

More than 40 million people worldwide are thought to be affected by Alzheimer´s disease (AD). Of these, estimated less than 10% develop symptoms usually well before the age of 65, due to familial (hereditary) AD predisposition (Sierksma et al., 2020). AD is a multifactorial disorder, which includes a multitude of progressive degenerations in the brain parenchyma, but also in the vascular and hemostatic system. Currently, no drug is available or recognizable in the research pipeline for the effective treatment of this terrible disease. Therefore, the search for novel more effective drugs is an urgent task of pharmaceutical research. Already in the 1970s, clinical studies with small groups of senile dementia patients showed a positive effect of anticoagulant treatment on disease development (Grossmann, 2020). Particularly, results of basic research in the last 6 years on a contribution of cerebrovascular dysfunction to AD have brought back to light this idea. This perspective paper summarizes the role of toxic proteins of amyloid-beta (Aβ), thrombin and fibrin as key drivers in triggering vascular dysfunction and derived neurodegenerative changes, which leads to AD, and discuss how they can be treated with direct oral anticoagulants (DOACs).
Anticoagulants are drugs that are used prophylactically or therapeutically to inhibit blood clotting and thus, avoid the formation of thrombosis or embolisms (Grossmann, 2020). A medication based on anticoagulants for treatment of neurodegenerative brain amyloidoses, such as AD, is not yet in use. In the phase of blood clotting (coagulation), which leads to the closure of the wound and wound healing, a soluble protein from the blood, the fibrinogen, is converted into insoluble fibrin. Fibrin forms a fiber network with integrated erythrocytes and platelets, a fibrin clot (thrombus). The synthesis of the responsible serine protease thrombin is induced cascadingly by a variety of tissue and coagulation factors (e.g. factor Xa). A part of these factors is activated in a vitamin K-dependent process. Anticoagulants can inhibit blood clotting in different ways (Grossmann, 2020): Indirectly, such as in the case of vitamin K antagonists (VKAs, e.g. warfarin, with oral effect) and in heparins (e.g. enoxaparin, with parenteral effect), or directly, such as in the case of DOACs, which include the thrombin inhibitor dabigatran etexilate and blood clotting factor Xa-inhibitors. Oral anticoagulants are prescribed in Germany alone in nearly 3 million patients, who are predominantly over 70 years old, due to the indication areas for antithrombosis. To treat acute venous thrombosis and thrombosis prophylaxis in risk situations, e.g. after surgery, short-term antithrombosis is indicated. For the prophylaxis, e.g. of cardiac thrombosis and embolisms in patients with cardiac arrhythmias, such as atrial fibrillation, long-term permanent anticoagulation is prescribed. Anticoagulants reduce the risk of a fatal heart attack or stroke in vulnerable persons, but they also increase bleeding risks, including intracerebral hemorrhage (Grossmann, 2020).
Recent research revealed that the factors crucial for triggering AD pathogenesis are accumulation of misfolded, toxic proteins of Aβ in brain tissue (Zott et al. 2019; Greenberg et al., 2020). Sequentially acting secretase enzymes from α-, β- and γ-type release Aβ from the Aß-precursor protein AβPP. AβPP is anchored in the cell membrane of neurons and provides fission products of different lengths that are important for synapse function. In contrast, toxic Aβ accumulates as soluble dimers and oligomers, and as insoluble, deposited fibrils (Aβ plaques), which are localized between neurons, especially in neocortical and hippocampal brain parenchyma. Soluble Aβ aggregates hyperactivate and damage neurons and synapses (Zott et al., 2019), and are able to activate microglia cells (Venegas et al., 2017). Aβ is also actively transported into the blood system via the blood-brain barrier (BBB), and aggregates deposit around and in cerebral blood vessels (Strickland, 2018). All major gene modifications, leading to an increased risk for AD, are related to Aβ generation, aggregation, and clearance, and associated microglia responses (Sierksma et al., 2020). In addition, intraneural deposits and spread of tau-protein fibrils, disruption of BBB, neuroinflammation, and ultimately, death of synapses and neurons are characteristic for disease progression. Especially, neuroinflammatory processes, initiated from activated microglia cells and their release of proinflammatory cytokines and protein complexes, trigger the production and spread of cerebral Aβ deposits (Venegas et al., 2017), via upregulation of γ-secretase activity for Aβ production (Hur et al., 2020), and synaptic loss in AD. It is estimated that cerebral Aβ and serum biomarkers for neuron cell death accumulate in people who develop AD, 10–20 years before the symptoms of the disease occur (Grossmann, 2020).
Also typical of the early development of AD, but nearly disregarded in therapeutic research, are Aβ-initiated pathological processes in the cerebral blood vessels, a disorder, known as cerebral amyloid angiopathy (CAA; Figure 1). In clinical studies, CAA and AD pathology co-occur in the same brain with an incidence of 82–98% (Jellinger, 2002). In the CAA, Aβ aggregates accumulate and deposit around and into the walls of cerebral arteries and capillaries, leading to cerebrovascular dysfunction (Jellinger, 2002; Greenberg et al., 2020). In particular, neocortical and hippocampal brain areas are disturbed in perfusion and no longer supplied adequately with blood and its constituents, such as oxygen and nutrients (Strickland, 2018). In addition, perivascular clearance of Aβ from the interstitial brain fluid into the blood is hindered by the damaged blood vessels. This boosts Aβ accumulation in the brain tissue and thus, the progression of AD. Investigations in AD patients and transgenic mouse lines with gene alterations in Aβ for human AD risk (AD mouse models; Sierksma et al., 2020) revealed that CAA is caused by deposition of Aβ in the cerebral blood vessels (Jellinger, 2002; Greenberg et al., 2020). The consequences are that the cortical CBF decreases by about 25%, and therefore, blood circulation suffers in certain areas of the brain (Grossmann, 2020). This hypoperfusion leads to undersupply of the brain tissue, especially with oxygen (hypoxia) and nutrients. Hypoxia induces Aß synthesis, inflammatory and neurodegenerative processes, and ultimately, cognitive decline (Figure 1). Studies involving living brain tissue showed that deposits of Aß, especially around cortical capillaries, cause pericytes on the outer wall of the vessel to contract (Nortley et al., 2019). This diminishes the diameter of the capillaries and leads to vasoconstriction. CBF declines and a chronic hypoperfusion with hypoxia is caused. Hypoperfusion is considered to be an essential pathophysiological factor that stimulates AD, together with additional effects of vascular dysfunction in CAA. These include, besides vasoconstriction, microvascular infarctions and hemorrhages, which injure BBB and stimulate inflammatory and degenerative changes in brain tissue (Figure 1). Moreover, investigations in AD mouse models and AD patients showed that, in parallel with Aβ, fibrin and thrombin accumulate in the brain (Strickland, 2018). Aβ is able to bind to fibrinogen and fibrin, thereby triggering the formation of Aβ-containing fibrin clots. These clots have an abnormal structure of the fibrin mesh, which makes them resistant to clot-dissolving enzymes in plasmin fibrinolysis (Strickland, 2018). They were detected in blood vessels of CAA, as well as in brain parenchyma (Strickland, 2018; Figure 1), where formation is facilitated by the fact that BBB becomes increasingly permeable for plasma proteins like thrombin and fibrin(ogen) in AD (Grossmann, 2020). In total, these Aβ-containing fibrin clots can hamper cerebral blood flow, evoke inflammatory and degenerative alterations in brain parenchyma, and can ultimately lead to the death of synapses and neurons. In addition, Aβ was shown to activate the blood clotting factor FXII in the coagulation cascade for the production of thrombin (Strickland, 2018). As a result, proinflammatory thrombin, as well as the formation of fibrin and fibrin-Aβ deposits increases in vascular tissue and in brain parenchyma. Thrombin and fibrin are recognized as key mediators of a multitude of inflammatory processes in the vessel walls and parenchymal tissue in the AD brain, including direct activation of microglia cells and astrocytes, and neuronal injury (Strickland, 2018; Grossmann, 2020; Iannucci et al., 2020; Figure 1). Activation of the blood clotting factor FXII by Aβ additionally induces the synthesis of proinflammatory bradykinin, which appears to be causally linked to neuroinflammatory, degenerative and cognitive changes in AD (Strickland, 2018). These findings suggest a key role of Aβ, fibrin and thrombin in CAA, with dramatic implications for AD. Due to increasing vascular dysfunction particularly in neocortical and hippocampal brain areas, CBF and consequently, brain perfusion and tissue supply with oxygen and nutrients decrease. As a result, neuroinflammatory and neurodegenerative processes are intensified by hypoxia-induced synthesis of Aβ and fibrin-Aβ deposits, disruption of BBB, reduced perivascular Aβ clearance, fibrin and thrombin accumulation, activation of microglia cells, Aβ-induced neuronal hyperactivation, and neuroinflammation (Figure 1). This circulus vitiosus from steadily deteriorating vascular disorders and resulting degenerative effects promotes death of synapses and neuron cells and ultimately, contributes to cognitive decline.
Figure 1.
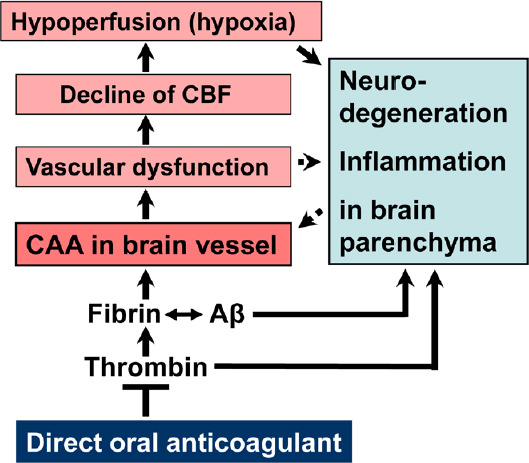
Cerebral amyloid angiopathy (CAA) and triggered cascade of brain alterations, present in Alzheimer´s disease, and proposed mechanism of action of direct oral anticoagulant for therapy.
Direct (solid lines) and indirect (dotted lines) stimulatory effects, reported in literature, and hypothesized central point of anticoagulant intervention. Aβ: Amyloid-beta proteins; CBF: cerebral blood flow.
A therapy against AD with anticoagulants is based on the hypothesis that the drugs counteract the formation and effects of key factors in CAA and neuroinflammation, contributing to AD (Figure 1). By inhibiting thrombin (synthesis or enzymatic activity) and formation of fibrin, anticoagulants can block thrombin- and fibrin-triggered inflammatory processes, as well as progressive fibrin-Aβ deposition in brain parenchyma and vascular system. In the latter case of CAA, fibrin-Aβ clots reduce regional CBF and brain perfusion. Treatment with anticoagulants can maintain CBF and therefore, can avoid shortage with oxygen and nutrients, and resulting neurodegenerative changes in brain tissue. Given complete cerebral perfusion and low Aβ-, thrombin- and fibrin-piling and inflammatory milieu, anticoagulants could decrease vascular-driven progression in neurodegenerative and cognitive changes. In this respect, previous studies with AD mouse models suggested that thrombin inhibitors can alleviate symptoms in AD, because of their inhibitory effect on proinflammatory thrombin and cortical Aβ deposition (Iannucci et al., 2020). Meanwhile, Cortes-Canteli et al. (2019) have reported results in AD mouse model, which support a therapy with anticoagulants. Long-term anticoagulation with dabigatran etexilate prevented toxic fibrin deposition in the brain, hypoperfusion and memory decline. The extent of cortical Aβ oligomers and plaques, and neuroinflammatory activity decreased, while function of BBB was preserved. No hemorrhages or intracranial bleeding was observed (Cortes-Canteli et al., 2019). With respect of their application, oral anticoagulants are an advantage for therapy of vascular disturbance in AD. However, VKAs, such as warfarin, have undesirable effects, which include bleeding complications and pharmacodynamic interactions (Grossmann, 2020). Specific acting DOACs, with the direct thrombin inhibitor dabigatran etexilate and the blood clotting factor Xa-inhibitors rivaroxaban, apixaban, edoxaban, and betrixaban, are much more suitable. Of these, dabigatran etexilate could be preferred for therapy. Dabigatran etexilate is the prodrug form that releases in vivo the active drug dabigatran. In contrast to factor Xa-inhibitors, which block the production of thrombin, dabigatran binds directly to existing thrombin and inactivates enzymatic thrombin for fibrin formation, but also soluble thrombin for proinflammatory effects (Grossmann, 2020; Iannucci et al., 2020). Compared to the VKA warfarin, dabigatran has a shorter half-life in humans, and, observed in elderly patients with atrial fibrillation, a reduced risk of ischaemic stroke, intracranial brain haemorrhage by up to 66%, and death (Graham et al., 2015). The latter study by the U.S. Food and Drug Administration found an incidence rate for harmful intracranial brain hemorrhage per 1000 person-years of 3.3 after anticoagulation with dabigatran etexilate, whereas a rate of 9.6 was observed for warfarin treatment (Graham et al., 2015). Dabigatran leads to predictable and reliable anticoagulation, which can be neutralized by a specific antidote within minutes and therefore, reduces bleeding risk (Grossmann, 2020). Likewise, specific reversal agents for factor Xa-inhibiting DOACs are available since recently. Nevertheless, permanent therapy with DOACs in AD patients that are more prone to bleeding, given the causal link of CAA and vascular fragility, must be carefully evaluated for bleeding risk, using e.g. clinical HAS-BLED-Score criteria and imaging methods.
DOAC-type anticoagulants offer the possibility to treat vascular dysfunction and resulting neurodegenerative processes in AD. Provided, they are applied early, therapeutically or, in the case of genetic predisposition, prophylactically. The listed DOACs are clinically approved and over years prescribed medicaments with known safety profile. Hence, approval for this new indication should be faster and more cost effective to achieve. If further preclinical studies strengthen this therapeutic approach, clinical studies would be highly recommended. Then, it would be desirable, if clinical institutes, supported by pharmaceutical companies and funding programs, are stimulated by this research impulse to study the therapeutic value of DOACs against AD.
Footnotes
C-Editors: Zhao M, Song LP; T-Editor: Jia Y
Copyright license agreement: The Copyright License Agreement has been signed by the author before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
References
- 1.Cortes-Canteli M, Kruyer A, Fernandez-Nueda I, Marcos-Diaz A, Ceron C, Richards AT, Jno-Charles OC, Rodriguez I, Callejas S, Norris EH, Sanchez-Gonzalez J, Ruiz-Cabello J, Ibanez B, Strickland S, Fuster V. Long-term dabigatran treatment delays Alzheimer´s disease pathogenesis in the TgCRND8 mouse model. J Am Coll Cardiol. 2019;74:1910–1923. doi: 10.1016/j.jacc.2019.07.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Graham DJ, Reichman ME, Wernecke M, Zhang R, Southworth MR, Levenson M, Sheu T-C, Mott K, Goulding MR, Houstoun M, MaCurdy TE, Worrall C, Kelman JA. Cardiovascular, bleeding, and mortality risks in elderly medicare patients treated with dabigatran or warfarin for nonvalvular atrial fibrillation. Circulation. 2015;131:157–164. doi: 10.1161/CIRCULATIONAHA.114.012061. [DOI] [PubMed] [Google Scholar]
- 3.Greenberg SM, Bacskai BJ, Hernandez-Guillamon M, Pruzin J, Sperling R, van Veluw SJ. Cerebral amyloid angiopathy and Alzheimer disease -one peptide, two pathways. Nature Rev Neurol. 2020;16:30–42. doi: 10.1038/s41582-019-0281-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grossmann K. Anticoagulants for treatment of Alzheimer´s disease. J Alzheimers Dis. 2020;77:1373–1382. doi: 10.3233/JAD-200610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hur JY, Frost GR, Wu X, Crump C, Pan SJ, Wong E, Barros M, Li T, Nie P, Zhai Y, Wang JC, TCW J, Guo L, McKenzie A, Ming C, Zhou X, Wang M, Sagi Y, Renton AE, Esposito BT, Kim Y, Sadleir KR, Trinh I, Rissmann RA, Vassar R, Zhang B, Johnson DS, Masliah E, Greengard P, Goate A, Li YM. The innate immunity protein IFITM3 modulates γ-secretase in Alzheimer´s disease. Nature. 2020;586:735–740. doi: 10.1038/s41586-020-2681-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iannucci J, Renehan W, Grammas P. Thrombin, a mediator of coagulation, inflammation, and neurotoxicity at the neurovascular interface: implications for Alzheimer´s disease. Front Neurosci. 2020;14:762. doi: 10.3389/fnins.2020.00762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jellinger KA. Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm. 2002;109:813-836. doi: 10.1007/s007020200068. [DOI] [PubMed] [Google Scholar]
- 8.Nortley R, Korte N, Izquierdo P, Hirunpattarasilp C, Mishra A, Jaunmuktane Z, Kyrargyri V, Pfeiffer T, Khennouf L, Madry C, Gong H, Richard-Loendt A, Huang W, Saito T, Saido TC, Brandner S, Seti H, Attwell D. Amyloid ß oligomers constrict human capillaries in Alzheimer´s disease via signaling to pericytes. Science. 2019;365:eaav9518. doi: 10.1126/science.aav9518.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sierksma A, Escott-Price V, De Stopper B. Translating genetic risk of Alzheimer´s disease into mechanistic insight and drug targets. Science. 2020;370:61–66. doi: 10.1126/science.abb8575. [DOI] [PubMed] [Google Scholar]
- 10.Strickland S. Blood will out: vascular contributions to Alzheimer´s disease. J Clin Invest. 2018;128:556–563. doi: 10.1172/JCI97509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, Vieira-Saecker A, Schwartz S, Santarelli F, Kummer MP, Griep A, Gelpi E, Beilharz M, Riedel D, Golenbock DT, Geyer M, Walter J, Latz E, Heneka MT. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer´s disease. Nature. 2017;552:355–361. doi: 10.1038/nature25158. [DOI] [PubMed] [Google Scholar]
- 12.Zott B, Simon MM, Hong W, Unger F, Chen-Engerer HJ, Frosch MP, Sakmann B, Walsh DM, Konnerth A. A vicious cycle of β amyloid-dependent neuronal hyperactivation. Science. 2019;365:559–565. doi: 10.1126/science.aay0198. [DOI] [PMC free article] [PubMed] [Google Scholar]