

Klotho is one of a number of well-known longevity-associated genes. Its depletion in aging or disease may promote several neuropathologies associated with the central nervous system, including hypomyelination and phosphorylation of neurofilaments, synaptic loss and modulation of their plasticity, behavioral impairments, neuroinflammation, and finally neurodegeneration. Therefore, the complexity of neuronal physiology and function raises some fundamental questions about the molecular mechanisms involved in Klotho action. To date, the evidence is summarized that the common link between Klotho and neuropathological changes are perturbations in oxidative homeostasis leading to the irreversible accumulation of molecular damage in DNA, protein, and lipids fractions, and finally to cell death or senescence. However, recent studies shed more detailed light on a possible protective mechanism of Klotho in neuronal cells through modulation of endoplasmic reticulum (ER) stress pathways, autophagic process, and inflammatory reaction in response to misfolded protein accumulation.
It is well established that the formation of misfolded protein aggregates causing cellular toxicity is one of the fundamental hallmarks of neurodegenerative proteinopathies. In general, the misfolding process results in the formation of a protein with a different conformation from its native fold. It can occur by several reasons: somatic mutations, mistakes on the processes of transcription or translation, errors during post-translational modifications, structural changes induced by environmental factors and/or induction of protein misfolding by seeding and cross-seeding mechanisms. In response, cells developed multiple methods for preventing and addressing these problems. The most described mechanism associated with ER stress is known as the unfolded protein response (UPR). Initially, UPR machinery acts as the first-line defense mechanism leading proteins to become folded in response to small perturbations. However, when it becomes clear that the damage cannot be restored, inflammation-accompanied systems such as the proteasome, autophagy, and ER-associated degradation are activated to degrade these misfolded proteins. Furthermore, a strong line of evidence suggests that ER and mitochondria cooperate in maintaining the control of prosurvival/pro-death pathways through the regulated transfer of Ca2+ from ER to mitochondria. According to literature, the role of Klotho protein in ER stress associated with stress-initiated UPR was demonstrated for the first time by Banerjee et al. (2013). During studies, authors used several cell lines, including neuroblastoma cells, and confirmed that Klotho can counteract ER stress and that loss of Klotho is linked to ER stress-induced apoptosis. Further analysis revealed that Klotho is involved in all three branches of UPR mediated by proximal sensors, namely the kinase and endoribonuclease IRE1 (α and β), the PERK kinase, and the basic leucine-zipper transcription factor ATF6 (α and β) (Banerjee et al., 2013). Being inspired by this research, we have recently characterized the role of Klotho protein in the protection of neuronal hippocampal cells. We have shown that Klotho acts as a signal transducer between prosurvival and proapoptotic crosstalk mediated by ER stress. For the purpose of this study, Klotho silencing in hippocampal neurons was obtained by siRNA strategy and then the cells were treated with lipopolysaccharide (LPS) to induce inflammation. In control hippocampal neurons, the LPS challenge-initiated activation of IRE1α-mediated pathway and in turn resulted in upregulated secretion of a powerful anti-inflammatory cytokine interleukin (IL)-10 and ensured activation of adaptation response leading to a prosurvival outcome. On the other hand, Klotho deficiency switched ER response to eukaryotic translation initiation factor 2α (eIF2α)-mediated branch of UPR (PERK-independent), which significantly exacerbated neuroinflammation and induced apoptotic cell death. In this case, the PERK-independence seems to be extremely crucial since the PERK-eIF2α arm of UPR is considered as pro-survival via the translational upregulation of ATF4. In Klotho-deficient neurons, the status of ATF4 expression and phosphorylation upon stress remained unaffected, which further explains why the apoptotic pathway was not associated with CHOP activation (Rusinek et al., 2020). On the other hand, when acting alone, p-eIF2α may drive inflammation as well as control the cell cycle course. These results find confirmation in in vivo studies where authors confirmed the role of Klotho protein in modulation of ASK1 and p38 MAPK signaling in dopaminergic neurons treated with a neurotoxin (Brobey et al., 2015). Therefore, this line of Klotho-ER stress crosstalk seems to play a pivotal role in the protection of neuronal cells against the accumulation of misfolded proteins, factors promoting inflammation, oxidative stress, senescence and finally leading to increase in the severity of cognitive impairments and dementia. At this point, one must remember that Klotho protein is considered as a regulator of oxidative stress as well as antioxidant enzymes and ER stress-mediated accumulation of misfolded proteins in neurons is dependent on several peroxiredoxins. Also, Klotho protein is associated with protection against reactive oxygen species (ROS)-induced mitochondrial damage (Kim et al., 2017). Thus, Klotho’s action on ER stress could be considered not only as direct action but also as an indirect effect mediated by antioxidation and this possibility is further studied by our group.
Continuing, it is well established that the UPR influences inflammatory response on many levels, from the regulation of cytokine transcription factors and stimulation of pattern recognition receptors to modulation of inflammatory signaling pathways. ER stress has been additionally found to affect NLRP3 inflammasome activation through multiple effects including UPR (Vanaja et al., 2015). More importantly, inflammatory factors also have been found to trigger ER stress and UPR activation, suggesting mutual regulation of these two pathways. The role of Klotho protein in the neuroinflammatory process is a little bit mysterious. Teocchi et al. (2013) reported that in patients with temporal lobe epilepsy associated with hippocampal sclerosis the levels of tumor necrosis factor α are elevated and correlated with activation of the nuclear factor-κB (NF-κB) signaling pathway accompanied by downregulated levels of Klotho. In contrast, retinoic-acid-inducible gene-I expression, NF-κB activation, and secretion of proinflammatory cytokines could be inhibited by Klotho overexpression. Similarly, Klotho supplementation suppresses thioredoxin-interacting protein-dependent activation of the NLRP3 inflammasome through enhancement of fibroblast growth factor 23 signaling. A more detailed understanding of this topic has been brought by our studies. As mentioned in the previous paragraph, Klotho-depletion augmented inflammatory response in hippocampal neuronal cells. We detected upregulated levels of proinflammatory cytokines, tumor necrosis factor α and IL-1β, accompanied by reduced secretion of anti-inflammatory cytokines, IL-10, IL-2, and IL-3, and inhibited surface deposition of CD62L molecule in response to LPS treatment. According to literature data, the inflammatory response could be driven by ER stress mostly through NF-κB activation. Further, Klotho was also shown to act as a modulator of peroxisome proliferator-activated receptor gamma (PPARγ) signaling, a negative regulator of inflammation. When cells are Klotho-silenced, the activation of PPARγ is blocked and thus inflammation becomes accelerated (Chen et al., 2018). Besides, PPARγ activity is also modulated by ER stress. Due to the regulation of PPARγ action, ER stress can enhance proinflammatory NF-κB activation and maintain increased levels of cytokines. Also, there is one more aspect that needs to be indicated at this point. Neuronal inflammation is driven by the insulin/insulin-like growth factor 1 (IGF-1) signaling pathway and Klotho is considered as a negative regulator of this pathway. Since IGF-1 significantly augments the adaptive capacity of the ER by enhancing compensatory mechanisms (Novosyadlyy et al., 2008), Klotho-mediated inhibition of IGF-1 prevents the restoration of ER homeostasis, and promotes apoptosis. Therefore, apparent Klotho anti-inflammatory action could be associated with its direct modulation of the main transcription factors such as NF-κB or with its indirect modulation through ER stress pathways.
Furthermore, ER stress and autophagy are generally assumed as mechanisms independent of each other and serving distinct functions. However, increasing pieces of evidence indicate that ER stress triggers autophagy, which disposes of unfolded proteins in cells. In general, autophagy is considered as a process involved in maintaining the general cellular homeostasis under physiological and pathological conditions by facilitating the lysosomal degradation pathway and affecting the recycling and elimination of intracellular defective macromolecules and organelles such as misfolded proteins in neuronal cells. However, recent development reveals a key role of autophagy in inhibition of inflammatory response by clearance of danger signals such as mitochondrial ROS and DNA that promote the activation of the inflammasome. It is also highly possible that autophagy affects inflammation through regulation of transcription factors such as already mentioned NF-κB. Although in this context, the autophagy process focuses on its pro-survival outcome, autophagy is also known as a multifaceted regulator of cell death and may lead to both, cell survival as well as demise. However, autophagy often accompanies cell death and the requirement of autophagic machinery for cell death execution is highly contextual. Klotho was shown to be engaged in autophagy course. A comprehensive study done by Fernandez et al. revealed that Klotho deficiency results in the upregulated binding of Beclin 1-BCL2 and finally leads to a significant reduction of autophagy (Fernandez et al., 2018). Our experiments with hippocampal neuronal cells partially confirmed this conclusion (Rusinek et al., 2020). We have also identified Klotho as a molecular switch between apoptosis and autophagy. As said, Klotho-silencing inhibits expression of Atg13, prevents the formation of the ULK1 complex, and leads cells towards apoptotic death instead of autophagy (Mytych et al., 2020). However, our different studies, also in hippocampal neuronal cells, brought the opposite results (Mytych et al., 2019). Firstly, as mentioned above, stimulation of neuronal cells with LPS triggers adaptive response mediated by mild ER stress, and Klotho depletion results in the activation of a pro-death branch of UPR with accompanied inhibition of autophagy (Rusinek et al., 2020). Secondly, amitriptyline treatment activates pro-survival autophagy and Klotho depletion results in the initiation of pro-death autophagy leading to apoptotic cell death (Mytych et al., 2019). We believe that the outcome strictly depends on the type of insult and the extent/type of accompanied DNA damage. Amitriptyline induces micronuclei formation and telomere damage in Klotho-deficient cells, while LPS is known as a mild genotoxic factor. As autophagy is a critical mechanism for efficient removal of double-strand DNA breaks (Gillespie and Ryan, 2016), it is primarily activated when damage occurs. Nonetheless, the cells enter the apoptotic pathway and this fact suggests the irreparable damage of DNA. Continuing, the crosstalk on the line Klotho-autophagy in hippocampal neuronal cells seems to be also dependent on the course of ER signaling and/or inflammatory reaction and our study strongly confirms this opinion. Amitriptyline is known as a potent anti-inflammatory agent with no effect on ER condition leading towards autophagy with Klotho’s role as a switch between pro-survival and pro-death autophagy. On the other hand, LPS triggers an inflammatory response leading towards ER stress and autophagy reduction with Klotho’s role as a switch between pro-survival and pro-death UPR. Therefore, we believe that the activation of specific pathways in hippocampal neurons is strictly dependent on insult type, while the outcome on the line pro-survival and pro-death is defined by Klotho activity (Figure 1).
Figure 1.
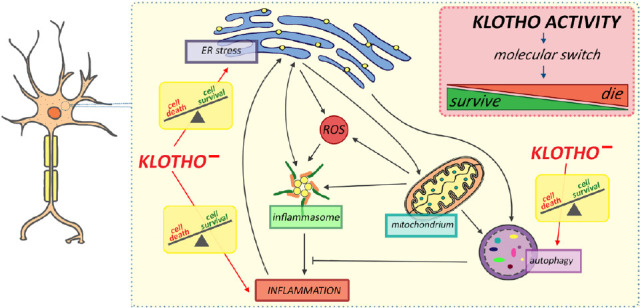
Schematic representation of the crosstalk between autophagy, endoplasmic reticulum, and inflammation in neurons mediated by Klotho depletion.
Under endoplasmic reticulum (ER) stress, the inflammatory response is activated by mitochondria-released reactive oxygen species (ROS) and enhanced secretion of proinflammatory cytokines. As feedback, ER stress could be further accelerated by an inflammatory response. On the other hand, inflammation could be inhibited by autophagy triggered by ER. In response, autophagy-mediated degradation of inflammatory-related proteins and mitochondrial turnover could inhibit ER stress. Although Klotho seems to be engaged in all processes, its role is probably limited to being a molecular switch on the line “die” (cell death) or “survive” (cell survival). Klotho–: Klotho depletion.
Undoubtedly, Klotho is a powerful neuroprotective factor with a central impact on hippocampal neuronal cells. However, whether Klotho-dependent neuroprotection and neurodegeneration are strictly associated with Klotho-mediated activation of specific pathways or Klotho action is limited to the decision “survive” or “die” deserves further study. This gives us a powerful tool for Klotho utility in clinical practice in the future. Since Klotho expression declines with age, maintaining its level by stimulating endogenous production or exogenous administration may be a potential therapeutic target to slow down the neurodegeneration process.
Footnotes
C-Editors: Zhao M, Wang L; T-Editor: Jia Y
Copyright license agreement: The Copyright License Agreement has been signed by the author before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
References
- 1.Banerjee S, Zhao Y, Sarkar PS, Rosenblatt KP, Tilton RG, Choudhary S. Klotho ameliorates chemically induced endoplasmic reticulum (ER) stress signaling. Cell Physiol Biochem. 2013;31:659–672. doi: 10.1159/000350085. [DOI] [PubMed] [Google Scholar]
- 2.Brobey RK, German D, Sonsalla PK, Gurnani P, Pastor J, Hsieh CC, Papaconstantinou J, Foster PP, Kuro-o M, Rosenblatt KP. Klotho protects dopaminergic neuron oxidant-induced degeneration by modulating ASK1 and p38 MAPK signaling pathways. PLoS One 10. 2015:e0139914. doi: 10.1371/journal.pone.0139914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen J, Hu J, Liu H, Xiong Y, Zou Y, Huang W, Shao M, Wu J, Yu L, Wang X, Wang X, Lin L. FGF21 protects the blood-brain barrier by upregulating PPARgamma via FGFR1/beta-klotho after traumatic brain injury. J Neurotrauma. 2018;35:2091–2103. doi: 10.1089/neu.2017.5271. [DOI] [PubMed] [Google Scholar]
- 4.Fernández ÁF, Sebti S, Wei Y, Zou Z, Shi M, McMillan KL, He C, Ting T, Liu Y, Chiang WC, Marciano DK, Schiattarella GG, Bhagat G, Moe OW, Hu MC, Levine B. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature. 2018;558:136–140. doi: 10.1038/s41586-018-0162-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gillespie DA, Ryan KM. Autophagy is critically required for DNA repair by homologous recombination. Mol Cell Oncol 3. 2016:e1030538. doi: 10.1080/23723556.2015.1030538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim SJ, Cheresh P, Eren M, Jablonski RP, Yeldandi A, Ridge KM, Budinger GRS, Kim DH, Wolf M, Vaughan DE, Kamp DW. Klotho, an antiaging molecule, attenuates oxidant-induced alveolar epithelial cell mtDNA damage and apoptosis. Am J Physiol Lung Cell Mol Physiol. 2017;313:L16–26. doi: 10.1152/ajplung.00063.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mytych J, Solek P, Tabecka-Lonczynska A, Koziorowski M. Klotho-mediated changes in shelterin complex promote cytotoxic autophagy and apoptosis in amitriptyline-treated hippocampal neuronal cells. Mol Neurobiol. 2019;56:6952–6963. doi: 10.1007/s12035-019-1575-5. [DOI] [PubMed] [Google Scholar]
- 8.Mytych J, Solek P, Bedzinska A, Rusinek K, Warzybok A, Tabecka-Lonczynska A, Koziorowski M. Klotho-mediated changes in the expression of Atg13 alter formation of ULK1 complex and thus initiation of ER- and Golgi-stress response mediated autophagy. Apoptosis. 2020;25:57–72. doi: 10.1007/s10495-019-01579-z. [DOI] [PubMed] [Google Scholar]
- 9.Novosyadlyy R, Kurshan N, Lann D, Vijayakumar A, Yakar S, LeRoith D. Insulin-like growth factor-I protects cells from ER stress-induced apoptosis via enhancement of the adaptive capacity of endoplasmic reticulum. Cell Death Differ. 2008;15:1304–1317. doi: 10.1038/cdd.2008.52. [DOI] [PubMed] [Google Scholar]
- 10.Rusinek K, Solek P, Tabecka-Lonczynska A, Koziorowski M, Mytych J. Focus on the role of Klotho protein in neuro-immune interactions in HT-22 cells upon LPS stimulation. Cells. 2020;9:1231. doi: 10.3390/cells9051231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teocchi MA, Ferreira AE, da Luz de Oliveira EP, Tedeschi H, D’Souza-Li L. Hippocampal gene expression dysregulation of Klotho, nuclear factor kappa B and tumor necrosis factor in temporal lobe epilepsy patients. J Neuroinflammation. 2013;10:53. doi: 10.1186/1742-2094-10-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vanaja SK, Rathinam VA, Fitzgerald KA. Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol. 2015;25:308–315. doi: 10.1016/j.tcb.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]