

Abstract
Cyclic dinucleotides (CDNs), such as c-di-GMP (CDG), are agonists for stimulator of interferon genes (STING) and are promising for cancer immunotherapy. Yet, the therapeutic efficacy of CDNs has been limited by poor delivery and biostability. Here, STING-activating DNA nanovaccines (STING-NVs) are developed, which biostabilize, deliver, and conditionally release CDG in the endosome of immune cells, elicit potent antitumor immune responses in murine and human immune cells, ameliorate immunosuppression in vitro and in the tumor microenvironment, and mediate potent cancer immunotherapy in a murine melanoma model. STING-NVs have PLA-b-PEG in the core and cytosine (C)-rich i-motif DNA on the surface. i-Motif DNA undergoes characteristic pH-responsive conformational switch, allowing efficient CDG loading via C:G base pairing at physiological pH, and CDG release in sensitive response to acidic environment such as cell endosome. STING-NVs protect CDG from enzymatic degradation. STING-NVs facilitate cell delivery. Remarkably, STING-NVs promote the endosome escape of CDG by ninefold, and potentiate antitumor immunity. STING-NVs repolarize immunosuppressive M2-like macrophages into antitumor M1-like macrophages in vitro and in the tumor microenvironment of melanoma. In a poorly immunogenic murine melanoma model, intralesional STING-NVs outperform liposomal CDG and fluoride-CDG for melanoma immunotherapy. These results suggest the great potential of STING-NVs for cancer immunotherapy.
Keywords: cancer immunotherapy, DNA engineering, i-motif, nanovaccines, STING agonists
1. Introduction
The past decade has witnessed the historical breakthrough of cancer immunotherapy.[1] However, current immunotherapy approaches, especially immune checkpoint blockade, have only benefited a small subset of cancer patients.[2] Immunostimulants hold great potential to elicit or augment antitumor immune responses, ameliorate immunosuppression systemically and locally in the tumor microenvironment (TME), and synergize or complement current immunotherapy approaches.[3] Stimulator of interferon genes (STING) agonists are a novel class of immunostimulants.[4] STING, also known as transmembrane protein 173 (TMEM173), is a cytosolic DNA sensor and an adaptor protein that primarily resides in the endoplasmic reticulum of a wide variety of immune cells, and STING activation has been involved in microbial infection, autoimmune diseases, and cell senescence.[3a] Natural STING agonists that are directly or indirectly derived from viruses, bacteria, protozoa, and dying self-cells can activate the STING signaling pathway and hence upregulate the expression of a variety of proinflammatory cytokines and chemokines, including type I interferons (IFNs), in a wide range of cells such as macrophages, dendritic cells (DCs), monocytes, myeloid derived suppressor cells (MDSCs), as well as cancer cells.[4a, b] STING agonists, such as CDNs, have been developed and tested for the immunotherapy of cancer and infectious diseases caused by coronavirus and influenza[5] Naturally derived CDNs are particularly attractive as candidates for immunotherapy, such as cyclic dimeric guanosine monophosphate (CDG), cyclic dimeric adenosine monophosphate (CDA), and cyclic GMP-AMP (cGAMP).[6] In addition to activate innate immune responses that are important for cancer immunotherapy, STING agonists have been reported to elicit adaptive immune responses by promoting cancer antigen presentation.[5d,e,f] Moreover, recent studies demonstrated that STING activation stimulated immunogenic cell death, and elicited antitumor CD4+ and CD8+ T cells, which promoted the cancer immunotherapeutic efficacy when combined with immune checkpoint blockade.[5g] However, the development of CDN immunotherapeutics, typically administered intralesionally, has been hampered by the poor pharmacokinetics given their hydrophilicity, small molecular sizes, negative charges, and susceptibility to enzymatic degradation.[3c,4c] Current approaches to address these complications include chemical modifications that enhance the biostability of CDNs,[7] and drug delivery systems based on passive loading or electrostatic loading into liposomes,[5f,8] polymer nanoparticles (NPs),[5d] polymersomes,[5e,9] inorganic NPs,[10] and hydrogels.[11]
Herein, by leveraging a novel CDG loading and release mechanism, we report STING-activating DNA nanovaccines (STING-NVs) that protect CDG from enzymatic degradation, promote CDG delivery, enable conditional CDG release in the acidic endosome, and facilitate endosome escape of CDG in immune cells to remodel the tumor immune microenvironment for cancer immunotherapy. Specifically, CDG was loaded into i-motif DNA nanoparticles to form STING-NVs under physiological pH via hydrogen bonding (i.e., G:C base pairing) between the guanosine (G) in CDG and the C in i-motif DNA that was coated on the surface of polymer NPs; when delivered into the acidic endosome of immune cells, CDG was released from STING-NVs in response to acidic pH to modulate the tumor immune microenvironment for cancer immunotherapy (Scheme 1). i-Motifs are DNA with multiple domains of consecutive C. i-Motifs are featured with pH-responsive conformational reconfiguration between 1) linear strands under neutral or physiological pH, and 2) intermolecular or intramolecular four-stranded C-quadruplexes that are formed via protonated C:C+ base pairing under a dynamic pH range of 5–7.[12] Taking advantage of this feature of i-motifs, we modified i-motif DNA on the surfaces of poly(d,l-lactide)-block-poly(ethylene glycol) (PEG-b-PLA) NPs to enable 1) CDG loading by binding with i-motifs at physiological pH, and 2) conditional CDG release upon the formation of C-quadruplexes under acidic conditions that competed with CDG for C:C+ base pairing. We optimized the lengths of i-motifs for CDG loading into NPs, and validated conditional CDG release from the resulting STING-NVs under a range of acidic conditions. STING-NVs protected CDG from enzymatic degradation. STING-NVs were efficiently delivered into immune cells, and facilitated the endosome escape of CDG into cytosol where CDG can interact with STING. STING-NVs dramatically potentiated the immunostimulatory efficacy of CDG in both murine and human immune cells. STING-NVs elicited potent antitumor immune responses in vitro and in vivo, via, for example, repolarizing immunosuppressive M2-like macrophages to antitumor M1-like macrophages. Consequently, in a poorly immunogenic melanoma mouse model, STING-NVs showed superior immunotherapeutic efficacy relative to free CDG, fluoride-modified CDG (F-CDG), which is a current gold standard of biostable CDG, and liposomal CDG (Lipo-CDG).
Scheme 1.
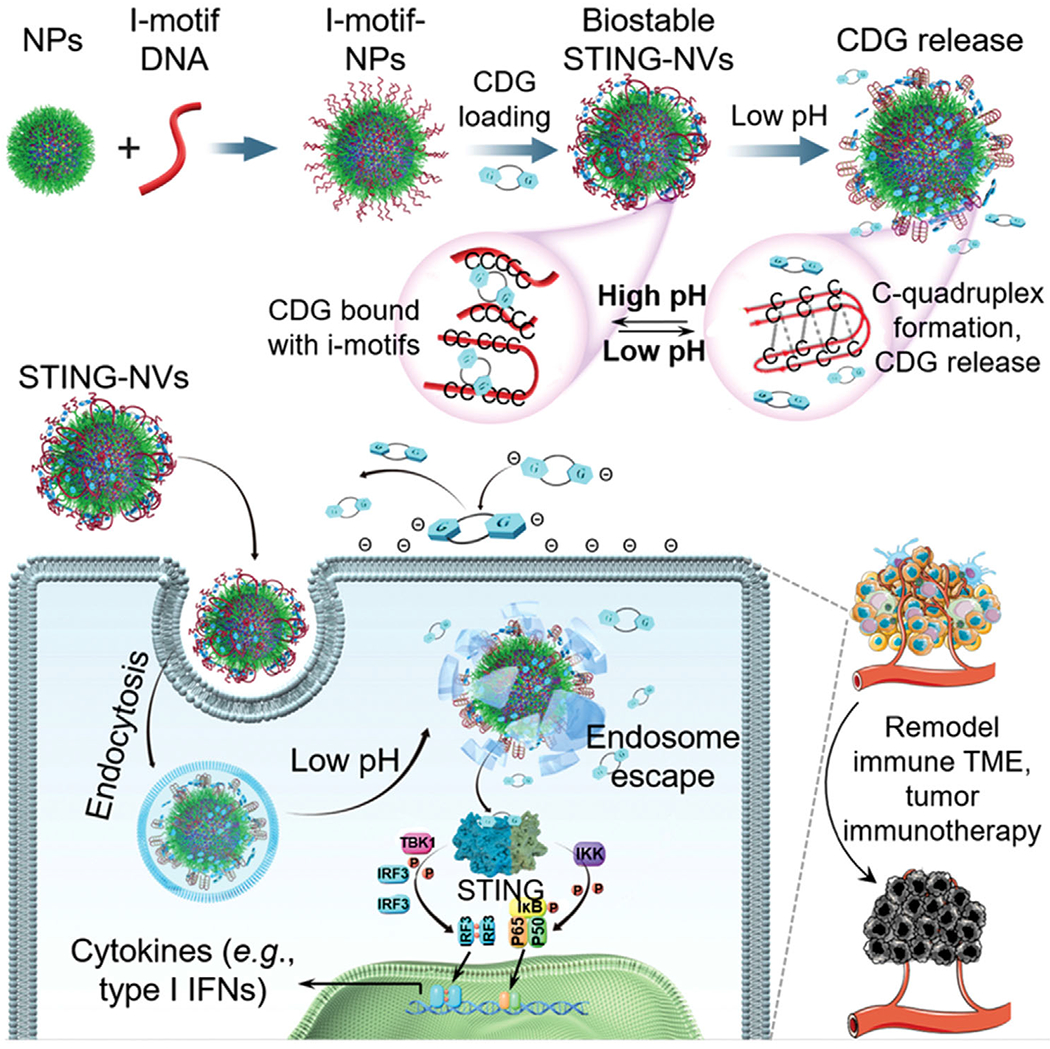
Schematic illustration of pH-responsive STING-NVs that efficiently load CDG at physiological pH, stabilized CDG, delivered CDG to immune cells, conditionally release CDG in the acidic endosome, and facilitated endosome escape of CDG for cancer immunotherapy. CDG was loaded in DNA i-motif-coated PEG-b-PLA NPs by hydrogen bonding (i.e., G:C base pairing) under physiological pH (linear i-motifs). Under acidic conditions such as immune cell endosome, i-motifs form C-quadruplexes through protonated C:C+ base-pair formation, leading to dissociation of CDG from i-motifs and hence CDG release from STING-NVs. TBK1: TANK-binding kinase 1; IRF3: interferon regulatory factor 3; IKK: Iκ B kinase.
2. Results and Discussion
2.1. Characterization of STING-NVs
To synthesize the core of STING-NVs, we used biocompatible amphiphilic diblock copolymer PEG-b-PLA to self-assemble micellular NPs with hydrodynamic diameters of 78 ± 35 nm in water solution as measured by dynamic light scattering (DLS) (Figure S1a, Supporting Information). Next, thiol-modified i-motif DNA, which consisted of 4 segregated domains of consecutive Cs (sequences in Table S1 in the Supporting Information), were conjugated with the terminal maleimide of PEG-b-PLA on the surfaces of the above NPs. To optimize the lengths of i-motifs for CDG loading, we synthesized NPs with four different i-motifs that respectively have 3, 5, 7, and 9 consecutive Cs in each C domain, as well as a scramble DNA as a control (Table S1, Supporting Information). UV–vis absorption spectra and gel electrophoresis verified the conjugation of DNA with PEG-b-PLA NPs (Figure 1a,b; Figure S2, Supporting Information). The resulting DNA-modified NPs have hydrodynamic sizes of 80–90 nm (Figure S1a, Supporting Information). The zeta potential of i-motif-NPs (≈−30 mV) decreased relative to PEG-b-PLA NPs (≈−20 mV) (Figure S3, Supporting Information), which further verified the conjugation of negatively charged DNA on NPs. To optimize i-motif lengths for CDG loading, CDG was incubated with the above five DNA-modified NPs, respectively, followed by purification via centrifuge filtration and quantification of loaded CDG in the NPs by UV–vis spectrometry. As a result, 5C-i-motif-modified NPs (5C-NPs) exhibited the highest loading efficiency (53% ± 1%) and loading capacity (2.93% ± 0.17% w/w) (Table S1, Supporting Information). Therefore, 5C-NPs and the corresponding CDG-loaded NPs (thereafter termed as STING-NVs) were used for the following studies. The hydrodynamic diameters of STING-NVs were 81 ± 36 nm (Figure 1c), as verified by transmission electron microscope (TEM) (Figure 1d). Due to the negative charges of i-motifs and CDG, the zeta potential of all CDG-loaded i-motif-NPs showed strong negative charge, whereas a control of CDG-loaded cationic liposomes (Lipo-CDG) was still positively charged (Figure 1e). Note that a 20:1 of PEG-b-PLA to 5C-i-motif molar ratio in the initial reaction yielded the highest loading efficiency (53% ± 1%), and this ratio was used for further studies (Table S2, Supporting Information). Interestingly, STING-NVs dramatically improved the biostability of CDG. After incubation in cell culture medium supplemented with 10% serum for 7 days, STING-NVs showed 91.4% intact CDG as determined by the characteristic absorption of CDG at 254 nm, in contrast to only 2.3% for free CDG (Figure 1f). The ability of STING-NVs to promote the biostability of CDG is presumably due to the sterical hindrance of i-motif-NPs that prevent the access of nucleases. Using HPLC and microscale thermophoresis (MST), we further studied the binding of CDG with 5C-i-motifs, which is the basis for CDG loading into i-motif-modified NPs. In HPLC, relative to free CDG and 5C-i-motif, the delayed elution time of physically mixed CDG and 5C-i-motif indicated the complexation of CDG and 5C-i-motif (Figure S4, Supporting Information). By MST, the binding affinity (Kd) of CDG with 5C-NPs was determined to be 47 × 10−9 m (Figure 1g,h), in contrast to negligible binding of CDG with scramble-NPs (Figure S5a,b, Supporting Information). The strong and specific binding between CDG and 5C-NPs provided the foundation for efficient CDG loading in 5C-NPs.
Figure 1.
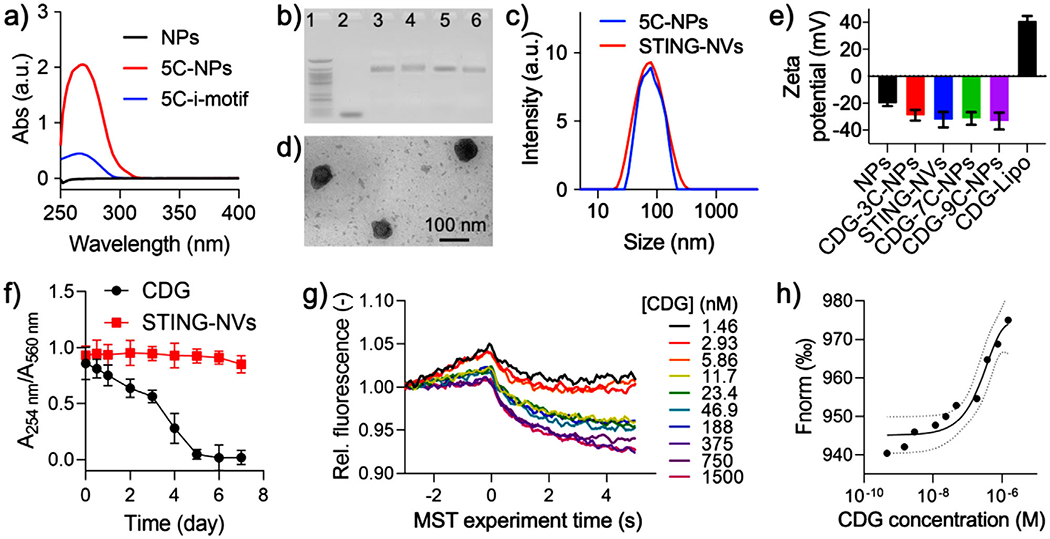
Characterization of STING-NVs. a) UV–vis absorbance spectra of PEG-b-PLA NPs, 5c-i-motif DNA, and 5c-NPs. b) An agarose electrophoresis image that showed conjugation of i-motifs on NPs. Lane legends: 1) 25 bp DNA ladder; 2) 9C-i-motif; 3) 3C-NPs; 4) 5C-NPs; 5) 7C-NPs; 6) 9C-NPs. c) DLS graphs showing the hydrodynamic diameters of 5C-NPs (79 ± 34 nm, PDI 0.157) and STING-NVs (81 ± 32 nm, PDI 0.131). d) A TEM image of STING-NVs. e) Zeta potential of blank NPs, CDG-loaded NPs, and Lipo-CDG. STING-NVs: CDG-loaded 5C-NPs. f) Stability of free CDG and CDG loaded in STING-NVs over 7 day incubation in 10% FBS-supplemented cell culture medium (37 °C). A254nm: 254 nm absorption from CDG; A560nm: 560 nm absorption from medium as an internal reference. g) MST traces and h) fitting curve of the kinetic interaction between 5C-NPs and CDG at pH 7.4. Data represent mean ± standard deviation (SD) (n = 3).
2.2. pH-Responsive Cumulative CDG Release from STING-NVs
Next, we evaluated the pH-responsive CDG release from STING-NVs. We hypothesize that under acidic condition, the formation of i-motif C-quadruplexes via protonated C:C+ base pairing competes with CDG binding with the Cs in i-motifs, leading to CDG release from the i-motifs in STING-NVs. By MST, we verified negligible binding of CDG with 5C-NPs at pH 5.0 (Figure S5c,d, Supporting Information). A release study in different pH at 37 °C showed that slightly acidic condition (pH 6.8) already significantly promoted CDG burst release from STING-NVs, and CDG release was further facilitated by lowering the pH down to pH 5.0 (Figure 2). Specifically, at pH 5.0, the CDG release half-lives from 3C-NPs, STING-NVs, 7C-NPs, and 9C-NPs are 0.10, 0.13, 0.14, and 0.16 h, respectively. CDG release from STING-NVs was increased by over five times within 1 h at pH 5.0 (≈65%) relative to pH 7.4 (≈12%). CDG release plateaued at 55–65%, presumably due to the equilibrium of CDG: i-motif binding and C-quadruplex formation. These results provide the basis for conditional CDG release from STING-NVs in the acidic endosome of immune cells.
Figure 2.
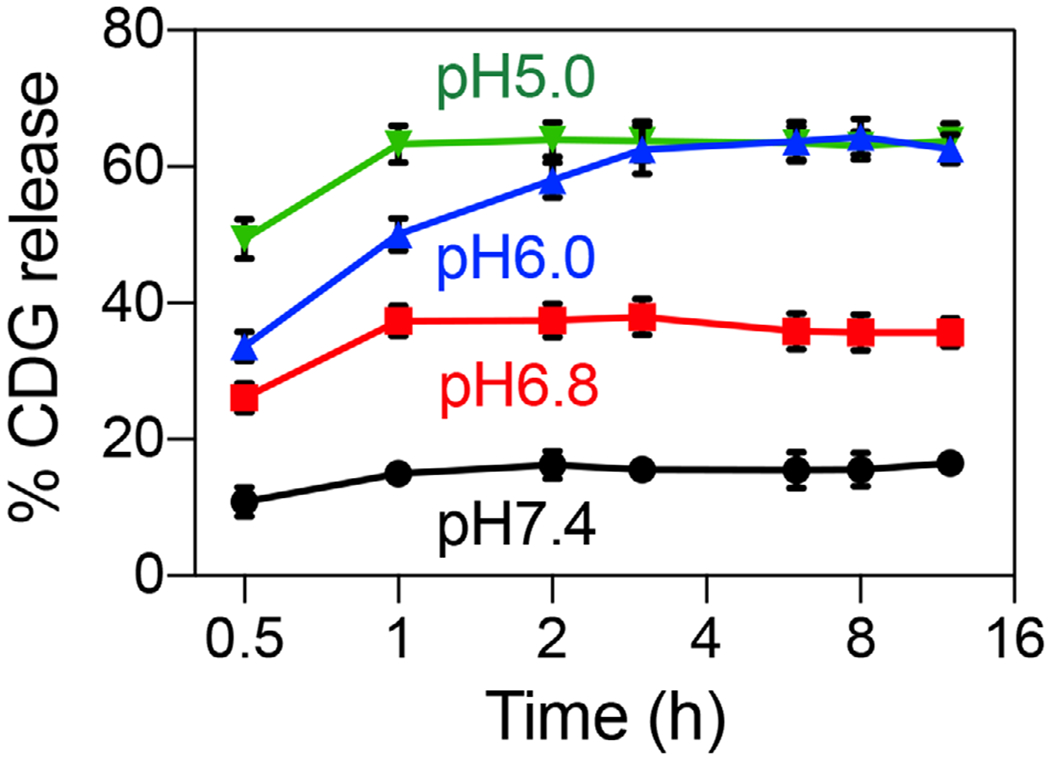
pH-responsive cumulative CDG release from STING-NVs. Data represent mean ± SD (n = 3).
2.3. STING-NVs Promoted the Intracellular Delivery of CDG and Facilitated Endosome Escape of CDG in Macrophages
We then studied the intracellular delivery of STING-NVs in macrophages, which are commonly involved in STING activation, with free CDG and Lipo-CDG[13] as controls. Fluorescein-conjugated CDG (FluoCDG) was used to monitor CDG uptake by flow cytometry and fluorescence confocal laser scanning microscopy (CLSM). RAW264.7 murine macrophages treated with free CDG showed only slight FluoCDG signal enhancement over 2 h; in contrast, cells treated with Lipo-CDG and STING-NVs showed significant FluoCDG fluorescence signal enhancement 0.5 h postincubation, which reached an order of magnitude higher FluoCDG fluorescence signal intensities than untreated cells within 1 h (Figure 3a). Interestingly, the intracellular FluoCDG signal of cells treated with STING-NVs outperformed cells treated with cationic Lipo-CDG that is typically efficient at intracellular delivery. This is presumably due to the less efficient release [only 10% CDG release from Lipo-CDG in 12 h (Figure S6, Supporting Information)] and less efficient endosome escape of CDG in Lipo-CDG than CDG in STING-NVs (Figure 3b,c). Indeed, CLSM revealed massive colocalization of endolysosome with STING-NVs as well as Lipo-CDG, respectively (Figure 3b). Remarkably, the ratio of intracellular FluoCDG fluorescence signal intensities outside over inside endolysosome is 0.52 for STING-NVs, which is nearly nine times of the ratio for Lipo-CDG (0.06) (Figure 3c). These results indicated that STING-NVs mediated efficient endosome escape of CDG, which is essential for CDG to activate STING that is located in the endothelium reticulum.
Figure 3.
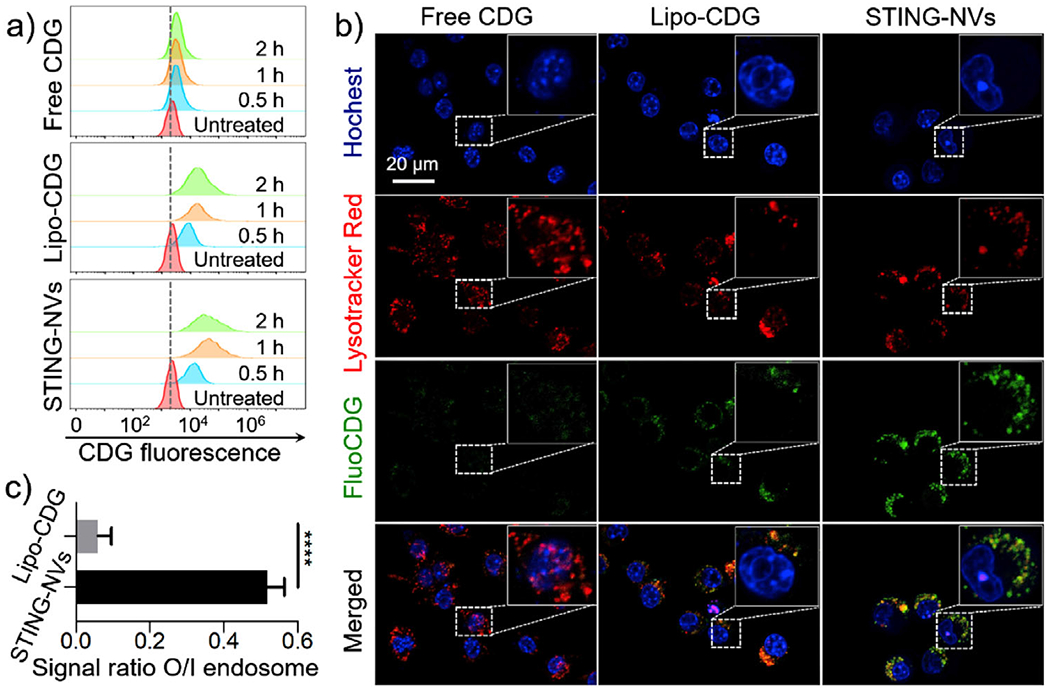
Efficient intracellular uptake and endosome escape of STING-NVs in macrophages. a) Flow cytometry results of RAW264.7 macrophages treated with free CDG, Lipo-CDG, and STING-NVs, respectively, for 0.5, 1, and 2 h. b) CLSM images of RAW264.7 cells treated with the above formulations for 1 h. Blue: nuclei stained with Hoechst33342. Red: endolysosome stained with LysoTracker Red DND-99. Green: FluoCDG. Insets: close-up views of single cells. c) The signal ratio of FluoCDG outside/inside (O/I) endolysosome, which was stained using Lysotracker Red in RAW264.7 cells after treatment for 1 h. FluoCDG: 0.5 μg mL−1 CDG equiv. Data represent mean ± SD (n = 10). ****p < 0.0001, by Student’s t-test.
2.4. Potent Immunostimulation by STING-NVs
Given efficient intracellular delivery and endosome escape of STING-NVs, we then evaluated STING-NVs for STING activation in macrophages and DCs, both of which are critical for tumor immunotherapy. Given the ability of CDNs to repolarize protumorigenic M2-like phenotype to antitumor M1-like phenotype in mouse and human macrophages,[8c] we studied STING-NVs for macrophage repolarization using a model of murine macrophage. Specifically, M2-like macrophages were generated by treating RAW264.7 murine macrophages with interleukin-4 (IL-4) (Figure 4a). As a control, M1-like macrophages were generated by treating RAW264.7 cells with lipopolysaccharide (LPS) and IFN-γ. In M2 cells, STING-NVs treatment for 24 h significantly enhanced the secretion of proinflammatory factors IL-6, IL-12p40, and IFN-α, compared to free CDG and F-CDG (Figure 4b). Moreover, immunostaining and flow cytometry revealed that, STING-NVs significantly upregulated the expression of M1 markers MHC class II (I-A/I-E) and costimulatory factor CD86, and downregulated the expression of M2 marker CD206 (Figure 4c; Figures S7–S9, Supporting Information). Consistently, as shown by quantitative reverse transcription PCR (RT-qPCR), compared to free CDG, F-CDG, and 5C-NPs, STING-NV treatment in M2 cells significantly decreased the mRNA levels of M2 gene markers Ym1, Fizz1, Arg1, and Mrc1 (Figure 4d), and significantly increased the mRNA levels of M1 gene markers Nos2, Tnf, IL6, and IL12b (Figure 4e). These results indicated that STING-NVs promoted the ability of CDG to repolarize M2-like macrophage to M1-like cells, which is expected to elicit innate and adaptive antitumor immune responses. Worth noting, STING-NVs outperformed Lipo-CDG, which is perhaps one of the current gold standards of CDN carriers, for immunostimulation (Figure 4).
Figure 4.
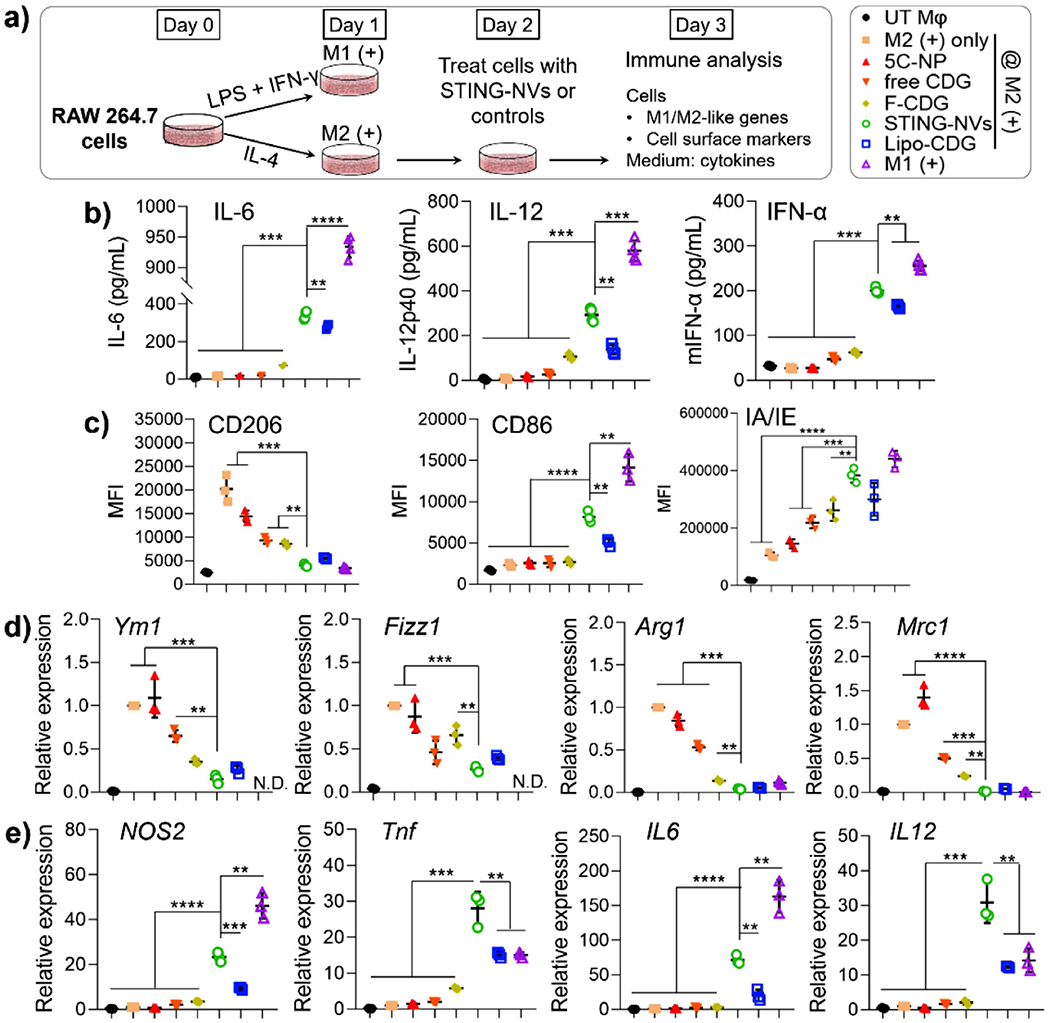
STING-NVs promoted the conversion of M2-like macrophages into M1-like macrophages. a) Murine RAW264.7 macrophages were treated with LPS and IFN-γ to induce an M1-like phenotype, and were treated with IL-4 to induce an M2-like phenotype. UT Mφ, untreated macrophage. On day 2, M2 cells were treated with free CDG, F-CDG, 5C-NPs, STING-NVs, or Lipo-CDG, respectively (1.5 μg mL−1 CDG equiv.) (n = 3). b) ELISA analysis of IL-6, IL-12, and IFN-α levels in supernatants collected on day 3. c) On day 3, treated cells were analyzed by flow cytometry for the levels of M1 markers (I-A/I-E and CD86) and M2 marker (CD206). d) On day 3, treated cells were lysed for the RT-qPCR analysis of mRNA levels of M2 markers (Ym1, Fizz1, Arg1, and Mrc1) and e) M1 markers (NOS2, Tnf, IL6, and IL12). N.D.: not determined. Data represent mean ± SD. b–e) **p < 0.01; ***p < 0.001; ****p < 0.0001, by one-way ANOVA with a Tukey’s post-hoc test (vs STING-NVs).
We further studied STING-NVs for immunostimulation in DCs using murine DC2.4 cells. Cells were again treated with STING-NVs for 24 h, followed by flow cytometric analysis of cell surface expression levels of MHC class II (IA/IE) and costimulatory factor CD80 and CD86, as well as ELISA analysis of IL-6, IL-12p40, and IFN-α in cell culture medium. Consistent with the studies in macrophages, compared with free CDG and F-CDG, STING-NV treatment in DC2.4 cells significantly promoted the secretion of IL-6, IL-12p40, and IFN-α (Figure S10a–c, Supporting Information). Further, STING-NVs elevated the expression levels of MHC-II, CD86, and CD80 on DC2.4 cells, relative to soluble CDG or CDG-F (Figure S10d, Supporting Information). Given the discrepancy of the responsiveness of human and murine STINGs to agonists, we also evaluated STING-NVs to activate type I IFN responses in human monocytes (Figure 5). Specifically, in human THP-1 cells, STING-NVs dramatically promoted IFN-α/β production in THP-1 cells, especially compared to F-CDG. Overall, STING-NVs showed potent immunostimulatory efficacy in mouse and human immune cells.
Figure 5.
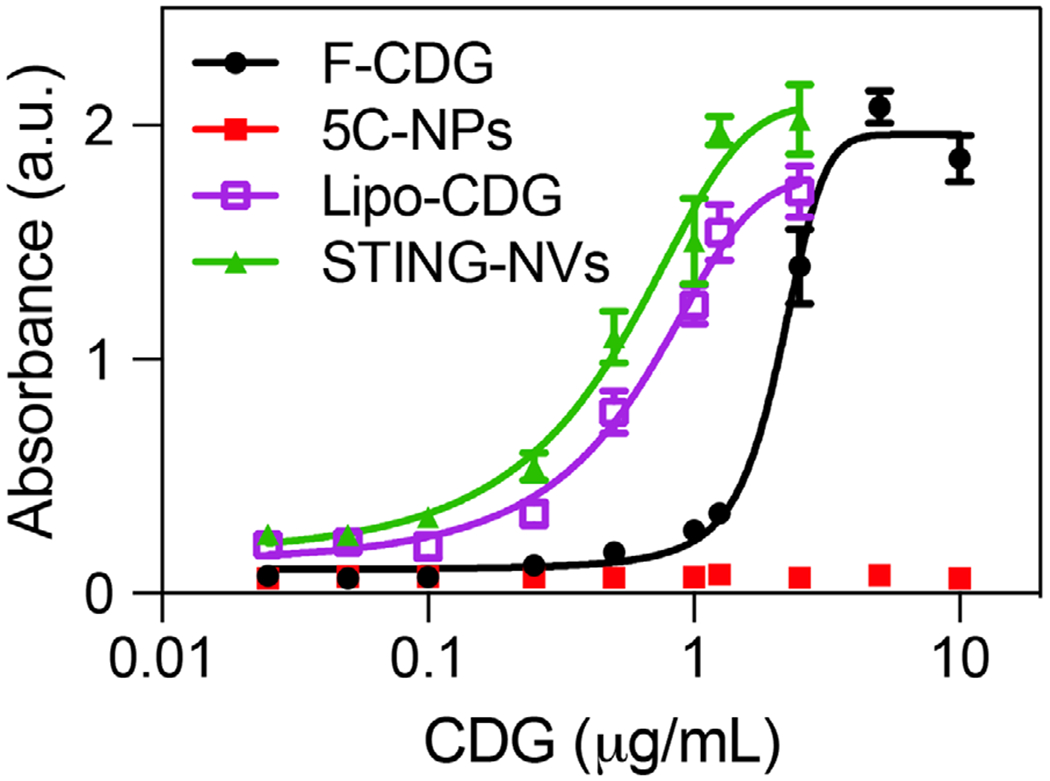
Potent immunostimulation by STING-NVs in human THP-1 monocytes. Shown is the dose-responsive IFN-α/β production after treatment with STING-NVs and controls for 24 h, as measured using HEK-Blue IFN-α/β reporter cells. Absorption: 630 nm.
2.5. STING-NVs Modulated the Immune Milieu in TME and Mediated Potent Immunotherapy
Encouraged by the above in vitro results, we then studied STING-NVs to modulate the tumor immune microenvironment in poorly immunogenic B16F10 melanoma tumors in syngeneic C57BL/6 mice. Subcutaneous tumor (≈60 mm3) were intratumorally administered with STING-NVs, with controls of PBS, CDG-F, 5C-NPs, and Lipo-CDG at a low dose of 3 μg every 3 days for five times (Figure 6a). 3 days after the last treatment, tumor tissues were harvested, homogenized, and lysed, followed by using RT-qPCR to analyze the tumor immune milieu related mRNA transcript levels in the TME. As a result, relative to all control regimens, STING-NVs significantly decreased M2-associated gene transcripts Ym1, Arg1, and Mrc1 (Figure 6b), and increased M1-associated gene transcripts Tnf, IL6, and Nos2 (Figure 6c). Moreover, STING-NVs also increased the expression of Ifnb1, Cxcl9, and Cxcl10 (Figure 6d), which are critical mediators of antitumor T cell activation and recruitment.[8a,9a,14] These results demonstrate the ability of STING-NVs to repolarize macrophages from an M2-like phenotype toward an M1-like phenotype and promote the activation of antitumor T cell responses in vivo. Overall, STING-NVs showed the ability to reprogram tumor immune milieu to reduce immunosuppression in vivo, which is critical for tumor immunotherapy.
Figure 6.
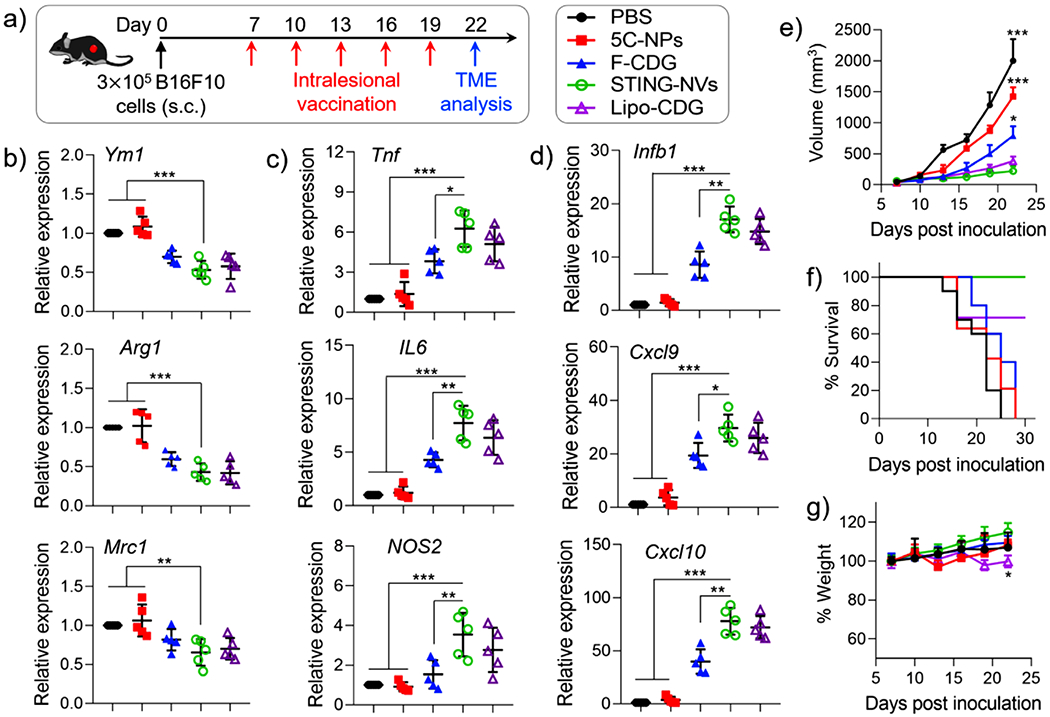
STING-NVs remodeled the tumor immune microenvironment for potent immunotherapy. a) Study scheme using B16F10 melanoma in syngeneic C57BL/6 mice. When tumor reached ≈60 mm3, mice were intratumorally administered with PBS, F-CDG, 5C-NPs, Lipo-CDG, or STING-NVs, respectively, every 3 days for five times (dose: 3 μg CDG equivalents). b–d) 3 days after the last treatment (day 22), RT-qPCR was used to analyze the transcript levels of b) M2 gene markers (Ym1, Arg1, and Mrc1), c) M1 gene markers (Tnf, Il6, and Nos2), and d) antitumor markers Ifnb1, Cxcl9, and Cxcl10 in B16F10 tumors (n = 5). After the same treatment as above, e) tumor volumes, f) mouse survival, and g) mouse body weights were monitored (n = 5–10 biologically independent samples, respectively). b–d) Data represent mean ± SD or e,g) mean ± s.e.m. b–g) *p < 0.05; **p < 0.01; ***p < 0.001, by one-way ANOVA with a Tukey’s post-hoc test (vs STING-NVs).
Given the ability of STING-NVs to remodel the TME, we studied STING-NVs for tumor immunotherapy in the above B16F10 melanoma mouse model in C57BL/6 mice. STING-NVs were administered intralesionally, as typically performed in current clinical studies of CDN-based immunotherapy. With the same treatment scheme as above (Figure 6a), STING-NVs significantly reduced tumor growth (Figure 6e) and prolonged mouse survival (Figure 6f), compared with blank 5C-NPs and F-CDG. Worth noting, STING-NVs showed the trend of superior therapeutic efficacy and survival benefit than Lipo-CDG, presumably due to the pH-responsive CDG release and efficient endosome escape. Remarkably, after STING-NV treatment, 5/10 melanoma tumors showed no significant tumor growth within the observation period (Figure S11, Supporting Information), whereas F-CDG only slowed tumor growth. Further, no significant body weight changes in STING-NVs treatment were observed over the study period of 22 days, indicating good biocompatibility of these STING-NVs (Figure 6g). By contrast, treatment with cationic Lipo-CDG led to significant weight lose despite its ability to slow down tumor growth. The ability of STING-NVs to significantly reduce tumor growth rate and prolong mouse survival without significant toxicity in a poorly immunogenic melanoma mouse model demonstrated the potential of intralesional STING-NVs for potent tumor immunotherapy.
3. Conclusion
In summary, by leveraging the pH-conditional structural reconfiguration of C-rich DNA i-motif and the resulting pH-responsive interaction between CDG and i-motifs, we developed pH-responsive STING-NVs for CDG delivery to modulate tumor immune microenvironment for cancer immunotherapy. STING-NVs efficiently loaded CDG at physiological pH, and dramatically protected CDG from enzymatic degradation. STING-NVs promoted CDG delivery into immune cells such as DCs and macrophages, and mediated pH-responsive CDG release in the acidic endosome upon C-quadruplex formation that led to CDG dissociation from i-motifs. Interestingly, STING-NVs facilitated the endosome escape of CDG into cytosol, which allow CDG to access and activate STING in the endothelium reticulum. In both mouse and human immune cells (macrophages, DCs, or monocytes), STING-NVs elicited potent immune responses, including type I IFN responses which are characteristic of STING activation. STING-NVs repolarized immunosuppressive M2-like macrophages into antitumor M1-like macrophages in a macrophage model in vitro. This was recapitulated in vivo, where STING-NVs repolarized M2-like macrophages toward M1-like macrophages in the TME of a poorly immunogenic melanoma model. Specifically, STING-NVs downregulated M2 gene markers and upregulated M1 gene markers in vitro and in vivo, and also promoted antitumor T cell activation and recruitment in vivo. In a poorly immunogenic melanoma model in syngeneic mice, STING-NVs showed superior immunotherapeutic efficacy relative to F-CDG and Lipo-CDG, which are perhaps the current gold standards of CDNs. Taken together, STING-NVs represent a novel CDG delivery system featured with efficient CDG loading presumably via G:C base pairing and conditional CDG release under acidic endosome of immune cells; STING-NVs exhibited potent immunostimulation in vitro and in vivo, resulting in potent immunotherapy with undetectable side effects in an aggressive melanoma mouse model. These results suggest the potential of STING-NVs to expand the armamentarium to promote the efficacy of cancer immunotherapy.
Supplementary Material
Acknowledgements
The authors thank Dr. Srinivas Sistla for assistance with MST, and Drs da Rocha, Hawkridge, McClay, and McRae for facility sharing. G.Z. acknowledges partial support from the Center for Pharmaceutical Engineering and Sciences-VCU School of Pharmacy, NIH KL2 Scholarship and Endowment Fund from VCU C. Kenneth and Dianne Wright Center for Clinical and Translational Research (UL1TR002649), American Cancer Society Institutional Research Grants (IRG-18-159-43), and VCU Presidential Research Quest Fund. Y.Z. and T.S. were supported by grant from the National Science Foundation for Young Scientists of China (21804038) and China Postdoctoral Science Foundation Funded Project (2019M663200). Microscopy was performed at the VCU Microscopy Facility, supported in part by funding from NIH-NINDS Center Core Grant 5 P30 NS047463 and, in part, by funding from the NIH-NCI Cancer Center Support Grant P30 CA016059.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
An invention disclosure has been filed via Virginia Commonwealth University, USA.
Contributor Information
Yu Zhang, Center for Translational Medicine, Precision Medicine Institute, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510080, China; Department of Pharmaceutics, Center for Pharmaceutical Engineering and Sciences, School of Pharmacy, Virginia Commonwealth University, Richmond, VA 23219, USA; Massey Cancer Center, Virginia Commonwealth University, Richmond, VA 23219, USA; Institute for Structural Biology, Drug Discovery and Development, Virginia Commonwealth University, Richmond, VA 23219, USA.
Tingting Shen, Department of Pharmaceutics, Center for Pharmaceutical Engineering and Sciences, School of Pharmacy, Virginia Commonwealth University, Richmond, VA 23219, USA; Massey Cancer Center, Virginia Commonwealth University, Richmond, VA 23219, USA; Institute for Structural Biology, Drug Discovery and Development, Virginia Commonwealth University, Richmond, VA 23219, USA; Molecular Sciences and Biomedicine Laboratory, State Key Laboratory for Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering and College of Biology, Collaborative Innovation Center for Molecular Engineering and Theranostics, Hunan University, Changsha 410082, China.
Shurong Zhou, Department of Pharmaceutics, Center for Pharmaceutical Engineering and Sciences, School of Pharmacy, Virginia Commonwealth University, Richmond, VA 23219, USA; Massey Cancer Center, Virginia Commonwealth University, Richmond, VA 23219, USA; Institute for Structural Biology, Drug Discovery and Development, Virginia Commonwealth University, Richmond, VA 23219, USA.
Weinan Wang, Department of Pharmaceutics, Center for Pharmaceutical Engineering and Sciences, School of Pharmacy, Virginia Commonwealth University, Richmond, VA 23219, USA; Massey Cancer Center, Virginia Commonwealth University, Richmond, VA 23219, USA; Institute for Structural Biology, Drug Discovery and Development, Virginia Commonwealth University, Richmond, VA 23219, USA.
Shuibin Lin, Center for Translational Medicine, Precision Medicine Institute, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510080, China.
Guizhi Zhu, Department of Pharmaceutics, Center for Pharmaceutical Engineering and Sciences, School of Pharmacy, Virginia Commonwealth University, Richmond, VA 23219, USA; Massey Cancer Center, Virginia Commonwealth University, Richmond, VA 23219, USA; Institute for Structural Biology, Drug Discovery and Development, Virginia Commonwealth University, Richmond, VA 23219, USA.
References
- [1].a) Couzin-Frankel J, Science 2013, 342, 1432; [DOI] [PubMed] [Google Scholar]; b) Mellman I, Coukos G, Dranoff G, Nature 2011, 480, 480; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ribas A, Wolchok JD, Science 2018, 359, 1350.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Ishikawa H, Barber GN, Nature 2008, 455, 674; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ishikawa H, Ma Z, Barber GN, Nature 2009, 461, 788; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Su T, Zhang Y, Valerie K, Wang X-Y, Lin S, Zhu G, Theranostics 2019, 9, 7759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Darvin P, Toor SM, Sasidharan Nair V, Elkord E, Exp. Mol. Med 2018, 50, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Barber GN, Nat. Rev. Immunol 2015, 15, 760; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G, Nat. Rev. Immunol 2015, 15, 405; [DOI] [PubMed] [Google Scholar]; c) Zhang Y, Lin S, Wang XY, Zhu G, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol 2019, 11, e1559; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Cen X, Zhu G, Yang J, Yang J, Guo J, Jin J, Nandakumar KS, Yang W, Yin H, Liu S, Cheng K, Adv. Sci 2019, 6, 1802042; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Chang H-C, Zou ZZ, Wang QH, Li J, Jin H, Yin QX, Xing D, Adv. Sci 2020, 7, 1900069; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Saeed AFUH, Ruan X, Guan H, Su J, Ouyang S, Adv. Sci 2020, 7, 1902599; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Mancini RJ, Tom JK, Esser-Kahn AP, Angew. Chem., Int. Ed 2014, 53, 189; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Angew. Chem 2014, 126, 193. [Google Scholar]
- [5].a) Baguley BC, Ching L-M, BioDrugs 1997, 8, 119; [DOI] [PubMed] [Google Scholar]; b) Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang SY, Tran JL, Moore P, Lehmann S, Eberl HC, Muelbaier M, Schneck JL, Clemens J, Adam M, Mehlmann J, Romano J, Morales A, Kang J, Leister L, Graybill TL, Charnley AK, Ye G, Nevins N, Behnia K, Wolf AI, Kasparcova V, Nurse K, Wang L, Li Y, Klein M, Hopson CB, Guss J, Bantscheff M, Bergamini G, Reilly MA, Lian Y, Duffy KJ, Adams J, Foley KP, Gough PJ, Marquis RW, Smothers J, Hoos A, Bertin J, Nature 2018, 564, 439; [DOI] [PubMed] [Google Scholar]; c) Danilchanka O, Mekalanos JJ, Cell 2013, 154, 962; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Chattopadhyay S, Liu Y-H, Fang Z-S, Lin C-L, Lin J-C, Yao B-Y, Hu C-MJ, Nano Lett. 2020, 20, 2246; [DOI] [PubMed] [Google Scholar]; e) Lin LC-W, Huang C-Y, Yao B-Y, Lin J-C, Agrawal A, Algaissi A, Peng B-H, Liu Y-H, Huang P-H, Juang R-H, Chang Y-C, Tseng C-T, Chen H-W, Hu C-MJ, Adv. Funct. Mater 2019, 29, 1807616; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Wang J, Li P, Yu Y, Fu Y, Jiang H, Lu M, Sun Z, Jiang S, Lu L, Wu MX, Science 2020, 367, eaau0810; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Wang-Bishop L, Wehbe M, Shae D, James J, Hacker BC, Garland K, Chistov PP, Rafat M, Balko JM, Wilson JT, J. Immuno Ther. Cancer 2020, 8, e000282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE, Nature 2011, 478, 515; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Woodward JJ, Iavarone AT, Portnoy DA, Science 2010, 328, 1703; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, Hopfner KP, Ludwig J, Hornung V, Nature 2013, 498, 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ, Metchette K, Dubensky TW Jr, Gajewski TF, Cell Rep. 2015, 11, 1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Koshy ST, Cheung AS, Gu L, Graveline AR, Mooney DJ, Adv. Biosyst 2017, 1, 1600013; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hanson MC, Crespo MP, Abraham W, Moynihan KD, Szeto GL, Chen SH, Melo MB, Mueller S, Irvine DJ, J. Clin. Invest 2015, 125, 2532; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Cheng N, Watkins-Schulz R, Junkins RD, David CN, Johnson BM, Montgomery SA, Peine KJ, Darr DB, Yuan H, McKinnon KP, Liu Q, Miao L, Huang L, Bachelder EM, Ainslie KM, Ting JP, JCI Insight 2018, 3, e120638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Shae D, Becker KW, Christov P, Yun DS, Lytton-Jean AKR, Sevimli S, Ascano M, Kelley M, Johnson DB, Balko JM, Wilson JT, Nat. Nanotechnol 2019, 14, 269; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lin LC, Chattopadhyay S, Lin JC, Hu CJ, Adv. Healthcare Mater 2018, 7, e1701395. [DOI] [PubMed] [Google Scholar]
- [10].An M, Yu C, Xi J, Reyes J, Mao G, Wei WZ, Liu H, Nanoscale 2018, 10, 9311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lee E, Jang HE, Kang YY, Kim J, Ahn JH, Mok H, Acta Biomater. 2016, 29, 271. [DOI] [PubMed] [Google Scholar]
- [12].a) Modi S, M GS, Goswami D, Gupta GD, Mayor S, Krishnan Y, Nat. Nanotechnol 2009, 4, 325; [DOI] [PubMed] [Google Scholar]; b) Liu H, Xu Y, Li F, Yang Y, Wang W, Song Y, Liu D, Angew. Chem., Int. Ed 2007, 46, 2515; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2007, 119, 2567; [Google Scholar]; c) Choi J, Kim S, Tachikawa T, Fujitsuka M, Majima T, J. Am. Chem. Soc 2011, 133, 16146; [DOI] [PubMed] [Google Scholar]; d) He D, He X, Wang K, Cao J, Zhao Y, Adv. Funct. Mater 2012, 22, 4704; [Google Scholar]; e) Dong Y, Yang Z, Liu D, Acc. Chem. Res 2014, 47, 1853. [DOI] [PubMed] [Google Scholar]
- [13].a) Midoux P, Pichon C, Expert Rev. Vaccines 2015, 14, 221; [DOI] [PubMed] [Google Scholar]; b) Mukalel AJ, Riley RS, Zhang R, Mitchell MJ, Cancer Le. 2019, 458, 102; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Colapicchioni V, Palchetti S, Pozzi D, Marini ES, Riccioli A, Ziparo E, Papi M, Amenitsch H, Caracciolo G, J. Mater. Chem. B 2015, 3, 7408; [DOI] [PubMed] [Google Scholar]; d) Wasungu L, Hoekstra D, J. Controlled Release 2006, 116, 255; [DOI] [PubMed] [Google Scholar]; e) Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, Meng M, Fritz D, Vascotto F, Hefesha H, Grunwitz C, Vormehr M, Husemann Y, Selmi A, Kuhn AN, Buck J, Derhovanessian E, Rae R, Attig S, Diekmann J, Jabulowsky RA, Heesch S, Hassel J, Langguth P, Grabbe S, Huber C, Tureci O, Sahin U, Nature 2016, 534, 396. [DOI] [PubMed] [Google Scholar]
- [14].Corrales L, McWhirter SM, Dubensky TW Jr, Gajewski TF, J. Clin. Invest 2016, 126, 2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.