in the ICH + vehicle and ICH + BzATP groups, 24 hours after collagenase injection Figure 7A, C, and D). These data showed that JSH-23 effectively inhibited NF-κB nuclear translocation, even when the activation of P2X7R was stimulated by BzATP. Furthermore, the expression of NOX2 was dramatically decreased in the ICH + JSH-23 and ICH + BzATP + JSH-23 groups (ICH + JSH-23 group vs. ICH + vehicle group, P < 0.001; ICH + BzATP + JSH-23 group vs. ICH + vehicle group, P < 0.01; ICH + BzATP + JSH-23 group vs. ICH + BzATP group, P < 0.001; Figure 7B and E). Consistently, the MDA levels were significantly reduced and the SOD, GSH, and GSH/GSSG levels were elevated in the ICH + JSH-23 (vs. ICH + vehicle group, P < 0.01) and ICH + BzATP + JSH-23 groups (vs. ICH + vehicle group, P < 0.05; vs. ICH + BzATP group, P < 0.001 Figure 7F–I). Finally, TUNEL staining showed that JSH-23 treatment significantly reduced the proportion of apoptotic cells in the tissues near the hematoma, 24 hours after collagenase injection, compared with those in the control groups (ICH + JSH-23 group vs. ICH + vehicle and ICH + BzATP groups, P < 0.001; ICH + BzATP + JSH-23 group vs. ICH + vehicle group, P < 0.01; Figure 7J and K). These results indicated that the inhibition of NF-κB activation was able to reduce the NOX2-mediated oxidative stress associated with P2X7R activation.
Figure 7.
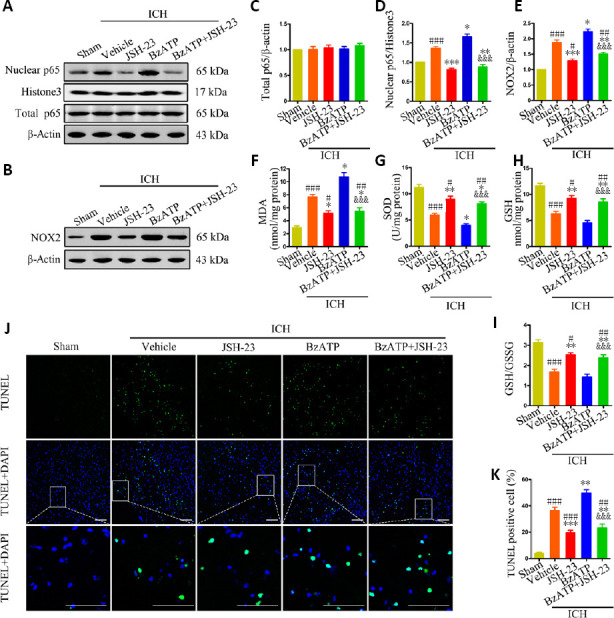
Inhibition of NF-κB reduced P2X7R-mediated upregulation of NOX2 expression, oxidative stress, and apoptosis, 24 hours after ICH.
(A, C, and D) Quantitative analysis of total NF-κB p65 and nuclear NF-κB p65 levels by western blot assay. (B and E) Quantitative analysis of NOX2 expression by western blot assay. (F-I) Quantitative analysis of MDA, SOD, GSH, and GSH/GSSG levels. (J) Apoptotic cells were detected by TUNEL staining. Low-magnification, full-field and high-magnification, partial visual fields (indicated by a white frame) are displayed. The ICH + JSH-23 treatment group displayed a significantly reduced proportion of apoptotic cells compared with the ICH + vehicle or ICH + BzATP groups. Scale bars: 100 µm. (K) Quantitative analysis of the percentage of apoptotic cells. Data are expressed as the mean ± SEM (n = 6). #P < 0.05, ##P < 0.01, ###P < 0.001, vs. sham group; *P < 0.05, **P < 0.01, ***P < 0.001, vs. ICH + vehicle group; &&&P < 0.001, vs. ICH + BzATP group (one-way analysis of variance followed by Tukey’s post hoc test). BzATP: P2X7 receptor agonist; DAPI: 4′,6-diamidino-2-phenylindole; ICH: intracerebral hemorrhage; JSH-23: nuclear factor kappa-B inhibitor; NF-κB: nuclear factor kappa-B; NOX2: NAPDH oxidase 2; P2X7R: P2X7 receptor; TUNEL: terminal deoxynucleotidyl transferase dUTP nick-end labeling.
Treatment with a NOX2 antagonist attenuates P2X7R-mediated oxidative stress, neurological damage, and brain edema after ICH
To further confirm that P2X7R-induced oxidative stress was mediated by NOX2 after ICH, mice were treated with the selective NOX2 inhibitor GSK2795039. Compared with the ICH + vehicle and ICH + BzATP groups, the MDA contents were significantly reduced and the SOD, GSH, and GSH/GSSG levels were significantly increased 24 hours after ICH in the ICH + GSK2795039 (vs. ICH + vehicle group, P < 0.01) and ICH + BzATP + GSK2795039 groups (vs. ICH + vehicle group, P < 0.05; vs. ICH + BzATP group, P < 0.001; Figure 8A–D). Treatment with GSK2795039 significantly improved the neurological function score 72 hours after collagenase injection (P < 0.05 or P < 0.01; Figure 8E–G) and alleviated brain edema, both 24 and 72 hours after collagenase injection (P < 0.01 or P < 0.001; Figure 8H), compared with the control groups. These results suggested that the inhibition of NOX2 attenuated P2X7R-mediated oxidative stress following ICH.
Figure 8.
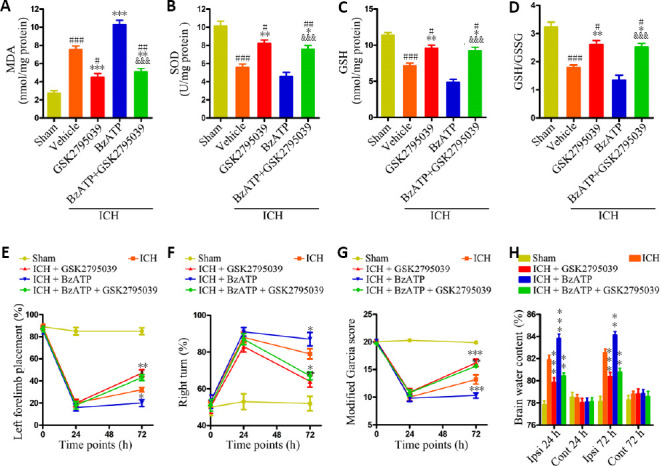
NOX2 antagonist attenuates the P2X7R-mediated increases in oxidative stress, brain edema, and neurological damage after ICH.
(A–D) Quantitative analysis of MDA, GSH, SOD, and GSH/GSSG levels (n = 6). (E) Forelimb placement test (n = 10). (F) Corner turn test (n = 10). (G) Modified Garcia score (n = 10). (H) Brain water contents (n = 6). Data are expressed as the mean ± SEM. #P < 0.05, ##P < 0.01, ###P < 0.001, vs. sham group; *P < 0.05, **P < 0.01, ***P < 0.001, vs. ICH + vehicle group; &&&P < 0.001, vs. ICH + BzATP group (oxidative stress and brain water contents: one-way analysis of variance, followed by Tukey’s post hoc test; neurobehavioral data: two-way repeated-measures analysis of variance, followed by Bonferroni’s post hoc test). BzATP: P2X7 receptor agonist; Cont: contralateral hemispheres; GSH: glutathione; GSK2795039: selective NAPDH oxidase 2 inhibitor; GSSG: oxidized glutathione; ICH: intracerebral hemorrhage; Ipsi: ipsilateral hemispheres; MDA: malondialdehyde; NOX2: NAPDH oxidase 2; P2X7R: P2X7 receptor; SOD: superoxide dismutase.
Discussion
The present study demonstrated that P2X7R activation increased NOX2 expression and aggravated brain edema, apoptosis, and neurological damage following ICH. Blocking NOX2 with GSK2795039 alleviated the oxidative stress associated with P2X7R activation after ICH. Furthermore, the data also indicated that the inhibition of ERK1/2 and NF-κB activation could reduce P2X7R-mediated increases in NOX2 expression. This study is the first to report that P2X7R activation aggravates NOX2-induced oxidative stress via the ERK1/2 and NF-κB pathways, after ICH in mice.
Oxidative stress is an important pathophysiological process that can cause secondary injury after ICH. ROS generation following ICH is primarily trigged by hematoma degradation products and plasma components (Hu et al., 2016). Under tender mechanical stimulation, almost all cell types, including dying and dead cells, release ATP (Lazarowski, 2012). Previous research has shown that P2X7R activation requires relatively high extracellular ATP concentrations, far beyond the concentrations required for other P2X receptor activation (Bartlett et al., 2014). Some scholars have reported that P2X7R-mediated oxidative stress may play a role in the occurrence and progression of neurodegenerative disorders (Ferrazoli et al., 2017; Cieślak and Wojtczak, 2018). Feng et al. (2015) reported that P2X7R induced the activation of the NOD, LRR- and pyrin domain-containing protein 3 inflammasome by promoting the NOX2-mediated generation of peroxynitrite (ONOO–) after ICH in rats. However, the mechanism through which P2X7R activation promoted NOX2 expression after ICH has not yet been explored. Our results showed that P2X7R expression was remarkably upregulated following ICH. Interestingly, we found that the application of a P2X7R antagonist, A438079, or agonist, BzATP, could regulate the expression of P2X7R after ICH. Therefore, we hypothesized that following the activation or inactivation of P2X7R, changes in the downstream signaling pathways might influence the transcription and translation of P2X7R.
NOX2, also known as gp91phox, transfers electrons to oxygen, producing superoxide, using nicotinamide adenine dinucleotide phosphate as an electron donor (Haslund-Vinding et al., 2017). As the primary source of ROS production in the central nervous system, NOX2 not only promotes the development of chronic neurodegenerative diseases but also aggravates brain injuries associated with various acute neurological diseases, such as ICH, traumatic brain injury, and acute ischemic stroke (Ma et al., 2017). A previous study reported that the NOX2 inhibitor apocynin attenuated oxidative stress induced by P2X7R activation in spinal cord dorsal horn neurons (Munoz et al., 2017). Similarly, our study suggested that P2X7R activation aggravated oxidative stress after ICH by inducing NOX2 expression. GSK2795039 is a novel, selective NOX2 inhibitor, with demonstrated bioavailability in the brain after intraperitoneal injection (Hirano et al., 2015). A recent study also confirmed that seizures caused by NOX2-induced oxidative stress could be suppressed by the intraventricular injection of GSK2795039 in rats (Malkov et al., 2019). Our results indicated that the inhibition of NOX2, using GSK2795039, reduced P2X7R-mediated oxidative stress following ICH. To date, our experiment is the first to evaluate whether GSK2795039 can play a beneficial role in an animal model of ICH.
Neuronal death in the tissues near the hematoma represents an important mechanism of neurological damage after ICH (Wasserman et al., 2008). Recent studies have shown that ROS can mediate many forms of neuronal death, including apoptosis, autophagy, and necrosis, after ICH (Duan et al., 2016). Our research also suggested that P2X7R activation aggravated cell apoptosis in the tissues near the hematoma, causing neurological damage after ICH, which may be due to NOX2-mediated oxidative stress.
Brain edema is an important sign of secondary injury after ICH. Neuroinflammation, oxidative stress, and blood-brain barrier destruction promote the occurrence of brain edema after ICH, which causes more lasting and severe brain injury (Chu et al., 2013; Zheng et al., 2016). Tang et al. (2005) found that NOX2 gene knockout reduced brain edema after ICH. Our data showed that P2X7R activation exacerbated brain edema, through the elevation of NOX2-mediated oxidative stress after ICH. Zhao et al. (2016) reported that P2X7R activates RhoA, which disrupts the blood-brain barrier after ICH, resulting in brain edema. Interestingly, some scholars have reported that RhoA activation by ROS can cause pulmonary artery smooth muscle contraction (MacKay et al., 2017). Continuing to explore the relationship between ROS and RhoA may help us clarify the mechanism through which P2X7R aggravates brain edema after ICH.
We further explored the possible mechanisms through which P2X7R activation promotes NOX2 expression. The MAPK pathway, a three-stage, enzymatic cascade, is involved in the regulation of extensive cellular activities. Many studies have shown that ERK1/2, JNK1/2, and p38 are important signaling molecules that regulate inflammation, apoptosis, and oxidative stress following ICH (Zhu et al., 2019). Our study found that P2X7R activation promoted ERK1/2 phosphorylation after ICH. Furthermore, the inhibition of ERK1/2 phosphorylation reduced the P2X7R-associated upregulation of NOX2 expression after ICH. Our data were consistent with previous evidence that P2X7R activation upregulates NOX2 expression by promoting ERK1/2 activation in a SOD1G93A mouse model of ALS (Apolloni et al., 2016). These results suggested that P2X7R activation may induce the upregulation of NOX2 expression by activating ERK1/2 after ICH.
NF-κB is a multi-subunit transcription factor, in which the different subunits form a variety of NF-κB dimers in the cytoplasm, which remain inactive due to the binding of the NF-κB inhibitory protein IκB (González-Ramos et al., 2012). Several studies have shown that P2X7R activation mediates inflammation, apoptosis, and oxidative stress, via the NF-κB pathway (Mariathasan et al., 2006; Savio et al., 2017). A previous study reported that NOX2 expression was inhibited in IκB overexpressing or RelA–/– (NF-κB p65) monocytes and microglia (Anrather et al., 2006). In addition, the same study identified the CYBB gene as a possible cis-acting element in the murine NOX2 promoter, controlled by an NF-κB-dependent regulation mechanism. NOX2 production disorder, which is caused by a CYBB gene mutation, is an important pathogenic cause of chronic granulomatous disease (Arnold and Heimall, 2017). Our study indicated that P2X7R activation resulted in the activation of NF-κB after ICH. Moreover, blocking NF-κB p65 translocation into the nucleus reduced the P2X7R-induced upregulation of NOX2 expression after ICH. These results suggested that P2X7R activation may induce the upregulation of NOX2 expression by activating NF-κB after ICH.
The present study had some limitations. First, we only explored the effects of P2X7R activation on NOX2 expression after ICH in mice but did not evaluate the membrane assembly of NOX2, which has also been shown to regulate its superoxide producing activity (Bedard and Krause, 2007). Second, the upstream molecular mechanisms through which P2X7R activates ERK1/2 and NF-κB after ICH remain unclear. Further research remains necessary to further clarify the mechanism.
In conclusion, our study suggested that P2X7R activation increased NOX2-induced oxidative stress via the ERK1/2 and NF-κB pathways following ICH in mice. These findings provided new preclinical evidence regarding potential intervention targets for the prevention of secondary injury after ICH.
Additional file:
Additional Table 1: Experimental grouping and animal mortality in this study.
Footnotes
C-Editor: Zhao M; S-Editors: Yu J, Li CH; L-Editors: Giles L, Yu J, Song CP; T-Editor: Jia Y
Funding: This study was supported by the National Natural Science Foundation of China, Nos, 81471201, 81873750, and the Science and Technology Plan Project of Wuhan of China, No. 2018060401011316 (all to ZPT).
Conflicts of interest: The authors declare that they have no conflict of interest.
Financial support: This study was supported by the National Natural Science Foundation of China, Nos, 81471201, 81873750, and the Science and Technology Plan Project of Wuhan, Hubei Province, China, No. 2018060401011316 (all to ZPT). The funding sources had no role in study conception and design, data analysis or interpretation, paper writing or deciding to submit this paper for publication.
Institutional review board statement: The study was approved by the Ethics Committee of Huazhong University of Science and Technology, China (approval No. TJ-A20160805) on August 26, 2019.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
References
- 1.Anrather J, Racchumi G, Iadecola C. NF-kappaB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J Biol Chem. 2006;281:5657–5667. doi: 10.1074/jbc.M506172200. [DOI] [PubMed] [Google Scholar]
- 2.Apolloni S, Fabbrizio P, Parisi C, Amadio S, Volonté C. Clemastine confers neuroprotection and induces an anti-inflammatory phenotype in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Mol Neurobiol. 2016;53:518–531. doi: 10.1007/s12035-014-9019-8. [DOI] [PubMed] [Google Scholar]
- 3.Apolloni S, Parisi C, Pesaresi MG, Rossi S, Carrì MT, Cozzolino M, Volonté C, D’Ambrosi N. The NADPH oxidase pathway is dysregulated by the P2X7 receptor in the SOD1-G93A microglia model of amyotrophic lateral sclerosis. J Immunol. 2013;190:5187–5195. doi: 10.4049/jimmunol.1203262. [DOI] [PubMed] [Google Scholar]
- 4.Arnold DE, Heimall JR. A review of chronic granulomatous disease. Adv Ther. 2017;34:2543–2557. doi: 10.1007/s12325-017-0636-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartlett R, Stokes L, Sluyter R. The P2X7 receptor channel: recent developments and the use of P2X7 antagonists in models of disease. Pharmacol Rev. 2014;66:638–675. doi: 10.1124/pr.113.008003. [DOI] [PubMed] [Google Scholar]
- 6.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 7.Butterfield DA, Halliwell B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci. 2019;20:148–160. doi: 10.1038/s41583-019-0132-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen S, Zhao L, Sherchan P, Ding Y, Yu J, Nowrangi D, Tang J, Xia Y, Zhang JH. Activation of melanocortin receptor 4 with RO27-3225 attenuates neuroinflammation through AMPK/JNK/p38 MAPK pathway after intracerebral hemorrhage in mice. J Neuroinflammation. 2018;15:106. doi: 10.1186/s12974-018-1140-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng JP, Li H, Li XJ. Extract of piper auritum can alleviate oxidative stress and inflammation of rat models of acute spinal cord injury. Zhongguo Zuzhi Gongcheng Yanjiu. 2019;23:5010–5016. [Google Scholar]
- 10.Chu H, Tang Y, Dong Q. Protection of vascular endothelial growth factor to brain edema following intracerebral hemorrhage and its involved mechanisms: effect of aquaporin-4. PLoS One 8. 2013:e66051. doi: 10.1371/journal.pone.0066051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cieślak M, Wojtczak A. Role of purinergic receptors in the Alzheimer’s disease. Purinergic Signal. 2018;14:331–344. doi: 10.1007/s11302-018-9629-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dang DK, Shin EJ, Nam Y, Ryoo S, Jeong JH, Jang CG, Nabeshima T, Hong JS, Kim HC. Apocynin prevents mitochondrial burdens, microglial activation, and pro-apoptosis induced by a toxic dose of methamphetamine in the striatum of mice via inhibition of p47phox activation by ERK. J Neuroinflammation. 2016;13:12. doi: 10.1186/s12974-016-0478-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 receptor in infection and inflammation. Immunity. 2017;47:15–31. doi: 10.1016/j.immuni.2017.06.020. [DOI] [PubMed] [Google Scholar]
- 14.Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10:453–471. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duan X, Wen Z, Shen H, Shen M, Chen G. Intracerebral hemorrhage, oxidative stress, and antioxidant therapy. Oxid Med Cell Longev. 2016;2016:1203285. doi: 10.1155/2016/1203285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feigin VL, Lawes CM, Bennett DA, Anderson CS. Stroke epidemiology: a review of population-based studies of incidence, prevalence, and case-fatality in the late 20th century. Lancet Neurol. 2003;2:43–53. doi: 10.1016/s1474-4422(03)00266-7. [DOI] [PubMed] [Google Scholar]
- 17.Feng L, Chen Y, Ding R, Fu Z, Yang S, Deng X, Zeng J. P2X7R blockade prevents NLRP3 inflammasome activation and brain injury in a rat model of intracerebral hemorrhage: involvement of peroxynitrite. J Neuroinflammation. 2015;12:190. doi: 10.1186/s12974-015-0409-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferrazoli EG, de Souza HD, Nascimento IC, Oliveira-Giacomelli Á, Schwindt TT, Britto LR, Ulrich H. Brilliant blue g, but not fenofibrate, treatment reverts hemiparkinsonian behavior and restores dopamine levels in an animal model of Parkinson’s disease. Cell Transplant. 2017;26:669–677. doi: 10.3727/096368917X695227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao L, Li PP, Shao TY, Mao X, Qi H, Wu BS, Shan M, Ye L, Cheng HW. Neurotoxic role of interleukin-17 in neural stem cell differentiation after intracerebral hemorrhage. Neural Regen Res. 2020;15:1350–1359. doi: 10.4103/1673-5374.272614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.González-Ramos R, Defrère S, Devoto L. Nuclear factor-kappaB: a main regulator of inflammation and cell survival in endometriosis pathophysiology. Fertil Steril. 2012;98:520–528. doi: 10.1016/j.fertnstert.2012.06.021. [DOI] [PubMed] [Google Scholar]
- 21.Gross BA, Jankowitz BT, Friedlander RM. Cerebral intraparenchymal hemorrhage: a review. JAMA. 2019;321:1295–1303. doi: 10.1001/jama.2019.2413. [DOI] [PubMed] [Google Scholar]
- 22.Haslund-Vinding J, McBean G, Jaquet V, Vilhardt F. NADPH oxidases in oxidant production by microglia: activating receptors, pharmacology and association with disease. Br J Pharmacol. 2017;174:1733–1749. doi: 10.1111/bph.13425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirano K, Chen WS, Chueng AL, Dunne AA, Seredenina T, Filippova A, Ramachandran S, Bridges A, Chaudry L, Pettman G, Allan C, Duncan S, Lee KC, Lim J, Ma MT, Ong AB, Ye NY, Nasir S, Mulyanidewi S, Aw CC, et al. Discovery of GSK2795039, a novel small molecule NADPH oxidase 2 inhibitor. Antioxid Redox Signal. 2015;23:358–374. doi: 10.1089/ars.2014.6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu X, Tao C, Gan Q, Zheng J, Li H, You C. Oxidative stress in intracerebral hemorrhage: sources, mechanisms, and therapeutic targets. Oxid Med Cell Longev. 2016;2016:3215391. doi: 10.1155/2016/3215391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hua Y, Schallert T, Keep RF, Wu J, Hoff JT, Xi G. Behavioral tests after intracerebral hemorrhage in the rat. Stroke. 2002;33:2478–2484. doi: 10.1161/01.str.0000032302.91894.0f. [DOI] [PubMed] [Google Scholar]
- 26.Keep RF, Hua Y, Xi G. Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurol. 2012;11:720–731. doi: 10.1016/S1474-4422(12)70104-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koo JW, Russo SJ, Ferguson D, Nestler EJ, Duman RS. Nuclear factor-kappaB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc Natl Acad Sci U S A. 2010;107:2669–2674. doi: 10.1073/pnas.0910658107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lazarowski ER. Vesicular and conductive mechanisms of nucleotide release. Purinergic Signal. 2012;8:359–373. doi: 10.1007/s11302-012-9304-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma MW, Wang J, Zhang Q, Wang R, Dhandapani KM, Vadlamudi RK, Brann DW. NADPH oxidase in brain injury and neurodegenerative disorders. Mol Neurodegener. 2017;12:7. doi: 10.1186/s13024-017-0150-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacKay CE, Shaifta Y, Snetkov VV, Francois AA, Ward JPT, Knock GA. ROS-dependent activation of RhoA/Rho-kinase in pulmonary artery: Role of Src-family kinases and ARHGEF1. Free Radic Biol Med. 2017;110:316–331. doi: 10.1016/j.freeradbiomed.2017.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malkov A, Ivanov AI, Latyshkova A, Bregestovski P, Zilberter M, Zilberter Y. Activation of nicotinamide adenine dinucleotide phosphate oxidase is the primary trigger of epileptic seizures in rodent models. Ann Neurol. 2019;85:907–920. doi: 10.1002/ana.25474. [DOI] [PubMed] [Google Scholar]
- 32.Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 33.Martel-Gallegos G, Casas-Pruneda G, Ortega-Ortega F, Sánchez-Armass S, Olivares-Reyes JA, Diebold B, Pérez-Cornejo P, Arreola J. Oxidative stress induced by P2X7 receptor stimulation in murine macrophages is mediated by c-Src/Pyk2 and ERK1/2. Biochim Biophys Acta. 2013;1830:4650–4659. doi: 10.1016/j.bbagen.2013.05.023. [DOI] [PubMed] [Google Scholar]
- 34.Miras-Portugal MT, Queipo MJ, Gil-Redondo JC, Ortega F, Gómez-Villafuertes R, Gualix J, Delicado EG, Pérez-Sen R. P2 receptor interaction and signalling cascades in neuroprotection. Brain Res Bull. 2019;151:74–83. doi: 10.1016/j.brainresbull.2018.12.012. [DOI] [PubMed] [Google Scholar]
- 35.Munoz FM, Gao R, Tian Y, Henstenburg BA, Barrett JE, Hu H. Neuronal P2X7 receptor-induced reactive oxygen species production contributes to nociceptive behavior in mice. Sci Rep. 2017;7:3539. doi: 10.1038/s41598-017-03813-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Donnell MJ, Chin SL, Rangarajan S, Xavier D, Liu L, Zhang H, Rao-Melacini P, Zhang X, Pais P, Agapay S, Lopez-Jaramillo P, Damasceno A, Langhorne P, McQueen MJ, Rosengren A, Dehghan M, Hankey GJ, Dans AL, Elsayed A, Avezum A, et al. Global and regional effects of potentially modifiable risk factors associated with acute stroke in 32 countries (INTERSTROKE): a case-control study. Lancet. 2016;388:761–775. doi: 10.1016/S0140-6736(16)30506-2. [DOI] [PubMed] [Google Scholar]
- 37.Pan C, Liu N, Zhang P, Wu Q, Deng H, Xu F, Lian L, Liang Q, Hu Y, Zhu S, Tang Z. EGb761 ameliorates neuronal apoptosis and promotes angiogenesis in experimental intracerebral hemorrhage via RSK1/GSK3β pathway. Mol Neurobiol. 2018;55:1556–1567. doi: 10.1007/s12035-016-0363-8. [DOI] [PubMed] [Google Scholar]
- 38.Pei L, Wang S, Jin H, Bi L, Wei N, Yan H, Yang X, Yao C, Xu M, Shu S, Guo Y, Yan H, Wu J, Li H, Pang P, Tian T, Tian Q, Zhu LQ, Shang Y, Lu Y. A novel mechanism of spine damages in stroke via DAPK1 and Tau. Cereb Cortex. 2015;25:4559–4571. doi: 10.1093/cercor/bhv096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sacco S, Marini C, Toni D, Olivieri L, Carolei A. Incidence and 10-year survival of intracerebral hemorrhage in a population-based registry. Stroke. 2009;40:394–399. doi: 10.1161/STROKEAHA.108.523209. [DOI] [PubMed] [Google Scholar]
- 40.Savio LEB, de Andrade Mello P, Figliuolo VR, de Avelar Almeida TF, Santana PT, Oliveira SDS, Silva CLM, Feldbrügge L, Csizmadia E, Minshall RD, Longhi MS, Wu Y, Robson SC, Coutinho-Silva R. CD39 limits P2X7 receptor inflammatory signaling and attenuates sepsis-induced liver injury. J Hepatol. 2017;67:716–726. doi: 10.1016/j.jhep.2017.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seminotti B, Amaral AU, da Rosa MS, Fernandes CG, Leipnitz G, Olivera-Bravo S, Barbeito L, Ribeiro CA, de Souza DO, Woontner M, Goodman SI, Koeller DM, Wajner M. Disruption of brain redox homeostasis in glutaryl-CoA dehydrogenase deficient mice treated with high dietary lysine supplementation. Mol Genet Metab. 2013;108:30–39. doi: 10.1016/j.ymgme.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 42.Shefa U, Jeong NY, Song IO, Chung HJ, Kim D, Jung J, Huh Y. Mitophagy links oxidative stress conditions and neurodegenerative diseases. Neural Regen Res. 2019;14:749–756. doi: 10.4103/1673-5374.249218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sies H, Berndt C, Jones DP. Oxidative stress. Annu Rev Biochem. 2017;86:715–748. doi: 10.1146/annurev-biochem-061516-045037. [DOI] [PubMed] [Google Scholar]
- 44.Tang J, Liu J, Zhou C, Ostanin D, Grisham MB, Neil Granger D, Zhang JH. Role of NADPH oxidase in the brain injury of intracerebral hemorrhage. J Neurochem. 2005;94:1342–1350. doi: 10.1111/j.1471-4159.2005.03292.x. [DOI] [PubMed] [Google Scholar]
- 45.Wang J. Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog Neurobiol. 2010;92:463–477. doi: 10.1016/j.pneurobio.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wasserman JK, Yang H, Schlichter LC. Glial responses, neuron death and lesion resolution after intracerebral hemorrhage in young vs. aged rats. Eur J Neurosci. 2008;28:1316–1328. doi: 10.1111/j.1460-9568.2008.06442.x. [DOI] [PubMed] [Google Scholar]
- 47.Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006;5:53–63. doi: 10.1016/S1474-4422(05)70283-0. [DOI] [PubMed] [Google Scholar]
- 48.Yang B, Wang S, Yu S, Chen Y, Li L, Zhang H, Zhao Y. C1q/tumor necrosis factor-related protein 3 inhibits oxidative stress during intracerebral hemorrhage via PKA signaling. Brain Res. 2017;1657:176–184. doi: 10.1016/j.brainres.2016.11.016. [DOI] [PubMed] [Google Scholar]
- 49.Zhang L, Li Z, Feng D, Shen H, Tian X, Li H, Wang Z, Chen G. Involvement of Nox2 and Nox4 NADPH oxidases in early brain injury after subarachnoid hemorrhage. Free Radic Res. 2017;51:316–328. doi: 10.1080/10715762.2017.1311015. [DOI] [PubMed] [Google Scholar]
- 50.Zhang Y, Deng H, Hu Y, Pan C, Wu G, Li Q, Tang Z. Adipose-derived mesenchymal stem cells stereotactic transplantation alleviate brain edema from intracerebral hemorrhage. J Cell Biochem. 2019;120:14372–14382. doi: 10.1002/jcb.28693. [DOI] [PubMed] [Google Scholar]
- 51.Zhao H, Zhang X, Dai Z, Feng Y, Li Q, Zhang JH, Liu X, Chen Y, Feng H. P2X7 receptor suppression preserves blood-brain barrier through inhibiting RhoA activation after experimental intracerebral hemorrhage in rats. Sci Rep. 2016;6:23286. doi: 10.1038/srep23286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao H, Pan P, Yang Y, Ge H, Chen W, Qu J, Shi J, Cui G, Liu X, Feng H, Chen Y. Endogenous hydrogen sulphide attenuates NLRP3 inflammasome-mediated neuroinflammation by suppressing the P2X7 receptor after intracerebral haemorrhage in rats. J Neuroinflammation. 2017;14:163. doi: 10.1186/s12974-017-0940-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zheng H, Chen C, Zhang J, Hu Z. Mechanism and therapy of brain edema after intracerebral hemorrhage. Cerebrovasc Dis. 2016;42:155–169. doi: 10.1159/000445170. [DOI] [PubMed] [Google Scholar]
- 54.Zhu H, Wang Z, Yu J, Yang X, He F, Liu Z, Che F, Chen X, Ren H, Hong M, Wang J. Role and mechanisms of cytokines in the secondary brain injury after intracerebral hemorrhage. Prog Neurobiol. 2019;178:101610. doi: 10.1016/j.pneurobio.2019.03.003. [DOI] [PubMed] [Google Scholar]