

Alzheimer’s disease (AD) is an irreversible disease that leads to neurodegeneration. The underpinning mechanisms of neuronal cell death are a matter of ongoing debate regarding the impact of accumulation of the amyloid beta (Aβ) peptide and post-translation modifications of the tau protein. However, a growing area of research and one that may provide a more rigorous account of the early changes seen in Alzheimer’s brains is inflammation. Brain inflammation is a coordinated immune response to subtle changes in the brain’s microenvironment indicative of injury and disease. Neuroinflammation can be either short lived or chronic and lead to changes in brain structure and its neurochemistry. In AD, the ability of the brain to self-regulate inflammatory signals is compromised and leads to a chronic and deleterious inflammatory response. Understanding the molecular entities that are involved in neuroinflammation in AD will open the door to much needed novel therapeutic strategies. However, this is a cautionary tale with respect to the timing and exact nature of the proposed therapeutic strategy. In this perspective article, we will briefly introduce the current view of the dual nature of Toll-like receptor (TLR) 2 and TLR4-driven inflammation in AD and highlight why we believe that modulating the inflammatory status within a defined time window might provide a better therapeutic concept than globally supressing it for a prolonged time.
Molecular studies as well as neuropathological studies of mouse tissue and post-mortem patient brains have provided striking evidence for a correlation between inflammation and AD. In addition, clinical studies have proposed that individuals with high levels of the inflammatory markers have an increased risk of developing dementia (Dziedzic, 2006). However, the exact nature of the relationship between AD and inflammation is still not well understood. While some studies claim that inflammation contributes to disease development and progression, others suggest that it is merely a result of ongoing neurodegeneration.
This controversy has growing relevance given the increase in anti-inflammatory drugs being proposed for managing complex neurological disorders, such as AD. In the context of dementia and AD, non-steroidal anti-inflammatory drugs (NSAIDs) have been trialed as a potential treatment option. However, despite promising initial epidemiological data, meta-analyses and clinical trials showed little to no therapeutic benefits and an increased frequency of adverse health effects. We postulate that this lack of robust effects is likely to be caused by an inappropriate timing of the intervention. Notably, most studies have investigated the impact of NSAIDS on the elderly when a more targeted approach within a finite time window may have proven more successful. We suggest that anti-inflammatory drugs should be administered before the chronic phase and diagnosis of AD through clinical presentation (Figure 1A). This could be achieved by using advanced neuroimaging technology to provide direct measurement of brain inflammation (e.g., ultra-high field imaging coupled with iron oxide nanoparticle injection), or an indirect measurement via blood-brain barrier permeability.
Figure 1.
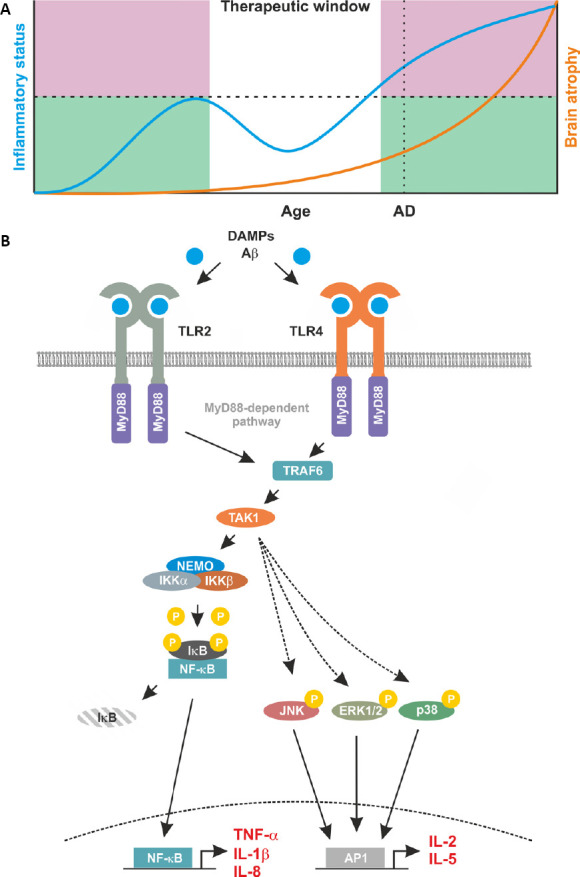
Proposed relationship between inflammation and brain atrophy and proinflammatory signaling induced by TLR2 and TLR4.
(A) A proposed temporal schematic of inflammatory status and disease progression in Alzheimer’s disease (AD). This profile indicates the progressive nature of a degeneration disease with age as indicated by brain atrophy (orange line, right axis), AD highlights a likely diagnosis of AD through clinical presentation. Alongside brain atrophy, inflammatory status is mapped (blue line, left axis). Here, an initial increase in inflammatory status is observed to counteract disease related damage. This increase is curtailed by the immune system and represents the ‘good’ inflammation (green shaded area). However, in patients, a secondary increase is observed. This escalates unchecked by the immune system and subsequently contributes to the degenerative processes. This ‘bad’ inflammation is indicated by the pink shaded area. This leaves a discrete therapeutic window, in which targeting the immune response may be beneficial for neurodegenerative diseases such as AD. (B) Proinflammatory feed forward loops mediated by TLR2 and TLR4. In AD, Aβ and DAMPs bind to and activate TLR2 and TLR4. Membrane-bound TLR2 and TLR4 recruit MyD88 and TRAF6 into to complex. This leads to the activation of TAK1 and in turn of the IKK complex composed of IKKα, IKKβ and NEMO. Once activated by upstream signals, IKKβ phosphorylates inhibitory IκB proteins thereby mediating their proteasomal degradation. Degradation of IκB liberates the nuclear localisation signals of DNA binding NF-κB subunits leading to their nuclear translocation and transcription of the pro-inflammatory target genes including TNF-α, IL-1β and IL-8. Once secreted, these proteins bind to their respective receptors further increasing NF-κB activity. This pro-inflammatory loop also involves a TAK1-mediated activation of the kinases JNK, ERK1/2, and p38. This results in activity of the transcription factor AP1 with additional pro-inflammatory targets including IL-2, and IL-5.
From neurobiochemical point of view, acute and chronic neuroinflammation is often attributed to an activation of TLRs. TLRs are type I integral membrane receptors composed of an extracellular domain, a transmembrane domain, and a cytoplasmic domain where the extracellular N-terminal end contains a leucine-rich binding domain for ligands.
The TLR family is involved in innate immune responses and it oversees the recognition of a broad range of pathogen-derived molecules (pathogen associated molecular patterns). In addition, TLRs also respond to a variety of other molecules that indicate tissue damage (danger-associated molecular patterns) including serum amyloid A and Aβ. Activation of TLRs via both danger-associated molecular patterns and pathogen associated molecular patterns culminates in activation of the prototypic, pro-inflammatory transcription factor nuclear factor-κB (NF-κB) and induces local inflammation. Notably, many target genes of NF-κB including tumour necrosis factor alpha, interleukin 1 beta (IL-1β) and IL-8 are mediators of inflammation thereby generating an inflammatory feed forward loop (Figure 1B). In addition, TLR-signaling also involves activation of c-Jun N-terminal kinases (JNK), extracellular signal-regulated kinases 1/2 (ERK1/2), and p38 that in turn activate activator protein 1 (AP1) with additional mediators of inflammation as target genes including but not limited to IL-2 and IL-5. Expression of TLRs is not limited to immune-competent cells with many TLRs expressed in neurons, microglia, astrocytes, and oligodendrocytes (Bsibsi et al., 2002). Importantly, microglia have been reported to express a wide repertoire of TLRs with TLR2 and TLR4 being expressed constitutively and at high levels. This is pertinent to the study of AD given the increasing evidence for a pivotal role of microglia in disease initiation, progression, and maintenance (Landreth and Reed-Geaghan, 2009). This microglia-mediated, and NF-κB-dependent regulation of neuronal homeostasis can at least partly be attributed to TLR2 and TLR4 (Kielian, 2006). In the context of brain homeostasis, physiological activity of microglia can induce NF-κB activation and this can contribute to neuroprotection (Kaltschmidt et al., 2005). Importantly, subtle changes in NF-κB activity can result in neurodegeneration (Kaltschmidt et al., 2005).
Thus, both the strength of the inflammatory molecular response and the contribution of individual cellular players evidently plays an important role in neuronal cell death.
Biochemical and genetic in vivo approaches alongside a multitude of in vitro studies provided strong evidence that Aβ can activate NF-κB and drive neuroinflammation in a TLR2 and TLR4 dependent manner (Song et al., 2011; Liu et al., 2012). It has been reported that TLR2 is expressed at mRNA and protein levels in both animal models of dementia and human samples, although these observations are contested. These discrepancies may indicate a coordinated timeline of TLR2 expression and activation from initiation to symptomatic presentation of AD. Functional studies have demonstrated that in vitro activation of TLR2 increases microglial uptake of exogenous Aβ. This contrasts with other reports indicating that inhibition of TLR2 activation in vitro and in vivo suppresses amyloid pathology. The in vivo datasets, suggestive of a beneficial TLR2 contribution, arise from work using mice that overexpress amyloid precursor protein and presenilin 1 (APP/PS1). These data highlight an impact of a 7-month intervention with an anti-TLR2 antibody administered prior to cognitive impairment in this model. Follow up studies using the same AD model and a TLR2–/– mouse revealed that cognitive decline is accelerated in the TLR2–/–/APP-PS1 mice compared to the APP/PS-1 mice. These deficits could be restored by viral induction of TLR2 expression prior to the cognitive decline (circa 3 months). Interestingly, TLR2-dependent JNK/NF-κB signaling has been linked to the inflammatory response to Aβ. However, singling out TLR2 as the main protagonist would be wrong as all TLRs can contribute to NF-κB signaling in a complementary manner.
Animal studies using transgenic AD mice coupled with modulation of TLR4 activity have reported a multitude of outcomes. TLR4 ablation through genetic manipulation led to enhanced Aβ deposition and worsening cognitive outcomes compared to control animals. Conversely, long-term mild stimulation of TLR4 via lipopolysaccharide administration for 3 weeks prior to onset of neurological deficits, improved cognitive performance in a tau mouse model of AD. This contrasts with studies where in vitro activation of TLR4 reduced microglial Aβ clearance through CD36 activity as well as with the increased expression of TLR4 in post-mortem tissue from AD patients. Collectively, the roles of TLR2/TLR4 are likely to be dynamically regulated and have considerable impact (both positive and negative) on cognitive outcomes. In this respect, TLR4 activation would be a favourable target in the acute phase in contrast to the chronic situation (Huang et al., 2017). Notably, signaling via TLRs is known to be a subject of considerable crosstalk (Tan et al., 2014) including a cross-coupling between TLR2 and TLR4. Thus, a similar strategy could be applied in modulating the activity of TLR2 signaling. Briefly, such therapeutic approach could involve activating TLR2 and TLR4 in the acute phase whilst suppressing it in the chronic phase.
As surface receptors, both TLR2 and TLR4 represent promising and easily accessible drug targets. This has been explored in a multitude of preclinical models of AD. Repeated systemic injections of the biased agonist of TLR4 monophosphorylic lipid A in the APPswe/PS1 mouse model of AD resulted in a significant reduction in Aβ load in the brain of and enhanced cognitive function. In addition, TAK-242, a specific inhibitor of TLR4 signaling, showed similar beneficial effects in the same mouse model. In addition to these well characterized drug candidates, several compounds believed to inhibit TLR4 signaling including α-mangostin, geraniin, osmotin, and berberine showed promising outcomes in lipopolysaccharide induced mouse models of AD. However, the exact mode of action of these compounds is still to be clarified. In contrast to TLR4, there is no specific chemical inhibitor of TLR2 signaling. Nevertheless, intranasal administration of a peptide blocking the TLR2-interacting domain of MyD88 resulted in a reduced microglial activation and improved memory and learning in a 5XFAD mouse model of AD.
Overall, a temporal profile for AD related inflammation is emerging that precedes the onset of clinical symptoms. For the early preclinical phase, our knowledge is limited and largely comes from animal studies. However, clinical imaging has begun to reveal some insights into human pathology. Late stage inflammation is predominantly associated with plaque deposition, whereby the hallmark Aβ plaques are sites of recurrent inflammation including an array of cellular phenotypes and associated inflammatory markers including high and constitutive activity of NF-κB.
Summary and conclusions: In summary, the research field of inflammation is rapidly expanding in the context of neurodegenerative diseases. The current evidence presents snapshots in time of the cellular players and their production of inflammatory biomarkers. This is an underestimation of the complexities of inflammatory processes within the brain involved in development and progression of neurodegenerative disorders that might have resulted in the failure of NSAID based prevention and treatment strategies in AD. We postulate that inflammation has an intrinsically dual role in AD. In the acute phase, TLR2 and TLR4-mediated inflammation in the brain represents a self-defence mechanism which affords protection. Therefore, a global inhibition of inflammatory signaling as a preventive measure during the acute phase might not be only ineffective but could even counteract regenerative processes. In contrast, uncontrolled and continuous high levels of inflammation resulting from the feed-forward inflammatory loop mediated by TLR2 and TLR4 are neurodegenerative and causative in the disease progression. Together this creates a therapeutic window aiming to prevent the switch from the acute to the chronic phase before clinical presentation of AD to slow down disease progression. The challenge for the Alzheimer’s research community now will be to accurately identify this window in a clinically relevant patient population.
Additional file: Open peer review report 1 (83.9KB, pdf) .
Footnotes
P-Reviewer: Coelho-Aguiar JM; C-Editors: Zhao M, Li JY; T-Editor: Jia Y
Copyright license agreement: The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Juliana M Coelho-Aguiar, Universidade Federal do Rio de Janeiro, Brazil.
References
- 1.Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61:1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- 2.Dziedzic T. Systemic inflammatory markers and risk of dementia. Am J Alzheimers Dis Other Demen. 2006;21:258–262. doi: 10.1177/1533317506289260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang NQ, Jin H, Zhou SY, Shi JS, Jin F. TLR4 is a link between diabetes and Alzheimer’s disease. Behav Brain Res. 2017;316:234–244. doi: 10.1016/j.bbr.2016.08.047. [DOI] [PubMed] [Google Scholar]
- 4.Kaltschmidt B, Widera D, Kaltschmidt C. Signaling via NF-kappaB in the nervous system. Biochim Biophys Acta. 2005;1745:287–299. doi: 10.1016/j.bbamcr.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 5.Kielian T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J Neurosci Res. 2006;83:711–730. doi: 10.1002/jnr.20767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Landreth GE, Reed-Geaghan EG. Toll-like receptors in Alzheimer’s disease. Curr Top Microbiol Immunol. 2009;336:137–153. doi: 10.1007/978-3-642-00549-7_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B, Rube CE, Walter J, Heneka MT, Hartmann T, Menger MD, Fassbender K. TLR2 is a primary receptor for Alzheimer’s amyloid beta peptide to trigger neuroinflammatory activation. J Immunol. 2012;188:1098–1107. doi: 10.4049/jimmunol.1101121. [DOI] [PubMed] [Google Scholar]
- 8.Song M, Jin J, Lim JE, Kou J, Pattanayak A, Rehman JA, Kim HD, Tahara K, Lalonde R, Fukuchi K. TLR4 mutation reduces microglial activation, increases Abeta deposits and exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. J Neuroinflammation. 2011;8:92. doi: 10.1186/1742-2094-8-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tan RS, Ho B, Leung BP, Ding JL. TLR cross-talk confers specificity to innate immunity. Int Rev Immunol. 2014;33:443–453. doi: 10.3109/08830185.2014.921164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.