

Stroke is the second leading cause of death in the world. A significant part of survived people become invalids, dependent on others and in need of outside care. This is a heavy burden for their families and society. The life of patients surviving ischemic stroke (~80% of all strokes) may be subdivided by three main phases: (a) The acute phase that lasts about first 24 hours, when the neurons may be saved and the neuroprotective therapy may be efficient; (b) The early recovery phase that lasts for several days and weeks, when the repair and regeneration processes develop, and partial recovery of brain functions occurs; and (c) The chronic phase (weeks, months and years), in which the organism state is stabilized and the patient gets the partial or full recovery. Different structural and functional modifications are possible throughout the whole post-stroke period, and the specific pharmacotherapy may be applied at each stage (Hankey, 2017).
In ischemic stroke, occlusion of cerebral vessels rapidly, during few minutes leads to local blood flow blockade, lack of oxygen and glucose, mitochondria failure, fall in ATP production, oxidative stress, damage to cell membranes, disruption of ionic homeostasis, excitotoxicity, and necrosis. It is impossible to save necrotic cells in the infarction core during this short period. Afterwards, the damage much slower, for several hours spreads to surrounding tissues and the transition zone, penumbra, is formed. The relatively long “therapeutic window” (3–6 hours and longer) allows saving cells in the penumbra, where apoptosis prevails (Hankey, 2017).
The pathobiochemical processes responsible for cell death and penumbra formation in the ischemic brain include energy deficit, release of glutamate and K+ from the damaged neurons, Ca2+accumulation, excitotoxicity, generation of reactive oxygen species, spreading depolarization, activation of some enzymes and early response genes, apoptosis, and edema. These primary events stimulate the signaling cascades that induce the secondary processes and systemic response of the cerebral tissue aimed both at cell death and cell protection. Some brain repair signals start to be generated in this phase. These processes are controlled by diverse signaling pathways and transcription factors such as c-Myc, p53, E2F1, NF-κB, hypoxia-inducible factor-1 (HIF-1), nuclear factor erythroid 2-related factor 2 (Nrf2), signal transducer and activator of transcription 3 (STAT3), cAMP response element-binding protein (CREB), etc. that control synthesis of specific proteins or protein groups that carry out complex cell functions (Puyal et al., 2013; Demyanenko, Uzdensky, 2017; Uzdensky, 2019). The signaling cascades and transcription factors that regulate the expression of different proteins in the ischemic brain are still poor known.
In addition to signaling pathways that regulate the expression of certain genes, and synthesis of specific proteins, the protein synthesis in the cell is globally regulated by epigenetic processes such as DNA methylation and covalent modifications of histones. Methylation, acetylation, phosphorylation, sumoylation, ubiquitination, or s-nitrosylation of histones regulate DNA packing and chromatin organization. Here we concentrate on the role of acetylation and deacetylation of histones H3 and H4 in the cerebral tissue response to ischemic damage. Histone acetylation induces decondensation of the nuclear chromatin and makes it more accessible for transcription factors. This stimulates protein synthesis in the cell. In contrast, histone deacetylation promotes chromatin condensation that inhibits gene expression and suppresses protein synthesis (Jhelum et al., 2017).
The recent proteomic study has shown the decrease in acetylation of lysine 9 in histone H3 (acH3K9) in the penumbra neurons during first 24 hours after photothrombotic stroke (PTS, a model of ischemic stroke) in the rat cerebral cortex. A possible reason of the histone H3 deacetylation could be downregulation of some histone acetyltransferases, which transfer the acetyl group from acetyl-coenzyme A to histone. However, in this stroke model, histone acetyltransferases HAT1 and PCAF (P300/CBP-associated factor) were shown to be upregulated (Demyanenko et al., 2020a). This could be a reason of the overexpression of dozens signaling and neuronal proteins, but not the downregulation of acH3K9 in the PTS-induced penumbra in the rat cerebral cortex (Demyanenko and Uzdensky, 2017). Interestingly, the overexpression of HAT1 and PCAF was greater in astrocytes than in neurons (Demyanenko et al., 2020a). Astrocytes play a significant role in the brain response to ischemia directed to functional restoration of the nervous system.
Another possible reason of decrease in the histone acetylation level in the ischemic penumbra was the upregulation of histone deacetylases (HDACs), which catalyze the removal of acetyl groups from the lysine residues in the histone tails. Four classes of histone deacetylases are distinguished in mammals according to their structure, functions, and localization. Class I consists of HDAC1, HDAC2, HDAC3 and HDAC8. Class II contains HDAC4, HDAC5, HDAC6, HDAC7, HDAC9 and HDAC10. Class IV -only HDAC11. All of them are zinc-dependent. On the contrary, the cofactor of sirtuins, class III histone deacetylases, is NAD+. Various HDAC isoforms can promote neurodegeneration, but others are protective. Some HDACs deacetylate not only histones, but also different cytoplasmic proteins (Ganai et al., 2016).
The deacetylation of histone H3 in the ischemic penumbra was shown to be associated with overexpression of HDAC1, HDAC2, HDAC6, and HDAC8 in the neuronal nuclei. HDAC1 was found both in the nuclei and cytoplasm of the brain cells; HDAC2 in the nuclei, and HDAC8 in the cytoplasm (Demyanenko et al., 2020b,c). The upregulation of HDAC1 in neurons 4 and 24 hours after PTS was accompanied by its redistribution from the nuclei to the cytoplasm. This evidences against the involvement of HDAC 1 in histone deacetylation after PTS. In contrast, the nuclear HDAC2 level increased not only in neurons, but also in astrocytes of the ischemic penumbra at the first 24 hours after PTS (Demyanenko et al., 2020b). Therefore, the acH3K9 decrease was likely associated with the upregulation of HDAC2, rather than HDAC1. The exact reasons for the overexpression of HDAC2 and HDAC1 in the PTS-induced penumbra are unknown. The overexpression of c-Myc, a possible inducer of HDAC expression, in the penumbra was previously observed 4 and 24 hours after PTS in the rat cerebral cortex (Demyanenko and Uzdensky, 2017). One can suggest that c-Myc connects the primary post-stroke processes that propagate ischemic damage and form penumbra (glutamate excitotoxicity, Ca2+ accumulation in the cytosol, K+-mediated depolarization, oxidative stress) with the upregulation of HDAC1 and HDAC2. Co-localization of HDAC2 with TUNEL-labeled cells indicated its involvement in apoptosis in the PTS-induced penumbra. Unlike HDAC1 and HDAC2, histone deacetylases HDAC3 and HDAC4 did not participate in apoptosis of the penumbra cell. They did not co-localize with apoptotic cells, and their inhibitors did not change the level of apoptosis and the volume of PTS-induced tissue infarct (Demyanenko et al., 2020c). In the early recovery period (3–7 days after photothrombotic stroke), the expression of HDAC2 increased in the damaged cerebral cortex, whereas the level of HDAC8 was shown to increase at 7–24 days post-stroke. Their levels in the damaged and contralateral hemispheres increased similarly that indicated the systemic response of the animal nervous system. So, HDAC2 and HDAC8 were suspected to be potential ischemic mediators in the early recovery period. Actually, the administration of α-phenyltroplon that inhibits both HDAC2 and HDAC8 reduced PTS-induced apoptosis of cortical cells in the PTS-induced penumbra and decreased the infarct volume (Demyanenko et al., 2020b). HDAC6 localized both in the cytoplasm and nuclei of most neurons, but not astrocytes in the cerebral cortex of mice or rats. The main cytoplasmic substrate of HDAC6 is α-tubulin. Its acetylation is necessary for stabilization of microtubules during axonal outgrowth and regeneration after ischemia. During the early recovery period from 1 to 14 days after PTS, HDAC6 was overexpressed in the neurons of the ischemic penumbra and the contralateral cortex in the mouse brain. The colocalization of HDAC6 with TUNEL-positive apoptotic cells in PTS-induced penumbra indicated its involvement in apoptosis (Demyanenko et al., 2019, 2020c).
Acetylation of histone H4 is known to promote genes associated with tissue regeneration and axonal growth. A high level of AcH4 is associated with the resistance of neurons to ischemia, which determines the outcome of stroke and contributes to the brain recovery. The significant increase in the level of acetylation of histone H4 (acH4) in the ischemic mouse hemisphere was observed later, in the early recovery period, at 3 days after PTS, and at 7 days in the contralateral cortex. The increased acetylation of histone H4 was not possibly the result of the downregulation of class I HDACs, but could be due to the downregulation of the II class HDACs. The histone H4 is known to be a direct target of HDAC4, and the relocation of HDAC4 from the cytoplasm into the nucleus is associated with survival of neurons after stroke. The dynamics of the acH4 level correlated with the downregulation of HDAC4 and its translocation into the neuronal nuclei (Demyanenko et al., 2019). Apoptosis of penumbra cells was not associated with the expression of HDAC3, HDAC4, or HDAC5 (Demyanenko et al., 2020b).
The involvement of different HDACs in pathological changes and death of the penumbra cells after ischemic stroke suggests that these histone deacetylases may be potential targets for anti-stroke therapy. Indeed, inhibitors of different HDACs protected the rodent brains against ischemic injury, excitotoxicity, oxidative stress, apoptosis, inflammation, and blood-brain barrier break. They also induced angiogenesis, neurogenesis, migration of the neural precursor cells to the damaged regions, and stimulated functional recovery after cerebral ischemia. Currently, the potential applications of various HDAC inhibitors for anti-stroke are thoroughly investigated (Ganai et al., 2016).
The first generation of HDAC inhibitors such as valproic acid, sodium butyrate, sodium 4-phenylbutyrate, suberoylanilide hydroxamic acid, and trichostatin A are non-selective pan HDAC inhibitors. They modulate the expression of 2–5% of all genes, not only promoting their activation, but also suppressing some pro-apoptotic and pro-inflammatory genes. These inhibitors are the promising neuroprotector agents for treatment of ischemic stroke. For example, administration of valproic acid during the early recovery phase alleviated brain injury in various stroke models. It increased the acetylation of histones H3 and H4, induced neurogenesis, enhanced microvessel density, promoted functional recovery, and improved the neurological outcome after ischemic stroke. Other pan-HDAC inhibitors such as sodium butyrate and sodium 4-phenylbutyrate showed the similar neuroprotective activity. Their neuroprotective effect was associated with inhibition of oxidative stress, reduction of the blood-brain barrier permeability, and anti-inflammatory action.
However, non-selective pan-HDAC inhibitors, which affect various HDACs with different cell localizations, targets and activities in various cell types, can also cause ineligible and long-lasting side effects. Some of these HDACs regulate apoptosis, whereas, others demonstrate the prosurvival activity. To overcome these problems, it is necessary to find more selective drugs that inhibit only those HDACs, which are associated with neuropathological events (Ganai et al., 2016). The second generation HDAC inhibitors are more selective, affecting only one HDAC isoform. For example, HDAC2 inhibitors MI192 or α-phenyltropolone, or HDAC6 inhibitors tubastatin A or HPOB decreased the PTS-induced infarct volume, reduced apoptosis of penumbra cells, and promoted the restoration of motor functions of ischemic mice (Demyanenko et al., 2020b,c). They are less toxic and may be used in lesser concentrations.
These data are summarized in Figure 1. Covalent modifications of histones are largely reversible. This provides the dynamic regulation of gene expression in response to external impacts. Different HATs and HDACs regulate tissue repair and cerebral functions after ischemic stroke. Some histone deacetylases such as HDAC2 and HDAC6 are involved in the regulation of apoptosis in the ischemic penumbra. Various HDAC inhibitors exert the beneficial effects during the early recovery period after ischemic stroke. This expands the “therapeutic window” from several hours to several days and weeks. Such a wider time frame may be a new strategy for functional recovery of stroke patients.
Figure 1.
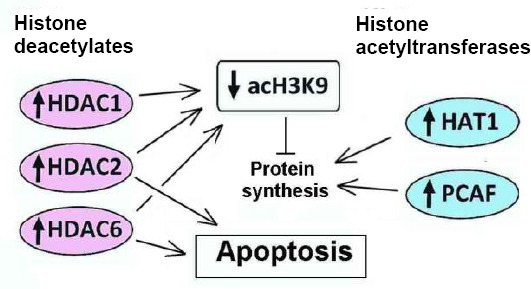
The involvement of histone acetyltransferases and histone deacetylases in regulation of protein synthesis and apoptosis in the ischemic brain.
acH3K9: Histone H3 acetylated on lysine 9; HAT1: histone acetyltransferase 1; HDAC1, HDAC2, HDAC6: histone deacetylases 1, 2 and 6; PCAF: P300/CBP-associated factor.
This work was funded by the grant of the Ministry of Science and Higher Education of the Russian Federation, No. 0852-2020-0028.
Footnotes
Copyright license agreement: The Copyright License Agreement has been signed by both authors before publication
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
C-Editors: Zhao M, Song LP; T-Editor: Jia Y
References
- 1.Demyanenko S, Berezhnaya E, Neginskaya M, Rodkin S, Dzreyan V, Pitinova M. Сlass II histone deacetylases in the post-stroke recovery period-expression, cellular, and subcellular localization-promising targets for neuroprotection. J Cell Biochem. 2019;120:19590–19609. doi: 10.1002/jcb.29266. [DOI] [PubMed] [Google Scholar]
- 2.Demyanenko S, Uzdensky A. Profiling of signaling proteins in penumbra after focal photothrombotic infarct in the rat brain cortex. Mol Neurobiol. 2017;54:6839–6856. doi: 10.1007/s12035-016-0191-x. [DOI] [PubMed] [Google Scholar]
- 3.Demyanenko SV, Dzreyan VA, Uzdensky AB. The expression and localization of histone acetyltransferases hat1 and pcaf in neurons and astrocytes of the photothrombotic stroke-induced penumbra in the rat brain cortex. Mol Neurobiol. 2020a;57:3219–3227. doi: 10.1007/s12035-020-01959-6. [DOI] [PubMed] [Google Scholar]
- 4.Demyanenko SV, Dzreyan VA, Neginskaya MA, Uzdensky AB. Expression of histone deacetylases HDAC1 and HDAC2 and their role in apoptosis in the penumbra induced by photothrombotic stroke. Mol Neurobiol. 2020b;57:226–238. doi: 10.1007/s12035-019-01772-w. [DOI] [PubMed] [Google Scholar]
- 5.Demyanenko SV, Dzreyan VA, Uzdensky AB. Overexpression of HDAC6, but not HDAC3 and HDAC4 in the penumbra after photothrombotic stroke in the rat cerebral cortex and the neuroprotective effects of α-phenyl tropolone, HPOB, and sodium valproate. Brain Res Bull. 2020c;162:151–165. doi: 10.1016/j.brainresbull.2020.06.010. [DOI] [PubMed] [Google Scholar]
- 6.Ganai SA, Ramadoss M, Mahadevan V. Histone deacetylase (HDAC) inhibitors - emerging roles in neuronal memory, learning, synaptic plasticity and neural regeneration. Curr Neuropharmacol. 2016;14:55–71. doi: 10.2174/1570159X13666151021111609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hankey GJ. Stroke. Lancet. 2017;389:641‐654. doi: 10.1016/S0140-6736(16)30962-X. [DOI] [PubMed] [Google Scholar]
- 8.Jhelum P, Karisetty BC, Kumar A, Chakravarty S. Implications of epigenetic mechanisms and their targets in cerebral ischemia models. Curr Neuropharmacol. 2017;15:815–830. doi: 10.2174/1570159X14666161213143907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Puyal J, Ginet V, Clarke PG. Multiple interacting cell death mechanisms in the mediation of excitotoxicity and ischemic brain damage: a challenge for neuroprotection. Prog Neurobiol. 2013;105:24–48. doi: 10.1016/j.pneurobio.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Uzdensky AB. Apoptosis regulation in the penumbra after ischemic stroke: expression of pro- and antiapoptotic proteins. Apoptosis. 2019;24:687–702. doi: 10.1007/s10495-019-01556-6. [DOI] [PubMed] [Google Scholar]