

Abstract
With an inwardly directed reactive center and a well-defined binding pocket, Au(I) functionalized resorcin[4]arene cavitands have been shown to catalyze molecular transformations. The reactivity profiles that emerge differ from other Au(I) catalysts. The added constraint of a binding pocket gives rise to the possibility that the substrates might have to fit into the resorcinarene pocket; our hypothesis is that substrates that match the available space have different reaction outcomes than those that do not. Herein we report on the intramolecular cyclization of alkyne-aromatic substrates with variable alkynes and aromatic composition. We see that scaffold size most drastically dictates reactivity, especially when the substrate’s features are particularly designed. The results of these experiments add to the veritable goldmine of information about the selectivity in catalysis that cavitands offer.
Keywords: Supramolecular, Catalysis, Gold, Friedel-Crafts, Host-Guest
Introduction
Gold cavitands [1] are an emerging catalytic species that have expanded on fundamental discoveries in the arena of C-Heteroatom and C-aryl bond forming reactions that originate from alkyne and alkene centers [2–5]. Cavitands are molecular cavities [6–8] and under certain circumstances fold into a ‘vase’ conformation. This state provides a binding pocket for supramolecular scientists to explore. Cavitands have a rich history of host–guest chemistry and now, with an inwardly directed coupled Au atom (1) [9], we have proposed that their potential to behave like biological catalysts will come to be [10–13].
Gold cavitands have a defined pocket that comes from their resorcinarene cavitand component. This pocket has been shown to select for certain sized guests, stabilize intermediates [14], bias the outcome of chemical transformations [15], or be inhibited by certain sized entities [11]. Under current study is cavitand 1 [9], that has three walls (Scheme 1) that create a small, defined pocket. The second salient feature relevant to biologically inspired catalysis is an inwardly directed reactive center, in our case Au. Other variations of 1 are easy to prepare including bis-Au two-walled analogs [16]. We are also learning that the walls are not benign in yne-yne coupling reactions, shortening the walls results in low or no reactivity [17,18].
Scheme 1.
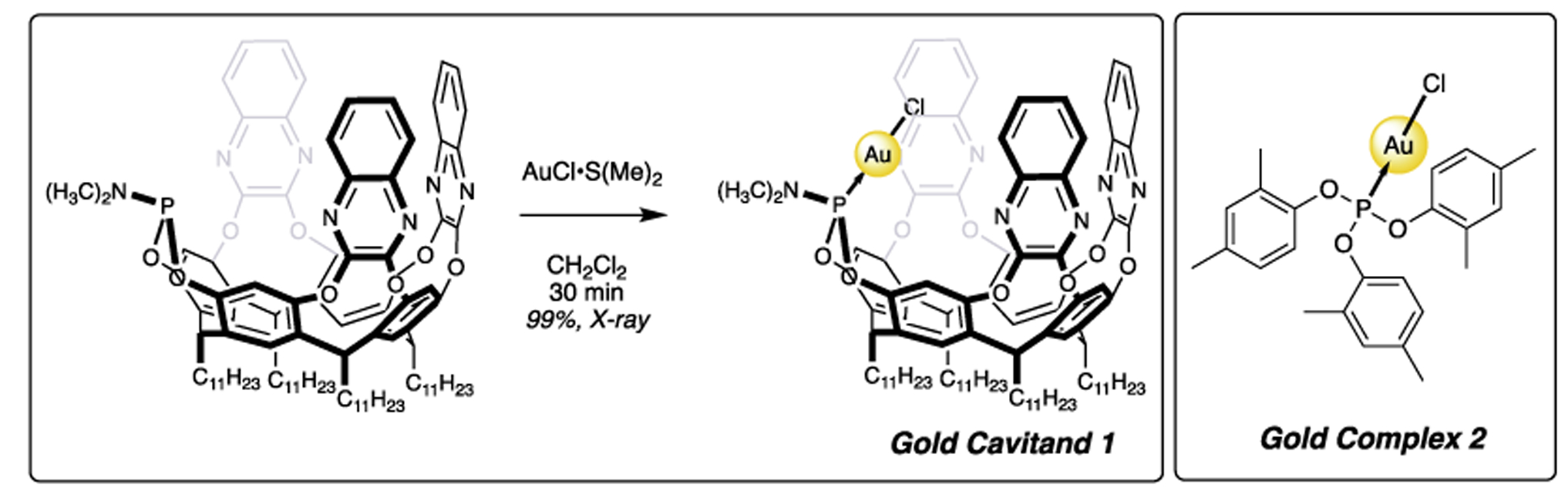
Inwardly directed Au cavitand 1 and electronically similar complex 2 that are used in this study.
Most recently we have explored the Au catalyzed cyclization of alkyne-acids, which already is an important method to prepare functionalized lactones 3 [19]. In an earlier work utilizing AuCl and mild reaction conditions, a variety of alkyne-acids underwent cyclization with 75–97% conversion [20]. The preparation of 3 is typical (Scheme 2). Work has been continued on this reaction [21].
Scheme 2.
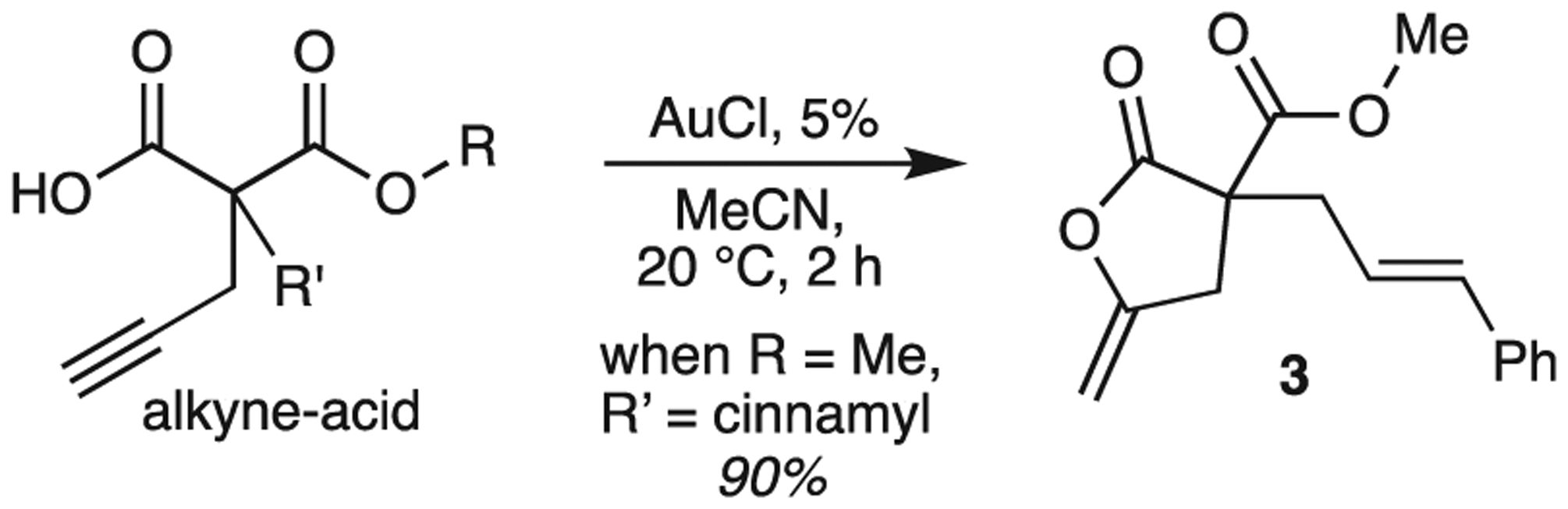
AuCl lactonization of alkyne-acids [20].
When we applied Au-cavitand 1 to a variety of alkyne-acid substrates we demonstrated that the R and R’ groups play an important role [11]. We reported that the R’ group, when matched to fit the cavitand, were potent in slowing down the reaction. Moreover, when R’ is benzyl, lactone 3 can serve to inhibit catalysis of other alkyne acids. Given these observations, the walls should provide an environment that can be used to select substrates or influence reaction outcomes.
The results encouraged us to explore other transformations where Au and alkynes react and where substrate variability could result in cavitand interaction. A series of reports that explored the cycloisomerization of alkyne 4 caught our attention [22,23]. When R = OMe and X = H, a variety of metals could affect the transformation of 4, most often to six-membered 6, but the 5-membered fluorene 5 could also be isolated (Scheme 3). For example, when PtCl2 or AuCl3 were employed (toluene 80 °C), 6 was the dominant species (5:6 5:95, in 75% or higher yield). Using InCl3, the yield plummeted to 44%, but the ratio of 5:6 inverted to 56:44. RuCl2 species were similar. Revisiting PtCl2, where R = OMe and X = COOMe resulted in favoring 5 over 6 (95:5), or with X = p-methoxyphenyl (40:60). A variety of X groups as well as R groups, including additional rings are well tolerated. In the case of X = halide they behaved as expected with a variety of metals (e.g. InCl3), but interestingly when AuCl was used 1,2 migration of the halide occurred [24].
Scheme 3.
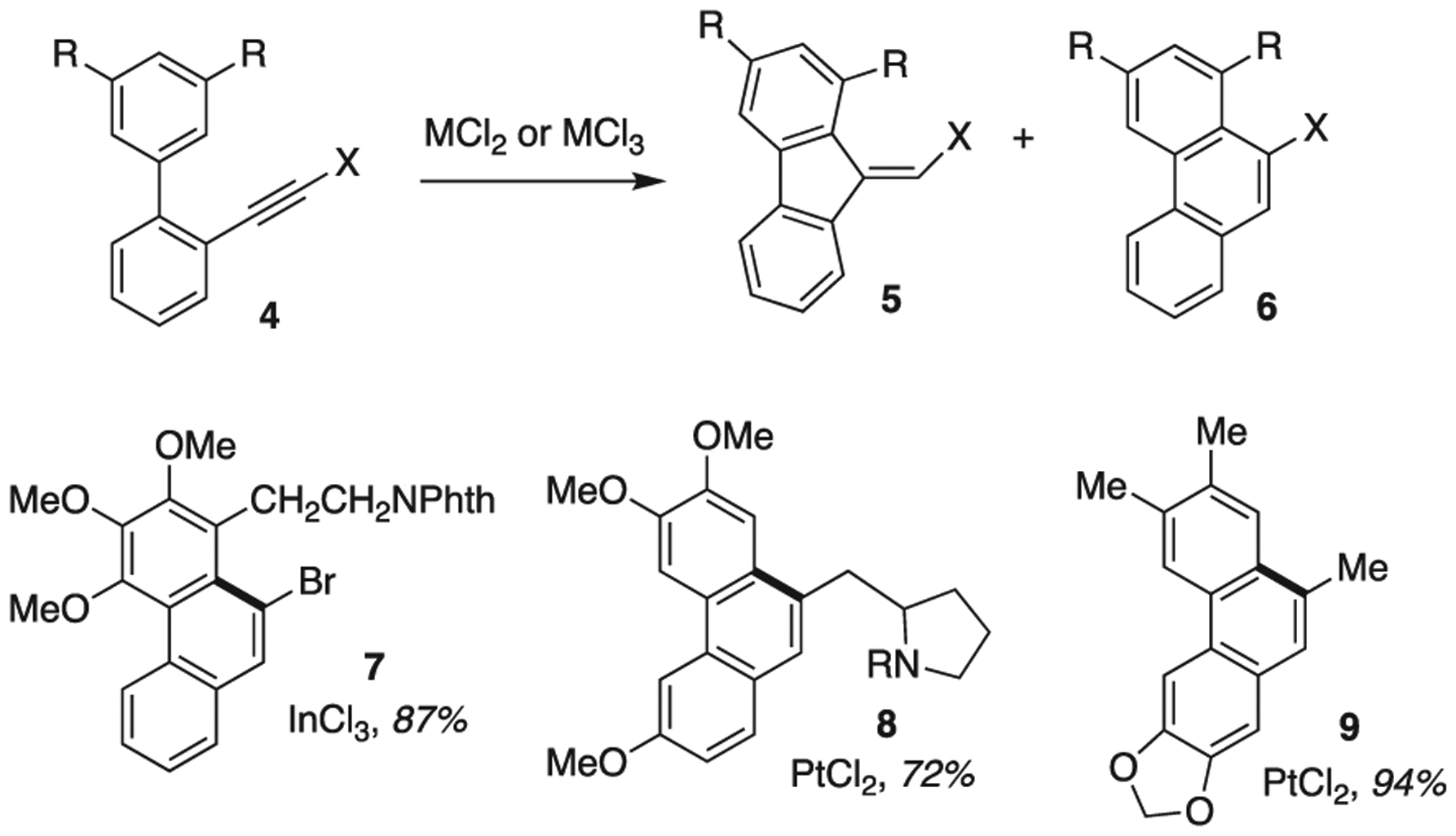
Metal catalyzed cycloisomerization of alkyne-arenes (4) and late-stage ring formation products (7–9) bolded bond indicates reaction locant (the right side of the bond originates from a functionalized alkyne).
Several alkaloid syntheses [22 24,25] have been facilitated by this methodology, where the metal directed isomerization takes place at a late stage (7 [22], 8 [26], 9 [24]). This arene-alkyne cycloisomerization has also played a central role in several reviews, where focused discussion on the mechanism has taken place and variable outcomes, say comparing AuCl3 vs AuCl, give mechanistic insight into plausible Au intermediates. [25,27–28] Variations where the alkyne is linked via ether [29] or ester [30] groups also exist, expanding on the application of this ring closing strategy. These reactions are putative Friedel-Crafts in nature and furans [31] (as well as other heterocycles) can replace phenyl as the nucleophilic partner. These three latter mentioned approaches use phosphorus ligated Au(I) as a mild and relatively stable catalyst. These substrates and this reaction were of great interest to us, given our experience activating alkynes with Au-1. The aromatic rings found in a variety of substrates provide a handle for us to work with in attempts to match size of guest with volume of cavity. We were attracted to the variable outcomes of the reaction of 4 to give different isomeric ratios of 5 and 6. Additionally, we wanted to know if halide rearrangement when 4 is converted to 6 (where X = halide) might change with 1. We believed Au-1 would indeed offer different reactivity compared to 2 and thus began a screening effort.
Results and discussion
In this work, we contrast the results of cavitand 1 with electronically similar complex 2 (Scheme 1). We began with the preparation of a simple alkyne functionalized biphenyl (Scheme 4). Following literature procedures [23], the Suzuki coupling of reaction partners provided aldehyde 10 in acceptable yield. We found that modifying the procedure using microwave irradiation [32] improved the outcome in our hands (see ESI). Continuing with the Corey-Fuchs protocol, we obtained the target alkyne 12 after isolation of dibromo compound 11. Replacing triphenylphosphine with triisopropyl phosphite gave an easier purification and higher yield of 11. After several repetitions, we also explored the conversion of aldehyde 10 to alkyne 12 using a variation of the Bestmann-Ohiray [33 34] reagent. When using azides or diazo compounds appropriate care should be taken. Azide 13 was prepared on a 1 g scale from the corresponding sulfonyl chloride and used within 48 h. Diazo 14 was prepared in situ following known protocols and was reacted with 10. These two variants (13 and 14) of many possible permutations were chosen specifically for their larger molecular mass as well as electronic composition (e.g. 13) to provide added measures of safety.
Scheme 4.
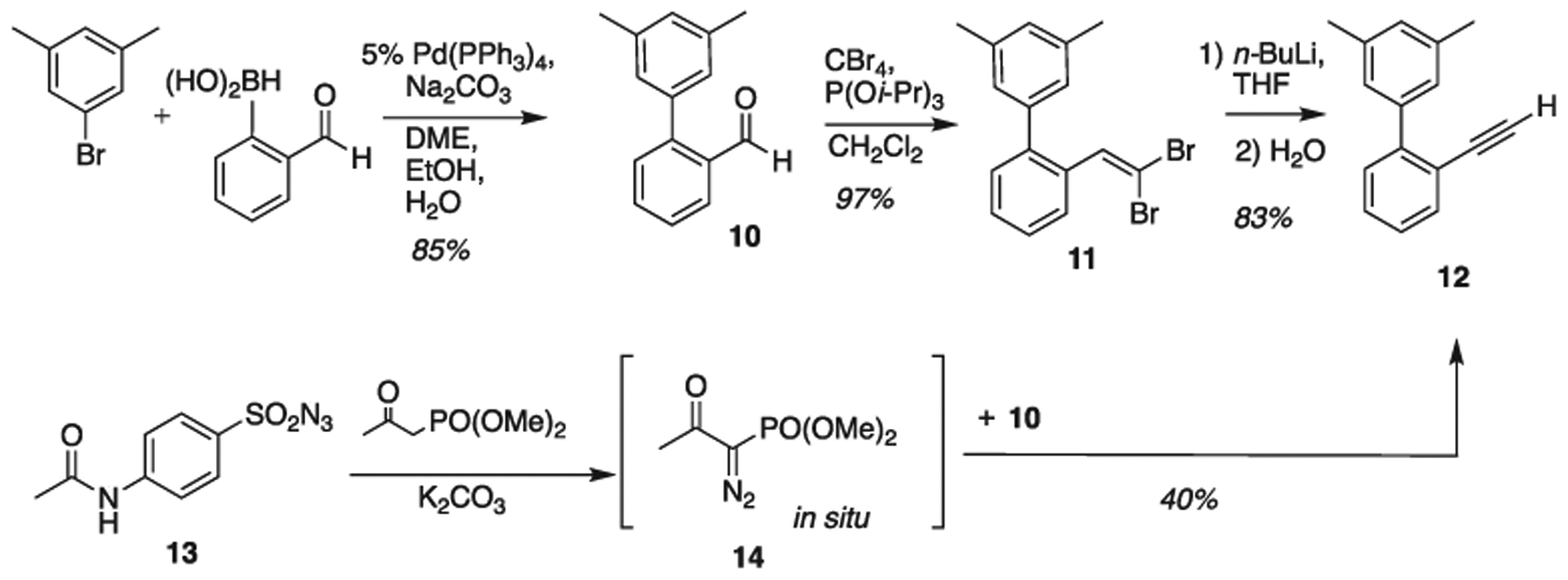
Preparation of alkyne functionalized biphenyl 12.
We next prepared a variety of substrates (Scheme 5) with which to compare the effects of 1 vs. 2. Halides 15, 16, 17 were prepared by treatment of 12 with the corresponding N-halidesuccinimide [35]. Internal alkynes (18, 19, 20) were prepared from reaction of dibromoalkyne 11 with n-butyllithium, followed by reaction with an appropriate electrophile [23]. Finally, variations on the aromatic scaffold (21, 22, 23) were obtained by altering the Suzuki-coupling reaction partners (see ESI).
Scheme 5.
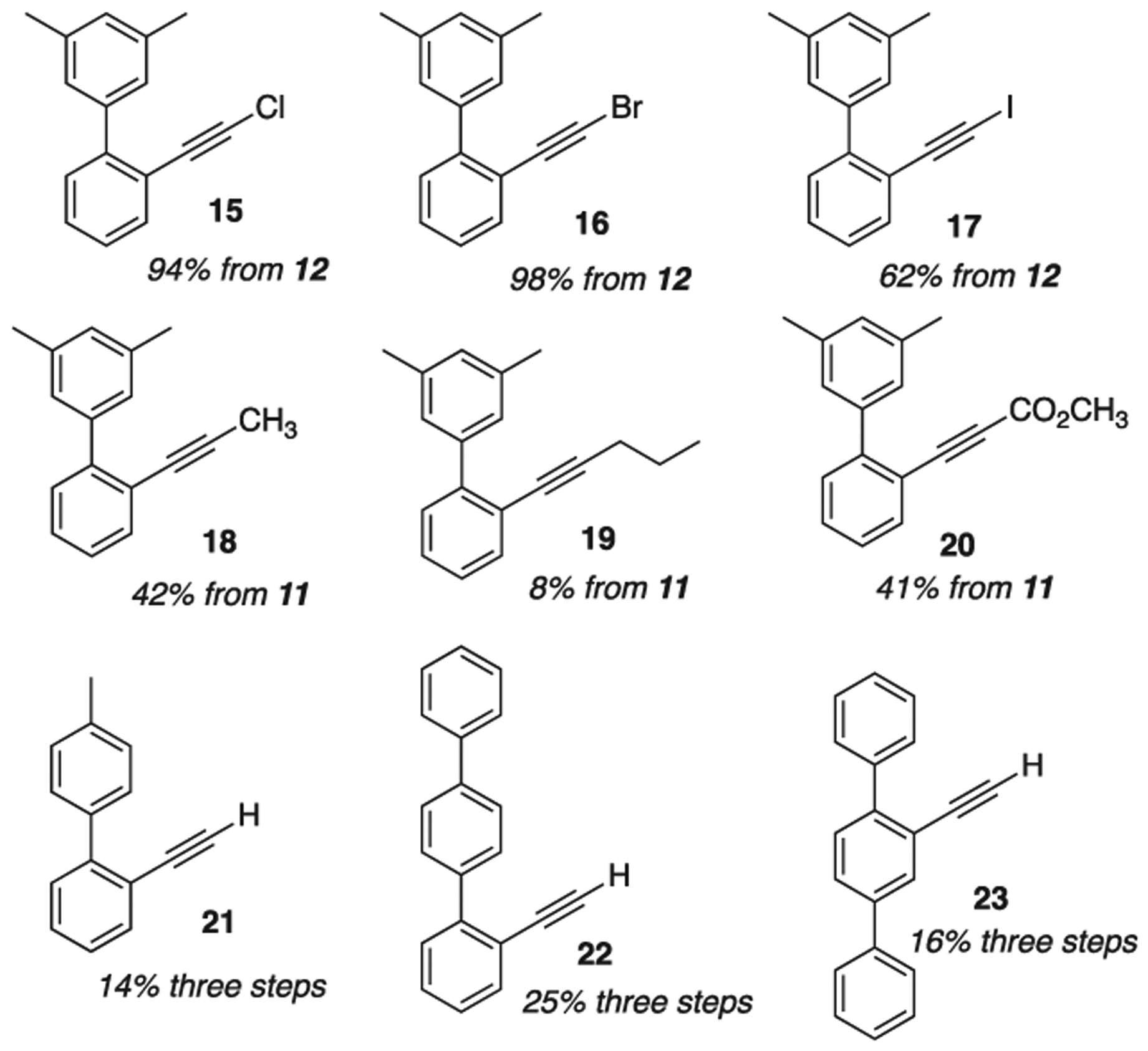
Alkyne variations on 12: 15–20, and aromatic scaffold variations on 12: 21–23.
With substrates in hand, we explored the Au catalyzed reaction (Scheme 6). Alkyne 12 was subjected to a variety of AuCl catalytic species in the presence of AgOTf. We found that without Ag, the reactions were unsuccessful; indeed with cavitand 1 (Table 1, entry 1), no reaction occurred even after extended heating as monitored by NMR. The same unresponsiveness resulted with AgOTf alone (Entry 2). The combination of the two gave appreciable turnover after 1 h with heating (48% conversion, entry 3). After 16 h, clean, quantitative conversion was achieved. Using a non-cavitand AuCl surrogate, namely (di-t-butylphenylO)3PAuCl 2, which is nearly isoelectronic at P and thus at Au compared to 1, we see similar results; no conversion on its own (Entry 4) and complete conversion after 16 h heating in the presence of AgOTf (Entry 5). Interestingly, AuCl was mildly reactive without an additive (Entry 6) and with AgOTf, produced trace amounts of 5 (Entry 7). We assume for now that Au/Ag synergistic/dependent effects are not at work [36,37], and that Ag is simply playing the role of Au activation through ligand replacement. We will explore this matter in a future report, and for the time being, the effect of substrate shape with cavitand will be our point of focus.
Scheme 6.
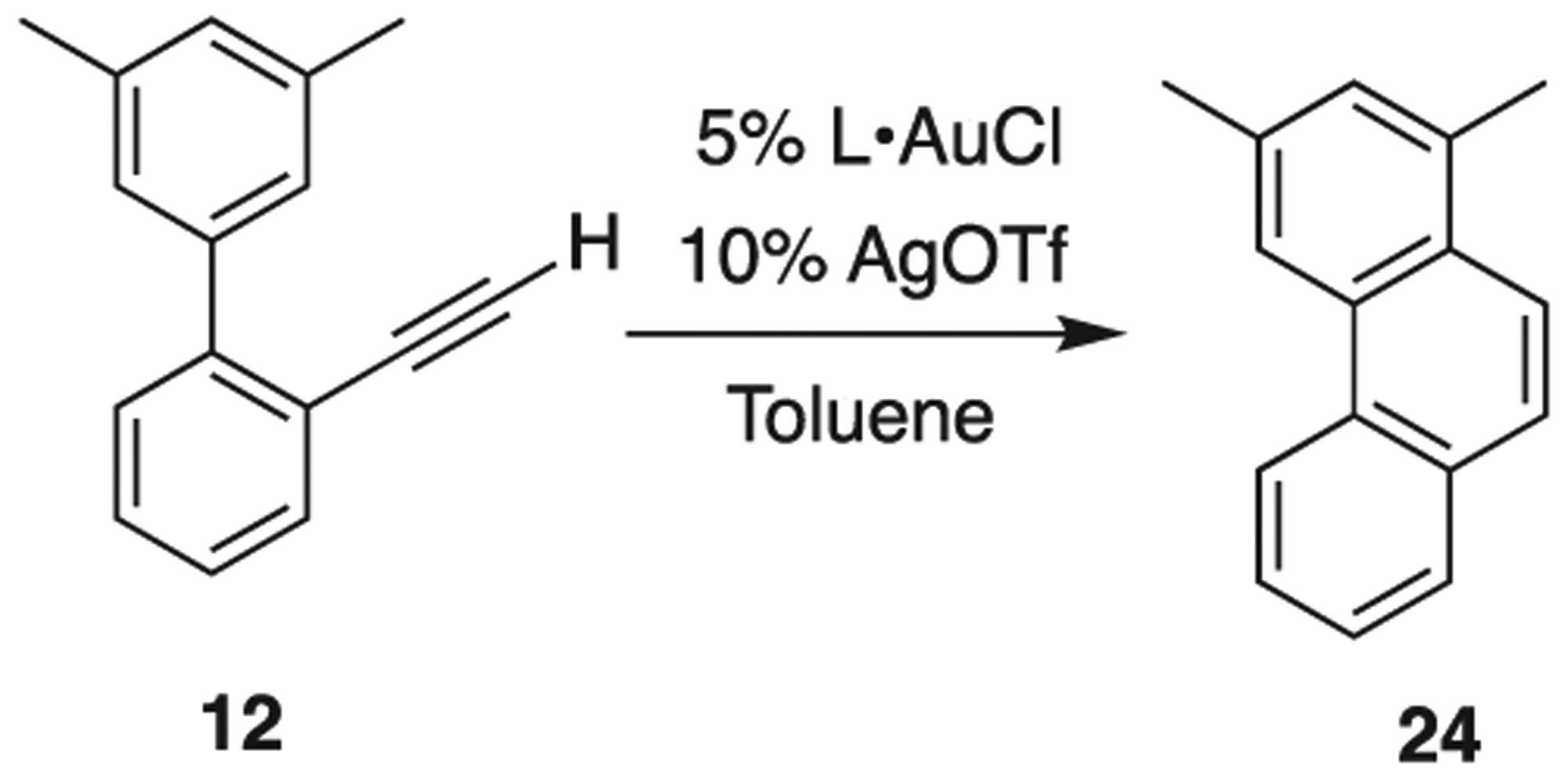
Catalytic screening of substrate 12.
Table 1.
Cycloisomerization of 12 under a variety of conditions.
| Entry | Conditionsa | Time | Conversionb |
|---|---|---|---|
| 1 | 1, 24 °C | 1 h | 0% |
| 1, 70 °C | 1 h | 0% | |
| 1, 70 °C | 16 h | 0% | |
| 2 | AgOTf, 70 °C | 1 h | 0% |
| AgOTf, 70 °C | 16 h | 0% | |
| 3 | 1, + AgOTf, 24 °C | 1 h | 0% |
| 1, + AgOTf, 70 °C | 1 h | 48% | |
| 1, + AgOTf, 70 °C | 16 h | 99% | |
| 4 | 2, 70 °C | 1 h | 0% |
| 2, 70 °C | 16 h | 0% | |
| 5 | 2 + AgOTf, 24 °C | 1 h | 0% |
| 2 + AgOTf, 70 °C | 1 h | 7% | |
| 2 + AgOTf, 70 °C | 16 h | 99% | |
| 6 | AuCl, 70 °C | 16 h | 11% |
| 7 | AuCl, + AgOTf, 70 °C | 16 h | 8%c |
[12] = 0.04 mM, [Au] = 0.002 mM (5 mol%), [additive] = 0.004 mM (10 mol%), reaction volume 0.60 mL.
As determined by NMR integration, all species were cleanly resolved and except for entry 7, the only observable compounds were starting 12 or product 24.
6% of fluorene 5 was detected.
Knowing that 12 is compatible with 1 and that this reaction is comparable with chloro[tris(2,4-di-tert-butylphenyl)phosphite] 2, we continued our inquiry looking for differences in reactivity. We continued with chloro (15), bromo (16) and iodo (17) terminated alkynes that gave 25 (Scheme 7). The cycloisomerization of 16 and 17 involving 1,2-migration of the halogen has been reported using AuCl [24]. Chloro alkyne (15) was resistant to cyclization in our hands with AuCl, AgOTf, or a combination, but cyclized readily when treated with 1 and AgOTf, giving 74% conversion after 16 h at 70 °C. Bromo alkyne (16), was completely converted to product 25 after 16 h, again with 1 + AgOTf, and iodo alkyne (17) was completely converted after only 1 h.
Scheme 7.
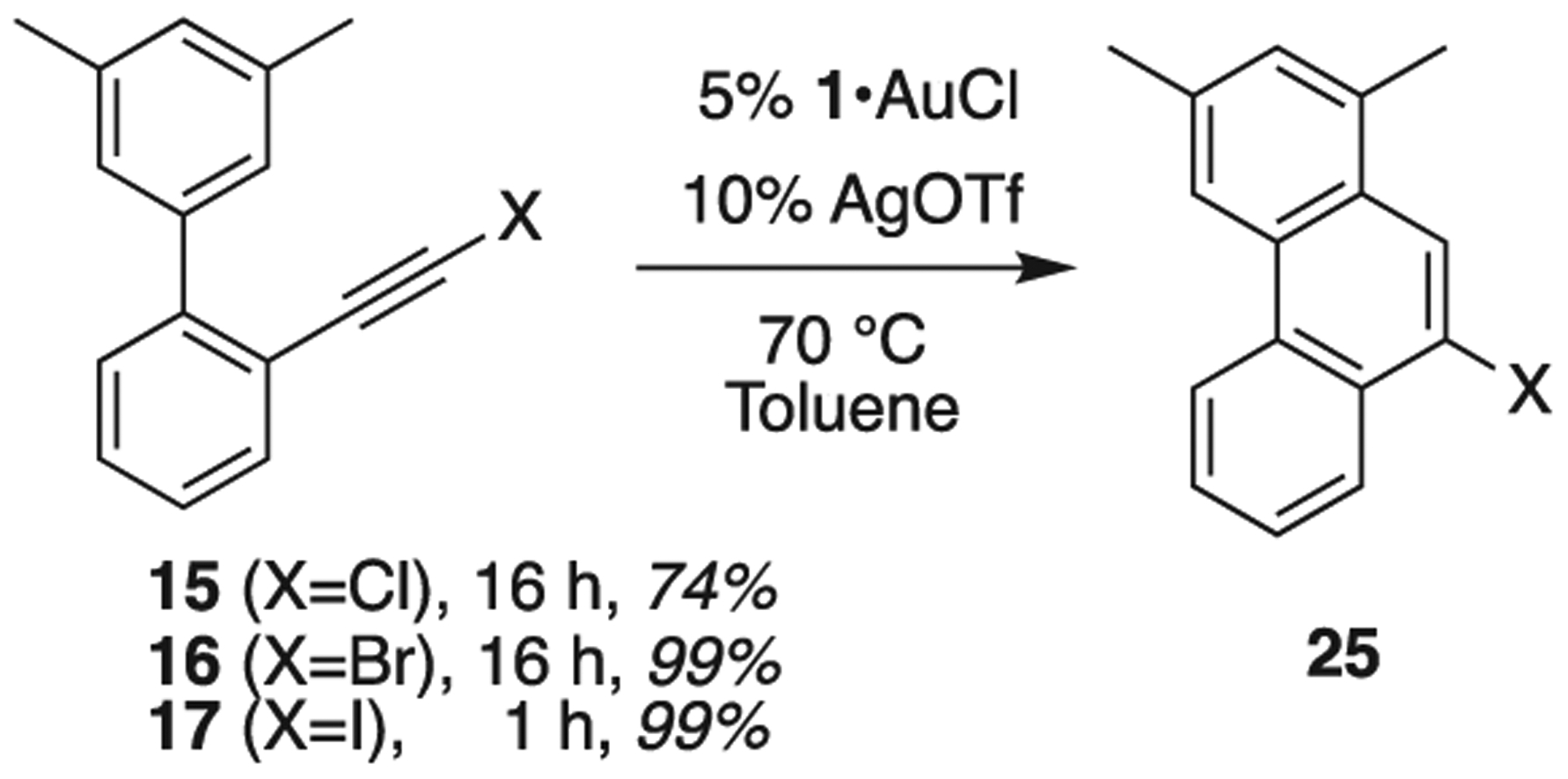
Catalytic screening of substrates 15–17. 1,2-Migration of X was observed in the product 25 (as previously reported).
We then explored substrates 18–20 (Scheme 8, Table 2) with terminal methyl, propyl, and ester groups. Methyl-terminated alkyne 18 readily converted to cycloadduct 26 under Au catalysis in the presence of AgOTf, with either 1 (Entry 1) or gold 2 (Entry 2), with complete conversion achieved after 16 h. The elongated propyl-terminated alkyne 19, had marginally higher reactivity with 1 vs. 2, resulting in 50% and 37% conversion, respectively. The ester-functionalized alkyne 20 was unreactive, whereas it was previously cyclized with PtCl2 to give fluorene 5 as the major product [24]. This sequence of experiments aimed to probe the effect of short vs. long alkyl groups on the alkyne in the reaction with 1; the hope was to find a permutation of cavitand volume and guest size that would alter reactivity. This was not found in experiments where the alkyne was modified, but when the aromatic scaffold was changed, something different happened.
Scheme 8.
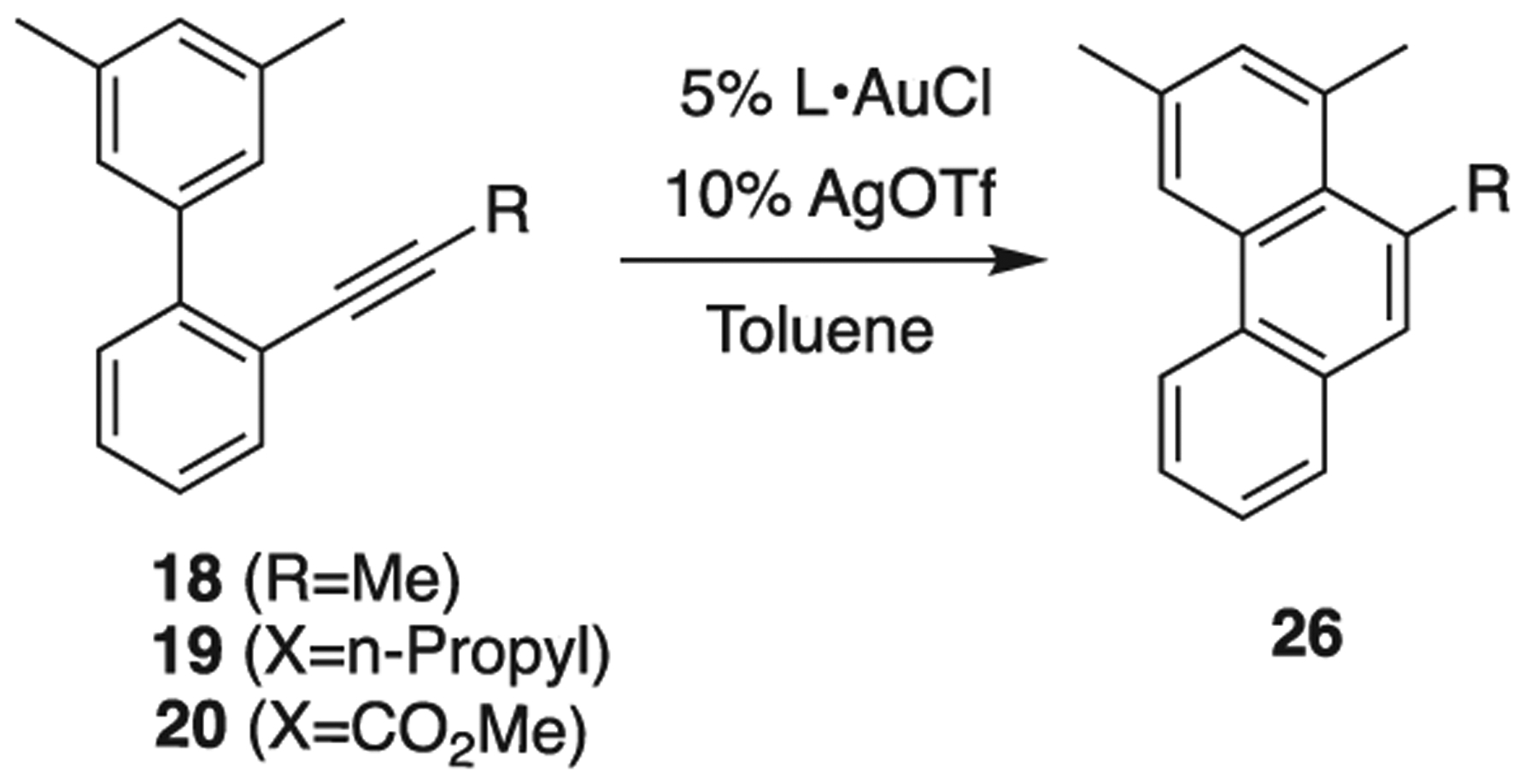
Catalytic screening of substrates 18–20, resulting in 26.
Table 2.
Cycloisomerization of 18–20: variation of R under a variety of conditions.
| Entry | Substrate | Conditionsa | Time | Conversionb |
|---|---|---|---|---|
| 1 | 18 | 1, + AgOTf, 70 °C | 1 h | 15% |
| 1, + AgOTf, 70 °C | 16 h | 99% | ||
| 2 | 18 | 2 + AgOTf, 70 °C | 1 h | 7% |
| 2 + AgOTf, 70 °C | 16 h | 99% | ||
| 3 | 19 | 1, + AgOTf, 70 °C | 1 h | 0% |
| 1, + AgOTf, 70 °C | 16 h | 50% | ||
| 4 | 19 | 2 + AgOTf, 70 °C | 1 h | 0% |
| 2 + AgOTf, 70 °C | 16 h | 37% | ||
| 5 | 20 | 1, + AgOTf, 70 °C | 16 h | 0% |
[Substrate] = 0.04 mM, [Au] = 0.002 mM (0.05 mol%) [additive] = 0.004 mM (0.1 mol%), reaction volume 0.60 mL.
As determined by NMR integration, all species were cleanly resolved, the only observable compounds were starting [Substrate] or product 26.
We prepared modified scaffold 21 where the xylyl group of 12 was replaced with tolyl (Scheme 9). Resorcinarene cavitands admit phenyl, benzyl, tolyl, and cyclohexyl sized groups with ease, but larger o-, m- xylyl or mesityl (1,3,5-trisubstituted) become too wide to access the interior. Alkynes 22 and 23 are further variations.
Scheme 9.
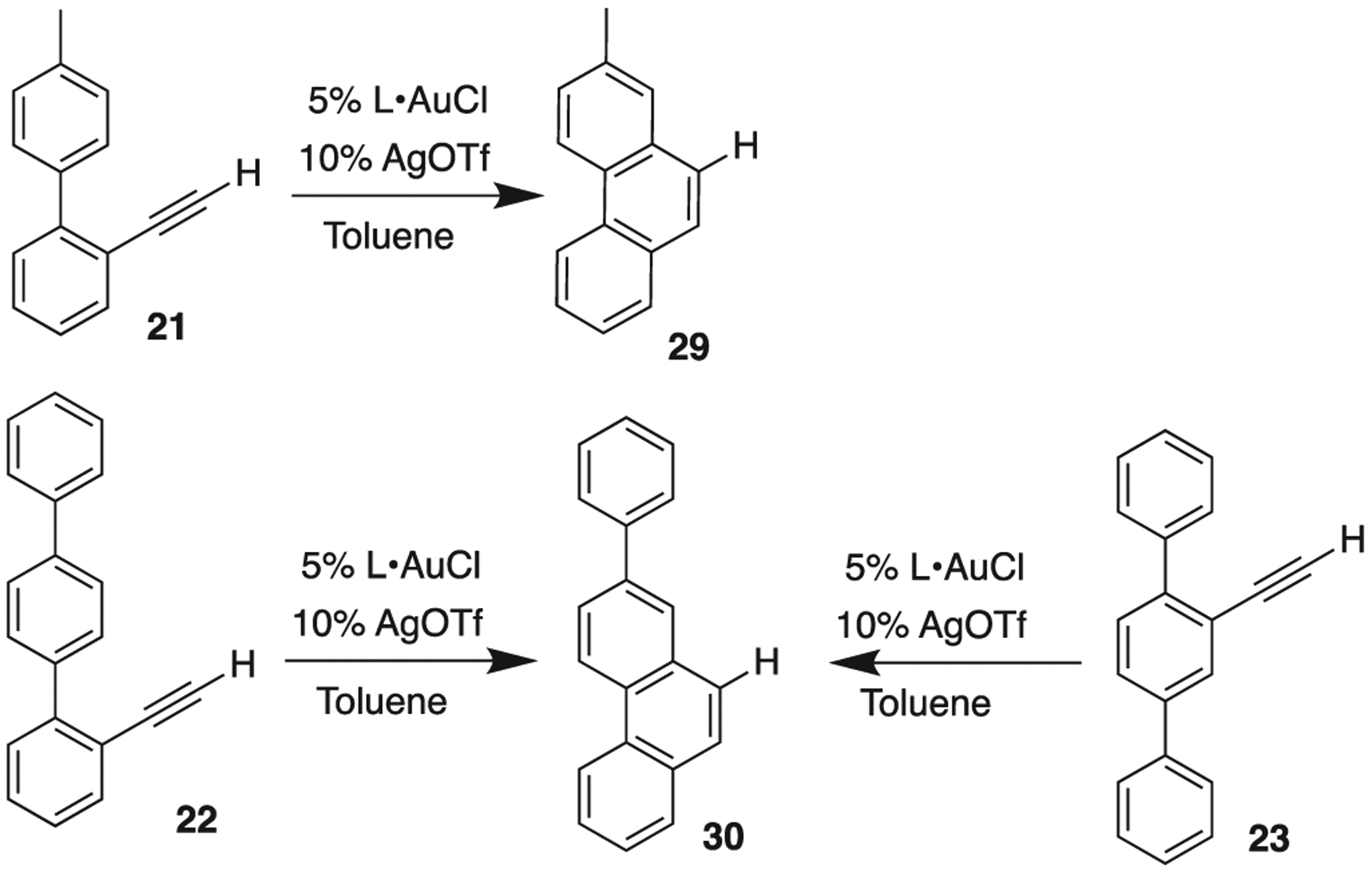
Catalytic screening of substrates 21–23: variation in the aromatic scaffold.
Immediately, a reactivity difference was noted between dimethyl substituted 12 and tolyl 21. Cavitand 1 was ineffective at producing measurable amounts of 29 (Table 3, Entry 1), even after multiple replicates. Using AuCl 2, 29 was produced with 76% conversion (Table 3, Entry 2). This is less than when 12 was reacted with AuCl 2 (Table 1, Entry 5, 99% conversion after 16 h), thus the difference with 2 could be electronic, as substrate 12 has a methyl group para to the cyclization center, while it is meta in 21. An electronic effect should be small enough in this case, that 1 should still give some turnover. None was observed, which we then interpret to be a size effect. Tentatively, the alkyne cannot effectively approach Au in 1; could this be caused by the tolyl group having a predilection for the cavity interior?
Table 3.
Cycloisomerization of 21–23: variation of aromatic scaffolds under a variety of conditions.
| Entry | Substrate | Conditionsa | Time | Conversionb |
|---|---|---|---|---|
| 1 | 21 | 1, + AgOTf, 70 °C | 1 h | 0% |
| 1, + AgOTf, 70 °C | 16h | 0% | ||
| 2 | 21 | 2 + AgOTf, 70 °C | 1 h | 0% |
| 2 + AgOTf, 70 °C | 16h | 76% | ||
| 3 | 22 | 1, + AgOTf, 70 °C | 1 h | 1% |
| 1, + AgOTf, 70 °C | 16h | 19% | ||
| 4 | 22 | 2 + AgOTf, 70 °C | 1 h | 13% |
| 2 + AgOTf, 70 °C | 16 h | 53% | ||
| 5 | 23 | 1, +AgOTf, 70 °C | 1 h | 0% |
| 1, + AgOTf, 70 °C | 16 h | 5% | ||
| 6 | 23 | 2 + AgOTf, 70 °C | 1 h | 20% |
| 2 + AgOTf, 70 °C | 16 h | 75% |
[Substrate] = 0.04 mM, [Au] = 0.002 mM (0.05 mol%) [additive] = 0.004 mM (0.01 mol%), reaction volume 0.60 mL.
As determined by NMR integration, all species were cleanly resolved, the only observable compounds were starting [Substrate] or product 26.
Thus far, binding studies have proven inconclusive with multiple substrates – even with bulky solvents that are excluded from the cavity (i.e. mesitylene-d12), we have not observed host–guest interactions with 1. These types of solvents have been used with great success to bias guest binding by removing competition with solvents (such as chloroform, benzene and toluene which all fit). For now we propose that some modicum of complementary binding is at work. This issue could be resolved at a later date.
With substrate 22 we expanded on this idea by giving the cavitand another ‘handle’ with which to bind in the form of an additional phenyl ring. With catalyst 1, we see very poor conversion to 30; with 2, modest conversion (Table 3, Entries 3–4). Again, there is a difference between 1 and 2, which could be ascribed simply to steric bulk of Au-1′s ‘ligand’; we think the elongated biphenyl has some degree of complementarity for 1, significant enough to prevent the alkyne from finding the reactive Au(I) center.
In our last substrate 23, the alkyne is positioned between two phenyl groups, and the reactivity difference between cavitand 1 and complex 2 (Table 3, Entries 5–6) was stark. Complex 2 resulted in 75% conversion of 23 to 30 after overnight heating, whereas 1 gave a minimally measurable amount of product.
Conclusion
As far as catalyst electronics are concerned, the selection of 2 very closely matches 1 at phosphorous, and thus Au. This gave us the ability to probe any limiting or enhancing features of the cavitand with respect to reactivity. As we continue our search for stabilizing events governed by the walls, we have successfully uncovered size-limiting or perhaps size-selection effects with shapes that are complementary for cavitand binding. We found that tolyl 21 and biphenyl 22 retained modest to good reactivity with Au-2, and all but lost reactivity with Au cavitand 1. This is peculiar as the first substrate in our study, xylyl 12 behaved identically between catalysts. Alkyne 12 is wider than 21 and herein we propose that the more slender 21 has potential for interaction with 1′s interior. Similar behavior was noted when 1 was deployed in the cyclization of alkyne acids to give lactones 3 (Scheme 2) [11]. In that work, auxiliary R’ = benzyl and p-tolyl groups resulted in sluggish reactivity, when compared to the wider R’ = 3,5-dimethyl, or naphthyl substituted substrates. For now, guests that can fit inside the cavitand behave differently, than those that can not – even in cases where larger size does not seem to interfere with access to the gold center (namely, 12).
Substrate 23 reinforces these observations – now two flanking phenyl groups which do not play a large role with catalyst 2, but with cavitand 1, conversion to 30 is abysmal. Taken together these examples of scaffold variation point to a size-effect that takes place when AuCl is mounted inwardly using a size-restrictive cavitand. The electronically similar 2 is much more promiscuous – taking almost all substrates in its stride, whereas 1 results in a restriction of sorts. Further investigation into the interactions of a substrate’s scaffold with a binding pocket will hopefully give rise to a better picture of these limits, directing us towards challenges which require size selectivity, perhaps with multiple reactivity centers. For now, the scaffold size of the substrate somehow interferes with the Au center of 1 from finding its otherwise nimble reaction partner, the alkyne.
Supplementary Material
Acknowledgments
The project described was supported by the generous support of the National Science Foundation (NSF RUI CHE-1708937). NMR instrumentation was provided by the National Science Foundation (MRI CHE-1337559). This project is supported in part by RISE (L.E.R.) (NIH 2R25GM071638-09A1). The content is solely the responsibility of the authors and does not necessarily represent the views of the National Science Foundation.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
The electronic supplementary material contains all procedures for the synthesis of new compounds including their characterization. Representative 1H NMR stack plots from which conversions were calculated are included. Supplementary data to this article can be found online at https://doi.org/10.1016/j.tetlet.2020.152333.
References
- [1].Jans ACH, Caumes X, Reek JNH, ChemCatChem 11 (2019) 287–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Corma A, Leyva-Pérez A, Sabater MJ, Chem. Rev 111 (2011) 1657–1712. [DOI] [PubMed] [Google Scholar]
- [3].Li Z, Brouwer C, He C, Chem. Rev 108 (2008) 3239–3265. [DOI] [PubMed] [Google Scholar]
- [4].Arcadi A, Chem. Rev 108 (2008) 3266–3325. [DOI] [PubMed] [Google Scholar]
- [5].Hashmi ASK, Chem. Rev 107 (2007) 3180–3211. [DOI] [PubMed] [Google Scholar]
- [6].Dalcanale E, Soncini P, Bacchilega G, Ugozzoli F, J. Chem. Soc., Chem. Commun (1989) 500–502. [Google Scholar]
- [7].Soncini P, Bonsignore S, Dalcanale E, Ugozzoli F, The Journal of Organic Chemistry 57 (1992) 4608–4612. [Google Scholar]
- [8].Rudkevich DM, Hilmersson G, Rebek J, J. Am. Chem. Soc 120 (1998) 12216–12225. [Google Scholar]
- [9].Schramm MP, Kanaura M, Ito K, Ide M, Iwasawa T, Eur. J. Org. Chem 2016 (2016) 813–820. [Google Scholar]
- [10].Inoue M, Fujii Y, Matsumoto Y, Schramm MP, Iwasawa T, Eur. J. Org. Chem 2019 (2019) 5862–5874. [Google Scholar]
- [11].Ho TD, Schramm MP, Eur. J. Org. Chem (2019) 5678–5684. [Google Scholar]
- [12].Inoue M, Ugawa K, Maruyama T, Iwasawa T, Eur. J. Org. Chem 2018 (2018) 5304–5311. [Google Scholar]
- [13].Endo N, Inoue M, Iwasawa T, Eur. J. Org. Chem 2018 (2018) 1136–1140. [Google Scholar]
- [14].Iwasawa T, Hooley RJ, Rebek J, Science 317 (2007) 493–496. [DOI] [PubMed] [Google Scholar]
- [15].Shenoy SR, Pinacho Crisóstomo FR, Iwasawa T, Rebek J, J. Am. Chem. Soc 130 (2008) 5658–5659. [DOI] [PubMed] [Google Scholar]
- [16].Endo N, Kanaura M, Schramm MP, Iwasawa T, Eur. J. Org. Chem 2016 (2016) 2514–2521. [Google Scholar]
- [17].Kanaura M, Endo N, Schramm MP, Iwasawa T, Eur. J. Org. Chem 2016 (2016) 4970–4975. [Google Scholar]
- [18].Endo N, Kanaura M, Schramm MP, Iwasawa T, Tetrahedron Lett. 57 (2016) 4754–4757. [Google Scholar]
- [19].Harkat H, Dembelé AY, Weibel J-M, Blanc A, Pale P, Tetrahedron 65 (2009) 1871–1879. [Google Scholar]
- [20].Genin E, Toullec PY, Antoniotti S, Brancour C, Genêt J-P, Michelet V, J. Am. Chem. Soc 128 (2006) 3112–3113. [DOI] [PubMed] [Google Scholar]
- [21].Gasperini D, Maggi L, Dupuy S, Veenboer RMP, Cordes DB, Slawin AMZ, Nolan SP, Adv. Synth. Catal 358 (2016) 3857–3862. [Google Scholar]
- [22].Fürstner A, Mamane V, Chemical Communications (2003) 2112–2113. [DOI] [PubMed] [Google Scholar]
- [23].Fürstner A, Mamane V, J. Organ. Chem 67 (2002) 6264–6267. [DOI] [PubMed] [Google Scholar]
- [24].Mamane V, Hannen P, Fürstner A, Chem. – A, Eur. J 10 (2004) 4556–4575. [DOI] [PubMed] [Google Scholar]
- [25].Bandini M, Emer E, Tommasi S, Umani-Ronchi A, Eur. J. Org. Chem 2006 (2006) 3527–3544. [Google Scholar]
- [26].Fürstner A, Kennedy JWJ, Chem. – A, Eur. J 12 (2006) 7398–7410. [DOI] [PubMed] [Google Scholar]
- [27].Jiménez-Núñez E, Echavarren AM, Chem. Rev 108 (2008) 3326–3350. [DOI] [PubMed] [Google Scholar]
- [28].Fürstner A, Davies PW, Angew. Chem. Int. Ed 46 (2007) 3410–3449. [DOI] [PubMed] [Google Scholar]
- [29].Menon RS, Findlay AD, Bissember AC, Banwell MG, J. Organ. Chem 74 (2009) 8901–8903. [DOI] [PubMed] [Google Scholar]
- [30].Aparece MD, Vadola PA, Org. Lett 16 (2014) 6008–6011. [DOI] [PubMed] [Google Scholar]
- [31].Chen Y, Lu Y, Li G, Liu Y, Org. Lett 11 (2009) 3838–3841. [DOI] [PubMed] [Google Scholar]
- [32].Sharma AK, Gowdahalli K, Krzeminski J, Amin S, J. Organ. Chem 72 (2007) 8987–8989. [DOI] [PubMed] [Google Scholar]
- [33].Pietruszka J, Witt A, Synthesis 2006 (2006) 4266–4268. [Google Scholar]
- [34].Roth GJ, Liepold B, Müller SG, Bestmann HJ, Synthesis 2004 (2004) 59–62. [Google Scholar]
- [35].Nie X, Wang G, J. Organ. Chem 71 (2006) 4734–4741. [DOI] [PubMed] [Google Scholar]
- [36].Zhdanko A, Maier ME, ACS Catalysis 5 (2015) 5994–6004. [Google Scholar]
- [37].Wang D, Cai R, Sharma S, Jirak J, Thummanapelli SK, Akhmedov NG, Zhang H, Liu X, Petersen JL, Shi X, J. Am. Chem. Soc 134 (2012) 9012–9019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.