

Abstract
Objective
To determine whether mutations reported for ZDHHC15 can cause mixed neurodevelopmental disorders, we performed both functional studies on variant pathogenicity and ZDHHC15 function in animal models.
Methods
We examined protein function of 4 identified variants in ZDHHC15 in a yeast complementation assay and locomotor defects of loss-of-function genotypes in a Drosophila model.
Results
Although we assessed multiple patient variants, only 1 (p.H158R) affected protein function. We report a patient with a diagnosis of hypotonic cerebral palsy, autism, epilepsy, and intellectual disability associated with this bona fide damaging X-linked variant. Features include tall forehead with mild brachycephaly, down-slanting palpebral fissures, large ears, long face, facial muscle hypotonia, high-arched palate with dental crowding, and arachnodactyly. The patient had mild diminished cerebral volume, with left-sided T2/FLAIR hyperintense periatrial ovoid lesion. We found that loss-of-function mutations in orthologs of this gene cause flight and coordinated movement defects in Drosophila.
Conclusions
Our findings support a functional expansion of this gene to a role in motor dysfunction. Although ZDHHC15 mutations represent a rare cause of neurodevelopmental disability, candidate variants need to be carefully assessed before pathogenicity can be determined.
Posttranslation addition of palmitate to cysteine residues by palmitoyl acyltransferases targets proteins to intracellular membrane–associated compartments and to synapses within the CNS.1 Many palmitoyl acyltransferases contain a 51-residue zinc finger domain with a DHHC motif crucial for enzymatic function. ZDHHC proteins were implicated in neurodevelopmental disorders (NDDs) when a balanced t(X;15) (q13.3;cen) translocation disrupting the ZDHHC15 gene was reported in association with intellectual disability.2 This patient showed skewed lyonization, with 100% inactivation of the normal X chromosome. Variants in the related ZDHHC9 have been associated with NDDs3 including motor delay, coordination difficulties, and cerebral palsy.4
However, the finding of normal intelligence (Wechsler Adult Intelligence Scale–III full-scale IQ 111) in a woman with a balanced translocation disrupting the gene5 suggests that ZDHHC15 loss of function may not be pathologic. Although X inactivation was not assessed in this report, the authors were unable to detect ZDHHC15 expression in peripheral blood necessitating validation studies of ZDHHC15's role in neurodevelopment. We identified individuals with predicted deleterious variants within a cohort of cryptogenic cerebral palsy patients who underwent whole-exome sequencing6 and through gene matching efforts (genematcher.org). We identify functional consequences of these variants and confirm a role in regulating movement in genetic models. Finally, we present clinical information from a patient harboring a verified deleterious, X-linked variant (p.H158R) that disrupts the core DHHC domain.
Methods
Patient Recruitment and Sequencing
Patients were recruited under the central IRB protocol (#15-080) approved by the Phoenix Children's Hospital IRB Committee (#1). Written informed consents for research were obtained for parents (and assent was obtained for children as appropriate) for families participating in the study. Whole-exome sequencing of patients with cerebral palsy identifying maternally inherited variants p.H158R and p. L13P reported in Jin et al.6 Depth of coverage for p.H158R at the variant position (X:74649792T<C) is 34 reads and p.L13P (X:74742822A<G) is 30 reads. Other variants were identified through GeneMatcher, and maternally inherited p.K115R was sequenced through GeneDx.
Clinical phenotypes for (p.H158R) were assessed by J.H. and M.C.K. This patient had negative testing for Fragile X, telomeric FISH, chromosomal microarray, and karyotyping. An epilepsy gene panel (Invitae) showed a single heterozygous predicted protein-damaging variant in ALDH7A1 (p.L246E), and WES found a de novo CPM (p.H369R) and X-linked AGTR2 (p.F320L) variant). These variants were thought to be unlikely to represent the underlying cause of his clinical phenotype because ALDH7A1 causes a recessive condition and no second mutation or genomic deletion was detected and the other genes are not predicted to contribute to the patient's phenotypes.
Yeast Complementation Studies
The BY4742 wild-type (MAT α his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0) and pfa3-Δ (MAT α his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 pfa3::KANMX) yeast strains were obtained from TransOMIC (Huntsville, AL). PFA3, ZDHHC15, and ZDHHC15 p.H158R cDNA sequences were synthesized (GenScript Inc.), confirmed by direct sequencing, and introduced into the p416GPD-URA3 vector using BamHI and EcoRI restriction sites, allowing us to express them under the strong promoter glyceraldehyde-3-phosphate dehydrogenase (GPD). The obtained plasmids were then used to transform yeast using the lithium acetate method.
Yeast cells were grown and labeled in YPD unless treated with dithiothreitol (DTT), in which case they were grown in minimal media. Rapidly growing cells were labeled with 20 μM FM 4–64 (Invitrogen) for 30 minutes and then collected and suspended in fresh media lacking FM 4–64 for 60 minutes. Cells were washed twice with PBS before imaging. For DTT-treated samples, 2 mM DTT was added to cultures at the start of FM 4–64 labeling 2 hours before microscopy. Two millimolar DTT was maintained in the media after the labeling period and in the PBS washes. Images were collected at room temperature with a Zeiss Axio Imager M2 inverted microscope with a ×100 objective, Axiocam 506 mono-camera, and NIH Image software.
Drosophila Movement Assays
Drosophila were reared on a standard cornmeal, yeast, sucrose food as previously reported.6 Two 5′ P-element insertions and a deficiency chromosome from Bloomington Drosophila stock center (NIH P40OD018537) were first outcrossed with balancer stocks for a single generation to replace the 1st chromosome with w+ and swap the 2nd chromosome balancer to include a larval marker. Experimental genotypes were CG1407MI00131/+ (control), CG1407063-G4/+ (control), and CG1407MI00131/063-G4 (compound heterozygote) for larval turning and w1118/+ (control), CG1407MI00131/063-G4 (compound heterozygote), and CG1407MI00131/Df(2R)X1 (hemizygote) for adult flight analysis.
Larval turning time was defined as the amount of time required to turn onto the ventral surface and initiate forward movement after rotation onto the dorsal surface. Time was measured for 48–50 larvae/genotype with 3 trials/larva, and the average time was calculated. A failed trial was when a larva did not move for 30 seconds after rotation onto dorsal surface. Differences in average time were compared with both genetic controls using the paired t test. The number of successes and failures was compared via χ2 analysis with 1 degree of freedom and was not significant.
We reanalyzed 9 videos with distance traveled impairments6 for flight behaviors for 7 seconds after animals were tapped to bottom of vials. The number of attempted flights was counted and categorized by net height. Up: net vertical distance was higher than the start point. Down: net vertical distance below the start point. Up to Down: flight was initially upward, but ended with a net vertical decrease. Center: no net vertical distance change. Movements where the flies jumped in a straight trajectory across the vial with no up or downward movement (i.e., no flight attempt) were excluded. The scorer was blinded to genotype. Comparisons of types of movements/fly between genotypes conducted using 2-tailed, paired t test and overall distribution via χ2 analysis with 3 degrees of freedom.
Data Availability
Variant information for p.H158R deposited in ClinVar (SCV001468640). Sequencing and clinical descriptions of p.L13P patient described in Jin et al.,6 doi.10.1038/s41588-020-0695-1. Any data not published within the article will be shared by request from any qualified investigator.
Results
ZDHHC15 (p.H158R) Causes Loss of Function
We investigated 4 patient variants in Saccharomyces cerevisiae and found no clear differences in protein abundance between variants and wild type (data not shown). We thus designed a complementation-based assay to discriminate deleterious and benign variants. The yeast ZDHHC15 ortholog, PFA3, encodes a palmitoyl transferase responsible for the palmitoylation of Vac8p, a protein involved in the fusion of vacuolar membranes in yeast. Palmitoylation of Vac8p is important for protein localization to the vacuolar membrane where it performs its function.7 Consequently, yeast cells lacking PFA3 (pfa3-Δ) show a vacuole fragmentation phenotype when stressed by low glucose or presence of the reducing agent DTT.
We designed a complementation assay where rescue of the vacuole fragmentation phenotype of the pfa3-Δ strain in the presence of DTT was analyzed after introducing a plasmid carrying either yeast PFA3 or human ZDHHC15 variants under the control of the yeast GPD strong promoter. We confirmed that pfa3-Δ cells develop vacuolar fragmentation in the presence of 2 mM DTT and reintroducing either wild type PFA3 or ZDHHC15 rescued this phenotype (Figure 1). In addition, we found that yeast cells expressing ZDHHC15 variants (p.L13P, p.K115R, and p.S330P) were indistinguishable from cells harboring the reference ZDHHC15 allele. However, expressing the ZDHHC15 (p.H158R) variant did not rescue the vacuolar fragmentation phenotype, indicating that this specific change significantly disrupted normal protein function, putatively by disrupting the core DHHC domain (Figure 1A).
Figure 1. Yeast Complementation Studies With Human ZDHHC15 Variants.

(A) Human ZDHHC15 protein depicting transmembrane, DHHC-CRD domains and the position of missense variants detected in individual cases. (B) Representative images of FM-464 vacuolar membrane staining in pfa3 yeast strain transformed with empty vector (pfa3), yeast PFA3 (PFA3), or human ZDHHC15 (ZDHHC15) after 2-hour incubation with 2 mM DTT. (C) Quantification of the number of cells with more than 6 vacuoles after incubation with DTT. ***Indicates significance determined by 2-tailed t test p < 0.0001 compared to ZDHHC15 control (grey bar). DTT = Dithiothreitol.
ZDHHC15 (p.H158R) Clinical Summary
The patient is an 18-year-old man with a history of hypotonic cerebral palsy, focal-onset epilepsy, cortical visual impairment, intellectual disability, autism spectrum disorder, anxiety, and aggressive behaviors. Physical examination revealed tall forehead with mild brachycephaly, down-slanting palpebral fissures, large ears, long face, and facial muscle hypotonia (Figure 2A). He has a high-arched palate with dental crowding. He has long, slender palms and fingers (arachnodactyly). He is nonverbal and was uncooperative with many portions of the examination.
Figure 2. Clinical Features of the Patient With Validated ZDHHC15 Mutation (p.H158R).
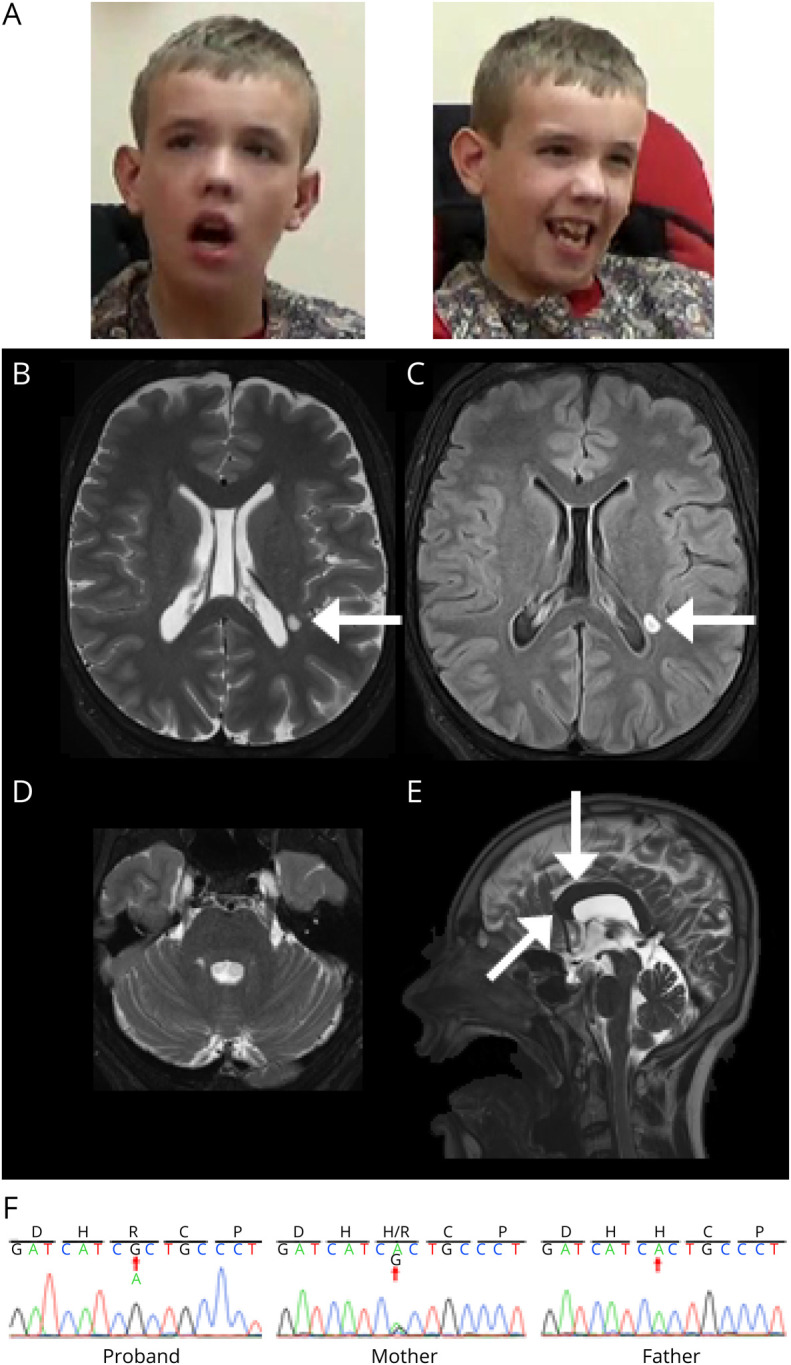
(A) Dysmorphic facial features include mild brachycephaly, down-slanting palpebral fissures, large ears, long face, and facial hypotonia. (B–E) Brain MRI at age 17 years. Axial T2 (B) and axial FLAIR (C) images demonstrate mild symmetric widening of the cerebral sulci and a small ovoid focus of T2/FLAIR signal prolongation in the left lateral periatrial white matter (white arrow). Axial FLAIR (D) image at the posterior fossa level demonstrates symmetric widening of the cerebellar folia. Sagittal T2 (E) image at midline shows a smaller-than-expected craniofacial ratio. In addition, the corpus callosum is mildly thickened and foreshortened (white arrow). All these findings have been stable over time on serial MRI. (F) Sanger traces of the proband, mother, and father confirming A to G base change inherited from the mother.
He was born at 39 weeks gestational age via spontaneous vaginal delivery. There were no complications reported with the pregnancy or delivery, and his neonatal course was unremarkable. He had global developmental delay. Previous cognitive testing established intellectual disability and autism diagnoses, which have been present and nonprogressive throughout his life, although those medical records were not available for review. He first walked independently at age 8 years, but currently requires 2 hands for support. His gait has deteriorated and become crouched from contractures of the gastrocnemius/soleus complex and hamstrings (Video 1). Gait changes are attributed to unbalanced biomechanical forces related to his hypotonia (no spasticity was apparent) despite previous orthopedic intervention (right pes planus reconstruction, right cavovarus foot reconstruction, and bilateral hamstring lengthening). There is no reported family history of intellectual disability, cerebral palsy, or epilepsy.
The patient exhibits crouch gait and hesitancy when walking.Download Supplementary Video 1 (9.1MB, m4v) via http://dx.doi.org/10.1212/000602_Video_1
His most recent routine EEG demonstrated poorly organized and poorly sustained awake background, and some sharp transients seen over the frontal regions during sleep. Polysomnogram demonstrated increased awakenings after sleep onset and reduced sleep efficiency, but no evidence of sleep disordered breathing, nocturnal movement disorders, or potentiation of epileptiform activity. Brain MRI was remarkable for mild cerebral sulci widening, corpus callosum foreshortening, and a left lateral periatrial white matter ovoid hyperintensity (Figure 2B–E). On laboratory testing, only transient hyperammonemia (attributed to valproic acid use) was noted. His seizures are currently well controlled on valproic acid and lamotrigine. He is receiving speech, physical, and occupational therapies.
ZDHHC15 Regulates Motor Control in a Genetic Model
We assessed the role of ZDHHC15 in motor control using the functional ortholog in D melanogaster (CG1407, DIOPT 12/15). We found an increase in average time to execute a coordinated axial twisting task in mutant larva (13.4 vs 10.3 and 9.6 seconds, p < 0.008, t test, Figure 3). We also examined movement strategies used by adult Drosophila in a negative geotaxis flight task. We characterized movement types by direction of flight path and net upward/downward travel. Wild-type flies often use upward flights to reach the top of the assay container. Mutant flies were less likely to initiate flights (2.4 and 2.2 vs 3.5 movements/fly, p < 0.04, t test). There was a change in the ratio of movements for the most severely impaired genotype compared with both the genetic control (p < 0.001) and the compound heterozygote (p < 0.05, χ2 test). This was attributable to a decrease in upward flight (1.7 vs 1.0 upward flights/fly, p < 0.02 t test). This suggests defects in coordinating and initiating movement as well as for effective flight. This is consistent with the decreased distance traveled in a locomotor assay we reported previously.6 Together, this shows a role of ZDHHC15 in the regulation of multiple motor activities across development in our genetic model.
Figure 3. Larval Turning and Adult Flight Defects Caused by Loss-of-Function Mutations in the ZDHHC15 Drosophila Ortholog.

(A) Time to turn is increased in compound heterozygotes compared with either heterozygote alone. (B) Number of flights normalized to number of flies in assay is decreased in compound heterozygous and hemizygous flies. (C) Types of movements used by each genotype. These could be up, center (no net change), up to down (initial up trajectory but net down movement), or down; center movements took place over the entire assay vial and were not limited to the bottom. -/Df flies had a significant change in types of flights compared with +/+ (p < 0.001) or −/− (p < 0.05). N = 48–50 larvae per genotype and 72-100 adult flies per genotype across 9 trials. *p < 0.05, **p < 0.008) by t test (A, B) or χ2 (C).
Discussion
We report a patient with a hemizygous X-linked mutation in the ZDHHC15 gene (p.H158R) with a mixed neurodevelopmental phenotype that included cerebral palsy, intellectual disability, autism spectrum disorder, and epilepsy. As cerebral palsy is a clinical description, not a specific pathogenic mechanism, the diagnosis of cerebral palsy was retained after identifying a genetic etiology in recognition of the expanding genetic landscape of cerebral palsy6 identifying it as a neurodevelopmental feature analogous to intellectual disability, autism, and epilepsy.
We used yeast and fly models to verify loss of function of the (p.H158R) variant and a role of this gene in coordinated motor tasks. We conducted the current studies in part to clarify whether ZDHHC15 mutations could lead to neurodevelopmental disorders. Previous work has shown that mutations within the conserved DHHC domain in ZDHHC9 impair protein function,8 and we show here that a similar disruption at residue 158 of ZDHHC15 also leads to a mixed neurodevelopmental disorder. However, most of the missense variants we detected in ZDHHC15 did not alter function. This has important implications for interpretation of these variants in the context of clinical sequencing studies, as borderline in silico prediction scores often pose problems in interpretation. This is particularly challenging for X-linked variants, which are typically inherited from an unaffected mother. The impact of heterozygous balanced translocations disrupting ZDHHC15 is currently unresolved. Possible explanations include variability in patterns of X inactivation across tissues or individuals as has been recently reported9 or epistatic influences.
ZDHHC15 regulates PSD95 palmitoylation and trafficking, with knockdown in rat neuron cultures reducing dendrite outgrowth and excitatory synapse maturation.10 Future work may enhance our understanding of the biological effects of ZDHHC15 mutations by cataloging other proteins that undergo palmitoylation by this enzyme, particularly in the developing brain. A palmitoylation profile could catalog protein posttranslational modification targets more comprehensively. This could represent an important first step toward the aim of ultimately developing targeted therapies.
Acknowledgment
The authors are grateful to the patients and their families for their gracious support of this work. These studies were supported by NIH 1R01NS106298 (M.C.K.), the Cure CP Foundation, and NIH R00HL143036-02 to S.C.J. The authors acknowledge Cathy Stevens for contributing information on variant p.K115R. They appreciate the Phoenix Children's Hospital clinicians for their expert care including Korwyn Williams, MD, PhD, Wendy Bernatacivious, MD, David Shafron, MD, Laura Wilner, MD, and Emily Andrisevic, MD.
Glossary
- DTT
dithiothreitol
- GPD
glyceraldehyde-3-phosphate dehydrogenase
- NDD
neurodevelopmental disorder
Appendix. Authors
Study Funding
NIH 1R01NS106298 (to M.C.K), the Cure CP Foundation, and NIH R00HL143036-02 (to S.C.J.).
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/NG for full disclosures.
References
- 1.Zarȩba-Kozioł M, Figiel I, Bartkowiak-Kaczmarek A, Włodarczyk J. Insights into protein S-palmitoylation in synaptic plasticity and neurological disorders: potential and limitations of methods for detection and analysis. Front Mol Neurosci 2018;11:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mansouri MR, Marklund L, Gustavsson P. Loss of ZDHHC15 expression in a woman with a balanced translocation t(X;15)(q13.3;cen) and severe mental retardation. Europ J Hum Genet 2005;13(8):970-977. [DOI] [PubMed] [Google Scholar]
- 3.Masurel-Paulet A, Kalscheuer VM, Lebrun N, et al. Expanding the clinical phenotype patients with a ZDHHC9 mutation. Am J Med Genet A 2014;164(3):789-795. [DOI] [PubMed] [Google Scholar]
- 4.van Eyk CL, Corbett MA, Frank MSB, et al. Targeted resequencing identifies genes with recurrent variation in cerebral palsy. NPJ Genom Med 2019;4:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moysés-Olivera M, Guilherme RS, Meloni VA, et al. X-linked intellectual disability related genes disrupted by balanced X-autosome translocations. Am J Med Genet 2015;168B(8):669-677. [DOI] [PubMed] [Google Scholar]
- 6.Jin SC, Lewis SA, Bakhtiari S, et al. Mutations disrupting neuritogenesis genes represent a major independent risk factor for cerebral palsy. Nat Genet, 2020. ;52(10):1046-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smotrys JE, Schoenfish MJ, Stutz MA, Linder ME. The vacuolar DHHC-CRD protein Pfa3p is a protein acyltransferase for Vac8p. J Cell Biol 2005;170(7):1091-1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mitchell DA, Hamel LD, Reddy KD, et al. Mutations in the X-linked intellectual disability gene, zDHHC9, alter autopalmitoylation activity by distinct mechanisms. J Bioll Chem 2014;289(26):18582-18592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tukiainen T, Villani AC, Yen A, et al. Landscape of X chromosome inactivation across human tissues. Nature 2017;550(7675):244-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shah BS, Shimell JJ, Bamji SX. Regulation of dendrite morphology and excitatory synapse formation by zDHHC15. J Cell Sci 2019;132(13):jcs230052. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The patient exhibits crouch gait and hesitancy when walking.Download Supplementary Video 1 (9.1MB, m4v) via http://dx.doi.org/10.1212/000602_Video_1
Data Availability Statement
Variant information for p.H158R deposited in ClinVar (SCV001468640). Sequencing and clinical descriptions of p.L13P patient described in Jin et al.,6 doi.10.1038/s41588-020-0695-1. Any data not published within the article will be shared by request from any qualified investigator.