

Abstract
Homologous recombination (HR)-deficient cancers are sensitive to inhibitors of Poly-ADP Ribose Polymerase (PARPi), which have shown clinical efficacy in the treatment of high-grade serous cancers (HGSC). However, the majority of patients will relapse, and acquired PARPi resistance is emerging as a pressing clinical problem. Here we generated seven single-cell clones with acquired PARPi resistance derived from a PARPi-sensitive, TP53−/− and BRCA1−/− epithelial cell line generated using CRISPR/Cas9. These clones showed diverse resistance mechanisms, and some clones presented with multiple mechanisms of resistance at the same time. Genomic analysis of the clones revealed unique transcriptional and mutational profiles and increased genomic instability in comparison to a PARPi-sensitive cell line. Clonal evolutionary analyses suggested that acquired PARPi resistance arose via clonal selection from an intrinsically unstable and heterogenous cell population in the sensitive cell line, which contained pre-existing drug tolerant cells. Similarly, clonal and spatial heterogeneity in tumor biopsies from a clinical BRCA1-mutant HGSC patient with acquired PARPi resistance were observed. In an imaging-based drug screening, the clones showed heterogenous responses to targeted therapeutic agents, indicating that not all PARPi-resistant clones can be targeted with just one therapy. Furthermore, PARPi-resistant clones showed mechanism-dependent vulnerabilities to the selected agents, demonstrating that a deeper understanding on the mechanisms of resistance could lead to improved targeting and biomarkers for HGSC with acquired PARPi resistance.
Introduction
Poly ADP Ribose Polymerase inhibitors (PARPi’s) are synthetically lethal in tumors which are deficient in homologous recombination (HR) DNA repair (1, 2). More than 50% of high-grade serous ovarian, fallopian tube or peritoneal cancers (HGSC) are deficient in HR most commonly via mutations in BRCA1 or BRCA2, or other genes of the Fanconi Anemia pathway (1). PARPi’s have been shown to improve progression-free survival as first-line maintenance therapy (3, 4) and in the treatment of relapsed ovarian cancer (5–7). However, as PARPi’s are entering wide clinical use, patients with acquired PARPi resistance are becoming a predominant clinical challenge.
Extensive in vivo and in vitro preclinical studies have identified several resistance mechanisms to PARPi’s. These mechanisms can be classified into four main categories including those that (i) lead to HR restoration, (ii) influence replication fork protection, (iii) directly impact on the abundance and activity of PAR chains, and (iv) influence cellular availability of the inhibitor (reviewed in (8)). Importantly, resistance to platinum-based chemotherapies is a strong predictor of PARPi resistance, suggesting that they probably share common mechanisms (9). Restoration of BRCA1/2 protein expression is the best characterized mechanism of platinum agent resistance mediated by HR in HGSC, which can be caused either by a reversion of the original deleterious mutation in the gene (10, 11), or by gaining the expression of a hypomorphic protein (12). Downregulation of Non-Homologous End Joining (NHEJ) factors, such as 53BP1(13), and REV7 (14) and proteins of the SHIELDIN complex (15) has been shown to restore HR via promoting end resection at the DNA double strand breaks, and thus contributing to PARPi resistance. Recently, emerging preclinical evidence has demonstrated that PARPi resistance can result from enhanced replication fork protection (16–19).
Previous studies on the mechanisms of PARPi resistance have been mostly performed in cell lines, derived from already drug-resistant patients’ samples (20, 21), and only individual mechanisms of PARPi resistance have been described using these cell lines. On the other hand, patient-derived samples have led to the detection of only reversion mutations in HR genes as mechanisms of resistance (11, 22). Detailed features of PARPi resistance, such as functional heterogeneity, genomic patterns, and the evolutionary trajectories of PARP inhibitor resistance are unknown.
In an effort to further dissect the DNA repair mechanisms leading to PARP inhibitor resistance we generated a TP53 deficient, BRCA1 deficient epithelial cell line model system. We herein show that BRCA1-deficiency gives rise to a chromosomally unstable cell population with intrinsic clonal heterogeneity which can contain pre-existing drug tolerant cells. Via clonal selection, these drug tolerant cells can give rise to multiple genomically and functionally heterogenous PARPi-resistant clones. The clonal evolutionary patterns are conserved in a clinical HGSC patient with germline BRCA1- mutation and acquired PARPi resistance. Importantly, we show that PARPi resistance is associated with cell cycle-related vulnerabilities, that can be targeted in a mechanism-depended manner.
Materials and Methods
Cell lines
We generated TP53 and BRCA1- deficient cells from RPE-1 hTERT cells purchased from ATCC as previously described (23). The cells were cultured using DMEM/F12 medium supplemented with 10% FBS and 1% Penicillin-Streptavidin. The cells were regularly tested negative for Mycoplasma and authenticated using whole-exome sequencing. The cells were treated cyclically with increasing concentrations of PARP inhibitor during three months (Figure S1A). After the generation of a resistant pool, single cell clones were isolated and propagated for the characterization assays for maximum of three months or 10 passages (Figure S1B). Details of the in vitro methods including cell viability and clonogenic assays, western blotting, immunofluorescence, flow cytometry and cytogenetics, are shown in Supplementary Materials and Supplemental Table 1.
DNA and RNA isolation and sequencing
The schematics and timelines of the sequencing is shown in Figure S1, and methods in Supplementary Materials. The RNAseq and WES data are publicly available in Synapse at doi:10.7303/syn22240938.
Differential gene expression analysis and pathway analysis
Details of the RNA-seq data analysis are shown in Supplemental Materials. Briefly, the multi-dimensional scaling (MDS) plots were generated using the plotMDS function of the limma R package (24). Differential gene expression analysis was performed using the edgeR package (25). Multiple testing correction to control the false discovery rate (FDR) was performed using the Benjamini-Hochberg procedure on the p-values. Differentially expressed genes (DEGs) were determined using an FDR of 2.5%. The Gene Set Enrichment Analysis (GSEA) (26) was performed using the GSEAPreranked module of the GSEA Java application (gsea-v3.0.jar).
Whole-exome sequencing analysis
Details of the WES analysis are presented in Supplemental Materials. In brief, the sequences were aligned to hg38 using BWA. Mutations were called using MuTect2 from GATK4.0.11 and CNVs using ASCAT and GATK4.1.2. Evolutionary analysis was performed from mutations and CNVs using PyClone0.13 and ClonEvol (27). Only subclones whose frequency was above 1% and at least 5 mutations after filtering were included in the analysis. The WES, mutational calls and CNVs from a BRCA1 mutated patient was accessed from Hill et al dbGap phs001685.v1.p1. LOH score was calculated as described in (28). Mutational signature 3 analysis was performed using SigMA R package (29) in which counts of different types of single nucleotide variants (SNV) were used to identify known patterns of mutational processes (30) and to determine the exposure of Sig 3.
Image-based drug sensitivity testing
Details of the drug sensitivity testing are shown in Supplemental Materials. The growth-rate-adjusted viabilities were calculated for each drug concentration as described previously (31). The GR50 values were calculated, normalized with respect to the B40 cell line, and visualized using hierarchical clustering heatmap with column normalization. The growth rate over 72 hours was calculated using a formula Gr=LN(N(72h)/N(0))/72h.
Statistics
All statistical analyses were performed by comparing a minimum of three independent measurements. The continuous variables were evaluated for normal distribution and analyzed with non-parametric tests upon non-normal distribution. Groups were compared using Student’s t-test or Mann-Whitney-U test. Categorical variables were compared using Fisher’s exact test. Linear regression was performed using Pearsons or Spearman’s correlation. Multiple comparisons were analyzed using Dunett’s or Sidak’s multiple comparison test.
Results
TP53 and BRCA1-deficient cells acquire resistance to multiple PARP inhibitors and are cross resistant to cisplatin.
To generate a model system to understand PARPi resistance mechanisms in BRCA1 deficient cells, and as immortalized human fallopian tube epithelial cells are not readily available for extensive genetic perturbations, we used the CRISPR/Cas9 system to delete the TP53 and BRCA1 genes in the retinal pigment epithelial (RPE) cell line (17), thus generating the RPETP53−/−BRCA1−/− cell line, hereafter referred as B40, which showed in vitro hypersensitivity to PARPi (Figure S1C, D).
The B40 cell line was cyclically exposed to increasing concentrations of Niraparib or Olaparib, and subjected along with six Niraparib-resistant cell clones and one Olaparib-resistant cell clone to functional and genomic profiling (Figure 1A). The selected cellular clones were resistant to multiple PARPi, including Niraparib (Figure 1B), Olaparib (Figure 1C), and Talazoparib (Figure 1D). Long-term clonogenic assay confirmed resistance to Niraparib (Figure 1E and Figure 1F). Further, 7-day survival assays demonstrated that most of the clones were also cross-resistant to cisplatin, except for two clones NA4 and NB1 (Figure S1E, F).
Figure 1. Niraparib-resistant RPETP53−/−BRCA1−/− cell clones are cross-resistant to Olaparib and Talazoparib.
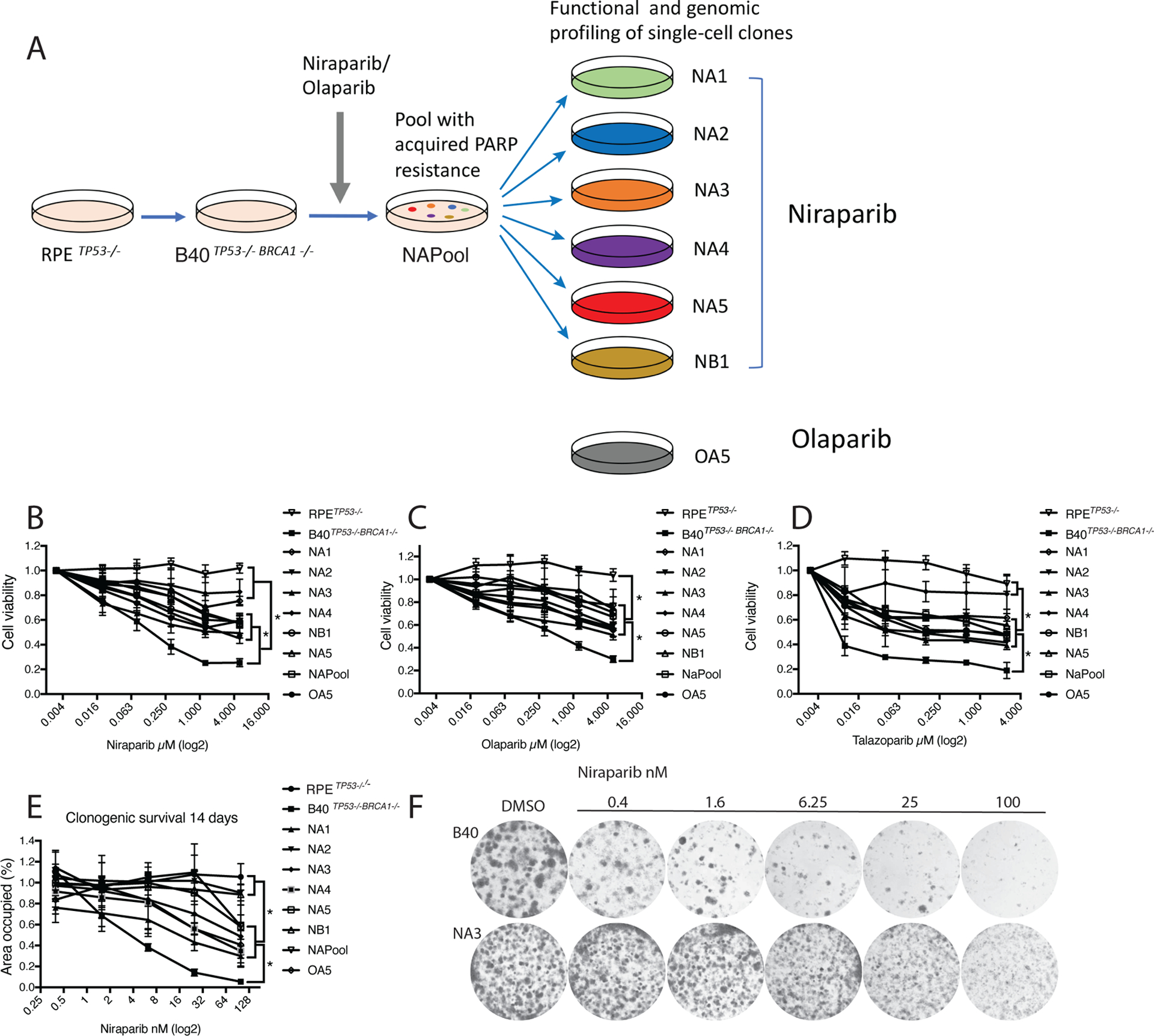
(A) RPETP53−/− and RPETP53−/−BRCA1−/− cells (B40) were generated using the CRISPR-Cas9 technology. A resistant cell line (NAPool) was generated by treating the B40 cell line with increasing concentrations of the PARP inhibitors Niraparib or Olaparib over three months. Next, we isolated single-cell clones from the Niraparib and Olaparib resistant pools for functional and genomic profiling.
(B) Resistance of these cell clones to Niraparib was confirmed in a 7-day CellTiter-Glo survival assay. The groups were compared using Oneway Anova and Tukeys multiple comparison test.
(C) Cell clones were also resistant to Olaparib, and (D) Talazoparib in a 7-day survival assay.
(E) Resistance to Niraparib was further confirmed in long-term 14-day clonogenic assays.
(F) Representative photographs showing the clonogenic growth of the NA3 resistant clone in presence of Niraparib in comparison to the B40 sensitive cell line. *p<0.05
Heterogenous mechanisms of resistance emerge from TP53 and BRCA1-deficient cells exposed to Niraparib
Using CRISPR/Cas9 we introduced a deleterious mutation in BRCA1 with the potential of reversion, therefore, we first excluded restoration of BRCA1 protein expression as a mechanism of resistance in the PARPi resistant clones (Figure S2A-B). Next, we tested the cells for 1) RAD51 foci formation, as an intermediate readout for HR-mediated DNA repair (32) and 2) DNA replication fork stabilization (reviewed in (33)). As expected, the B40 cell line lacked RAD51 foci formation both after irradiation (IR; Figure 2A) and after Niraparib treatment (Figure 2B). In contrast, most of the PARPi resistant clones, except for one (NB1), had restored RAD51 foci formation after IR (Figure 2A and Figure S2C). We next investigated the sensitivity of the cells to hydroxyurea (HU) – induced S-phase arrest and DNA replication fork destabilization as a mechanism of resistance. Interestingly, all of the PARPi resistant clones exhibited stabilized DNA replication forks when compared to the B40 cell line as quantified using the DNA fiber assay (Figure 2C).
Figure 2. PARP inhibitor resistant RPETP53−/− BRCA1−/− clones have heterogenous mechanisms of resistance.
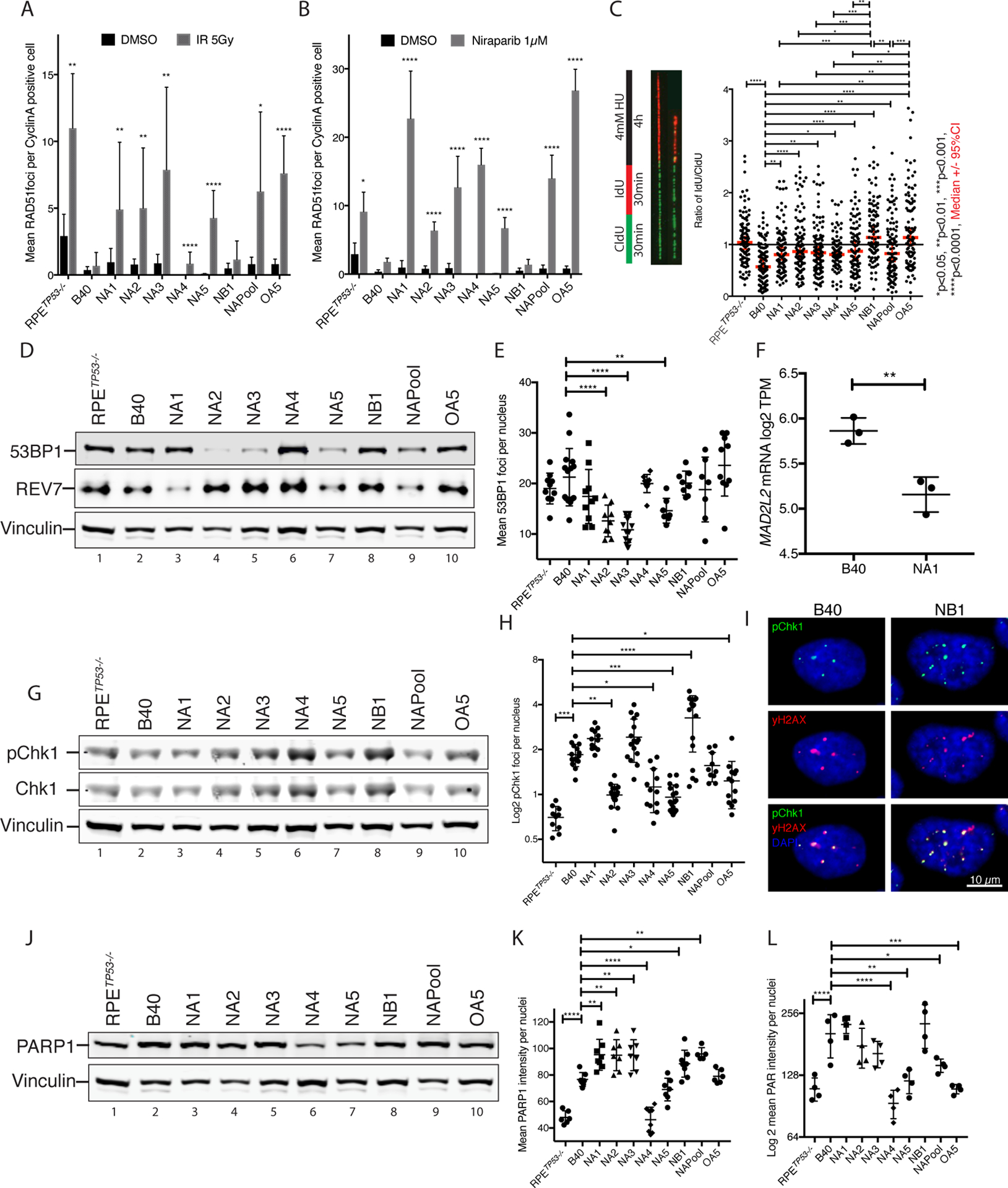
(A) The PARPi resistant clones, except for clone NB1, showed restored RAD51 foci formation 6 hours after 5Gy X-ray irradiation (IR). RAD51 foci were quantified as mean RAD51foci per CyclinA positive, S-phase cells in 5–10 40x images. Data are represented as mean and error bars represent the Standard Deviation (SD). Treatment group was compared to control using Mann-Whitney-U test for each cell line.
(B) Increased RAD51 foci formation in CyclinA positive cells after treatment with 1 μM Niraparib for 24hours in all PARPi resistant clones except for the NB1.
(C) Unstable DNA replication forks in the B40 cell line, denoted by decreased IdU incorporation after HU treatment, are stabilized in all the PARPi resistant clones as demonstrated in a DNA fiber assays. Thick red line indicates the Median, and thin lines represent the 95%CI.
(D) Western blot showing decreased 53BP1 protein levels in PARPi resistant clones NA2, NA3 and NA5, and reduced MAD2L2/REV7 protein levels in clone NA1.
(E) Decreased 53BP1 foci formation was validated in clones NA2, NA3 and in NA5 by quantification of nuclear 53BP1 foci using IF after inducing DNA double strand breaks with 5Gy IR. The data are plotted as mean 53BP1 foci per nucleus per image in 7–15 40x images.
(F) Decreased mRNA levels of MAD2L2/REV7 in clone NA1 with respect to the B40 cell line quantified using log2 TPM data from mRNA sequencing.
(G) PARPi resistant clone NB1 had increased baseline pChk1 protein expression in western blot.
(H) Quantified pChk1 foci using IF staining after 5GyI IR. The data was plotted as mean pChk1 foci per nucleus per image in 10–20 40x images. PARPi resistant clone NB1 had a significantly increased number of pChk1 foci after IR. pChk1 foci were increased in the B40 cells as compared to the RPETP53−/− cells, and decreased in NA2, NA4, NA5 and OA5 clones as compared to the B40 cells.
(I) Representative IF images of DNA double strand breaks visualized using γH2AX staining, and increased pChk1 foci in the sensitive cell line B40 and the resistant clone NB1.
(J) Western blot showing decreased PARP1 protein levels in the PARPi resistant clone NA4. PARP1 levels are also significantly higher in the B40 cells as compared to the RPETP53−/− cells.
(K) Quantification of baseline nuclear PARP1 nuclear staining in IF showed significantly reduced levels in the PARPi resistant clone NA4 compared to the B40 cell line. PARP1 levels were increased in B40 cells compared to RPETP53−/− cells, and in NA1–3, NB1 and NAPool when compared to the B40 cells.
(L) Decreased levels of PARylation after H2O2 treatment were observed using IF in clones NA4, NA5 and OA5, as well as the NAPool in comparison to the B40 cell line. PARylation levels were increased in the B40 cells compared to the RPETP53−/− cells. Data in E, F, H, J and K are plotted vertical line as the mean with whiskers representing SD. In C, E, F, H, J, K the groups were compared using Oneway Anova followed by Dunet’s test. * p<0.05, ** p<0.01, ***p<0.001, ****p<0.0001.
We next tested the cells for the presence of the other previously described mechanisms of PARPi resistance. First, we profiled the expression levels of NHEJ factors known to suppress HR. Decreased 53BP1 protein levels were observed in NA2, NA3, and NA5 clones (Figure 2D, upper panel), consistent with reduced number of 53BP1 foci (Figure 2E and Figure S2D). Further, we observed a decreased REV7 protein level in clone NA1 (Figure 2D, middle panel) consistent with reduced REV7/MAD2L2 gene expression levels (Figure 2F). Since both 53BP1 and REV7 are mediators of NHEJ, their downregulation is expected to improve HR via promoting RAD51 foci formation (13, 14), thus providing a mechanism of PARPi resistance in these clones.
The NB1 clone, which had not restored RAD51 foci, exhibited increased levels of phosphorylated Chk1 protein (pChk1) (Figure 2G) and increased pCHK1 foci (Figure 2H–I), a kinase that protects the replication fork and promotes PARPi resistance (18). This clone also had lower levels of pRPA foci, compared to B40, suggesting decreased levels of replication stress (Figure S3F). We next determined whether decreased PARP1 protein could account for the acquired PARPi resistance in the clones. We observed increased baseline levels of PARP1 in the B40 cells, compared to RPETP53−/− cells, and reduced expression in clone NA4 (Figure 2J), a finding that was confirmed by IF (Figure 2K), indicating NA4 could avoid PARPi action by downregulating PARP1 enzyme.
Consistently, an IF-based PARylation assay which measures H2O2 stimulation of PAR chains (34) verified significantly reduced PARylation levels in clone NA4 in comparison to the B40 cells (Figure 2L), suggesting that downregulation of PARP1, and avoidance of PARP trapping, may be the mechanism of resistance of this clone. In addition, clones NA5, OA5 and NAPool, had significantly reduced PARylation levels after H2O2, indicating that avoidance of PARP trapping could enhance PARPi resistance in these clones also (35). Niraparib effectively abolished PARylation in all clones, thus ruling out an efflux pump mechanism of PARPi resistance (Figure S2E). Consistently, the B40 cells showed enhanced PARylation after exposure to H2O2, in comparison to the RPETP53−/ (Figure 2 J, K), in line with the known dependency that HR deficient cells have on PARP1-mediated DNA repair (36).
Taken together, our cellular model recapitulates several of the known mechanisms described for PARPi resistance. Importantly, our results indicate that diverse resistance mechanisms can emerge from a BRCA1-deficient sensitive cell line, and that single clones, such as NA5, can simultaneously exhibit several resistance mechanisms.
PARP inhibitor resistant clones have unique gene expression profiles and are characterized by increased epithelial-to-mesenchymal transition
The PARPi resistant clones exhibited noticeable heterogeneity on resistance mechanisms, therefore we hypothesized that they also differ in their gene-expression profiles and performed RNA sequencing comparing the clones (Figure S1F). Hierarchical clustering showed that the RPETP53−/− and the B40 cells had the most similar gene expression profiles (Figure 3A), however after exposure to PARPi, the gene expression profiles of the Niraparib resistant clones formed unique clusters. Of note, the Olaparib resistant clone (OA5) clustered separately from the Niraparib resistant clones, suggesting that distinctive gene expression profiles can emerge from cells exposed to compounds with similar mechanisms of action (Figure 3A). A Multi-dimensional Scaling (MDS) plot of the total transcriptomes revealed that the most pronounced differences were among the RPETP53−/−, B40 and OA5 clones in the first two dimensions (Figure 3B, left panel). Furthermore, distinct transcriptomic profiles for the NA1, NA3 and NA4 clones were observed in the third and fourth dimensions (Figure 3B, right panel).
Figure 3. PARPi resistant clones have unique gene expression profiles.
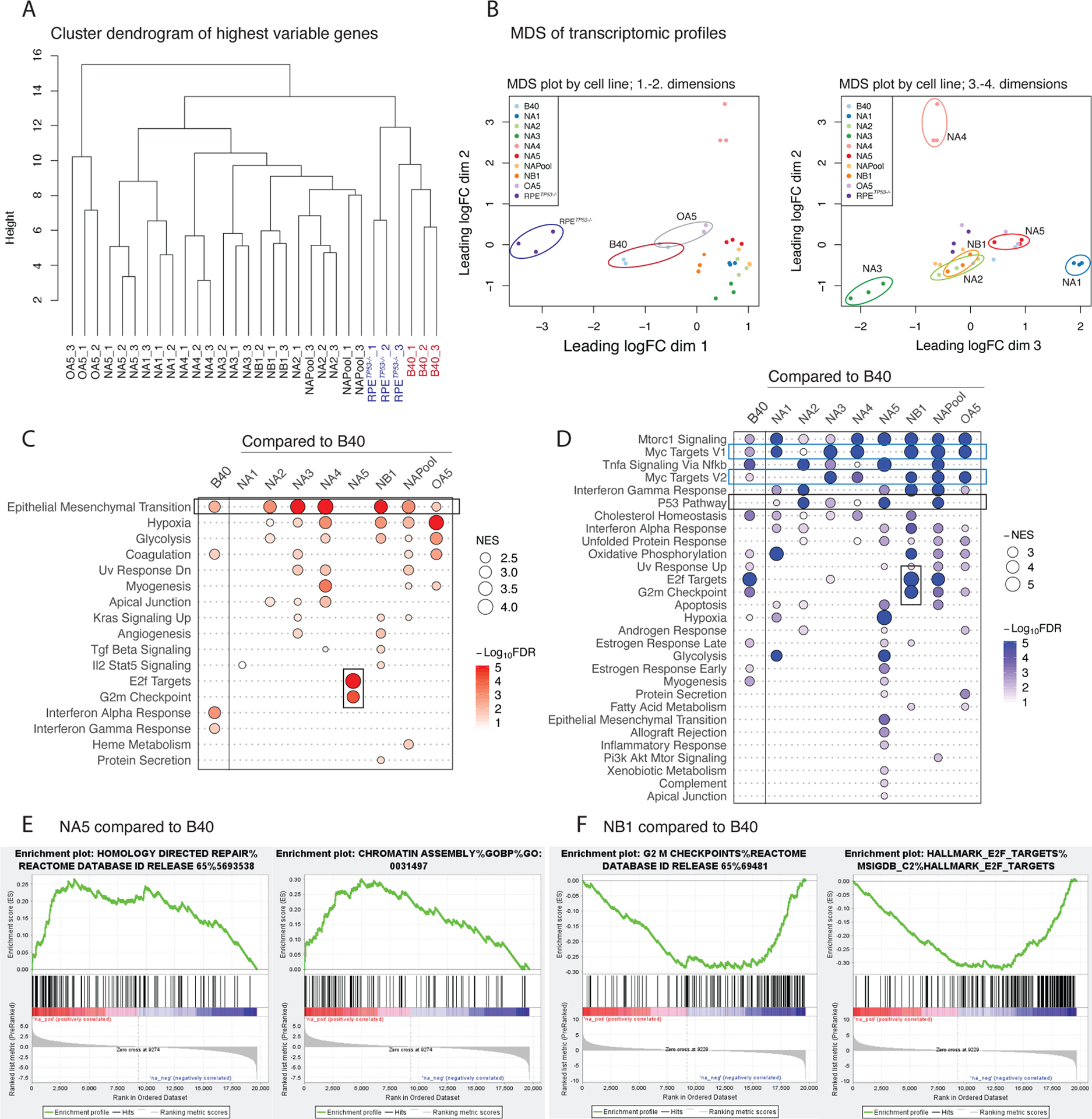
(A) Hierarchical clustering dendrogram of RNA expression profiles using the highest variable genes of the parental RPETP53−/− , B40 cells, NAPool, and the resistant clones in triplicates. RPETP53−/− and B40 cells, as well as the OA5 olaparib resistant cells form distinct clusters, whereas the Niraparib resistant clones separate later in the dendrogram.
(B) Multi-dimensional-scaling (MDS) plots of the mRNA expression profiles of the cells in triplicates. Over dimensions 1 and 2 (left panel), the RPETP53−/−, , B40, and OA5 separate from the Niraparib resistant clones. Over dimensions 2 and 3 (right panel), the resistant clones further present with distinct transcriptomic profiles. The distances in the MDS plots represent the leading log-fold-change between each pair of samples.
(C) Summary plot of the positively enriched genes of the Hallmark pathways in GSEA in B40 compared to the RPETP53−/− and in the resistant clones compared to the sensitive B40 cell line. Positive enrichment of epithelial to mesenchymal transition (EMT) was observed in the B40 cells as compared to the RPETP53−/− cells, and in six out of the seven PARPi resistant clones (upper black rectangle). In addition, NA5 showed enrichment of G2M Checkpoint and E2F target- genesets (lower black rectangle). The size of the dots represents the positive normalized enrichment score (NES) and red color intensity indicates the -Log10 FDR with <0.01 as the cut-off.
(D) Summary plots of the negative NES (-NES) of the Hallmark pathways in GSEA in B40 compared to the RPETP53−/− and in the resistant clones compared to the sensitive B40 cell line. Overall, a larger number of significant negatively enriched pathways were seen in the resistant clones compared to B40. Common downregulated pathways included MYC targets (blue rectangles) and p53 (upper black rectangle) pathways. NA5 clone had the largest number of significantly negatively enriched pathways, and in contrast to NA5, G2M checkpoint and E2F target pathways were negatively enriched in the NB1 clone (lower black rectangle). The size of the dots represents the -NES and blue color intensity indicates the -Log10 FDR with <0.01 as the cut-off.
(E) Enrichment plot of NA5 clone showed upregulation of HR- DNA repair genes (left panel) and Chromatin regulatory genes (right panel) as compared to the B40 cell line.
(F) Enrichment plot of NB1 clone showed downregulation of G2M checkpoint genes (left panel) and E2F target genes as compared to the B40 cell line.
Analysis of differentially expressed genes (DEGs) and gene-set enrichment analysis (GSEA, Table S2) showed that most of the clones had unique DEGs in comparison to the resistant pool, thus highlighting the transcriptional diversification across the resistant clones (Figure S3A). GSEA showed that the B40 cells had a significantly higher enrichment score for EMT compared to the RPETP53−/− cell line (Figure 3C). Further, the EMT pathway was significantly enriched in five of the seven PARPi-resistant clones (Figure 3C). This EMT phenotype was confirmed morphologically (Figure S3B) and by IF for the canonical EMT markers Vimentin (Figure S3C-D) and ZEB1 (Figure S3E).
Overall, the clones showed more downregulated than upregulated gene-sets (Figure 3D), indicating that gene downregulation may be a more common mechanism of acquired PARPi resistance. Common findings included downregulation of the mTORC, MYC and TNFα pathways, which were also downregulated in the B40 cells compared to the RPETP53−/−. Interestingly, the NA5 clone exhibited a significant upregulation of HR and chromatin regulatory pathways (Table S2, Figure 3E), suggesting that restoration of HR may result from changes in chromatin regulation and gene expression. On the other hand, the NB1 clone exhibited downregulation of E2F targets and G2/M cell cycle pathway genes (Figure 3F), indicating unique resistance mechanisms potentially related to cell cycle regulation. In summary, although EMT was common among all PARPi resistant clones, the individual clones exhibited unique gene expression programs, mainly resulting from downregulation of gene expression.
PARPi resistance correlates with a lower mutational burden and an increased level of genomic instability
To reveal the mutational profiles and patterns of genomic copy number variation (CNVs) of the PARPi resistant clones, we performed whole-exome sequencing (WES; Figure S1D). WES confirmed lack of BRCA1 reversion-mutation in the resistant clones. Mutation analysis and clustering showed that each of the PARPi resistant clones had unique mutational clusters (Figure 4A). In addition, analysis of three replicates from the same culture plate showed higher spontaneous heterogeneity of mutational clusters inside the B40 cell line, when compared to the PARPi resistant NAPool. Consistently, the resistant clones had, on average, fewer mutations than the B40 cell line, indicating that PARPi resistance is not associated with increased mutational events (Figure 4 B). We next quantified the mutational Signature 3 in the B40 and in the PARPi-resistant clones, using the SigMA algorithm. An enriched fraction of mutations belonging to Sig3 was evident in the B40 cell line, consistent with its HR-deficiency (Figure 4C). Interestingly, although six out of seven PARPi resistant clones had recovered the capacity for RAD51 foci formation (Figure 2A–B), most of the resistant clones still had a high fraction for mutational Signature 3, and only two clones (NA2 and NA4) had Signature 3 fractions lower than the B40 cell line (Figure 4C).
Figure 4. PARPi resistant clones have unique mutational profiles and are characterized by increased ploidy and LOH.
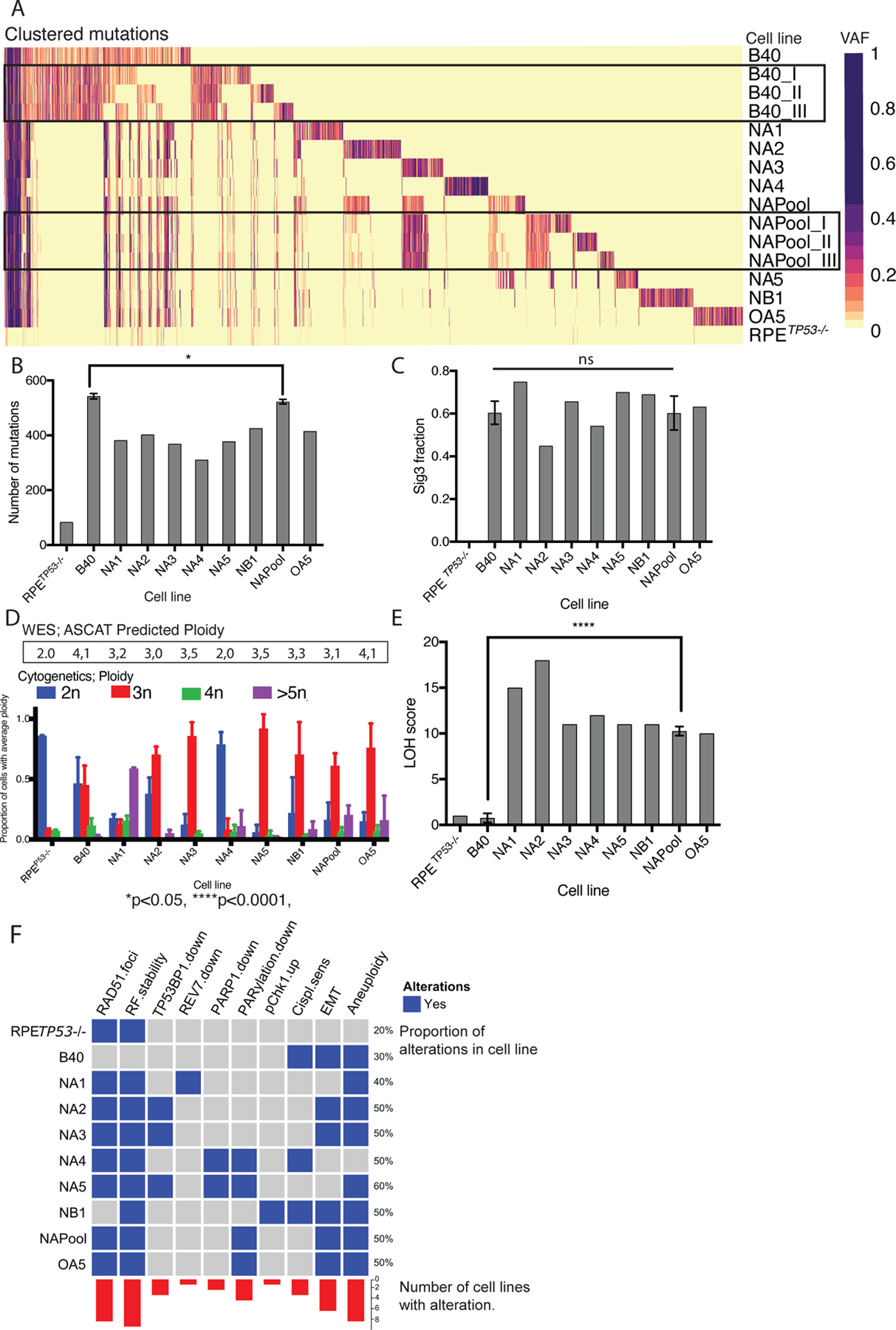
(A) Heatmap of mutational clusters by cell line. Mutational clustering revealed an a priori highly heterogenous mutational profile in the B40 cell line (three different regions sampled from the same tissue culture plate: B40_I, B40_II and B40_III), whereas the resistant NAPool showed more similar mutational clusters across regions (NAPool-I , NAPool-II and NAPool_III). Each single-cell derived resistant clone had unique mutational clusters. VAF; Variant allele frequency.
(B) The PARPi resistant clones had more mutations in comparison to the RPETP53−/− cell line, but less mutations in comparison to the B40 cell line. Bar graph of numbers of called mutations by cell line, error bars represent the SD. Groups of triplicate measurements (B40 and NAPool) were compared using Mann-Whitney U-Test. *p<0.05.
(C) The fraction of mutational Signature 3 was enriched in the B40 cell line. Two out of seven PARPi resistant clones had decreased fractions of Sig3, however there was no significant difference between the B40 cells and the NAPool. Error bars represent the SD. Groups of triplicate measurements (B40 and NAPool) were compared using Mann-Whitney U-Test. *p<0.05.
(D) WES predicted ploidy (black rectangle) was consistent with the cytogenetic analysis of ploidy using chromosome counting in metaphase spreads of the cells. The cytogenetic ploidy is shown as bar plots of proportion of cells with average ploidy (2n, 3n 4n or >5n). Error bars represent the SD.
(E) Barplot of Loss of Heterozygosity (LOH) scores showing higher scores in the PARPi resistant clones compared to the sensitive B40 cells. Error bars represent the SD. Groups of triplicate measurements (B40 and NAPool) were compared using Mann-Whitney U-Test. ****p<0.0001.
(F) Oncoprint figure summarizing the functional and genomic findings of the cell lines.
We next investigated the degree aneuploidy and genomic Copy number variations (CNVs) in the cell clones. The computationally predicted ploidy showed good concordance with the cytogenetics (Figure 4D). The RPETP53−/− cells were mostly diploid (2n), whereas the B40 cells showed diploid (2n) and triploid (3n) cells in equal proportions. Interestingly, PARPi resistant clones were all mostly polyploid (3n, 4n, and >5n), except for NA4, which showed a mostly diploid (2n) karyotype. Consistently, CNV profiles revealed few CNVs in the RPETP53−/− cell line and increased CNVs and allelic imbalances in the B40 cell line and in most of the resistant clones (Figure S4). The NA4 clone showed lower levels of genomic instability in comparison to all other resistant clones, consistent with its reduced levels of PARP1 expression as a distinct mechanism of resistance.
Genomic instability and loss-of heterozygosity (LOH) events have been proposed as biomarkers for PARPi sensitivity (7). Calculation of LOH scores showed, however, that all the resistant clones and the NAPool had significantly more LOH events than the B40 cells (Figure 4E). Taken together, our genomic analyses indicate that the genomes of these TP53−/− and BRCA1−/− PARPi resistant clones have unique mutational profiles, lower overall mutational burden, mostly triploid karyotypes and increased genomic instability in comparison to the PARPi-sensitive cell line. A summary of the functional and genomic findings in the clones are shown in Figure 4F.
PARPi resistance results from clonal selection in the cellular model and in BRCA1-mutated High Grade Serous Ovarian Cancer
We next investigated the clonal evolutionary patterns of acquired PARPi resistance, using WES data in our cellular model system, and also in tumor samples of a HGSC patient with BRCA1 germline mutation and acquired PARPi resistance (37).
In the evolutionary tracing of the cellular model we observed three subclones (clusters No. 1, No. 4 and No. 5), already present in the B40 cell line, indicative of early clonal heterogeneity in BRCA1-deficient cells (Figure 5A). After exposure to PARPi, the pool of acquired resistance (NAPool) had increased clonal heterogeneity, with five distinct subclones. Remarkably, cluster No. 5, already present in the B40 sensitive cell line, was selected for and enriched in the resistant NAPool. Importantly, cluster No. 5 was predicted to be the origin of six out of the seven PARPi resistant clones via clonal selection and genomic evolution. The time required for clonal evolution differed across the clones, with clones NA2 and NA3 arising earlier, and NA1, NA5 and NB1 arising later (Figure 5A), consistent with the varying growth rates of the cell lines (Figure S5A). By contrast, the only diploid resistant clone NA4, did not emerge from cluster No. 5, but rather directly from cluster No.1, which might partially explain its diploid karyotype and distinct mechanism of PARPi-resistance (i.e. PARP1 downregulation). Interestingly, a smaller subclone (cluster No. 4) was also present in the B40 cell line; however, this subclone was out-selected during PARPi exposure. Interestingly, three out of seven PARPi resistant clones showed lower risk rations for Sig3 in the mutations accumulated later in pseudotime in the evolutionary model (Figure S5B).
Figure 5. Acquired PARPi resistance is characterized by clonal heterogeneity and selection of pre-existing drug tolerant cells.
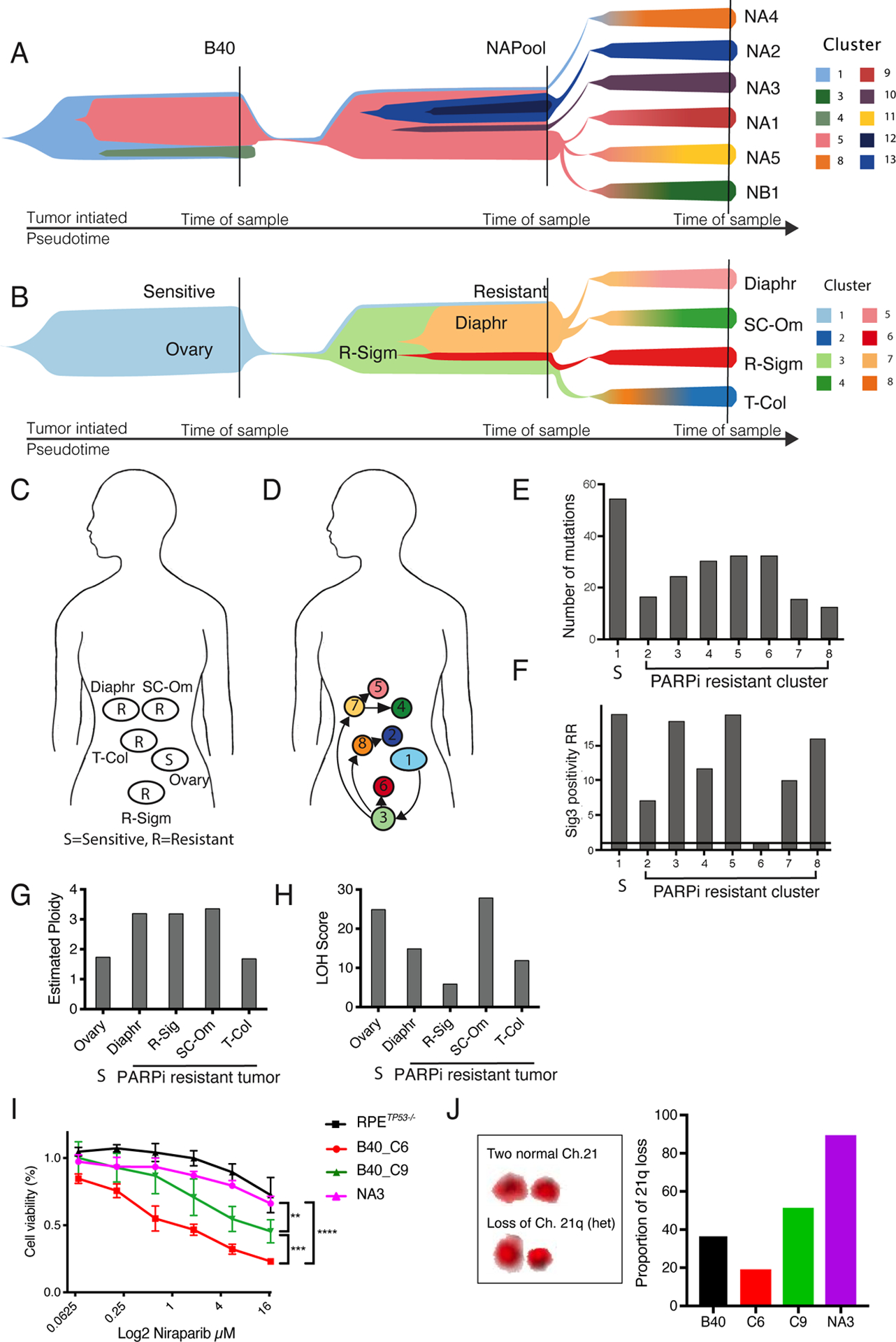
(A) Fish- plot of the clonal evolution of acquired PARPi resistance from the B40 sensitive cell line to the resistant cell line (NAPool) and to the resistant single-cell clones. Colors indicate mutational clusters modelled in pseudotime using using PyClone. Vertical lines indicate the sampling timepoints. Evolutionary modeling of the cells showed increased heterogeneity associated with PARPi resistance, and the enrichment of a subclone (cluster 5), already present in the sensitive B40 cell line is, in the resistant pool (NAPool) that then gave rise to most of the PARPi resistant subclones, except for NA4 that was derived directly from subclone 1.
(B) Fish- plot of the clonal evolution of acquired PARPi resistance in tumor samples from a patient with BRCA-1-mutated HGSC originating from the sensitive ovarian biopsy towards the resistant tumor clones. Similar to the cellular model, evolutionary tracing indicated increased heterogeneity and the emergence of the PARPi resistant clusters from a common precursor in the Recto-sigmoid sample (R-Sigm, cluster 3).
(C) Sites of tumor sampling from a patient with germline BRCA1 mutation who developed acquired PARPi resistance. The patient had received neoadjuvant carboplatin-paclitaxel and underwent interval debulking surgery at which a tumor biopsy was obtained from the ovary, followed by adjuvant carboplatin and paclitaxel, and maintenance treatment containing Olaparib. Four tumor biopsies were obtained during a secondary debulking surgery after Olaparib; from rectosigmoid area (R-Sigm), trans-colonic area (T-Col), diaphragm (Diaph) and supra-colic omentum (SC-Om). S= Sensitive, R= Resistant. After the surgery the patient was successfully treated with Olaparib/prexacertib (stable disease), and carboplatin gemicitabine (partial response), but eventually passed away due to the disease.
(D) Anatomical mapping of genomic clusters (subclones) indicate spatial heterogeneity in acquired PARPi resistance. From the PARPi sensitive ovarian biopsy (cluster 1), the first resistant subclone (cluster 3) emerged in the rectosigmoid area. From that subclone, subclone 7 emerged that spread to the upper abdomen giving rise to subclones 5 on the diaphragm and 4 in the supra-colic omentum. Subclone 8 and 6 evolved directly from the subclone 3, with subclone 2 emerging from subclone 8 in the trans-colonic biopsy.
(E) In comparison to the PARPi sensitive subclone (1), the number of mutations was lower in the clones PARPi resistant clusters 2–8. Bar plot of the number of mutations detected in each cluster. S= Sensitive.
(F) Sublones with acquired PARPi resistance differentially retain mutational signature 3 positivity. Bar plot of Sig3 positivity risk ratio (RR) called using SigMA. Subclones 3, 5 and 8 retained similar RR as compared to the sensitive subclone 1, whereas subclones 2, 4 and 7 showed intermediate decrease in RR, and subclone 6 was negative for Sig3.
(G) Barplot of estimated ploidy using ASCAT in the sensitive (S) and resistant tumor biopsies from the HGSC patient. The primary ovarian sample had an average ploidy pf 1.75, whereas three out of four resistant tumors had a triploid karyotype and one tumor had average of 1.7 predicted ploidy.
(H) Bar plot of the LOH scores in the sensitive (S) and resistant tumor biopsies. One biopsy showed similar or higher LOH scores compared to the sensitive tumor, and the score was lower in three samples.
(I) Differential sensitivity to Niraparib in two single cell clones from the B40 sensitive cell line showing intermediate resistance in the B40_C9 clone, and increased sensitivity in the B40_C6 clone compared the fully resistant NA3 clone. The difference was not significant between the to the RPETP53−/− cells and the NA3 clone. **p<0.01, **p<0.001, ****p<0.0001
(J) Quantification of this 21q loss as a genomic marker for subclone 5 via chromosome painting in the B40 cells, in the sensitive and drug tolerant singe-cell clones B40_C6 and B40_C9, respectively, and in the fully resistant NA3. The increasing proportions of 21q loss with resistance was consistent with our evolutionary model and confirmed that the drug tolerant B40_C9 clone could be identified as the subclone 5 genomically detected in the B40 cell line.
We next studied tumor samples from a patient with BRCA1-deficient HGSC and acquired resistance to PARPi (37), and explored whether clonal evolutionary patterns existed during the acquisition of PARPi resistance. A treatment-naïve sample from the primary ovarian tumor was sequenced from an archival biopsy (Figure 5B). However, likely due to the sample type (archival FFPE-derived DNA) we only detected one genomic clone in this sample (cluster No.1). At the time of clinical PARPi resistance, fresh tumor biopsies were obtained from four metastatic sites (Figure 5B,C). Seven unique PARPi resistant subclones with distinct mutational patterns were identified (Figure 5D, S5C). Evolutionary modeling of this HGSC predicted the emergence of all seven heterogenous PARPi resistant subclones from subclone No.3 that was detected in the rectosigmoid biopsy, and had further spread inside the abdominal cavity to different metastatic sites. Subclones No.6 and No.7 evolved early from subclone No.3 and followed distinct evolutionary trajectories; subclone No.6 remained in the rectosigmoid area without further evolution; subclone No.7 metastasized into the upper abdomen, giving rise to distinct subclone No.4 located in the supra-colic omentum and subclone No.5 located on the diaphragm. Further, emerging from subclone No. 3, subclone No.8 was detected in the trans-colonic biopsy, which further evolved into unique subclone No.2 (Figure 5D).
Consistently with our cellular model, the clinical PARPi resistant subclones exhibited lower numbers of mutations (Figure 5E) and had differentially retained mutational Signature 3 (Figure 5F). Further, we observed heterogeneity in Signature 3 among the clinical samples indicating that HR restoration occurred in some but not all clinical PARPi resistant clones. Interestingly, subclones No. 2 and No. 6, showed particularly lower risk ratios for Sig3 positivity (Figure 5F, S5D). Genomic prediction of ploidy also showed a predominantly triploid karyotype in three out of four PARPi resistant tumor samples (Figure 5G). Interestingly lower LOH scores were observed in three out of four tumor samples, especially in the R-Sigmoid tumor containing subclone 6 that also had lost Sig3 (Figure 5H), this subclone may have restored HR and acquired a different mechanism of resistance compared to the other subclones.
Acquired PARPi resistance results from clonal selection of pre-existing drug tolerant cells.
Our results suggested that PARPi resistance arose from a single clonal precursor that pre-existed in the overall-sensitive BRCA1-deficient cell population. To experimentally validate the pre-existence of this clone, we isolated ten single cell clones from the B40 sensitive cell line and exposed them to Niraparib. In this experiment we confirmed that cellular sub-clones with heterogeneous responses to Niraparib pre-existed in the B40 cell line (Figure S5E). In fact, two out of these 10 sub-clones (B40_C9 and B40_C5) had significantly increased IC50s in comparison to the B40 cell line, whereas sub-clone (B40_C6) was highly sensitive to Niraparib (Figure 5I). Interestingly, in functional experiments, the B40_C5 sub-clone showed increased RAD51 foci formation (Figure S5F), and decreased pRPA and γH2AX foci after IR (Figure S5G-H) indicative of increased HR capacity and decreased replication stress.
Based on WES data we identified chromosome 21q loss as a genomic marker for tracking the clonal evolution trajectory and predict the pre-existence of a drug tolerant sub-clone in the B40 cell line. We predicted that 21q loss is a genomic marker for subclone No. 5 and the emerging PARPi-resistant clones. Quantification of this genomic marker via 21q chromosome painting showed a high proportion of 21q loss in the drug tolerant subclone B40_C9, and enrichment of this genomic marker in the fully resistant clone NA3 (Figure 5J). These results suggest that B40_C9 functionally represents subclone No.5, detected in the B40 cells that was evolutionally selected and gave rise to all of the resistant clones. Consistently, 21q loss was under-represented in the PARPi-hypersensitive B40_C6 subclone (Figure 5J), and in the PARPi resistant clone NA4 that was not derived from subclone No.5 (Figure S5I). Cytogenetic analysis confirmed a mostly diploid karyotype in the sensitive B40_C6 sub-clone, an increasing amount of polyploid cells in the drug tolerant B40_C9 sub-clone, and triploid karyotype dominating the fully resistant NA3 clone (Figure S5I). The genomic profiling and evolutionary analyses thus suggested that BRCA1-deficiency gives rise to a chromosomally unstable cell population with intrinsic clonal heterogeneity that can contain pre-existing drug tolerant, partially HR-restored cells.
Heterogeneous PARP-inhibitor resistant clones have common and unique vulnerabilities
Due to the observed genetic and functional heterogeneity, we next sought rationale approaches for targeting PARPi resistant clones. Based on the transcriptomic analysis, cell cycle-related programs appeared to be actionable targets. To detect these potential vulnerabilities, we first performed flow-cytometry based cell cycle profiling at baseline and after Niraparib re-challenge (Figure 6A). At baseline, we observed a significantly increased proportion of cells in S-phase in the B40 cell line, and in three out of seven PARPi resistant clones, as compared to the RPETP53−/−. Further, the NB1 clone, which is deficient in RAD51foci, had a unique cell cycle profile, with increased proportion of cells in G2 and decreased proportion of cells in G1 phase. Noticeably, upon Niraparib re-challenge, the proportion of cells in G2 phase was increased in all the resistant clones, similar to the sensitive B40 cell line (Figure 6B). In addition, a shorter G1 phase was observed in two clones (NA5 and NB1) after Niraparib. These cell cycle aberrations indicated activation of DNA damage response in the PARPi resistant cells after Niraparib re-challenge, which was investigated by profiling the formation of pRPA, γH2AX, and pCHK1 foci before and after Niraparib. Consistent with restored replication fork stability, all clones had decreased levels of baseline nuclear pRPA foci, indicating decreased replication stress (Figure 6C). However, upon re-challenge with Niraparib, all resistant clones still exhibited increased nuclear foci for pRPA (Figure 6C) and pCHK1 (Figure S6A), indicative of an activated DNA damage response. Consistently, four out of seven resistant clones showed decreased DNA double strand breaks measured as γH2AX foci at baseline (Figure 6D). However, all clones, except for clone NA4 with decreased PARP1, exhibited increased γH2AX foci after PARPi re-challenge (Figure 6D). We next quantified the number of chromosomal aberrations in metaphase spreads from the clones after Niraparib treatment, interestingly, all clones still showed increased numbers of chromosomal aberrations (Figure 6E), indicating that homologous recombination was not fully restored even in the clones with RAD51 foci formation.
Figure 6. PARPi resistant clones show S/G2 arrest and DNA damage upon PARPi re-challenge and vulnerability to targeted agents.
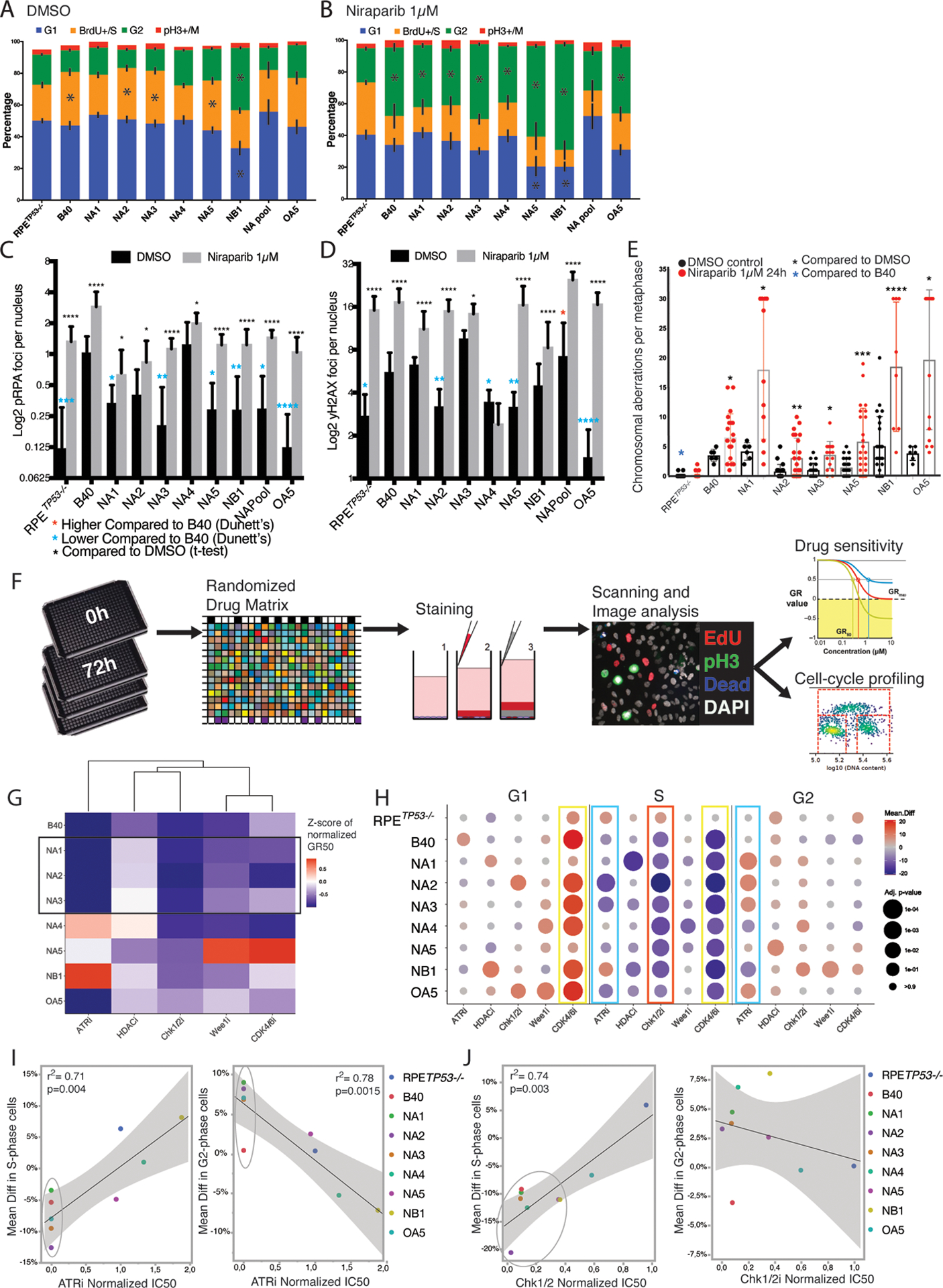
(A) Cell cycle analysis based on DNA content and BrdU incorporation in Flow-cytometry indicated prolonged S-phase at baseline in the sensitive B40 cell line and three out of seven PARPi resistant clones when compared to the RPETP53−/− cells. Black bars indicate SD of three independent measurements, asterisks indicated significant difference with p<0.05 (Mann-Whitney-U test) in comparison to the RPETP53−/−.
(B) Cell cycle analysis revealed a marked G2 arrest in response to Niraparib treatment (1μM for 24 hours) in the sensitive B40 and also in all of the PARPi resistant clones.
(C) Quantification of pRPA (S32) foci at baseline (black bars) and after Niraparib (1μM for 24 hours, grey bars) using IF showed baseline lower levels in the RPETP53−/− cells and in all resistant clones except NA4 in comparison to B40 (blue asterisks). In addition, all resistant clones showed an increase in pRPA after Niraparib treatment. Data are presented as mean foci per nucleus per image in 5–10 representative images and error bars represent the SD.
(D) Quantification of γH2AX foci using IF showed lower baseline DNA damage in comparison to B40 (blue asterisks) in four out of seven clones, however γH2AX foci were significantly higher after Niraparib treatment in all PARPi resistant clones, except for clones NA3 and NA4 (black asterisks).
(E) All PARPi resistant clones displayed increased chromosomal aberrations in metaphase spreads after treatment with Niraparib (1μM for 24 hours). At baseline, the resistant clones had on average similar numbers of chromosomal aberrations, whereas RPETP53−/− cells had lower numbers of aberrations compared to the B40. Bar graphs represent the Mean and error bars the SD.
(F) Schematic presentation of the workflow used in the drug screening. Cells were plated on 384-well plates in triplicates. On day 1 the control plates (0) were fixed and treated with increasing concentrations of the drugs on a randomized order using an automated drug dispenser. After 72 hours the cells were pulsed with EdU, fixed and stained using live-dead stain and a pH3 antibody for viability and cell cycle analysis. The plates were scanned, and the imaging data for growth-rate normalized survival and cell cycle profiles were analyzed using designed pipelines.
(G) Heatmap of IC50 values normalized to the RPETP53−/− cells of the selected drugs in the cell lines and resistant clones. Resistant clones with mechanisms related to increased end resection (NA1, NA2 and NA3) showed similar drug response profiles (black rectangle). Other clones had unique response profiles, with no single drug being as effective in all clones as in the as in the B40 cells.
(H) Cell cycle profiles were extracted from the drug screening image analysis and presented as the mean difference in the treated wells in comparison to the untreated wells (Mean difference in % of cells) for G1 (left panel), S (middle panel), and G2 (right panel) phases. The data are presented as dot plots with the mean difference calculated from three independent wells in color scale and p-value as the size of the dot (Sidak’s multiple comparison test and p-value <0.1). Note the increase in G1, and decrease in S phase as a response to CDK4/6 inhibition (yellow rectangles). ATR inhibition resulted in significant decrease in proportion of cells in S-phase (left blue rectangle) and increase in G2 (right blue rectangle) in the sensitive cell lines. Chk1/2 inhibition caused a decrease in S-phase cells (red rectangle).
(I) Linear regression of the mean differences in S (left panel) and G2 (right panel) phases after ATR inhibition. The sensitive cells (circles) showed a significant decrease in S and increase in G2.
(J) Linear regression of the mean differences in S (left panel) and G2 (right panel) phases after Chk1/2 inhibition. The sensitive cells (circles) showed a significant decrease in S but no significant correlation was observed for G2.
We next selected targeted therapeutics based on potentially actionable resistance mechanisms and gene expression programs of the PARPi resistant clones (Table S2). We performed a systematic drug sensitivity test using an automatized platform with an imaging-based readout that adjusts for the differential growth rate of the various clones (Figure 6F) (38). We tested targeted agents and correlated the sensitivity of PARPi resistant clones to drug-induced alterations in cell-cycle profiles. First, as the B40 cells and the PARPi resistant clones had different growth kinetics compared to the RPETP53−/− cells (Figure S5A), we confirmed the resistance to Niraparib using the growth-normalized assay (Figure S6B). Secondly, in the drug response profiles, resistant clones with downregulation of NHEJ (NA1, NA2, and NA3) had similar profiles of sensitivity to Prexacertib (Chk1/2i), Adavosertib (WEE1i) and Bertzosertib (ATRi) (Figure 6G). Interestingly NA1 and NA2 were more sensitive to Abemacyclib (CDK4/6i), when compared to B40. Similarly, NA4 was sensitive to Chk1/2i, CDK4/6i, and WEE1i, but resistant to ATRi, when compared to the B40 cells. In line with the observed chromatin regulatory pathway rewiring in the gene-expression profile, NA5 was the most sensitive clones to Belinostat (HDACi), but showed resistant to CDK4/6i and WEE1i. NB1, the only clone without RAD51 restoration, showed lower sensitivity to all of the agents compared to the B40 cell line, and was resistant to the ATRi with an almost 2-fold IC50 compared to the RPETP53−/− cells. OA5, the Olaparib resistant clone, was sensitive to CDK4/6i and ATRi, but had low sensitivities to Chk1/2i, WEE1i and HDACi.
CDK4/6i induced a robust G1 arrest in the B40 cells and in five out of six PARPi resistant clones (Figure 6H). G1 arrest was not observed in the CDK4/6i-resistant NA5 clone. Further, CDK4/6i induced a decrease in the proportion of cells in S-phase in all clones, except the RPETP53−/−. Interestingly, responses to ATRi significantly correlated with the drug-induced cell cycle aberrations; the PARPi resistant clones that responded (n=4 out of 7) to ATRi, showed a significantly decreased proportions of cells in S-phase and increased proportions of cells in G2 (Figure 6I). Similarly, PARPi resistant clones that were sensitive to Chk1/2i Prexacertib (n=4 out of 7) showed a significant decrease of cells in S-phase, but there was no significant correlation to increase in G2 (Figure 6J). This correlation of responses and cell cycle profiles is consistent with the mechanisms of action of ATRi and Chk1/2i in disrupting the DNA damage response, and indicates dependency of these clones to that checkpoint (18). HDACi and WEE1i did not cause cell cycle aberrations that were significantly associated with drug sensitivities. In summary, the PARPi resistant clones showed mechanism-dependent vulnerabilities to the selected agents, that for certain drugs associated with cell cycle aberrations. However the heterogeneity of the PARPi-resistant clones led to heterogeneous responses to our targeted screening and shows that clones could not be targeted with a single drug.
Discussion
As PARPi’s are entering wide clinical use, acquired PARPi resistance is an emerging clinical problem in the treatment of multiple cancer types, including HGSC. We engineered BRCA1- deficient cells that are resistant to PARPi and showed that clones derived from these cells, via evolutionary selection, have significant functional and genomic heterogeneity. Similar genomic features and evolutionary patterns were observed in a clinical BRCA1- mutated tumor with acquired PARPi resistance. Importantly, we show that these heterogenous PARPi resistant clones are vulnerable to mechanism-specific targeting.
The best-documented mechanism of resistance to PARPi or platinum agents in human tumors involves the restoration of HR via somatic reversion of the original mutation conferring HR deficiency. Multiple reports have confirmed the relevance of these somatic reversions in therapy resistant ovarian cancers (10, 11, 22). However, the prevalence of secondary mutations restoring BRCA1/2 function has varied from 0–21% (39, 40) in ovarian cancer to has high as 40%−50% in breast cancer (40, 41). In our study, we did not observe BRCA1 reversion mutations in the PARPi-resistant clones or in biopsy samples from a patient with BRCA1- germline HGSC with acquired PARPi resistance. Accordingly, PARPi resistance mechanisms other than reversion mutations are likely to exist also in the clinical setting.
Genomic cancer studies support a model in which drug resistance is driven by evolutionary selection of a pre-existing clone rather than acquisition of new mutational events ((42–45), and reviewed in (46)). Consistently, we show that a PARPi-tolerant subclone pre-existed within a heterogenous BRCA1-deficient cell population and was clonally selected during the evolutionary pressure imposed by the PARPi treatment. In our study, PARPi-resistant clones displayed increased chromosomal instability, modifying the cellular karyotype, the cellular system of inheritance, affecting the topology of the genome and the transcriptomes (47). Here, we propose that the chromosomal instability in the BRCA1-deficient cells, amplified by the effects of PARPi on further disrupting genome integrity (48), provides an avenue for improved fitness, increased clonal heterogeneity, and selection of pre-existing drug-resistant cells leading to PARPi resistance. As HGSC is among the tumors with highest levels of clonal heterogeneity (49), the existence of intrinsically PARPi resistant cells at tumor presentation is likely also in the clinical setting.
Previous studies on PARPi resistance have been performed in commercially available BRCA1 deficient cell lines derived from drug resistant tumors (20, 21) and have focused on single mechanism of resistance (13, 14). By contrast, our genetically engineered model system suggested that acquired PARPi resistance involves the adoption of several distinct, potentially simultaneous resistance mechanisms. Consistently, the PARPi resistant clones showed molecular heterogeneity with unique transcriptomic and mutational profiles. This heterogeneity was recapitulated at the clonal level in a BRCA1- mutated tumor with acquired PARPi resistance, further supporting the roles of clonal and functional heterogeneity as molecular underpinnings of acquired PARPi resistance.
Measurements of genomic instability, such as LOH and mutational signature 3 resulting from error-prone DNA repair processes, have been suggested as biomarkers for PARP inhibitor sensitivity (50).
Regardless of the fact that the majority of the PARPi resistant clones had restored RAD51foci formation, most of the resistant clones still retained mutational signature 3, DNA damage, and chromosomal aberrations after re-treatment with Niraparib. In line with previous observations from cell lines and mouse models (13), these findings suggest that restoration of HR in the clones via the mechanisms described herein is only partial.
Despite their highly heterogeneous nature, the PARPi resistant clones shared vulnerabilities to targeted agents. Most of the clones exhibited cell cycle abnormalities upon re-exposure to Niraparib, indicating residual vulnerability to PARPi’s. However, the clones showed heterogenous responses for targeted chemotherapeutic agents, and a single drug was not able to efficiently target all PARPi resistant clones. Some clones (NA1, NA2 and NA3) with a resistance mechanism involving suppression of the NHEJ pathway had a similar drug sensitivity profile, whereas other clones (NA4, NA5 and NB1) with distinct mechanisms of resistance were overall less drug sensitive. NA4 and NB1, with distinct mechanisms were the only two clones with persistent cisplatin sensitivity. Taken together, the PARPi resistant clones exhibit mechanism-dependent vulnerabilities that can potentially be exploited in combination therapies. The optimal way of targeting the heterogenous PARPi resistant clones in combinations or sequentially, should be addressed in future in vitro and in vivo experiments or via e.g. mathematical modeling.
We acknowledge certain limitations of this study. First, we used an epithelial cell line not originating from fallopian tube and generated a deleterious BRCA1 mutation not easily reversible to explore some of the previously described mechanisms for PARPi resistance. Thus, it is possible that additional mechanisms for PARPi resistance, including reversion mutations could exist in the clones if different approaches were taken. Secondly, we only had access to sequencing data from one HGSC patient with acquired PARPi resistance. In this sample set, the original PARPi sensitive ovarian tumor was sequenced from formalin-fixed tissue, compared to the PARPi resistant samples which were analyzed from fresh frozen tissue. We did not observe multiple clones in the PARPi sensitive sample, potentially due to the lower sequencing quality and resolution in variant calling (50). Moreover, we did not have access to additional fresh clinical patient samples, which could be analyzed with similar functional studies as in the cellular model. Therefore, we were not able to verify the pre-existing resistant subclones in the PARPi sensitive patient sample, that were in fact verified to pre-exist in the cellular model. Therefore, future studies should focus on fresh tumor biopsies taken before and after PARPi treatment. These studies will be critical for revealing the mechanisms, development of biomarkers, and clinical targeting of HGSC with acquired PARPi resistance.
Supplementary Material
Statement of significance;
This study shows that BRCA1-deficient cells can give rise to multiple genomically and functionally heterogenous PARPi-resistant clones, which are associated with various vulnerabilities that can be targeted in a mechanism-specific manner.
Acknowledgements
This work was supported by OCRA, Sigrid Jusélius Foundation, and Biomedicum Helsinki Foundation to AF, and NIH Grant U54-HL127365 to PKS. We wish to thank André Madeira for comments.
Footnotes
The authors declare no conflicts of interest.
References
- 1.Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D’Andrea AD. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015;5(11):1137–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MI, et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature. 2015;518(7538):258–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moore K, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med. 2018;379(26):2495–505. [DOI] [PubMed] [Google Scholar]
- 4.González-Martín A, Pothuri B, Vergote I, DePont Christensen R, Graybill W, Mirza MR, et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med. 2019;381(25):2391–402. [DOI] [PubMed] [Google Scholar]
- 5.Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014;15(8):852–61. [DOI] [PubMed] [Google Scholar]
- 6.Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N Engl J Med. 2016;375(22):2154–64. [DOI] [PubMed] [Google Scholar]
- 7.Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18(1):75–87. [DOI] [PubMed] [Google Scholar]
- 8.Francica P, Rottenberg S. Mechanisms of PARP inhibitor resistance in cancer and insights into the DNA damage response. Genome Med. 2018;10(1):101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28(15):2512–9. [DOI] [PubMed] [Google Scholar]
- 10.Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol. 2011;29(22):3008–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451(7182):1116–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson N, Johnson SF, Yao W, Li YC, Choi YE, Bernhardy AJ, et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc Natl Acad Sci U S A. 2013;110(42):17041–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013;3(1):68–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature. 2015;521(7553):541–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gupta R, Somyajit K, Narita T, Maskey E, Stanlie A, Kremer M, et al. DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell. 2018;173(4):972–88.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. 2016;535(7612):382–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lim KS, Li H, Roberts EA, Gaudiano EF, Clairmont C, Sambel LA, et al. USP1 Is Required for Replication Fork Protection in BRCA1-Deficient Tumors. Mol Cell. 2018;72(6):925–41.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK, et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017;31(3):318–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rondinelli B, Gogola E, Yücel H, Duarte AA, van de Ven M, van der Sluijs R, et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat Cell Biol. 2017;19(11):1371–8. [DOI] [PubMed] [Google Scholar]
- 20.Beaufort CM, Helmijr JC, Piskorz AM, Hoogstraat M, Ruigrok-Ritstier K, Besselink N, et al. Ovarian cancer cell line panel (OCCP): clinical importance of in vitro morphological subtypes. PLoS One. 2014;9(9):e103988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DelloRusso C, Welcsh PL, Wang W, Garcia RL, King MC, Swisher EM. Functional characterization of a novel BRCA1-null ovarian cancer cell line in response to ionizing radiation. Mol Cancer Res. 2007;5(1):35–45. [DOI] [PubMed] [Google Scholar]
- 22.Kondrashova O, Nguyen M, Shield-Artin K, Tinker AV, Teng NNH, Harrell MI, et al. Secondary Somatic Mutations Restoring. Cancer Discov. 2017;7(9):984–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clairmont CS, Sarangi P, Ponnienselvan K, Galli LD, Csete I, Moreau L, et al. TRIP13 regulates DNA repair pathway choice through REV7 conformational change. Nat Cell Biol. 2020;22(1):87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dang HX, White BS, Foltz SM, Miller CA, Luo J, Fields RC, et al. ClonEvol: clonal ordering and visualization in cancer sequencing. Ann Oncol. 2017;28(12):3076–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abkevich V, Timms KM, Hennessy BT, Potter J, Carey MS, Meyer LA, et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br J Cancer. 2012;107(10):1776–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gulhan DC, Lee JJ, Melloni GEM, Cortés-Ciriano I, Park PJ. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat Genet. 2019;51(5):912–9. [DOI] [PubMed] [Google Scholar]
- 30.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hafner M, Niepel M, Chung M, Sorger PK. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat Methods. 2016;13(6):521–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tumiati M, Hietanen S, Hynninen J, Pietilä E, Färkkilä A, Kaipio K, et al. A Functional Homologous Recombination Assay Predicts Primary Chemotherapy Response and Long-Term Survival in Ovarian Cancer Patients. Clin Cancer Res. 2018;24(18):4482–93. [DOI] [PubMed] [Google Scholar]
- 33.D’Andrea AD. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair (Amst). 2018;71:172–6. [DOI] [PubMed] [Google Scholar]
- 34.Jungmichel S, Rosenthal F, Altmeyer M, Lukas J, Hottiger MO, Nielsen ML. Proteome-wide identification of poly(ADP-Ribosyl)ation targets in different genotoxic stress responses. Mol Cell. 2013;52(2):272–85. [DOI] [PubMed] [Google Scholar]
- 35.Pettitt SJ, Krastev DB, Brandsma I, Dréan A, Song F, Aleksandrov R, et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat Commun. 2018;9(1):1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gottipati P, Vischioni B, Schultz N, Solomons J, Bryant HE, Djureinovic T, et al. Poly(ADP-ribose) polymerase is hyperactivated in homologous recombination-defective cells. Cancer Res. 2010;70(13):5389–98. [DOI] [PubMed] [Google Scholar]
- 37.Hill SJ, Decker B, Roberts EA, Horowitz NS, Muto MG, Worley MJ, et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov. 2018;8(11):1404–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hafner M, Heiser LM, Williams EH, Niepel M, Wang NJ, Korkola JE, et al. Quantification of sensitivity and resistance of breast cancer cell lines to anti-cancer drugs using GR metrics. Sci Data. 2017;4:170166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ang JE, Gourley C, Powell CB, High H, Shapira-Frommer R, Castonguay V, et al. Efficacy of chemotherapy in BRCA1/2 mutation carrier ovarian cancer in the setting of PARP inhibitor resistance: a multi-institutional study. Clin Cancer Res. 2013;19(19):5485–93. [DOI] [PubMed] [Google Scholar]
- 40.Weigelt B, Comino-Méndez I, de Bruijn I, Tian L, Meisel JL, García-Murillas I, et al. Diverse. Clin Cancer Res. 2017;23(21):6708–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waks AG, Cohen O, Kochupurakkal B, Kim D, Dunn CE, Buendia Buendia J, et al. Reversion and non-reversion mechanisms of resistance to PARP inhibitor or platinum chemotherapy in BRCA1/2-mutant metastatic breast cancer. Ann Oncol. 2020;31(5):590–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Christie EL, Fereday S, Doig K, Pattnaik S, Dawson SJ, Bowtell DDL. Reversion of BRCA1/2 Germline Mutations Detected in Circulating Tumor DNA From Patients With High-Grade Serous Ovarian Cancer. J Clin Oncol. 2017;35(12):1274–80. [DOI] [PubMed] [Google Scholar]
- 43.Yates LR, Gerstung M, Knappskog S, Desmedt C, Gundem G, Van Loo P, et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med. 2015;21(7):751–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, Burleigh A, et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009;461(7265):809–13. [DOI] [PubMed] [Google Scholar]
- 45.Kozłowska E, Färkkilä A, Vallius T, Carpén O, Kemppainen J, Grénman S, et al. Mathematical Modeling Predicts Response to Chemotherapy and Drug Combinations in Ovarian Cancer. Cancer Res. 2018;78(14):4036–44. [DOI] [PubMed] [Google Scholar]
- 46.Cooke SL, Brenton JD. Evolution of platinum resistance in high-grade serous ovarian cancer. Lancet Oncol. 2011;12(12):1169–74. [DOI] [PubMed] [Google Scholar]
- 47.Heng HH, Bremer SW, Stevens JB, Horne SD, Liu G, Abdallah BY, et al. Chromosomal instability (CIN): what it is and why it is crucial to cancer evolution. Cancer Metastasis Rev. 2013;32(3–4):325–40. [DOI] [PubMed] [Google Scholar]
- 48.Li ML, Greenberg RA. Links between genome integrity and BRCA1 tumor suppression. Trends Biochem Sci. 2012;37(10):418–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Raynaud F, Mina M, Tavernari D, Ciriello G. Pan-cancer inference of intra-tumor heterogeneity reveals associations with different forms of genomic instability. PLoS Genet. 2018;14(9):e1007669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De Paoli-Iseppi R, Johansson PA, Menzies AM, Dias KR, Pupo GM, Kakavand H, et al. Comparison of whole-exome sequencing of matched fresh and formalin fixed paraffin embedded melanoma tumours: implications for clinical decision making. Pathology. 2016;48(3):261–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.