

Abstract
Next-generation genomic sequencing has identified multiple novel molecular alterations in cancer. Since the identification of DNA methylation and histone modification, it has become evident that genes encoding epigenetic modifiers that locally and globally regulate gene expression play a crucial role in normal development and cancer progression. The histone methyltransferase enhancer of zeste homolog 2 (EZH2) is the enzymatic catalytic subunit of the polycomb repressive complex 2 (PRC2) that can alter gene expression by trimethylating lysine 27 on histone 3 (H3K27). EZH2 is involved in global transcriptional repression, mainly targeting tumor suppressor genes. EZH2 is commonly overexpressed in cancer and shows activating mutations in subtypes of lymphoma. Extensive studies have uncovered an important role for EZH2 in cancer progression and have suggested that it may be a useful therapeutic target. In addition, tumors harboring mutations in other epigenetic genes such as ARID1A, KDM6, and BAP1 are highly sensitive to EZH2 inhibition, thus increasing its potential as a therapeutic target. Recent studies also suggest that inhibition of EZH2 enhances the response to tumor immunotherapy. Many small molecule inhibitors have been developed to target EZH2 or the PRC2 complex, with some of these inhibitors now in early clinical trials reporting clinical responses with acceptable tolerability. In this review, we highlight the recent advances in targeting EZH2, its successes and potential limitations, and we discuss the future directions of this therapeutic subclass.
Epigenetic regulators in cancer
The field of cancer epigenetics has recently gained considerable interest due to a greater appreciation of the role of epigenetic genes in the progression of cancer, as well as our increasing ability to pharmacologically target these gene products. The term epigenetics was characterized by Conrad Hal Waddington in 1942, who described epigenetics as the heritable changes in phenotype without genotype alterations (1, 2). While initially applied only to “heritable” changes, this definition has loosened over time to include the study of all modifications of chromatin or DNA that affect gene transcription independent of mutations in the genetic sequence.
Chromatin, the macro complex of DNA and histone proteins, is generally categorized into hetero- and euchromatin. Heterochromatin (aka restrictive chromatin) is the highly condensed form which prevents active transcription, whereas euchromatin (aka permissive chromatin) is open in configuration and amenable to active transcription. The functional unit is the nucleosome, which is composed of a histone octamer (two copies of each H2A, H2B, H3, and H4 proteins) with 145–147 base pairs of DNA wrapped around it. Chromatin/nucleosomes can be modified by chromatin-remodeling complexes, resulting in changes in gene accessibility at promoters and enhancers, and resultant transcriptional downstream effects (3–9). These complexes include the switch/sucrose non-fermentable (SWI/SNF) complex and the chromodomain helicase DNA-binding (CHD) protein family, and are frequently mutated in human cancers both at the germline as well as somatic level. The SWI/SNF complex is composed of a central ATPase (BRG1 or BRM) as well as other proteins termed BRG1/BRM associated factors (BAFs) that are necessary for DNA and protein interactions (ARID1A, ARID1B, ARID2, PBRM1, SMARCD1, SMARCE1). Many of these genes functionally interact with EZH2 (described in detail later).
DNA methylation is another mechanism of epigenetic regulation. Human cancers often exhibit abnormal methylation patterns with promoter hypermethylation leading to gene suppression (in tumor suppressor genes) and genome-wide hypomethylation resulting in instability and activation of oncogenes (10). DNMT1, DNMT3A and DNMT3B are examples of DNA methyltransferases, and DNA demethylases include TET1, TET2 and TET3. Mutations in all of these genes have been identified in various human cancers (11).
Histone modifying enzymes in cancer
In addition to chromatin remodeling complexes and DNA methyltransferases/demethylases, chromatin structure and function can be regulated by histone modifying enzymes (Supplementary Figure 1). These enzymes catalyze a variety of post-translational modifications including methylation, acetylation, phosphorylation, ubiquitination, and sumoylation (12). Both loss-of-function and gain-of-function mutations have been described in genes encoding for histone acetyltransferases/deacetylases and histone methyltransferases/demethylases.
Over 30 histone lysine acetyltransferases (HAT) are known. Truncating (inactivating) mutations in p300 and CBP (CREB-binding protein) occur frequently in hematological malignancies like diffuse large B-cell lymphoma or acute myeloid leukemia. As histone acetylation generally leads to more open chromatin and active transcription, loss of acetyltransferase activity is associated with general gene repression, which also involve many tumor suppressor genes. Histone deacetylases (HDAC) on the other hand are frequently overexpressed in cancers and facilitate removal of histone acetyl groups and transcriptional repression of tumor suppressor genes (13–15). Numerous HDAC inhibitors including belinostat, panobinostat, and vorinostat have been approved as anti-cancer therapeutics in a variety of hematological malignancies (16–18). Importantly, histone acetylation and methylation can be functionally competitive and antagonistic as described below.
Histone methylation occurs on lysine and arginine residues in mono-, di-, and trimers with trimethylation generally regarded as the most mechanistically effective mark. Lysine methyltransferases (KMTs) or protein arginine methyltransferases (PRMTs) are the responsible enzyme classes and use S-adenosyl-L-methionine (SAM) as the methyl donor. Methylation of different amino acid residues on histone 3 are associated with distinct transcriptional effects. H3K4me2/3 is generally linked to transcriptional activation, whereas H3K9me2/3 and H3K27me2/3 are associated with transcriptional repression (19, 20). Importantly, histone methylation may prevent other marks such as acetylation, with resultant additive functional antagonism. Forty-nine histone methyltransferases are currently known in the human genome, and include the H3K4 methyltransferase MLL and the H3K27 methyltransferase EZH2 (4, 21). Thus, the global and focal epigenetic transcriptional output depends on the end sum of the various activities of these various epigenetic modifiers (in addition to transcriptional activators and co-activators). It is critical to note, however, that the mechanisms that determine genetic locus specificity to these modifications are an area of intense active investigation and are likely to vary by cellular context.
Aberration of EZH2 signaling in cancer
EZH2 is overexpressed in numerous tumor entities including melanoma, ovarian, breast, endometrial, bladder, renal cell, lung, and liver cancer, and is associated with aggressive disease, leading to its classification as an oncogene (22–31). EZH2 overexpression leads to increases in H3K27me3 with repression of tumor suppressor genes as well as genes which drive cellular differentiation including p16 and E-cadherin (among numerous others)(32). In prostate cancer, EZH2 is significantly overexpressed in metastatic disease compared to localized cancer and benign prostatic tissue at both the transcript and protein levels. Furthermore, EZH2 plays a role in prostate cancer cell proliferation and depending on its phosphorylation status acts as a coactivator for transcription factors like the androgen receptor. Interestingly, this effect is independent of its function as a transcriptional repressor (33, 34).
The mechanisms by which EZH2 is overexpressed (or activated) in cancer have been carefully evaluated and include transcriptional and post-transcriptional mechanisms. Critical cancer-related transcription factors have been shown to activate EZH2 transcription including E2F, ELK1, HIF1α and NF-κB (35–38). Furthermore EZH2 is regulated by different miRNAs at the post-transcriptional level. miR-101 and miR-26a have been shown to decrease EZH2 expression and therefore downregulation of these miRNAs leads to EZH2 overexpression in different cancer types (39, 40). Finally, the activity of EZH2 has been shown to be inhibited by AKT-mediated phosphorylation, which can be repressed by MYC-mediated PTEN upregulation and resultant PI3K/AKT pathway inhibition (41) (Figure 1).
Figure 1. Aberration of EZH2 signaling in cancer.
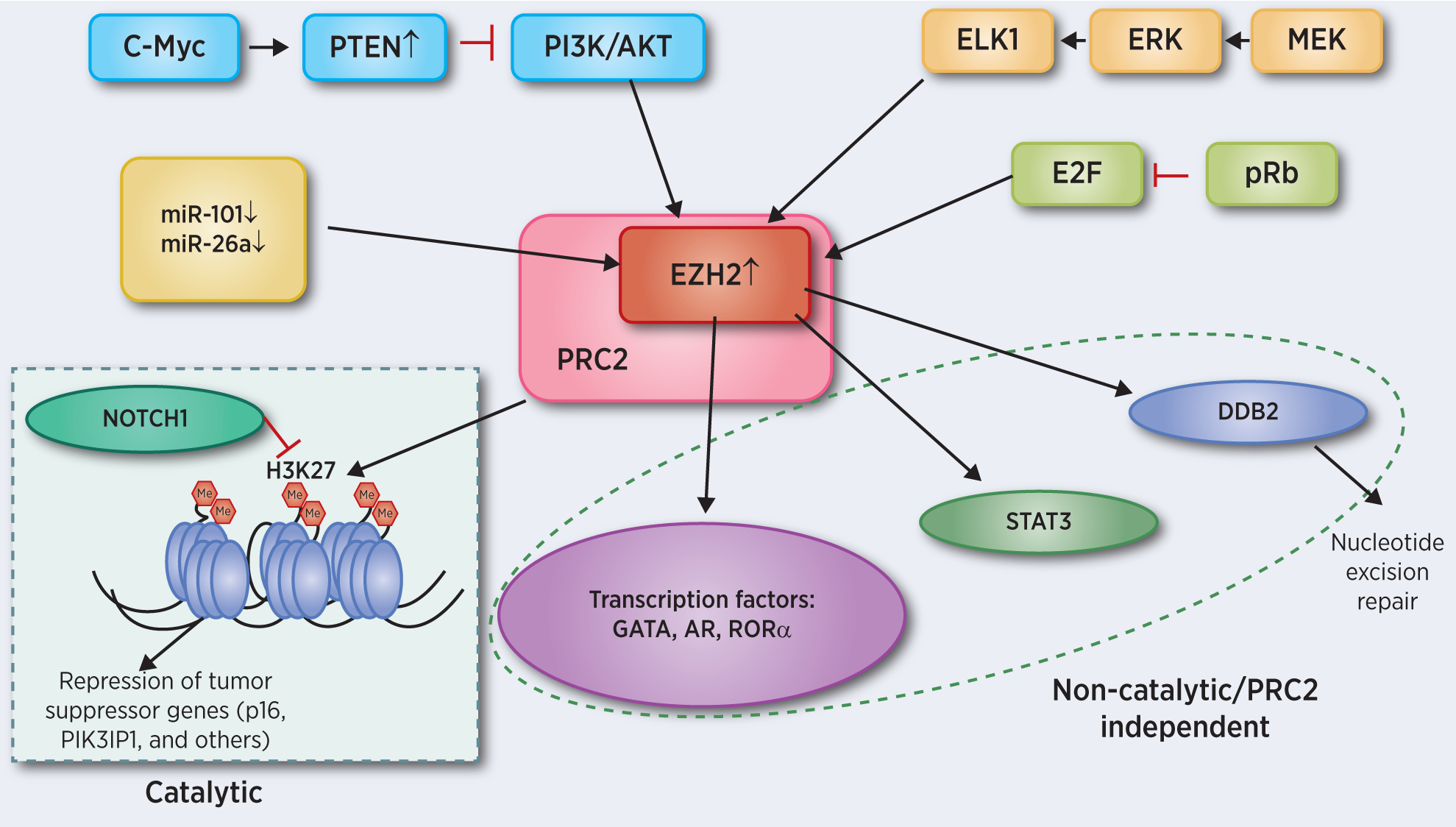
EZH2 transcription is activated by transcription factors like E2F or ELK1. On the post-transcriptional level miR-101 and miR-26a have been shown to decrease EZH2 and therefore their down regulation also leads to EZH2 overexpression. Activity of EZH2 is regulated by AKT-mediated phosphorylation. C-myc leads to PTEN upregulation inhibiting the PI3K/AKT signaling pathway resulting a decreased inhibition of EZH2. Increased activity of EZH2 then leads to upregulated H3K27 methylation resulting in repression of tumor suppressor genes. This catalytic function is antagonized by NOTCH1. Non-catalytic or PRC2-independent roles for EZH2 include enhancing STAT3 activity, interaction with transcription factors, or stabilization of DDB promoting nucleotide excision repair.
In a subset of hematological malignancies on the other hand, EZH2 is predominantly altered by somatic genetic mutation leading to either gain of function (mostly in lymphomas) or loss of function (predominantly in myeloid malignancies). Gain of function mutations have been associated with downregulation of tumor suppressor and differentiation genes (42). Seven percent of follicular lymphomas and 22% of the diffuse large B-cell lymphomas of germinal-center origin harbor a somatic point mutation in the SET domain of EZH2 (mainly Y641) leading to enhanced H3K27me3 levels and silencing of genes important for B-cell differentiation and cell cycle control (43, 44).
Loss of function mutations in EZH2 occur in 6% of myelodysplastic syndromes, 3–13% of myeloproliferative neoplasms, and 25% of T-cell acute lymphatic leukemia suggesting a unique tumor suppressor role for EZH2 in this subset of hematologic malignancies (45–47). As NOTCH1 is a main driver of this malignancy, NOTCH1 was found to antagonize the methyltransferase function of EZH2, and inhibition of EZH2 in the setting of NOTCH1 activation resulted in increased tumor progression (47). Studies are ongoing to better understand why EZH2 functions as a tumor suppressor in the setting of T-cells. Perhaps T-cells harbor a unique H3K27 repressive gene profile due to a unique epigenome, or the downstream signaling effects in T-cells are fundamentally different. Regardless, these possible oncogenic effects of EZH2 inhibition in T-cells will need to be carefully considered in clinical trials. In myelodysplastic syndromes RUNX1, a transcription factor critical for hematopoiesis, is frequently mutated (10–20%) and its mutations are significantly associated with −7/7q− chromosome anomalies. The EZH2 gene is located at 7q36, thus the RUNX1 alterations can lead to EZH2 loss of function (48, 49). In mouse models, EZH2 loss significantly promotes the transformation of hematopoietic stem cells expressing the RUNX1S291fs mutant into myelodysplastic cells (50). These findings suggest that EZH2 may act as an oncogene in most solid tumors and lymphomas, while acting as a tumor suppressor in limited minority of T-cell leukemias and myelodysplastic syndromes.
Targeting EZH2 in cancer
PRC2 is a complex composed of either EZH1 or 2, EED (embryonic ectoderm development), SUZ12 (suppressor of zeste 12), and RbAP46/48 (retinoblastoma binding protein 46/48). The PRC2 complex catalyzes the methylation of H3K27 leading to chromatin compaction and gene repression (51). The mammalian genome encodes for two different EZH orthologs, EZH1 and EZH2. They are the SET domain (Su(var)3–9, enhancer of zeste, trithorax) containing subunits and therefore responsible for the catalytic methyltransferase activity. EZH1/2 are in an inactive state unless associated with EED and SUZ12 (52). The comparative methyltransferase activities of EZH1 and 2 have been intensely studied with the possibility of functional redundancy and compensation in mind. In embryonic stem cells EZH2 knock down results in reduced but not complete absence of global H3K27me3. In addition, inhibition of EZH2 via small molecule inhibitors diminishes H3K27me3 to a great extent, but residual tri-methylation can still be detected (53). Knockdown of EZH1 enzyme in EZH2 null cells results in elimination of any residual tri-methylation due to EZH1, showing a role for EZH1 as well in H3K27 tri-methylation (54). On the other hand, PRC2-EZH1 shows a lower histone methyltransferase activity and its knockdown alone does not lead to global reduction in of H3K27 methylation (55). Finally, EZH1 is present in dividing and differentiated cells, while EZH2 is only found in actively proliferating tissues (56). The preponderance of the evidence suggests that EZH2 is the predominant H3K27 methyltransferase in malignant cells, and EZH2-specific inhibitors have been developed with this in mind. However, the residual H3K27me3 after EZH2 inhibition provides a rationale for pursuing dual EZH2 and EZH1 inhibition.
How exactly PRC2 targets certain genes and not others is an area of active investigation. PRC2 seems to be present on CpG island promoters of untranscribed genes (57). Several groups have tried to identify proteins modulating PCR2 function and gene targeting. PHF1, MTF2, PHF19, Polycomb-like (PCL) 1, −2 and −3, PALI1 (C10ORF12), EPOP (C17ORF96), JARID2, and AEBP2 are some of these proteins. However further research is needed to illuminate these complex processes (58, 59).
Furthermore, non-catalytic and PRC-independent roles for EZH2 have been described. EZH2 interacts with a variety of transcription factors including GATA4, RORα and androgen receptor in a PRC2-independent manner (34, 60, 61). EZH2 can directly methylate the NKT cell lineage defining transcription factor PLZF.(62) In stem-like glioblastoma multiforme cells phosphorylation of EZH2 at serine 21 (pS21 EZH2) by AKT signaling facilitates methylation of STAT3 by EZH2, which enhances STAT3 activity (63). In small cell lung cancer (SCLC) EZH2 stabilizes DDB2 and promotes nucleotide excision repair. Consistent with this, EZH2 depletion but not catalytic inhibition sensitizes SCLC cells to cisplatin (64). In breast cancer cells EZH2 interacts with the RelA/RelB complex co-regulating a subset of NF-κB targets increasing the aggressiveness of breast cancer cells (65). In triple negative breast cancer (TNBC), EZH2 protein binds to p38 MAP kinase leading to accumulation in the cytoplasm of TNBC cells. In breast cancer tissue samples, cytoplasmic EZH2 phosphorylated at Thr 367 (pT327 EZH2) was associated with low H3K27me3 levels and a triple negative phenotype. The pT367 EZH2 protein binds to cytoplasmic regulators of cell migration and invasion, and promotes metastasis (66). Taken together, a variety of non-catalytic functions of EZH2 have been described, but many are still not completely characterized. These non-catalytic functions support the rationale of identifying EZH2 small molecule inhibitors which function by inhibiting EZH2 protein interactions, without necessarily inhibiting catalytic activity.
Several EZH2 inhibitors have been developed within the last decade. 3-Deazaneplanocin A (DZNep) is an S-adenosylhomocysteine (SAH) hydrolase inhibitor leading to accumulation of SAH. SAH is the byproduct after methyl transfer from S-adenosyl-methionine (SAM), and buildup of this byproduct inhibits further SAM-mediated methyl transfer. DZNep induces cell cycle genes and induces apoptosis in primary AML cells (67). It also depletes EZH2 protein levels which results in a global decrease in H3K27me3. However, because other methyltransferases are dependent on SAM as a methyl donor, it also decreases other histone methylation marks including H3K4me3, H3K9me1/2/3, H3K36me3, H3K79me3, H3R2me2, and H4K20me3, and is thus not specific for EZH2 (68, 69). Other more specific inhibitors including GSK126 and EI1 have been tested in cell culture or xenografts. GSK126, a SAM-competitive, small molecule inhibitor with a Ki value of ~0.5 nM, was shown to be more than 1,000-fold more selective for EZH2 versus 20 other human methyltransferases (70, 71). GSK126 inhibits proliferation of multiple myeloma cells lines, lymphoma cells, and gastric cancer cells (among others).(72–74) GSK126 also suppresses anti-tumor immunity by increasing myeloid-derived suppressor cells and decreasing CD4+ and INF-γ+CD8+ cells (75). In mouse breast cancer models of BRCA1-deficient mice, GSK126 combination with cisplatin shows decreased cell proliferation and improves survival (76). EI1 is also an SAM-competitive inhibitor, which decreases H3K27 methylation without affecting other histone H3 methylation marks. Diffuse large B-cell lymphoma cells harboring the Y641 activating EZH2 mutation showed decreased proliferation, cell cycle arrest, and apoptosis upon treatment with EI1 (77). Tazemetostat (EPZ-6438) another SAM-competitive inhibitor shows a good oral bioavailability (78). It inhibits H3K27 methylation in EZH2 wild type and mutant lymphoma cells (79). Other EZH2 SAM-competitive inhibitors including PF-06821497 (80), SHR2554, and CPI-1205 are currently in clinical trials. The latter has displayed potent antitumor effects in preclinical models (81).
In an attempt to identify small molecules which inhibit PRC2 through a different mechanism, protein-protein interaction inhibitors have been developed. MAK683 binds to the domain of EED which interacts with H3K27me3 thus disrupting the interaction with EZH2 and decreasing methyltransferase activity. This inhibitor is currently under investigation in a phase I/II trial of patients with advanced malignancies including nasopharyngeal carcinoma, gastric cancer, ovarian cancer, prostate cancer, sarcoma and diffuse large B-cell lymphoma (clinicaltrials.gov NCT02900651).
Furthermore, dual EZH1 and EZH2 inhibitors have been developed. UNC1999 is an orally bio-available EZH1/2 inhibitor with a 10-fold selective towards EZH2 (compared to >150 fold with GSK126). In MLL-rearranged leukemia it suppresses tumor growth (82, 83). Valemetostat Tosylate (DS-3201b) is another EZH1/2 dual inhibitor which has demonstrated synthetic lethality in malignancies overexpressing EZH2 or harboring mutations to histone-modifying genes in preclinical models (53, 84). (Supplementary Figure 2)
EZH2 and synthetic lethality
Synthetic lethality describes the relationship between two genes or pathways in which loss of either one alone causes no or minimal phenotype, but loss of both leads to cell death. As loss of function defects in tumor suppressor genes are difficult to therapeutically target, investigators have sought to identify biologic vulnerabilities conferred by inactive tumor suppressors that could in turn be targeted through a synthetic lethality approach.
The SWI/SNF chromatin-remodeling complex modulates transcription and is essential for differentiation, proliferation and DNA repair. Loss of function mutations in nearly every component of this 12–15 protein complex have been identified across numerous cancers (85). In fact, mutations in subunits of the SWI/SNF complex occur in 20% of all human cancers, rivaling the prevalence of other tumor suppressors such as p53 (86). The functionally antagonistic roles of the SWI/SNF complex and PRC2 are well known and evolutionarily conserved. Thus, targeting PRC2 complexes in SWI/SNF mutant cancer cells could result in synthetic lethality (Supplementary Figure 3). This concept was first tested in SMARCB1 (aka SNF5 or INI1) deficient tumors, including pediatric rhabdoid tumors. Genetic loss of SMARCB1 leads to highly penetrant rhabdoid tumors and lymphomas in mice, and this phenotype can be rescued by genetic or pharmacologic inhibition of EZH2 (78, 87). These studies have led to the current phase I trial investigating the EZH2 inhibitor tazemetostat in pediatric rhabdoid tumors with SMARCB1 mutations. (clinicaltrials.gov NCT02601937).
Mutations in other subunits of the SWI/SNF complex have been found to confer sensitivity to EZH2 inhibitors as well. Notably, ARID1A is the DNA binding component of the SWI/SNF complex, and is the most frequently mutated protein in the complex. In fact, it is the most commonly mutated epigenetic gene in all of human cancer with nearly 50% of ovarian clear cell carcinoma (OCCC), 20% of gastric cancer, 20% of bladder cancer, 14% of hepatocellular, 12% of melanoma, and 10% of colorectal cancers harboring inactivating ARID1A mutations (88). Notably, OCCC cells in culture that are normally resistant to EZH2 inhibition with GSK-126, become sensitive upon ARID1A knockdown (89). These cells’ dependence on EZH2 was found to be related to EZH2-mediated transcriptional repression of PIK3IP1 (which decreases proliferation and promotes apoptosis by inhibiting PI3K-AKT signaling) (89). In addition, a potential resistance mechanism was found in cells harboring concomitant activating mutations in the Ras pathway (90).
Furthermore, mutations in BRG1 (SMARCA4), an ATPase subunit of SWI/SNF, have been shown to confer sensitivity to EZH2 and Topo II inhibition in lung cancer cell lines (90). All of these preclinical data have led to the phase 1 and 2 investigations of EZH2 inhibitors (Tazemetostat) in solid tumors harboring somatic mutations in SWI/SNF components (91). Kadoch et al have offered convincing molecular data describing the mechanisms of this process And showed that SWI/SNF machinery actively evicts PRC2 and EZH2 via its ATPase activity, thus decreasing the H3K27Me3 mark (presumably through histone turnover and competing demethylase activity) to allow active transcription (92). Thus, mutations in SWI/SNF members allow uninhibited PRC2-mediated gene silencing, which in cancer cells may predominantly consist of tumor suppressors (but may also orchestrate tumor immunity as described below). Inhibition of EZH2 then allows accumulation of tumor suppressors and cell death. Of course, the downstream transcriptional effects of this interplay are likely highly context dependent and can be affected by the presence and activity of demethylases, nucleosome turnover, and functionally redundant histone methyltransferases.
The histone lysine demethylase KDM6A (aka UTX) directly antagonizes EZH2/PRC2 by removing methyl marks on H3K27. Thus, it would be hypothesized to act as a tumor suppressor, to be mutated/inactivated in tumors cells, and to confer sensitivity to EZH2 inhibitors in its absence. KDM6A is found to harbor mutations in multiple tumor types including bladder cancer, multiple myeloma, pancreatic, and esophageal cancers (93, 94). In multiple myeloma, cell lines harboring mutations in KDM6A are exquisitely sensitive to EZH2 inhibition, while wild-type KDM6A cells are not. Importantly, KDM6A knockdown in wild type cells or KDM6A reconstitution in KDM6A mutant cells can alter EZH2 inhibitor sensitivity, both in vitro and in mouse xenografts (95). Similar findings were identified in bladder cancer cells.(96)
Finally, mesothelioma cells lacking the H2aK119Ub deubiquitinase BAP1 are sensitive to EZH2 pharmacologic inhibition (97). The authors suggest that this is due to upregulation of EZH2 transcription via BAP1-mediated deubiquitination of a chromatin modulator that regulates the levels of H4K20me1 at the EZH2 locus.
Taking all the data above together, it is evident that many different cancer cell types are dependent on a dysregulation of one or more components of the epigenetic machinery, which in turn leads to dependence on PRC2/EZH2 activity that can be targeted pharmacologically via a synthetic lethality approach.
EZH2 and immunotherapy combination
In addition to functioning as an oncogene in tumor cells, EZH2 is also critical in coordinating the immune response to the tumor via transcriptional modulation both in tumor cells and immune cells.
EZH2 immunomodulation in tumor cells
As EZH2 is a master epigenetic regulator of transcription, its effects are dependent upon the transcriptional program that it modulates, which can be highly context- and cell type-dependent. In cancer cells, not only does EZH2 work to repress the expression of tumor suppressor genes as described above, but can also modulate the expression of genes involved in orchestrating the tumor immune microenvironment (98). Peng et al determined that EZH2-mediated transcriptional silencing of the cytokines CXCL9 and CXCL10 prevents efficient T-cell trafficking in ovarian cancer cell xenograft models, and can be reversed by EZH2 inhibition in combination with anti-PD-L1 checkpoint blockade (99). Furthermore, ovarian cancer patient outcomes negatively correlate with EZH2 expression and tumor infiltrating CD8+ T cells. Similar findings have been described for colon cancer (100). The same group recently discovered that ARID1A mutations result in higher EZH2-dependent immunosilencing via CXCL9/10, which is resistant to PD-L1 blockade in colon and ovarian cancer xenograft models.(101) However, the effect of combination therapy with EZH2 inhibitors and immunotherapy was not reported. Zingg et al uncovered an interesting interplay between infiltrating tumor immune cells and melanoma cancer cell EZH2 expression (102). They showed that CD8+ T-cells in the immune infiltrate secrete TNFα, leading to upregulation of EZH2 in tumor cells. This EZH2 upregulation results in transcriptional silencing of genes involved in immunogenicity and antigen presentation, which could be reversed by EZH2 inhibition in conjunction with CTLA-4 and IL-2 immunotherapy. Importantly, since PD-L1 is downregulated in these cells, combination therapy with anti-PD-L1 produced no additive response.
EZH2 immunomodulation in immune cells
In addition to its role in cancer cells, EZH2 has been shown to play a critical role in immune cell differentiation and function, and specifically in anti-tumor immunity (98). While EZH2 has been investigated in all immune cell lineages including natural killer cells, dendritic cells, B-lymphocytes, and macrophages, its role in T-cell biology has been the most well-characterized (103). Notably, while EZH2 appears to prevent naive CD4 cells from differentiating into Th1/Th2 effector cells, it also appears to promote CD8+ effector cell survival and Treg development. Since naïve CD4 cells and Tregs generally function to repress cancer immunity, while CD8+ effector T-cells promote it, the implications of systemic EZH2 inhibition for cancer immunotherapy could be highly context dependent.
Recently, the role of EZH2 in maintaining the cancer immune-inhibitory phenotype of Tregs has been intensively studied. Goswami et al showed that EZH2 inhibition with CPI-1205 slowed bladder tumor xenograft growth in immunocompetent mice, and this was therapeutically additive with anti-CTLA4 therapy. Specifically, mice with EZH2-deficient Tregs were more sensitive to anti-CTLA4 therapy than wild-type mice, and Rag1−/− mice (lymphocyte deficient) did not respond to EZH2 inhibition (104). Similar results were found by Wang et al, who showed that Treg specific EZH2 deletion was sufficient to inhibit tumor growth in immunocompetent mice with colon, prostate, and melanoma tumors (105).
However, Zhao et al focused on EZH2 expression in CD8+ effector T-cells and found EZH2 to be necessary for their survival and anti-tumor immunity (106). In both melanoma and ovarian cancer cell models, mice with T-cells with pharmacologically or genetically inhibited EZH2 showed higher tumor burden. Furthermore, correlative studies in humans showed that patients with increased EZH2+CD8 T-cells in their tumors had favorable clinical outcomes, presumably due to a more active tumor immune microenvironment. These divergent functions of EZH2 in different T-cell subsets will need to be considered in design of any human clinical trials. Notably, there are currently at least 3 phase I clinical trials investigating the synergy of EZH2 inhibition with immunotherapies.
Ongoing clinical trials targeting EZH2 and outcomes
On January 23, 2020 the EZH2 inhibitor tazemetostat (Tazverik; Epizyme) was approved by the FDA for treatment of patients aged ≥ 16 years with metastatic or locally advanced epithelioid sarcoma not eligible for complete resection. In preclinical studies tazemetostat showed antitumor activity in cell culture and xenograft models (78, 79). The accelerated FDA-approval of tazemetostat was based on a single arm clinical trial (NCT02601950). The study included 62 patients with 9 patients (15%) showing a response with 1 (1.6%) complete response, and 8 (13%) partial responses. Of these 9 patients, 6 had responses that lasted 6 months or longer (107, 108).
The first clinical trial of tazemetostat (NCT01897571) in patients with B-cell non-Hodgkin lymphoma (21 patients), and advanced solid tumors (43 patients) conducted between June 13, 2013, and Sept 21, 2016 revealed tolerable side effects including asthenia (21 [33%]), anemia (9 [14%]), anorexia (4 [6%]), muscle spasms (9 [14%]), nausea (13 [20%]), and vomiting (6 [9%]). Durable objective responses, including complete responses, were observed in 8 (38%) of 21 patients with B-cell non-Hodgkin lymphoma and 2 (5%) of 43 patients with solid tumors. None of the patients died of treatment-related causes, 7 (11%) patients experienced non-treatment-related deaths (91).
An open-label, multicenter, phase 2 study evaluated tazemetostat in patients with relapsed or refractory diffuse large B-cell lymphoma or follicular lymphoma (Grade 1–3b) harboring either wild-type EZH2 or mutationally hyperactive EZH2. Interim data as of February 1, 2019 on the 95 follicular lymphoma patients were as follows. Forty-one patients harbored EZH2 active-mutant tumors and 54 were EZH2 wild-type. The objective response rate in the EZH2 mutant groups with 39 patients being evaluable was 74% (95% CI: 57.9, 87.0) with a complete response rate of 10%, partial response of 64% and stable disease of 26%. None of the patients showed progressive disease. The median progression-free survival was 60 weeks (95% CI: 46.7, 83.9). In patients with EZH2 wild-type tumors 53 were evaluable. The objective response rate was 34% (95% CU: 21.5, 48.3) with a complete response rate of 6%, partial response of 28% and stable disease of 30%. Twenty-eight percent of patients showed progressive disease. The median progression free survival was 24.6 weeks (95% CI: 15.1, 47.9). Response duration was greater than 24 weeks in 83% of patients, and 50% of patients had a response duration greater than one year. Treatment-emergent adverse events (TEAEs) leading to dose reductions occurred in 8% of patients and study drug discontinuation due to TEAEs occurred in 9% (109). Interim data as of May 2018 on 226 DLBCL patients were as follows. Thirty-six patient with EZH2 mutant tumors were treated with tazemetostat monotherapy. The objective response rate was 17 % (95% CI: 6, 33) with a complete response rate of 3% and a partial response rate of 14%. Progressive disease was seen in 39% of patients. The median progression free survival was 15.9 weeks (range: 0.1–84.1 weeks). In the 121 patients containing EZH2 wild-type cancers who received tazemetostat monotherapy the objective response rate was 17% (95% CI: 10, 25) with a complete response rate of 9% and a partial response rate of 7%. Progressive disease was seen in 60% of patients. The median progression free survival was 8 weeks (range: 0.1–103.9 weeks) (110). Taken together, tazemetostat demonstrated enhanced activity in lymphomas harboring EZH2 activating mutations, with acceptable tolerability.
Another Phase 2, multicenter study evaluated tazemetostat monotherapy in adults with relapsed or refractory malignant mesothelioma with BAP1 inactivation (NCT02860286). Seventy-four patients were enrolled. All had at least one prior systemic therapy. Thirty-one patients (51%) achieved disease control at 12 weeks and 15 patients (25%) sustained disease control (CR or PR+SD) at 24 weeks. Two of 61 patients had a confirmed partial response. None of the patients discontinued due to adverse events, however, 5 patients underwent dose reductions due to adverse events. The most frequently observed adverse events include fatigue (32%), decreased appetite (28%), dyspnea (28%), and nausea (27%) (111) (completed clinical trials are summarized in Supplementary Table 1).
In addition to monotherapy, tazemetostat is being evaluated in combination with other drugs including PD-L1 and PD-1 inhibitors, anti-androgens, or antibodies against CD20. One example includes a phase 1b clinical trial on patients with relapsed/refractory DLBCL treated with atezolizumab (anti-PD-L1-antibody) and tazemetostat (NCT02220842). As of August 28 2018, 43 patients were enrolled. Median progression-free survival was 1.9 months (95% CI 1.8, 2.8). Best overall response rate was 16% (complete response, n=2 [5%]; partial response, n=5 [12%]). Eight (19%) patients had stable disease; 19 (44%) had progressive disease. In 5 patients (17%) EZH2 mutations were identified. At least one adverse event occurred in 95% of patients with anemia (26%) and fatigue (23%) being the most frequent ones. Six patients (14%) had adverse events leading to discontinuation of either study drug (atezolizumab, n=3; tazemetostat, n=3). Two grade 5 adverse events occurred: sepsis, deemed unrelated to treatment; and hyponatremia, related to both study treatments (112).
Further ongoing clinical trials are summarized in Table 1. They include the evaluation of tazemetostat as a single agent in B-cell lymphomas, advanced solid tumors, malignant mesotheliomas, rhabdoid tumors, and sarcomas. Further, tazemetostat is being investigated in combination with antiandrogens, checkpoint inhibitors, chemotherapy and targeted therapeutics like rituximab. Other EZH2 inhibitors including CPI-1205, PF-06821497, SHR2554, and Valemetostat Tosylate are likewise currently being investigated in clinical trials. The preliminary data above suggest that patient selection using molecular biomarkers such as EZH2 activating mutations, and/or synthetically lethal mutations like those described above will be crucial to optimize clinical outcomes.
Table 1.
On-going clinical trials on EZH2 and EED inhibitors
| Drug(s) | Indication | Phase | Status | Identifier | Estimated Enrollment |
|---|---|---|---|---|---|
| Tazemetostat, atezolizumab, obinutuzumab | Relapsed/refractory Follicular Lymphoma and Diffuse Large B-cell Lymphoma | I | Completed | NCT02220842 | 96 participants |
| Tazemetostat | Relapsed or refractory INI1-negative tumors or Synovial Sarcoma, rhabdoid tumors, malignant rhabdoid tumor of ovary | I | Recruiting | NCT02601937 | 82 participants |
| Tazemetostat, abiraterone/ prednisone, enzalutamide | Metastatic prostate cancer | I | Recruiting | NCT04179864 | 48 participants |
| Tazemetostat | B-cell Lymphomas, Advanced Solid Tumors, Diffuse Large B-cell Lymphoma, Follicular Lymphoma, Transformed Follicular Lymphoma, Primary Mediastinal Large B-Cell Lymphoma | I/II | Active, not recruiting | NCT01897571 | 420 participants |
| Tazemetostat, pembrolizumab | Locally advanced or metastatic urothelial carcinoma, Stage III-IVB Bladder Cancer AJCC v8 | I/II | Recruiting | NCT03854474 | NCI 30 participants |
| Tazemetostat | Relapsed or refractory malignant mesothelioma,
BAP1-deficient relapsed or refractory malignant
mesothelioma |
II | Completed | NCT02860286 | 74 participants |
| Tazemetostat | Malignant Rhabdoid tumors, rhabdoid tumors of the kidney, atypica teratoid rhabdoid tumors, NI1-negative tumours, relapsed/refractory Synovial Sarcoma | II | Recruiting | NCT02601950 | 250 participants |
| Tazemetostat | Diffuse Large B-cell Lymphoma, Follicular Lymphoma, Malignant Rhabdoid Tumors (MRT), Rhabdoid Tumors of the Kidney (RTK), Atypical Teratoid Rhabdoid Tumors (ATRT), Synovial Sarcoma, Epitheliod Sarcoma, Mesothelioma, Advanced Solid Tumors | II | Recruiting | NCT02875548 | 300 participants |
| Tazemetostat | Relapsed or refractory advanced solid tumours, Non-Hodgkin’s Lymphoma or histiocytic disorders with EZH2, SMARCB1, or SMARCA4 gene mutations | II | Recruiting | NCT03213665 | 49 participants |
| Tazemetostat, doxorubicin, HCI, placebo | Advanced soft-tissue sarcoma or epitheloid sarcoma | III | Recruiting | NCT04204941 | 154 participants |
| Tazemetostat, lenalidomide, placebo, rituximab | Relapsed/refractory follicular Lymphoma | III | Recruiting | NCT04224493 | 518 participants |
| Tazemetostat | Relapsed or Refractory B-cell Non-Hodgkin’s Lymphoma | II | Active, not recruiting | NCT03456726 | 21 participants |
| Tazemetostat | Relapsed or Refractory B-cell Non-Hodgkin’s Lymphoma | I | Active, not recruiting | NCT03009344 | 6 participants |
| Tazemetostat | Hepatic Impairment Advanced Malignant Solid Tumor |
I | Recruiting | NCT04241835 | 24 participants |
| Tazemetostat and [14C] Tazemetostat | Diffuse Large B Cell Lymphoma Primary Mediastinal Lymphoma Mantle-Cell Lymphoma Follicular Lymphoma Marginal Zone Lymphoma Advanced Solid Tumors |
I | completed | NCT03010982 | 3 participants |
| Tazemetostat, Fluconazole, Omeprazole, Repaglinide | Diffuse Large B Cell Lymphoma Primary Mediastinal Lymphoma Mantle Cell Lymphoma Advanced Solid Tumor Marginal Zone Lymphoma |
I | Active, not recruiting | NCT03028103 | 28 participants |
| CPI-1205 | B-Cell Lymphoma | I | completed | NCT02395601 | 41 participants |
| CPI-1205, Enzalutamide, Abiraterone/Prednisolone | Metastatic Castration Resistant Prostate Cancer (mCRPC) | I/II | Active, not recruiting | NCT03480646 | 242 participants |
| CPI-1205, ipilimumab | Advanced Solid Tumors | I/II | Active, not recruiting | NCT03525795 | 24 participants |
| Valemetostat Tosylate | Adult T-cell Leukemia/Lymphoma | II | Recruiting | NCT04102150 | 25 participants |
| MAK683. | Advanced malignancies like Diffuse Large B-cell Lymphoma, nasopharyngeal carcinoma or other adcanced solid tumors | I/II | Recruiting | NCT02900651 | 203 participants |
| SHR2554 | Relapsed or Refractory Mature Lymphoid Neoplasm | I | Recruiting | NCT03603951 | 42 participants |
| SHR3680, SHR2554 | Prostate Cancer Castration-resistant Prostate Cancer |
I/II | Recruiting | NCT03741712 | 100 participants |
| PF-06821497 | Small Cell Lung Cancer (SCLC), Follicular Lymphoma (FL), Castration Resistant Prostate Cancer (CRPC), Diffuse Large B-Cell Lymphoma (DLBCL) | I | Recruiting | NCT03460977 | 172 participants |
Future perspective/Discussion.
EZH2 overexpression and mutation plays a critical role in the development, progression, and metastasis of many types of cancer. Extensive research in the last 20 years on the histone methyltransferase EZH2 in cancer has led to the development of different inhibitors that target both wild type and mutant EZH2, as well as some that target the PRC2 complex member EED. EZH2 now serves as a target for precision medicine and many studies have shown that EZH2 is a target for synthetic lethality and hence serves as a valuable drug in cancers with specific mutations in trithorax/SWI/SNF family members like BAP1 and ARID1A (88–91, 111).
EZH2 inhibition alone may not be highly effective in certain tumors, however, and combination with other drugs and immunotherapies may prove beneficial in cancers that are not sensitive to EZH2 inhibition alone. For example, activation of IGF-1R, PI3K, and MEK pathways can lead to resistance to EZH2 inhibitors. Furthermore, some mutations in the EZH2 gene prevents small molecular inhibitors from binding to EZH2. Other acquired resistance mechanisms include inhibiting proapoptotic proteins TNFSF10 and BAD via a FOXO3A-dependent mechanism (113).
Therefore identification of additional mutations that confer EZH2 dependence to tumors could increase patient and tumor eligibility. While our ability to predict EZH2 sensitivity is improving, more work is required to develop highly predictive biomarkers for EZH2 therapeutic response. In addition, further combination trials will be crucial to maximize therapeutic benefit, while keeping toxicity tolerable. Overall, EZH2 is one molecular target that has elicited tremendous interest both in tumor biologists with a focus on epigenetics, and in clinical oncologists. With increased testing and trials, the future of targeting EZH2 to treat specific type of cancers either as a single agent or in combination is highly promising.
Supplementary Material
Acknowledgments:
Dr. SV is supported by Department of Defense funding (W81XWH1910588). Part of this work is supported by supplemental funding from U54CA118948 and startup funding from UAB to SV. SV is also supported in part by R01ES026219 from NIH.
Footnotes
Conflict of Interest: No conflict of interest
References
- 1.Waddington CH. Canalization of development and genetic assimilation of acquired characters. Nature. 1959;183(4676):1654–5. [DOI] [PubMed] [Google Scholar]
- 2.Waddington CH. The epigenotype. 1942. Int J Epidemiol. 2012;41(1):10–3. [DOI] [PubMed] [Google Scholar]
- 3.Pfister SX, Ashworth A. Marked for death: targeting epigenetic changes in cancer. Nat Rev Drug Discov. 2017;16(4):241–63. [DOI] [PubMed] [Google Scholar]
- 4.Albert M, Helin K. Histone methyltransferases in cancer. Semin Cell Dev Biol. 2010;21(2):209–20. [DOI] [PubMed] [Google Scholar]
- 5.Savas S, Skardasi G. The SWI/SNF complex subunit genes: Their functions, variations, and links to risk and survival outcomes in human cancers. Crit Rev Oncol Hematol. 2018;123:114–31. [DOI] [PubMed] [Google Scholar]
- 6.Euskirchen GM, Auerbach RK, Davidov E, Gianoulis TA, Zhong G, Rozowsky J, et al. Diverse roles and interactions of the SWI/SNF chromatin remodeling complex revealed using global approaches. PLoS Genet. 2011;7(3):e1002008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwon H, Imbalzano AN, Khavari PA, Kingston RE, Green MR. Nucleosome disruption and enhancement of activator binding by a human SW1/SNF complex. Nature. 1994;370(6489):477–81. [DOI] [PubMed] [Google Scholar]
- 8.Lu C, Allis CD. SWI/SNF complex in cancer. Nat Genet. 2017;49(2):178–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muchardt C, Yaniv M. ATP-dependent chromatin remodelling: SWI/SNF and Co. are on the job. J Mol Biol. 1999;293(2):187–98. [DOI] [PubMed] [Google Scholar]
- 10.Timp W, Feinberg AP. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer. 2013;13(7):497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bennett RL, Licht JD. Targeting Epigenetics in Cancer. Annu Rev Pharmacol Toxicol. 2018;58:187–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zentner GE, Henikoff S. Regulation of nucleosome dynamics by histone modifications. Nat Struct Mol Biol. 2013;20(3):259–66. [DOI] [PubMed] [Google Scholar]
- 13.Sobulo OM, Borrow J, Tomek R, Reshmi S, Harden A, Schlegelberger B, et al. MLL is fused to CBP, a histone acetyltransferase, in therapy-related acute myeloid leukemia with a t(11;16)(q23;p13.3). Proc Natl Acad Sci U S A. 1997;94(16):8732–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ogiwara H, Sasaki M, Mitachi T, Oike T, Higuchi S, Tominaga Y, et al. Targeting p300 Addiction in CBP-Deficient Cancers Causes Synthetic Lethality by Apoptotic Cell Death due to Abrogation of MYC Expression. Cancer Discov. 2016;6(4):430–45. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Iwasaki H, Krivtsov A, Febbo PG, Thorner AR, Ernst P, et al. Conditional MLL-CBP targets GMP and models therapy-related myeloproliferative disease. EMBO J. 2005;24(2):368–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee HZ, Kwitkowski VE, Del Valle PL, Ricci MS, Saber H, Habtemariam BA, et al. FDA Approval: Belinostat for the Treatment of Patients with Relapsed or Refractory Peripheral T-cell Lymphoma. Clin Cancer Res. 2015;21(12):2666–70. [DOI] [PubMed] [Google Scholar]
- 17.Assouline SE, Nielsen TH, Yu S, Alcaide M, Chong L, MacDonald D, et al. Phase 2 study of panobinostat with or without rituximab in relapsed diffuse large B-cell lymphoma. Blood. 2016;128(2):185–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25(21):3109–15. [DOI] [PubMed] [Google Scholar]
- 19.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13(5):343–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hublitz P, Albert M, Peters AH. Mechanisms of transcriptional repression by histone lysine methylation. Int J Dev Biol. 2009;53(2–3):335–54. [DOI] [PubMed] [Google Scholar]
- 21.Liu H, Takeda S, Kumar R, Westergard TD, Brown EJ, Pandita TK, et al. Phosphorylation of MLL by ATR is required for execution of mammalian S-phase checkpoint. Nature. 2010;467(7313):343–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, Cai Q, Godwin AK, Zhang R. Enhancer of zeste homolog 2 promotes the proliferation and invasion of epithelial ovarian cancer cells. Mol Cancer Res. 2010;8(12):1610–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu C, Bonome T, Li Y, Kamat AA, Han LY, Schmandt R, et al. Gene alterations identified by expression profiling in tumor-associated endothelial cells from invasive ovarian carcinoma. Cancer Res. 2007;67(4):1757–68. [DOI] [PubMed] [Google Scholar]
- 24.Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A. 2003;100(20):11606–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou J, Roh JW, Bandyopadhyay S, Chen Z, Munkarah AR, Hussein Y, et al. Overexpression of enhancer of zeste homolog 2 (EZH2) and focal adhesion kinase (FAK) in high grade endometrial carcinoma. Gynecol Oncol. 2013;128(2):344–8. [DOI] [PubMed] [Google Scholar]
- 26.Jia N, Li Q, Tao X, Wang J, Hua K, Feng W. Enhancer of zeste homolog 2 is involved in the proliferation of endometrial carcinoma. Oncol Lett. 2014;8(5):2049–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang H, Qi J, Reyes JM, Li L, Rao PK, Li F, et al. Oncogenic Deregulation of EZH2 as an Opportunity for Targeted Therapy in Lung Cancer. Cancer Discov. 2016;6(9):1006–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kondo Y, Shen L, Suzuki S, Kurokawa T, Masuko K, Tanaka Y, et al. Alterations of DNA methylation and histone modifications contribute to gene silencing in hepatocellular carcinomas. Hepatol Res. 2007;37(11):974–83. [DOI] [PubMed] [Google Scholar]
- 29.Wagener N, Macher-Goeppinger S, Pritsch M, Husing J, Hoppe-Seyler K, Schirmacher P, et al. Enhancer of zeste homolog 2 (EZH2) expression is an independent prognostic factor in renal cell carcinoma. BMC Cancer. 2010;10:524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang SH, Huang HS, Wu HU, Tsai YT, Chuang MJ, Yu CP, et al. Pharmacologic down-regulation of EZH2 suppresses bladder cancer in vitro and in vivo. Oncotarget. 2014;5(21):10342–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bachmann IM, Halvorsen OJ, Collett K, Stefansson IM, Straume O, Haukaas SA, et al. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J Clin Oncol. 2006;24(2):268–73. [DOI] [PubMed] [Google Scholar]
- 32.Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C, et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007;21(5):525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419(6907):624–9. [DOI] [PubMed] [Google Scholar]
- 34.Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012;338(6113):1465–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fujii S, Tokita K, Wada N, Ito K, Yamauchi C, Ito Y, et al. MEK-ERK pathway regulates EZH2 overexpression in association with aggressive breast cancer subtypes. Oncogene. 2011;30(39):4118–28. [DOI] [PubMed] [Google Scholar]
- 36.Chang CJ, Yang JY, Xia W, Chen CT, Xie X, Chao CH, et al. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-beta-catenin signaling. Cancer Cell. 2011;19(1):86–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fujikawa D, Nakagawa S, Hori M, Kurokawa N, Soejima A, Nakano K, et al. Polycomb-dependent epigenetic landscape in adult T-cell leukemia. Blood. 2016;127(14):1790–802. [DOI] [PubMed] [Google Scholar]
- 38.Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22(20):5323–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Varambally S, Cao Q, Mani RS, Shankar S, Wang X, Ateeq B, et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science. 2008;322(5908):1695–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sander S, Bullinger L, Klapproth K, Fiedler K, Kestler HA, Barth TF, et al. MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood. 2008;112(10):4202–12. [DOI] [PubMed] [Google Scholar]
- 41.Kaur M, Cole MD. MYC acts via the PTEN tumor suppressor to elicit autoregulation and genome-wide gene repression by activation of the Ezh2 methyltransferase. Cancer Res. 2013;73(2):695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lue JK, Amengual JE. Emerging EZH2 Inhibitors and Their Application in Lymphoma. Curr Hematol Malig Rep. 2018;13(5):369–82. [DOI] [PubMed] [Google Scholar]
- 43.Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42(2):181–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM, et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci U S A. 2010;107(49):20980–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42(8):722–6. [DOI] [PubMed] [Google Scholar]
- 46.Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tonnissen ER, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010;42(8):665–7. [DOI] [PubMed] [Google Scholar]
- 47.Ntziachristos P, Tsirigos A, Van Vlierberghe P, Nedjic J, Trimarchi T, Flaherty MS, et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med. 2012;18(2):298–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harada H, Harada Y, Niimi H, Kyo T, Kimura A, Inaba T. High incidence of somatic mutations in the AML1/RUNX1 gene in myelodysplastic syndrome and low blast percentage myeloid leukemia with myelodysplasia. Blood. 2004;103(6):2316–24. [DOI] [PubMed] [Google Scholar]
- 49.Niimi H, Harada H, Harada Y, Ding Y, Imagawa J, Inaba T, et al. Hyperactivation of the RAS signaling pathway in myelodysplastic syndrome with AML1/RUNX1 point mutations. Leukemia. 2006;20(4):635–44. [DOI] [PubMed] [Google Scholar]
- 50.Sashida G, Harada H, Matsui H, Oshima M, Yui M, Harada Y, et al. Ezh2 loss promotes development of myelodysplastic syndrome but attenuates its predisposition to leukaemic transformation. Nat Commun. 2014;5:4177. [DOI] [PubMed] [Google Scholar]
- 51.Schwartz YB, Pirrotta V. Polycomb silencing mechanisms and the management of genomic programmes. Nat Rev Genet. 2007;8(1):9–22. [DOI] [PubMed] [Google Scholar]
- 52.Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ 3rd, et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. 2009;461(7265):762–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamagishi M, Hori M, Fujikawa D, Ohsugi T, Honma D, Adachi N, et al. Targeting Excessive EZH1 and EZH2 Activities for Abnormal Histone Methylation and Transcription Network in Malignant Lymphomas. Cell Rep. 2019;29(8):2321–37 e7. [DOI] [PubMed] [Google Scholar]
- 54.Shen X, Liu Y, Hsu YJ, Fujiwara Y, Kim J, Mao X, et al. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol Cell. 2008;32(4):491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, et al. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell. 2008;32(4):503–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469(7330):343–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Riising EM, Comet I, Leblanc B, Wu X, Johansen JV, Helin K. Gene silencing triggers polycomb repressive complex 2 recruitment to CpG islands genome wide. Mol Cell. 2014;55(3):347–60. [DOI] [PubMed] [Google Scholar]
- 58.Smits AH, Jansen PW, Poser I, Hyman AA, Vermeulen M. Stoichiometry of chromatin-associated protein complexes revealed by label-free quantitative mass spectrometry-based proteomics. Nucleic Acids Res. 2013;41(1):e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van Mierlo G, Veenstra GJC, Vermeulen M, Marks H. The Complexity of PRC2 Subcomplexes. Trends Cell Biol. 2019;29(8):660–71. [DOI] [PubMed] [Google Scholar]
- 60.He A, Shen X, Ma Q, Cao J, von Gise A, Zhou P, et al. PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes Dev. 2012;26(1):37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee JM, Lee JS, Kim H, Kim K, Park H, Kim JY, et al. EZH2 generates a methyl degron that is recognized by the DCAF1/DDB1/CUL4 E3 ubiquitin ligase complex. Mol Cell. 2012;48(4):572–86. [DOI] [PubMed] [Google Scholar]
- 62.Vasanthakumar A, Xu D, Lun AT, Kueh AJ, van Gisbergen KP, Iannarella N, et al. A non-canonical function of Ezh2 preserves immune homeostasis. EMBO Rep. 2017;18(4):619–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell. 2013;23(6):839–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koyen AE, Madden MZ, Park D, Minten EV, Kapoor-Vazirani P, Werner E, et al. EZH2 has a non-catalytic and PRC2-independent role in stabilizing DDB2 to promote nucleotide excision repair. Oncogene. 2020;39(25):4798–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee ST, Li Z, Wu Z, Aau M, Guan P, Karuturi RK, et al. Context-specific regulation of NF-kappaB target gene expression by EZH2 in breast cancers. Mol Cell. 2011;43(5):798–810. [DOI] [PubMed] [Google Scholar]
- 66.Kleer C Abstract OI: Novel non-canonical functions of EZH2 in triple negative breast cancer. 2020;80(4 Supplement):OI–OI. [Google Scholar]
- 67.Fiskus W, Wang Y, Sreekumar A, Buckley KM, Shi H, Jillella A, et al. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood. 2009;114(13):2733–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yamagishi M, Uchimaru K. Targeting EZH2 in cancer therapy. Curr Opin Oncol. 2017;29(5):375–81. [DOI] [PubMed] [Google Scholar]
- 69.Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK, et al. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther. 2009;8(6):1579–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492(7427):108–12. [DOI] [PubMed] [Google Scholar]
- 71.Schubert HL, Blumenthal RM, Cheng X. Many paths to methyltransfer: a chronicle of convergence. Trends Biochem Sci. 2003;28(6):329–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Adamik J, Silbermann R, Lontos K, Zhang P, Sun Q, Galson DL, et al. EZH2 Inhibitor GSK126 Exhibits Osteo-Anabolic Properties in MM Bone Disease and Synergizes with Bortezomib to Inhibit MM Cell Viability. Blood. 2016;128(22):3247–. [Google Scholar]
- 73.Lue JK, Prabhu SA, Liu Y, Gonzalez Y, Verma A, Mundi PS, et al. Precision Targeting with EZH2 and HDAC Inhibitors in Epigenetically Dysregulated Lymphomas. Clin Cancer Res. 2019;25(17):5271–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang Y, Zhu F, Wang Q, Ding Y, Ying R, Zeng L. Inhibition of EZH2 and EGFR produces a synergistic effect on cell apoptosis by increasing autophagy in gastric cancer cells. Onco Targets Ther. 2018;11:8455–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Huang S, Wang Z, Zhou J, Huang J, Zhou L, Luo J, et al. EZH2 Inhibitor GSK126 Suppresses Antitumor Immunity by Driving Production of Myeloid-Derived Suppressor Cells. Cancer Res. 2019;79(8):2009–20. [DOI] [PubMed] [Google Scholar]
- 76.Puppe J, Opdam M, Schouten PC, Jozwiak K, Lips E, Severson T, et al. EZH2 Is Overexpressed in BRCA1-like Breast Tumors and Predictive for Sensitivity to High-Dose Platinum-Based Chemotherapy. Clin Cancer Res. 2019;25(14):4351–62. [DOI] [PubMed] [Google Scholar]
- 77.Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci U S A. 2012;109(52):21360–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013;110(19):7922–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, Xiao Y, et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther. 2014;13(4):842–54. [DOI] [PubMed] [Google Scholar]
- 80.Kung PP, Bingham P, Brooun A, Collins M, Deng YL, Dinh D, et al. Optimization of Orally Bioavailable Enhancer of Zeste Homolog 2 (EZH2) Inhibitors Using Ligand and Property-Based Design Strategies: Identification of Development Candidate (R)-5,8-Dichloro-7-(methoxy(oxetan-3-yl)methyl)-2-((4-methoxy-6-methyl-2-oxo-1,2- dihydropyridin-3-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (PF-06821497). J Med Chem. 2018;61(3):650–65. [DOI] [PubMed] [Google Scholar]
- 81.Vaswani RG, Gehling VS, Dakin LA, Cook AS, Nasveschuk CG, Duplessis M, et al. Identification of (R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1 -(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), a Potent and Selective Inhibitor of Histone Methyltransferase EZH2, Suitable for Phase I Clinical Trials for B-Cell Lymphomas. J Med Chem. 2016;59(21):9928–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Konze KD, Ma A, Li F, Barsyte-Lovejoy D, Parton T, Macnevin CJ, et al. An orally bioavailable chemical probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem Biol. 2013;8(6):1324–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xu B, On DM, Ma A, Parton T, Konze KD, Pattenden SG, et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood. 2015;125(2):346–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Maruyama D, Tobinai K, Makita S, Ishida T, Kusumoto S, Ishitsuka K, et al. First-in-Human Study of the EZH1/2 Dual Inhibitor DS-3201b in Patients with Relapsed or Refractory Non-Hodgkin Lymphomas — Preliminary Results. Blood. 2017;130(Supplement 1):4070–. [Google Scholar]
- 85.Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11(7):481–92. [DOI] [PubMed] [Google Scholar]
- 86.Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45(6):592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wilson BG, Wang X, Shen X, McKenna ES, Lemieux ME, Cho YJ, et al. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell. 2010;18(4):316–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hodges C, Kirkland JG, Crabtree GR. The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer. Cold Spring Harb Perspect Med. 2016;6(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AV, et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat Med. 2015;21(3):231–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fillmore CM, Xu C, Desai PT, Berry JM, Rowbotham SP, Lin YJ, et al. EZH2 inhibition sensitizes BRG1 and EGFR mutant lung tumours to TopoII inhibitors. Nature. 2015;520(7546):239–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Italiano A, Soria JC, Toulmonde M, Michot JM, Lucchesi C, Varga A, et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol. 2018;19(5):649–59. [DOI] [PubMed] [Google Scholar]
- 92.Kadoch C, Williams RT, Calarco JP, Miller EL, Weber CM, Braun SM, et al. Dynamics of BAF-Polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat Genet. 2017;49(2):213–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016;22(2):128–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.van Haaften G, Dalgliesh GL, Davies H, Chen L, Bignell G, Greenman C, et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet. 2009;41(5):521–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ezponda T, Dupere-Richer D, Will CM, Small EC, Varghese N, Patel T, et al. UTX/KDM6A Loss Enhances the Malignant Phenotype of Multiple Myeloma and Sensitizes Cells to EZH2 inhibition. Cell Rep. 2017;21(3):628–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ler LD, Ghosh S, Chai X, Thike AA, Heng HL, Siew EY, et al. Loss of tumor suppressor KDM6A amplifies PRC2-regulated transcriptional repression in bladder cancer and can be targeted through inhibition of EZH2. Sci Transl Med. 2017;9(378). [DOI] [PubMed] [Google Scholar]
- 97.LaFave LM, Beguelin W, Koche R, Teater M, Spitzer B, Chramiec A, et al. Loss of BAP1 function leads to EZH2-dependent transformation. Nat Med. 2015;21(11):1344–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang X, Brea LT, Yu J. Immune modulatory functions of EZH2 in the tumor microenvironment: implications in cancer immunotherapy. Am J Clin Exp Urol. 2019;7(2):85–91. [PMC free article] [PubMed] [Google Scholar]
- 99.Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527(7577):249–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nagarsheth N, Peng D, Kryczek I, Wu K, Li W, Zhao E, et al. PRC2 Epigenetically Silences Th1-Type Chemokines to Suppress Effector T-Cell Trafficking in Colon Cancer. Cancer Res. 2016;76(2):275–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li J, Wang W, Zhang Y, Cieslik M, Guo J, Tan M, et al. Epigenetic driver mutations in ARID1A shape cancer immune phenotype and immunotherapy. J Clin Invest. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zingg D, Arenas-Ramirez N, Sahin D, Rosalia RA, Antunes AT, Haeusel J, et al. The Histone Methyltransferase Ezh2 Controls Mechanisms of Adaptive Resistance to Tumor Immunotherapy. Cell Rep. 2017;20(4):854–67. [DOI] [PubMed] [Google Scholar]
- 103.Karantanos T, Chistofides A, Barhdan K, Li L, Boussiotis VA. Regulation of T Cell Differentiation and Function by EZH2. Front Immunol. 2016;7:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Goswami S, Apostolou I, Zhang J, Skepner J, Anandhan S, Zhang X, et al. Modulation of EZH2 expression in T cells improves efficacy of anti-CTLA-4 therapy. J Clin Invest. 2018;128(9):3813–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang D, Quiros J, Mahuron K, Pai CC, Ranzani V, Young A, et al. Targeting EZH2 Reprograms Intratumoral Regulatory T Cells to Enhance Cancer Immunity. Cell Rep. 2018;23(11):3262–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L, et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat Immunol. 2016;17(1):95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mullard A FDA approves an inhibitor of a novel ‘epigenetic writer’. Nat Rev Drug Discov. 2020;19(3):156. [DOI] [PubMed] [Google Scholar]
- 108.Stacchiotti S, Schoffski P, Jones R, Agulnik M, Villalobos VM, Jahan TM, et al. Safety and efficacy of tazemetostat, a first-in-class EZH2 inhibitor, in patients (pts) with epithelioid sarcoma (ES). (NCT02601950). Journal of Clinical Oncology. 2019;37(15_suppl):11003–. [Google Scholar]
- 109.Morschhauser F, Tilly H, Chaidos A, Phillips T, Ribrag V, Campbell P, et al. INTERIM UPDATE FROM A PHASE 2 MULTICENTER STUDY OF TAZEMETOSTAT, AN EZH2 INHIBITOR, IN PATIENTS WITH RELAPSED OR REFRACTORY FOLLICULAR LYMPHOMA. Hematological Oncology. 2019;37(S2):154–6. [Google Scholar]
- 110.Ribrag V, Morschhauser F, McKay P, Salles GA, Batlevi CL, Schmitt A, et al. Interim Results from an Ongoing Phase 2 Multicenter Study of Tazemetostat, an EZH2 Inhibitor, in Patients with Relapsed or Refractory (R/R) Diffuse Large B-Cell Lymphoma (DLBCL). Blood. 2018;132(Supplement 1):4196–. [Google Scholar]
- 111.Zauderer MG, Szlosarek P, Moulec SL, Popat S, Taylor P, Planchard D, et al. Phase 2, multicenter study of the EZH2 inhibitor tazemetostat as monotherapy in adults with relapsed or refractory (R/R) malignant mesothelioma (MM) with BAP1 inactivation. Journal of Clinical Oncology. 2018;36(15_suppl):8515–. [Google Scholar]
- 112.Palomba ML, Cartron G, Popplewell L, Ribrag V, Westin J, Chitra S, et al. SAFETY AND CLINICAL ACTIVITY OF ATEZOLIZUMAB IN COMBINATION WITH TAZEMETOSTAT IN RELAPSED OR REFRACTORY DIFFUSE LARGE B-CELL LYMPHOMA: PRIMARY ANALYSIS OF A PHASE 1B STUDY. Hematological Oncology. 2019;37(S2):517–9. [Google Scholar]
- 113.Bisserier M, Wajapeyee N. Mechanisms of resistance to EZH2 inhibitors in diffuse large B-cell lymphomas. Blood. 2018;131(19):2125–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Justin N, Zhang Y, Tarricone C, Martin SR, Chen S, Underwood E, et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat Commun. 2016;7:11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.