

SUMMARY
The major cap-binding protein eukaryotic translation initiation factor 4E (eIF4E), an ancient protein required for translation of all eukaryotic genomes, is a surprising yet potent oncogenic driver. The genetic interactions that maintain the oncogenic activity of this key translation factor remain unknown. In this study, we carry out a genome-wide CRISPRi screen wherein we identify more than 600 genetic interactions that sustain eIF4E oncogenic activity. Our data show that eIF4E controls the translation of Tfeb, a key executer of the autophagy response. This autophagy survival response is triggered by mitochondrial proteotoxic stress, which allows cancer cell survival. Our screen also reveals a functional interaction between eIF4E and a single anti-apoptotic factor, Bcl-xL, in tumor growth. Furthermore, we show that eIF4E and the exon-junction complex (EJC), which is involved in many steps of RNA metabolism, interact to control the migratory properties of cancer cells. Overall, we uncover several cancer-specific vulnerabilities that provide further resolution of the cancer translatome.
Graphical Abstract

In brief
Kuzuoglu-Ozturk et al. identify more than 600 genetic interactions that sustain oncogenic activity of the major cap-binding protein eIF4E by a genome-wide CRIPSRi screen. Their data reveal interactions among distinct cellular processes and eIF4E, uncovering several cancer-specific vulnerabilities.
INTRODUCTION
Genomic sequencing has revolutionized our ability to identify oncogenic drivers and has established the basis for targeted, personalized cancer treatments (Berger and Mardis, 2018). While genomic-driven cancer therapies hold great promise, they have also exposed several limitations. This includes the evolution of drug resistance and incomplete knowledge of genetic interactions supporting cancer cell fitness (Vasan et al., 2019). Interestingly, in the last decade, there has been a greater appreciation of the broad action of several oncogenes in manipulating the cancer proteome at the post-genomic step for sustained cancer cell fitness, particularly at the step of translation control (Barna et al., 2008; Faller et al., 2015; Furic et al., 2010; Hsieh et al., 2010; Truitt and Ruggero, 2016; Wolfe et al., 2014). In this respect, the most frequently mutated oncogenes, Myc, Ras, and phosphatidylinositol 3-kinase (PI3K)/AKT, all regulate the expression or activity of the major cap-binding protein, eukaryotic translation initiation factor 4E (eIF4E) (Truitt and Ruggero, 2016). While Myc enhances the transcription of eIF4E (Jones et al., 1996; Rosenwald et al., 1993), Ras activates the phosphorylation of eIF4E by regulating mitogen-activated protein kinase (MAPK)-interacting serine/threonine kinase 1 (MNK1) (Furic et al., 2010; Waskiewicz et al., 1997), resulting in eIF4E hyperactivation. Moreover, eIF4E is also regulated via the mTOR pathway through eIF4E-binding protein (4EBPs) suppressors, which inhibit eIF4E activity (Haghighat et al., 1995; Hsieh et al., 2012; Pourdehnad et al., 2013). Our previous findings surprisingly revealed that a 50% reduction in eIF4E expression (eIF4E+/−) is compatible with normal development and global protein synthesis, but it is essential for cancer cells to maintain a selective translatome that promotes oncogenic transformation (Truitt et al., 2015). Cells that are grown in an anchorage-independent manner have shown that eIF4E+/− cells cannot be transformed in vitro even upon the overexpression of undruggable oncogenes, including Myc and Ras, as they fail to translate a specific network of mRNAs that are selective to cancer cells but not normal cells. Reducing eIF4E expression to 50% showed a significant decrease in Kras-driven lung cancer tumor growth in vivo and revealed that eIF4E inhibitors can be used in combinational therapy (Truitt et al., 2015).
Interestingly, inhibitors that target specific translation factors in cancer and distinct diseases have been on the rise. Inhibitors that block the ability of eIF4E to recruit the pre-initiation complex, 4EGI-1, 4E1RCat, and 4E2RCat, have revealed anti-tumor effects in preclinical trials (Chen et al., 2012). Moreover, MNK1 inhibitors that block eIF4E phosphorylation, and hence the activity of eIF4E, such as cercosporamide and eFT508 (also named tomivosertib) suppress tumor progression and metastasis in both xenograft and genetically engineered mouse models (Konicek et al., 2011; Xu et al., 2019; Xu and Ruggero, 2020). Importantly, eFT508 is currently in phase II clinical trials (ClinicalTrials.gov: NCT03616834 and NCT04622007).
Although eIF4E expression is widely accepted as a druggable oncogenic event, the genetic interacting partners of eIF4E, which may act downstream or upstream of translation control to maintain cancer cell fitness, remain completely unknown. In this study, we characterized synthetic lethal partners of eIF4E in cells expressing Myc and Ras. This approach unveiled not only combinational therapies, which can selectively target cancer cells at the post-genomic level, but also previously unknown functional connections between eIF4E and unexpected cellular processes. We focused on one of the most surprising connections between eIF4E and the mitochondria in cancer. This was characterized by a specific interaction between eIF4E with a selected anti-apoptotic protein as well as a mitochondrial protein important for activating an adaptive stress response. Moreover, the screen revealed a genetic interaction with the exon-junction complex (EJC), a key complex involved in several steps of mRNA post-transcriptional processing, which is critical for the expression of transcripts driving cancer cell migration. Overall, our data highlight the importance of the cap-binding protein as a hub to connect distinct cellular processes in cancer and uncover several cancer-specific vulnerabilities that require the activity of eIF4E for cancer cell fitness and survival.
RESULTS
The anti-apoptotic protein Bcl-xL is identified as a synthetic lethal partner of eIF4E
To identify critical, yet unknown, genetic interaction partners of eIF4E, we performed a genome-wide CRISPRi screen in mouse embryonic fibroblasts (MEFs). The screen was performed in MEFs expressing either 100% (eIF4E+/+) or 50% (eIF4E+/−) eIF4E together with two undruggable oncogenes, Myc and Ras (Figures S1A and S1B). Myc and Ras overexpressing cells were engineered to express a stable inactive form of the Cas9 protein (dCas9) that is fused to the Krüppel-associated box (KRAB) transcriptional repressor domain. Cells were infected with a genome-scale CRISPRi library containing five single-guide RNAs (sgRNAs) targeting genes (Horlbeck et al., 2016) (Figure 1A). To determine the genes that are synthetic lethal partners of eIF4E, sgRNA enrichment of each gene was compared in both conditions (p < 0.05 and Δlog2[fold change (FC; eIF4E+/− − eIF4E+/+)] < −0.5). The analysis revealed 630 genes that are significantly more lethal in eIF4E+/− compared to eIF4E+/+ (Figure 1B; Table S1). Among these genes, 139 of them were not essential for eIF4E+/+ cells (|log2FC[eIF4E+/+]| < 1) but promote lethality specifically in the eIF4E+/− condition (log2FC[eIF4E+/−] < −1) (Figure 1B, green dots; Table S1).
Figure 1. Genome-wide CRISPRi screen reveals anti-apoptotic protein Bcl-xL as synthetic lethal partner of eIF4E.
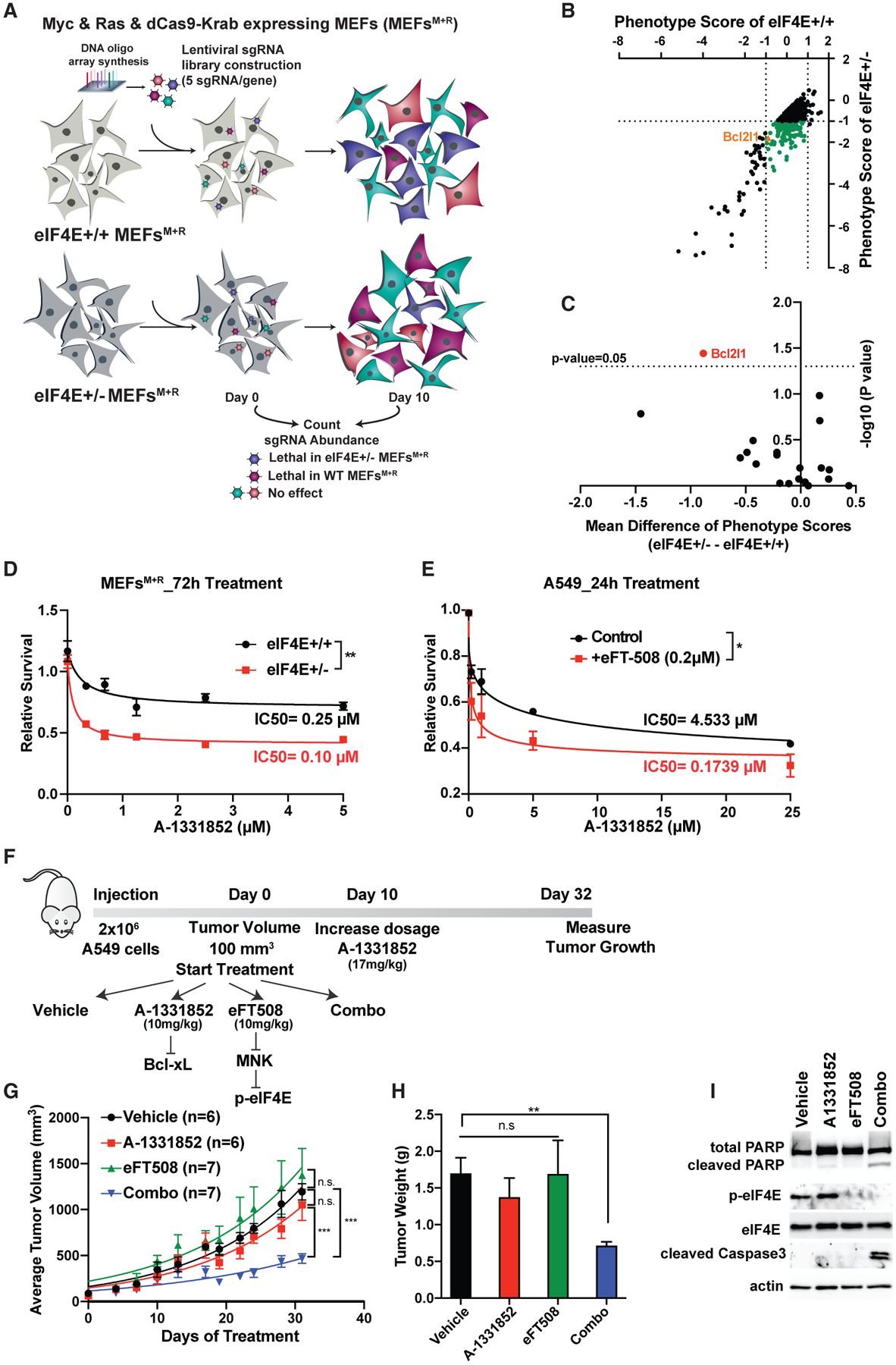
(A) Schematic representation of genome-wide CRISPRi screen in MEFs expressing Myc and Ras. Two independent screens were performed for each condition.
(B) Synthetic lethal partners of eIF4E. Phenotype scores of each gene are calculated based on the mean value of the activity of five sgRNAs on day 10 versus day 0. The green dots represent genes that promote specific lethality in eIF4E+/− cells (|log2[eIF4E+/+]| < 1 and log2[eIF4E+/−] < 1).
(C) Identification of Bcl2l1 as the only Bcl2 family gene that significantly promotes lethality in eIF4E+/− cells. The mean difference is calculated by subtracting the phenotype scores of each gene in eIF4E+/− − eIF4E+/+ cells. For each gene, the p value was calculated using a paired Wilcoxon test.
(D) Treatment of MEFs expressing Myc and Ras with different concentrations of the Bcl-xL inhibitor A-1331852 for 72 h. A CellTiter Glo assay was performed and relative survival for each genotype was plotted normalized to cells treated with DMSO (n > 3).
(E) Treatment of A549 lung cancer cells with A-1331852 together with eIF4E inhibitor eFT508 (0.2 μM) for 24 h. A two-way ANOVA test was performed to determine statistical significance (n = 3).
(F) Schematic representation of in vivo injection of A549 cells into NSG mice.
(G) Measurement of tumor volumes over 32 days of treatment. “n” represents number of mice used in each group. A one-way ANOVA with covariates followed by a false discovery rate (FDR) adjustment was performed.
(H) Measurement of tumor weight on day 32. A Kruskal-Wallis test and then a Wilcoxon rank-sum test were performed followed by FDR adjustment.
(I) Western blot analysis of representative tumor samples collected on day 32.
All values represent the mean + SEM. *p < 0.01, **p < 0.001, ***p < 0.0001. See also Figures S1 and S2.
Evasion of apoptosis is one of the hallmarks of cancer (Hanahan and Weinberg, 2011); therefore, targeting anti-apoptotic factors is one of the most significant avenues for cancer therapies. Among the 139 genes that are synthetic lethal with eIF4E, one of the hits is the Bcl2l1 gene, which encodes the anti-apoptotic protein Bcl-xL (Figure S2A). To test the specificity of this genetic interaction, we carefully analyzed all of the Bcl-2 family protein members and compared the mean difference of the phenotype score between eIF4E+/+ and eIF4E+/− cells. Among the 20 members of the Bcl-2 family, it is notable that Bcl-xL was the only family member that shows a significant phenotype in eIF4E+/− (Figure 1C), indicating the specificity of this interaction. Moreover, it is the only functionally significant apoptotic factor that interacts with eIF4E to promote cancer cell survival. We then proceeded to validate the phenotype from the screen by performing a fluorescence-based competitive growth assay. We transduced eIF4E+/+ and eIF4E+/− cells overexpressing Myc and Ras with individual sgRNAs (BFP+) targeting Bcl-xL. eIF4E+/− cells were found to be more sensitive to Bcl-xL depletion than were eIF4E+/+ cells (Figures S2B–S2D). Moreover, the specificity of the eIF4E and Bcl-xL genetic interaction was evident when we knocked down another Bcl2 family protein, Bcl2l2, and performed a competitive growth assay. Depletion of Bcl2l2 together with eIF4E did not reveal any significant change in cell proliferation (Figure S2E and S2F).
We next used a potent Bcl-xL inhibitor, A-1331852, which has been shown to specifically inhibit Bcl-xL activity with oral bioavailability (Leverson et al., 2015). eIF4E+/+ and eIF4E+/− cells expressing Myc and Ras were treated with different concentrations of the inhibitor for 72 h and cell viability was assessed. The eIF4E+/− cells were significantly more sensitive to the Bcl-xL inhibitor (Figure 1D), in agreement with both the CRISPRi screen results and the genetic validation. We also confirmed that A-1331852 promotes cell death more efficiently in eIF4E+/−, which shows higher expression of cleaved Parp and cleaved caspase-3, key markers of apoptosis (Figure S2G). In addition, annexin V and propidium iodide (PI) staining also confirmed that eIF4E+/− cells treated with A-1331852 promote more apoptosis compared to eIF4E+/+ cells (Figure S2H).
We next used the human A549 lung cancer cell line, which endogenously expresses high levels of Myc and possesses the hyperactivating KRAS G12S point mutation. We treated A549 with two different inhibitors targeting eIF4E and Bcl-xL to investigate this combinational therapy. In order to block the activity of eIF4E, we used a potent and highly selective dual MNK1/2 kinase inhibitor, eFT508, a compound in phase 2 clinical trials, which has shown to inhibit eIF4E function by inhibiting its phosphorylation (Konicek et al., 2011; Xu et al., 2019; Xu and Ruggero, 2020). To inhibit Bcl-xL we used A-1331852 in combination with eFT508. A549 cells are significantly more sensitive to A-1331852 when combined with eFT-508 (Figure 1E), which indicates a strong synergy of these compounds on cell growth in vitro. To investigate the effect of combinational therapy targeting Bcl-xL and eIF4E in vivo, we used xenograft mouse models where A549 cells were subcutaneously injected into immune-compromised mice (NSG) (Figure 1F). Mice were treated with a lower concentration of A-1331852 (10 mg/kg) for the first 10 days; as no side effect was observed during this period, the dose was increased to 17 mg/kg. Strikingly, the tumors of mice treated with eFT508 and A-1331852 together grow significantly less compared to single agents or control groups (Figure 1G), and the average tumor weight from the combo group was also significantly lower (Figure 1H). Western blot analysis of the tumor samples from each group demonstrated an increase in the expression of cleaved caspase-3 and Parp only in the combo group (Figures 1I and S2I), indicating that eFT508 and A-1331852 synergistically promote cancer cell death in vivo. The identification of Bcl-xL as a synthetic lethal partner of eIF4E from the screen uncovered a functional connection between the translation machinery and selective anti-apoptotic factors, which holds promise in combinatorial cancer therapies.
Mitochondrial peptidase Pmpcb depletion triggers an adaptive stress response that requires eIF4E activity
Mitochondria play a key role in the maintenance of cancer cell viability, and it has been recently appreciated that in addition to functioning as metabolic organelles, they also regulate protein homeostasis (Senft and Ronai, 2016; Vyas et al., 2016; Wallace, 2012). In addition to Bcl-xL, which functions in the mitochondria to regulate apoptosis, unexpectedly, Pmpcb, which encodes the catalytic subunit of the mitochondrial processing peptidase (MPP) complex, another mitochondrial protein, was found among the top hits of the eIF4E synthetic lethal partners. Most mitochondrial proteins are first translated in the cytoplasm and then imported to the mitochondria. These proteins are first processed by Pmpcb and its non-protease homolog Pmpca, which are localized in the mitochondria and are responsible for the cleavage of the mitochondrial entry signal that primes these proteins to undergo additional modifications depending on their final localization (Gakh et al., 2002; Quirós et al., 2015) (Figure 2A). Discovering two distinct mitochondrial proteins, Bcl-xL and Pmpcb, as synthetic lethal partners of eIF4E highlighted a previously unappreciated molecular connection between translational regulation and mitochondrial function.
Figure 2. Mitochondrial Pmpcb depletion activates a specific adaptive stress response that requires eIF4E.

(A) Schematic representation of the PMPCB’s function in mitochondria. TOM, translocase of the outer membrane; TIM, translocase of the inner membrane; MTS, mitochondrial targeting sequence.
(B) Validation of the synthetic lethal interaction between eIF4E and Pmpcb and the rescue of the phenotype by overexpressing GFP-tagged Pmpcb. A competitive growth assay was performed using a specific Pmpcb sgRNA in MEF expressing either GFP or GFP-Pmpcb. A two-way ANOVA test was performed to determine statistical significance (n = 3).
(C) Annexin V staining of eIF4E+/+ and eIF4E+/− MEFs cells expressing either scramble or Pmpcb sgRNA after 2 days of dCas9-Krab induction with doxycycline (Dox). A two-way ANOVA test was performed to determine statistical significance (n = 3).
(D) Schematic representation of UPRmt transcription factor activation profiling plate array.
(E) Heatmap of transcription factor activities in different conditions.
(F) Western blot analysis of Tfeb in Pmpcb-depleted MEFs after 2 days of dCas9-Krab induction with Dox.
(G) Overexpression of Tfeb partially rescues the synthetic lethal phenotype Pmpcb depletion in eIF4E+/− MEFs. A competitive growth assay was performed in cells expressing either GFP or GFP-Tfeb. A two-way ANOVA test was performed to determine statistical significance (n = 3).
All values represent the mean + SEM. *p < 0.01, **p < 0.001. See also Figure S3.
First, we validated this interaction by showing that although Pmpcb knockdown (KD) has a small effect on cell growth in Myc and Ras overexpressing eIF4E+/+ cells, eIF4E+/− cells are significantly more sensitive to Pmpcb depletion (Figures S3A and S3B). Moreover, we rescued the KD by overexpressing Pmpcb (Figure 2B). We also demonstrated that Pmpcb KD results in more apoptosis in eIF4E+/− cells compared to eIF4E+/+ cells (Figure 2C). Next, we compared protein levels of Pmpcb in eIF4E+/+ and eIF4E+/− cells and did not observe any significant differences at the protein level, indicating that eIF4E +/− sensitivity to Pmpcb KD is not due to the expression levels of Pmpcb (Figure S3C). Furthermore, Pmpcb depletion has no effect on global protein synthesis by performing an S35 methionine-incorporation assay (Figure S3D).
The mitochondrial unfolded protein response (UPRmt) is a conserved transcriptional response activated by multiple forms of mitochondrial dysfunction and regulated by mitochondrial-to-nuclear communication (Melber and Haynes, 2018). UPRmt activation promotes lifespan extension and protects against bacterial pathogens that perturb mitochondrial function (Shpilka and Haynes, 2018). As cancer cells need to detect faulty mitochondria in order to increase cell survival, we hypothesized that this may be facilitated through the activation of an UPRmt response elicited by Pmpcb depletion. Interestingly, recent studies showed that UPRmt can be activated in breast cancer (Papa and Germain, 2011; Chen et al., 2020) and can promote invasion and metastasis (Kenny et al., 2017; Zhu et al., 2021). Therefore, we investigated the activity of several transcription factors (TFs) that are critical in activating the UPRmt machinery. We used biotinylated probes made against consensus sequences of TF DNA-binding sites, which are hybridized with nuclear lysates, and the activity of individual TFs was measured in different cellular conditions (Figure 2D). Although few studies have focused on UPRmt in mammals, and not all the TFs tested in the assays are shown to be directly connected to UPRmt, we discovered a very surprising activation of a subset of these specific TFs upon Pmpcb depletion. Among 16 TFs that we have tested, Tfeb, Atf4, and Xbp1 all showed an increase in activity when Pmpcb is depleted specifically in Myc and Ras overexpressing eIF4E+/+, but not in eIF4E+/− cells (Figure 2E). These data indicated that these proteins may be essential to promote a UPRmt-like adaptive stress response as a consequence of Pmpcb depletion and eIF4E is required to trigger this response. To understand this adaptive response upon Pmpcb depletion and the role of eIF4E in this pathway, we examined the expression level of Tfeb, Atf4, and Xbp1 by western blot (Figures 2F and S3E). We observed a specific upregulation of these proteins in Pmpcb KD cells where eIF4E is expressed at a 100% level, but not eIF4E+/− cells, which indicates that eIF4E activity is required for the expression of proteins that trigger a mitochondria-driven adaptive response. Moreover, the overexpression of Tfeb, in itself, partially rescued the proliferation phenotype in eIF4E+/− cells upon Pmpcb depletion (Figure 2G).
eIF4E regulates the translation of Tfeb through its 5′ UTR
To understand whether Tfeb is regulated at the translational or transcriptional level, we first compared the transcript levels of Tfeb upon Pmpcb depletion in eIF4E+/+ and eIF4E+/− cells. We did not observe any difference in mRNA levels in both conditions (Figure 3A). Next, to investigate whether Tfeb is translationally regulated, we performed a polysome sucrose gradient assay to examine the translation efficiency of Tfeb in Pmpcb-depleted cells where eIF4E levels are either 100% or 50% (Figure 3B). Interestingly, qPCR analysis of the Tfeb mRNA in polysome fractions showed a significant shift from low polysome to high polysome levels as a response to Pmpcb KD in eIF4E+/+ cells, demonstrating that Tfeb is translationally upregulated when Pmpcb is depleted. However, the same shift was not observed in eIF4E+/− cells, suggesting that eIF4E is required for translation of Tfeb (Figure 3C).
Figure 3. eIF4E regulates translation of Tfeb mRNA through its 5′ UTR.
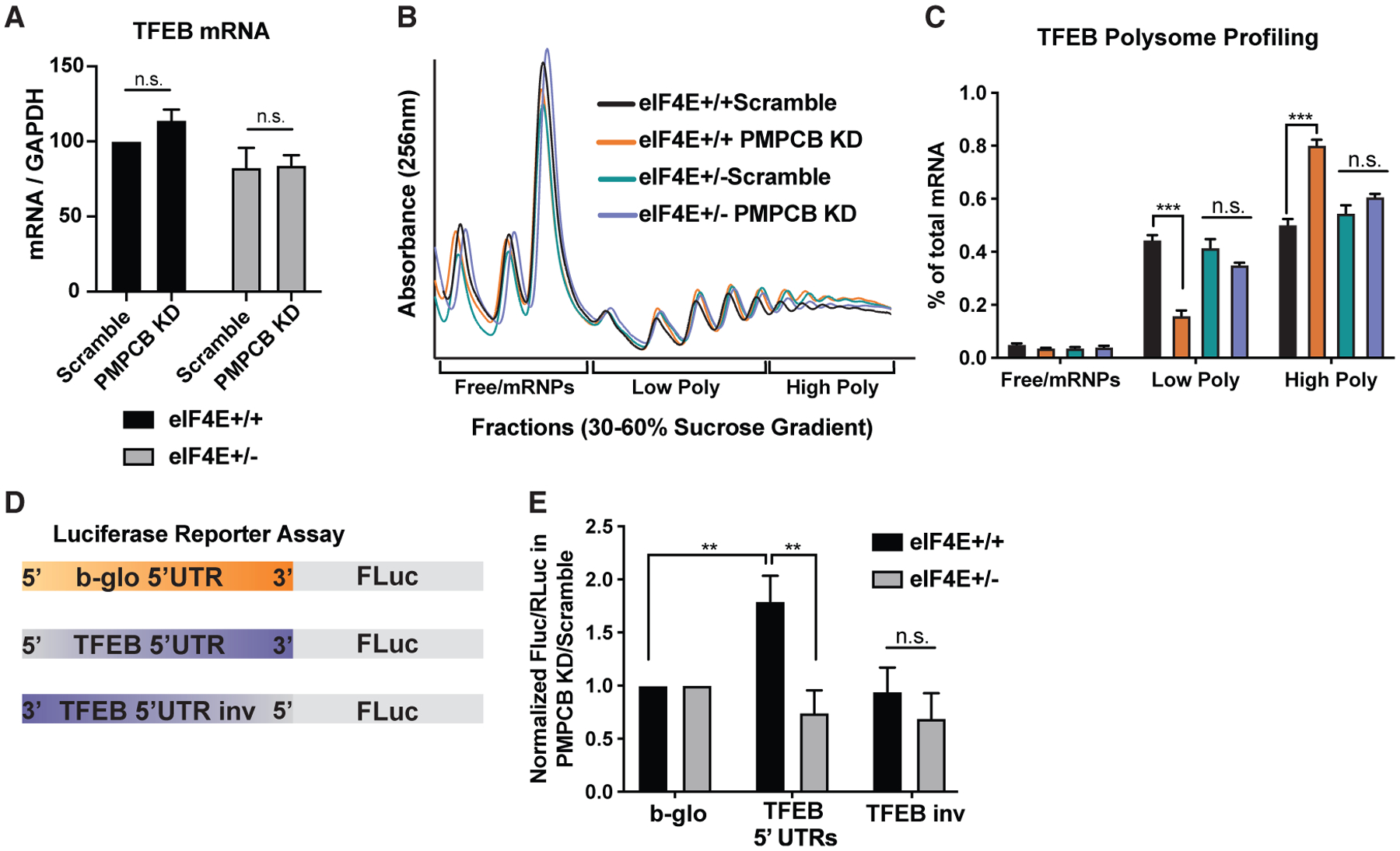
(A) qPCR analysis of Tfeb mRNA after 2 days of dCas9-Krab induction with Dox using scramble and Pmpcb sgRNAs in eIF4E+/+ and eIF4E+/− MEFs expressing Myc and Ras (n = 6).
(B) Representative polysome profiles of Pmpcb-depleted eIF4E+/+ and eIF4E+/− MEFs on a sucrose gradient.
(C) qPCR analysis of Tfeb mRNA levels isolated from 30%–60% sucrose gradient fractions. Pooled fractions corresponding to free ribosomal subunits and messenger ribonucleoprotein (mRNPs) (Free/mRNPs), low-molecular-weight polysomes (Low Poly), and high-molecular-weight polysomes (High Poly) are displayed. Color keys are the same as indicated in (B). A two-way ANOVA test was performed to determine statistical significance (n = 3).
(D) Diagram of the 5′ UTR luciferase reporter assay.
(E) eIF4E is required for 5′ UTR-mediated translation of Tfeb mRNA. Luciferase activity measures in Pmpcb depleted cells over control cells are shown in eIF4E+/+ and eIF4E+/− MEFs. A two-way ANOVA test was performed to determine statistical significance (n > 3).
All values represent the mean + SEM. *p < 0.01, **p < 0.001.
We next performed 5′ UTR luciferase reporter assays to investigate whether eIF4E regulates Tfeb translation through its 5′ UTR. We generated a reporter where the 5′ UTR of Tfeb is cloned upstream of the firefly luciferase open-reading frame. As control, we used a housekeeping 5′ UTR from the β-globin mRNA and the Tfeb 5′UTR, which was inversed (Figure 3D). These results demonstrated that the Tfeb 5′ UTR contains regulatory elements that are regulated by eIF4E dosage (Figure 3E). Taken together, these findings reveal that eIF4E is essential for the translation of Tfeb, which is activated as a response to Pmpcb depletion.
Tfeb mediates an autophagy survival response that is translationally regulated by eIF4E
Tfeb upregulation is associated with deadly cancers, such as pancreatic ductal adenocarcinoma (Perera et al., 2015) and colorectal cancer (Liang et al., 2018); therefore, it is critical to activate an autophagic survival pathway in cancer (Levy et al., 2017; White, 2012). In order to understand whether Pmpcb depletion promotes autophagy due to impairments in protein homeostasis in the mitochondria, we further performed immunofluorescence staining of the autophagosome marker LC3I/II in Myc and Ras overexpressing cells. Pmpcb KD specifically increased the formation of autophagosomes in eIF4E+/+ cells, but not in eIF4E+/− cells (Figures 4A and 4B), and this difference was no longer evident upon Tfeb overexpression (Figures 4C and 4D). These findings reveal a previously unreported link between mitochondrial protein processing and an autophagy survival pathway in cancer cells that is under translational control.
Figure 4. Tfeb mediates an autophagy survival response that is translationally regulated by eIF4E.
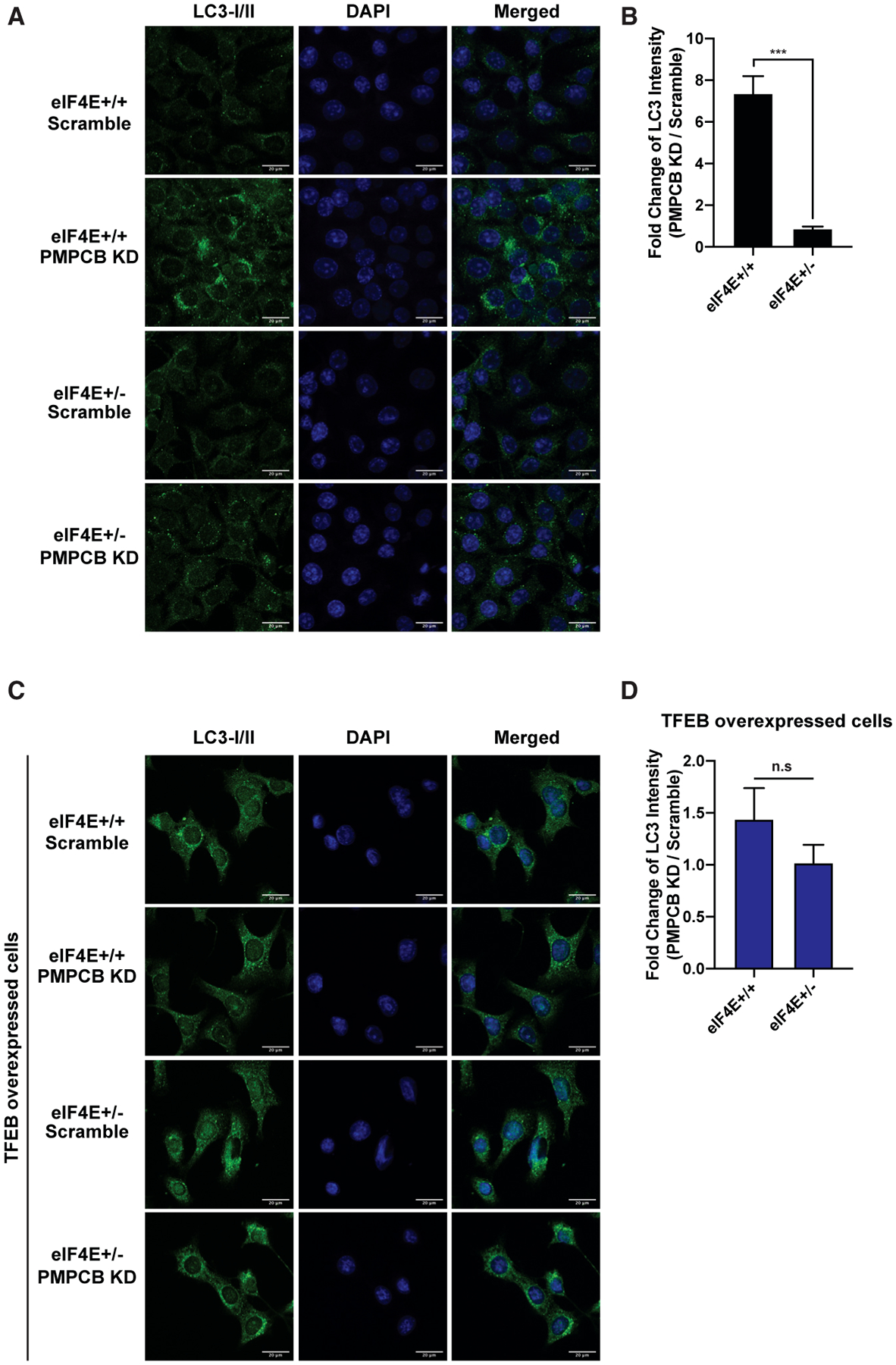
(A) Autophagosomes, shown in green, are accumulated specifically in eIF4E+/+ Pmpcb KD cells. Representative confocal microscopy images of eIF4E+/+ and eIF4E+/− MEFs expressing either scramble or Pmpcb sgRNAs, immunostained with anti-LC3I/II (green, first column) are shown. Nuclei of cells are stained with DAPI (blue, second column). Scale bars, 20 μm.
(B) The bars represent fold change of LC3 intensity in Pmpbc KD cells normalized to scramble controls in eIF4E+/+ and eIF4E+/− MEFs. ***p < 0.001, by multiple t tests.
(C) Tfeb overexpression rescues the difference in the accumulation of autophagosome between eIF4E+/+ and eIF4E+/− Pmpcb KD cells. The colors are the same as described in (A). Scale bars, 20 μm.
(D) The bars represent fold change of LC3 intensity in Pmpbc KD cells normalized to scramble controls in eIF4E+/+ and eIF4E+/− MEFs where Tfeb was overexpressed.
For each panel, at least 30 different images were quantified. All values represent the mean + SEM.
Cooperation between the activities of eIF4E and Pmpcb sustains tumor growth in vivo
Gliomas are known to be highly dependent on mitochondrial function, and numerous therapeutic compounds targeting mitochondria were shown to be efficient in glioma therapies (Guntuku et al., 2016). We therefore used the glioma U251 cell line wherein we knocked down Pmpcb and subcutaneously injected these cells into NSG mice (Figure 5A). Once tumors developed, we started treatment with either vehicle or the eIF4E inhibitor, eFT508 (Figure 5B). The tumors of mice injected with U251 expressing Pmpcb short hairpin RNA (shRNA) treated with eFT508 grow significantly less compared to other groups (Figure 5C). Importantly, when we isolated the tumor samples from each group, we detected the upregulation of Tfeb expression at the protein level in tumors where Pmpcb is depleted; however, the eFT508-treated group did not show a similar upregulation (Figures 5D and 5E). These data demonstrate that Pmpcb depletion can also trigger upregulation of Tfeb expression in an eIF4E-dependent manner in vivo and that inhibition of eIF4E expression in combination with Pmpcb depletion can block tumor growth. Overall, our data uncovered an uncharacterized adaptive stress response, which cancer cells utilize upon Pmpcb depletion, and highlight the importance of eIF4E activity in this process. This translationally regulated adaptive response serves as a point of vulnerability for cancer cells that represents a previously unknown target for potential cancer therapies.
Figure 5. Cooperation between the activities of eIF4E and Pmpcb sustains tumor growth in vivo.
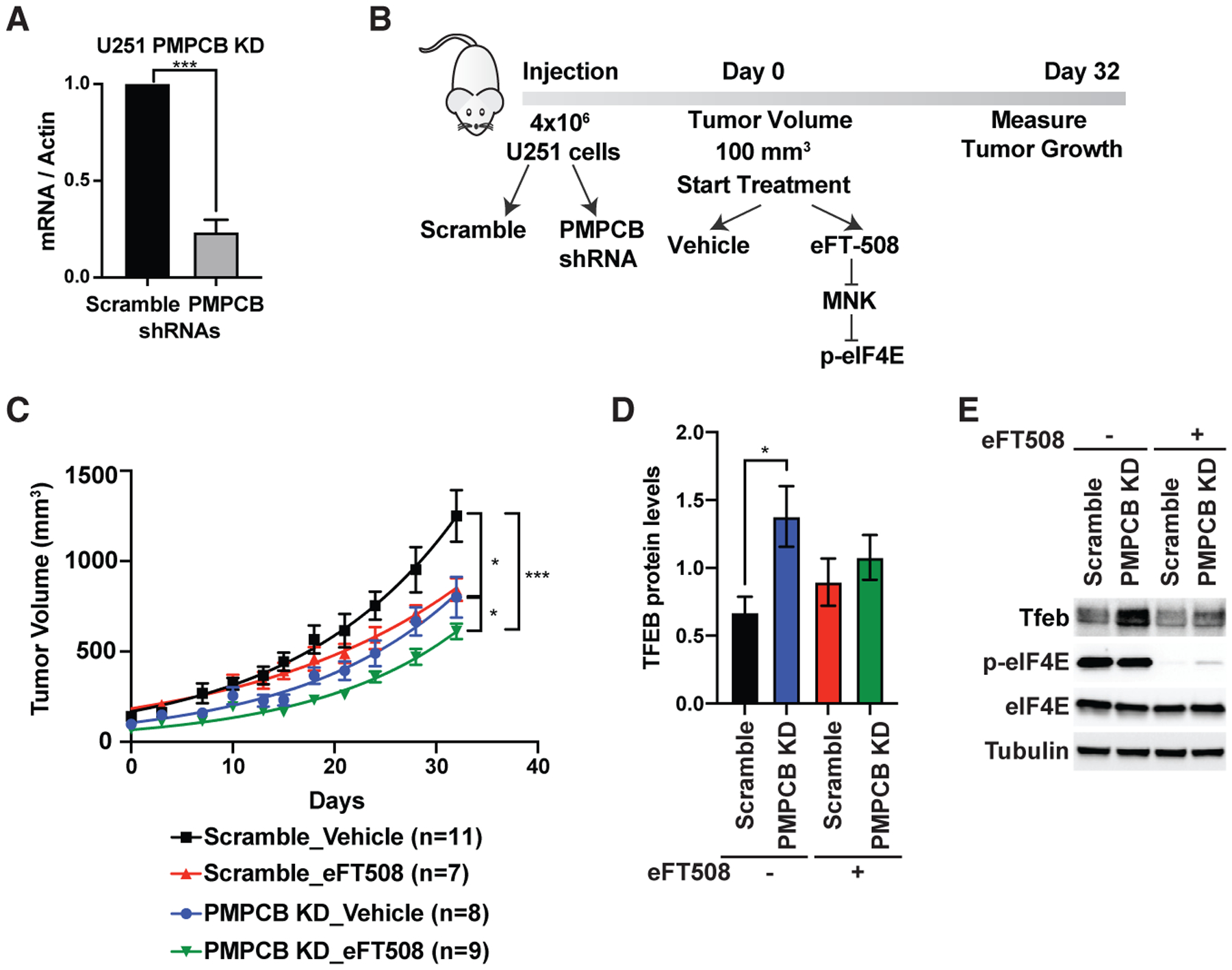
(A) qPCR analysis of U251 cells expressing scramble and PMPCB shRNAs. Multiple t tests were performed to determine statistical significance (n > 3).
(B) Schematic representation of in vivo injection of U251 cells into NSG mice.
(C) Measurement of tumor volumes over 32 days of treatment. “n” represents number of mice used in each group. A one-way ANOVA with covariates followed by a FDR adjustment was performed.
(D) Quantification of Tfeb protein levels in tumor samples collected at the end of the experiment on day 32. A two-way ANOVA test was performed to determine statistical significance. Replicate numbers are indicated in ©.
(E) A representative western blot analysis showing upregulation of Tfeb expression upon Pmpcb depletion in vivo.
All values represent the mean + SEM. *p < 0.01, **p < 0.001, ***p < 0.0001.
The synthetic lethal interaction between EJC and eIF4E is essential for cancer cell migration
The eIF4E interactome provides a rich resource for exploring additional, unexpected functional interactions (Figure 6A). For example, the positive regulation of two main signaling pathways, Erk1/2 and Wnt, which have been shown to be crucial for cancer cell progression and survival (Anastas and Moon, 2013; Little et al., 2013; Roskoski, 2012), were also identified by Gene Ontology (GO) and pathway enrichment analyses. During embryonic development, it has been shown that there is pervasive translational regulation of these signaling pathway components between different tissues (Fujii et al., 2017), although a previous link to eIF4E has not been established. Connections between specific genes involved in Erk1/2 and Wnt signaling pathways and eIF4E, which may be therapeutically targeted, represent interesting cancer-specific vulnerabilities. Moreover, two different organelles, peroxisome and Golgi, were enriched in the GO term analysis, indicating a potential role for eIF4E in either peroxisome organization or intra-Golgi vesicle-mediated transport. Additionally, the known role of eIF4E in translational control in neurons is reflected by “synaptic plasticity” and “neuromuscular process controlling balance,” which are significantly enriched among the eIF4E synthetic lethal genes.
Figure 6. The synthetic lethal interaction between EJC and eIF4E is essential for cancer cell migration.

. (A) Representation of GO terms enriched in synthetic lethal partners of eIF4E in a cellular context. Each circle represents a gene, and the size of the circle corresponds to the −log10(p value) from a paired Wilcoxon test to show the significance of the mean difference of the phenotype between eIF4E+/− and eIF4E+/+ MEFs in the screen.
(B) Cartoon of the exon-junction complex.
(C) Schematic representation of the RNA-seq experiment.
(D) Heatmap representation of differentially expressed genes in comparison to eIF4E or Rbm8a depletion. Although the alterations were minor when Rbm8a or eIF4E was depleted (left and middle columns, respectively), more significant changes were observed when Rbm8a was depleted in eIF4E+/− MEFs (right column). Each line represents the value calculated based on log2(FC) × −log10(p value).
(E) Enriched GO terms of differentially expressed genes in eIF4E+/− Rbm8a KD versus eIF4E+/− Scramble MEFs (right column of Figure 4D). The size of the dots represents the number of genes in each category.
(F) qPCR analysis of mRNA targets (Itga5, Mmp2, Snail1) that are specifically downregulated in eIF4E+/− Rbm8a KD. Multiple t tests were performed to determine statistical significance (n > 3).
(G) Schematic representation of transwell migration assay and a representative confocal image of migrated cells. Migrated cells were counted for each group (n > 30). The bottom right graph represents the normalized value of Rbm8a KD over the scramble control in eIF4E+/+ and eIF4E+/− MEFs. Multiple t tests were performed to determine statistical significance.
(H) Progression-free survival probability in liver cancer using TCGA data. The p value represents the interaction score between eIF4E and Rbm8a expression. A two-way ANOVA test was performed to determine statistical significance.
All values represent the mean + SEM. *p < 0.01, **p < 0.001, ***p < 0.0001. See also Figure S4.
GO terms, such as splicing and mRNA processing, point to previously uncharacterized connections between RNA metabolism and translation. In particular, Magoh, a subunit of the EJC, was one of the top hits within the GO terms for mRNA processing and splicing. The EJC is involved in the regulation of splicing, mRNA transport, translation, and decay, and it is constituted of four main subunits, including Magoh, Rbm8a, eIF4A3, and Casc3, as well as other accessory proteins (Figure 6B) (Boehm and Gehring, 2016; Le Hir et al., 2016). First, we compared the phenotype score of these four main subunits from the screen and showed that in addition to Magoh, eIF4A3 and Rbm8a also promote more lethality in eIF4E+/− cells compared to eIF4E+/+ cells (Figure S4A). Indeed, our data showed that eIF4E+/− cells were significantly more sensitive to the depletion of each subunit (Figures S4B and S4C). Two subunits of the EJC, Rbm8a and eIF4A3, selectively targeted eIF4E+/− cells without affecting eIF4E+/+ cells (Figure S4B). Given the known roles of the EJC in RNA metabolism, we analyzed the transcriptome of cells where EJC subunits (Magoh, eIF4A3, or Rbm8a) were depleted individually either in Myc and Ras overexpressing eIF4E+/+ or eIF4E+/− background (Figure 6C). We carried out RNA sequencing (RNA-seq) analysis at very early time points when cells do not show any difference in cell proliferation (Figures S4D and S4E), in order to detect the early response of EJC depletion.
We selected Rbm8a for further analysis, as Rbm8a KD shows the strongest phenotype specific to the eIF4E+/− background (Figure S4B). We first analyzed the genes that are specifically downregulated in Rbm8a KD eIF4E+/− MEFs (Figures 6D and S4F; Table S2; each group was first compared to the scramble control as indicated). Surprisingly, the most significant GO term in this condition was “the positive regulator of cell migration,” suggesting that the EJC may play a role in cell migration that is dependent on eIF4E (Figure 6E). Additionally, Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis also showed that gene clusters are enriched in categories, such as the ECM receptor, focal adhesion, and proteoglycans (Figure S4G). First, we validated the gene expression alterations by performing qPCR analysis for several mRNAs involved in cell migration, including Itga5, Mmp2, and Snail1 (Figure 6F). The qPCR results showed that although Rbm8a KD has no effect on the expression of these mRNAs in eIF4E+/+ cells, they are all specifically downregulated upon Rbm8a KD in eIF4E+/− cells. We therefore performed an in vitro transwell assay, where we monitored the effect of Rbm8a KD on cell migration. After 4 h, migrated cells were stained with DAPI and counted. The results demonstrated that although Rbm8a KD had no effect on cell migration in the eIF4E+/+ background, it significantly reduced the number of cells migrating in eIF4E+/− cells, showing a cooperation between eIF4E and Rbm8a that facilitates cancer cell migration (Figure 6G).
It has been reported that a high expression of Rbm8a promotes tumor progression and metastasis in hepatocellular carcinoma (Liang et al., 2017). Therefore, we asked whether human tumor samples expressing both high Rbm8a and eIF4E mRNAs have increased tumor progression compared to low Rbm8a/ eIF4E samples by using The Cancer Genome Atlas (TCGA) and Pan-Cancer Atlas datasets. We calculated the interaction score for progression-free survival probability in three different cancer types: liver hepatocellular carcinoma, lung adenocarcinoma, and pancreas adenocarcinoma (Figures 6H and S4H). In each cancer type, a significant interaction between the expression of Rbm8a and eIF4E was observed. We could also detect a strong correlation between high Rbm8a/eIF4E expression and low survival probability, indicating that Rbm8a overexpression together with eIF4E increases lethal disease progression. In summary, our findings show that EJC is a synthetic lethal partner of eIF4E and this interaction is important for cancer cell migration, uncovering a surprising functional interaction between the different components involved in RNA metabolism and eIF4E in cancer development.
DISCUSSION
In this study, we focused on synthetic lethal interactions of eIF4E and showed previously uncharacterized functions of eIF4E with distinct and important cellular processes. Each interaction represents an undiscovered point of vulnerability that can be therapeutically targeted. Moreover, we identified more than 600 genes to be synthetic lethal with eIF4E, which represents a very rich resource for future studies. Our current study discovered previously unknown connections between eIF4E and distinct mitochondrial processes, such as apoptosis and UPRmt-like adaptive stress response, highlighting the involvement of translation control in linking the mitochondria to tumorigenesis. One of the most fascinating connections we have identified is the role of eIF4E in activating a UPRmt-like adaptive stress response, which triggers autophagy for cancer cell survival. The UPRmt was first described in mammals (Martinus et al., 1996; Zhao et al., 2002) and then further characterized in C. elegans where it extends lifespan (Durieux et al., 2011; Houtkooper et al., 2013, Shpilka and Haynes, 2018). Cancer cells rely on mitochondrial function for their survival and therefore they need to sense and respond to mitochondrial dysfunctions (Vyas et al., 2016). Therefore UPRmt, triggered by mitochondrial alterations, may be a critical pathway for cancer development (Zhu et al., 2021). Our data showing that Pmpcb depletion triggers an UPRmt-like response to activate autophagy highlights a previously unknown role of Pmpcb as a key regulatory component for mitochondria protein homeostasis. Moreover, our mechanistic studies identified the role of eIF4E in this mitochondria response by translationally regulating one of the major TFs required for autophagy, Tfeb. Thus, it is intriguing that cancer cells are dependent on translation control to activate autophagy. Interestingly, it has been recently reported that Pmpcb (Mas1 in yeast) deletion activates an early UPRmt-like stress response as a survival mechanism in yeast (Poveda-Huertes et al., 2020). It will be very interesting to investigate whether combinatorial therapies targeting eIF4E together with autophagy inhibitors can be beneficial to block tumor growth and to overcome drug resistance.
Our studies further highlight the importance of translation control in regulating apoptosis, another cancer survival pathway. Importantly, the synthetic lethal interaction between eIF4E and Bcl-xL is in line with a previous study where the inhibition of Bcl-xL together with the mTOR pathway, which functions upstream of eIF4E, has been shown to induce apoptosis in PIK3CA mutant breast cancer cells (Anderson et al., 2016). Inhibitors targeting different Bcl-2 family proteins have been used in clinical trials to block tumor progression; for example, a dual inhibitor of Bcl-2 and Bcl-xL, navitoclax, shows high promise in clinical trials (Lessene et al., 2008). Our data demonstrating that Bcl-xL and eIF4E inhibitors synergistically promote cell death point to a vulnerability that can open unexplored avenues for cancer therapy.
Our study further points to the coupling of unexpected cellular processes such as the EJC, which is involved in multiple steps of RNA metabolism and translation control. Such interactions may reflect presently unknown nodes of post-transcriptional regulation, which may be hijacked by cancer cells to sustain distinct cancer cell behaviors, such as cell migration. Therefore, the 630 genes found to be synthetically lethal with eIF4E activity reflects one of the most comprehensive interaction maps required to establish the cancer translatome. This provides previously uncharacterized insight into cancer vulnerabilities at the post-genomic level.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PARP | Cell Signaling Technology | Cat# 9542; RRID:AB_2160739 |
| Celaved Caspase-3 (D175) | Cell Signaling Technology | Cat# 9661; RRID:AB_2341188 |
| eIF4E | BD | Cat# 610270; RRID:AB_397665 |
| p-eIF4E Ser209 | Cell Signaling Technology | Cat# 9721; RRID:AB_561095 |
| beta-Actin | Sigma-Aldrich | Cat# A5316; RRID:AB_476743 |
| Pmpcb | Protein Tech | Cat# 16064-1-AP; RRID:AB_2167122 |
| Tfeb | Bethyl Laboratories | Cat# A303-673A; RRID:AB_11204751 |
| a-Tubulin | Sigma-Aldrich | Cat# T8203; RRID:AB_1841230 |
| LC3A/B | Cell Signaling Technology | Cat# 4108; RRID:AB_2137703 |
| c-Myc (D84C12) | Cell Signaling Technology | Cat# 5605; RRID:AB_1903938 |
| Ras | Cell Signaling Technology | Cat# 3965; RRID:AB_2180216 |
| Atf4 (D4B8) | Cell Signaling Technology | Cat# 11815; RRID:AB_2616025 |
| Xbp-1 | Abcam | Cat# Ab37152; RRID:AB_778939 |
| Magoh | Protein Tech | Cat# 12347-1-AP; RRID:AB_2265988 |
| eIF4AIII/eIF4A3 | Bethyl Laboratories | Cat# A302-981A; RRID:AB_10748369 |
| Rbm8a | Bethyl Laboratories | A301-033A; RRID:AB_2300943 |
| Bcl-xL (H5) | Santa Cruz | Cat# sc-8392; RRID:AB_626739 |
| Bacterial and virus strains | ||
| MegaX DH10B T1R Electrocomp Cells | Thermo Fisher Scientific | C640003 |
| Chemicals, peptides, and recombinant proteins | ||
| A-1331852 | MedChemExpress | HY-19741 |
| eFT508 | eFFECTOR Therapeutics | |
| Trizol | Sigma-Aldrich | T9424-200ML |
| Trizol LS | Invitrogen | 10296010 |
| Matrigel | Corning | CB40234 |
| PhosSTOP | Roche | 4906837001 |
| Complete Mini proteosome inhibitors | Roche | 11836170001 |
| Glycogen | Roche | 10901393001 |
| Paraformaldehyde | ||
| Poly-lysine | Sigma | P4707-50ML |
| Critical commercial assays | ||
| FITC-Annexin V | Thermo Fisher Scientific | A13199 |
| UPRmt TF Activation Profiling | Signosis | FA-1010 |
| Cell Titer Glo | Promega | G9241 |
| Polybrene | Millipore Sigma | TR-1003-G |
| Lipofectamine 2000 | Thermo Fisher Scientific | 11662019 |
| Lipofectamine LTX | Thermo Fisher Scientific | 15338100 |
| TOPO-TA | Invitrogen | 450641 |
| Super Signal West Pico Plus Chemiluminescent substrate | Thermo Scientific | 34580 |
| NE-PER Nuclear and Cytoplasmic Extraction Reagents | Thermo Scientific | 78835 |
| Direct-zol RNA Microprep | Zymo Research | R2061 |
| High-Capacity cDNA Reverse transcription kit | Applied Biosystems | 4368814 |
| PowerUP SYBR Green master mix | Applied Biosystems | A25741 |
| TurboDNA-free kit | Invitrogen | AM1907 |
| Dual-luciferase Reporter Assay System | Promega | E1910 |
| SuperScript III first-strand synthesis kit | Invitrogen | 18080044 |
| Prolong Diamond Antifade mountant with DAPI | Invitrogen | P36971 |
| Ribo-Zero | Illumina | 20040526 |
| ScriptSeq v2 RNA-seq library preparation kit | Epicenter | SSV21124 |
| Deposited data | ||
| RNA seq Data | This paper | GEO: GSE175417 |
| Experimental models: Cell lines | ||
| MEFs eIF4E+/− | This paper | |
| Oligonucleotides | ||
| See Table S3 | This paper | |
| Recombinant DNA | ||
| pBabe-Neo-LargeT | Hahn et al., 2002 | Addgene #1780 |
| pCL-Eco | Naviaux et al., 1996 | Addgene #12371 |
| pX458 | Ran et al., 2013 | Addgene #48138 |
| human HRASV12 | Serrano et al., 1997 | Addgene #18749 |
| human c-Myc | Boehm et al., 2005 | Addgene #10674 |
| psPAX2 | Didier Trono | Addgene #12260 |
| pMD2.G | Didier Trono | Addgene #12259 |
| pLKO.1 | Stewart et al., 2003 | Addgene #8453 |
| pMSCV | Clontech | 634401 |
| pGL3 | Promega | E1751 |
| pRL | Promega | E2231 |
| pLG15 | Luke Gilbert | |
| sgRNA expressing vectors | This paper | |
| pMSCV-GFP-Tfeb | This paper | |
| pMSCV-GFP-Pmpcb | This paper | |
| TFEB 5’UTR containing pGL3 reporters | This paper | |
| mouse CRIPSRi library expressing 5 sgRNAs for each gene | IGI CRISPR screening core at UCSF | |
| Software and algorithms | ||
| ImageJ | https://imagej.nih.gov/ij/ | |
| Fiji | https://imagej.net/software/fiji | |
| ImageLab software | BioRad | |
| Prism | GraphPad | Version 9.1.0 |
| R Studio | https://www.rstudio.com/ | |
| HISAT v2.0.5 | http://daehwankimlab.github.io/hisat2/ | |
| RSEM v1.3.0 | https://github.com/deweylab/RSEM/releases | |
| DAVID v6.8 | https://david.ncifcrf.gov/ | |
| ‘survminer’ R package | https://github.com/kassambara/survminer | |
| AdobePhotoshop | Adobe | |
| Adobe Illustrator | Adobe | |
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Davide Ruggero (davide.ruggero@ucsf.edu)
Materials availability
Plasmids generated in this study are available from the lead contact, Davide Ruggero (davide.ruggero@ucsf.edu) upon request.
Data and code availability
Original data for Figures 6C and 6D (RNA Seq) in the paper is available at GEO with accession number GEO: GSE175417.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Male immunodeficient mice (NSG) at the age of 12–16 weeks were used for injection. A549 or U251 cells were grown in tissue culture and harvested just prior to injection. Cells were resuspended in 50% Matrigel (Corning) and subcutaneously injected to the right flank of each mouse (2 million A549 cells and 4 million U251 cells). Once the tumors reached 100 mm3 volume (2 weeks after injection), we started the treatment of different groups: Vehicle, A-1331852 (10mg/kg), eFT508 (10mg/kg), and the combo group. A-1331852 was dissolved in 60% Phosal 50 PG (Lipoid), 27% PEG 400, 10% Ethanol and 2.5% DMSO as described (Leverson et al., 2015). eFT508 was dissolved in 90% PG and 10% NMP. 10 days after starting the treatment, the concentration of A-1331852 was increased to 17mg/kg. Each compound was orally administrated 5 days/week and tumor growth was monitored at least twice every week.
Mice were maintained under specific pathogen-free conditions and all experiments were performed in compliance with guidelines approved by the Institutional Animal Care and Use Committee of UCSF.
Cell lines
Primary mouse embryonic fibroblasts (MEFs) were isolated from E13.5 embryos, cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum, 10% glutamax (GIBCO) and Penicillin/Streptomycin (GIBCO). Primary MEFs were immortalized by retroviral particles produced by transfecting HEK293T cells with pBabe-Neo-LargeT and pCI-Eco plasmids using Lipofectamine 2000 (Invitrogen). The supernatants of transfected HEK293T cells were filtered through 0.22uM filter and mixed with 10μg/ml polybrene (Millipore) before incubating with the MEFs. Infected cells were selected with neomycin and single clones were grown to have a monoclonal population.
Immortalized monoclonal MEFs were transfected with pX458 plasmids expressing either control or eIF4E sgRNAs using Lipofectamine LTX with plus reagent (Invitrogen). 24h post transfection, GFP positive cells were sorted with BD FACS Aria cell sorter and plated on a 96-well plate. Single colonies were grown and eIF4E levels were measured by western blotting. Once the colonies showing 50% eIF4E expression were detected, the region where eIF4E sgRNA binds were amplified by PCR and cloned into the Topo-TA (Invitrogen) vector for sequencing. At least 12 minis were sequenced to be sure that the cells are originated from the same single clone. Once the clones were selected, they were transformed by retroviral infection with human HRasV12 and human c-Myc vectors followed by hygromycin and blasticidin selection, respectively.
A549 non-small-cell lung cancer cell line and U251 human glioblastoma astrocytoma (a kind gift from William Weiss, UCSF) were cultured in DMEM supplemented with 10% Fetal Bovine Serum, 10% glutamax (GIBCO) and Penicillin/Streptomycin (GIBCO) at 37°C with 5% CO2.
All cell lines used in this study were found to be negative of mycoplasma contamination using a MycoAlert mycoplasma detection kit (Lonza, Allendale, NJ, USA).
METHOD DETAILS
Genome-wide CRISPRi screen in MEFs
Myc and Ras overexpressed eIF4E+/+ and eIF4E+/− MEFs were first transduced with a plasmid expressing dCas9-Krab and BFP positive cells were sorted with BD FACS Aria. The library containing 5 sgRNAs for each gene in the mouse genome was obtained from the IGI CRISPR screening core at UCSF. First, a small-scale transfection and infection is tested to optimize the conditions to get a 30%–40% infection rate, as measured by %BFP positive cells. Cells were grown at minimum library coverage of 1,000 for the genome-wide screen on 15-cm dish plates. HEK293T cells were transfected with the library and psPAX2 packaging and pMD2.G envelope plasmids. 125 million eIF4E+/+ and eIF4E+/− MEFs were infected with fresh virus harvested and filtered from the HEK293T cells. 48h after infection, cells were selected with 2 μg/ml puromycin for one day. 24h post-selection, 100 million cells were harvested as Day 0 cells were cultured for 10 more days by splitting them every 48h for 1:4. eIF4E+/+ and eIF4E+/− cells were treated exactly the same way and at the end of 10 days, 100 million cells were harvested again. The library was prepared as described previously (Bassik et al., 2013; Gilbert et al., 2014) and sequenced by the Center for Advance Technology core at UCSF. A Wilcoxon paired test was performed to identify significant lethal partners of eIF4E. The mean difference between the phenotype scores of eIF4E+/+ and eIF4E+/− MEFs was compared for each gene. 630 genes were identified by applying these criteria: p value < 0.05 and Δlog2[Fold change (eIF4E+/− − eIF4E+/+)] < −0.5.
Competitive growth assay
To validate the phenotype obtained from the screen, MEFs were first transduced with an inducible dCas9-Krab-mCherry plasmid. The single sgRNAs (at least 2 sgRNA/gene) were cloned in a plasmid under mouse U6 promoter. A nonspecific sgRNA was used for all of the experiments as a negative control for normalization. eIF4E+/+ and eIF4E+/− MEFs were transduced with the lentiviral particles expressing the sgRNAs and selected with puromycin. On Day 0 of the growth experiment, sgRNA expressing cells were counted and mixed at a 1:1 ratio with parental MEFs that do not express any sgRNA (BFP negative). The BFP% was detected by Attune NxT flow cytometer (Invitrogen). Mixed cells were seeded on 24-well plates as triplicates and dCas9-Krab-mCherry expression is induced by 0.4μg/ml doxycycline treatment. Cells were monitored for at least 5 days and their mCherry and BFP percentiles were measured. The phenotype score is calculated as described in Extended Data Figure 2C.
Western blot analysis
The western blot analysis for tissue culture samples was performed by lysing cell pellets in a RIPA buffer (50 mM Tris pH 8, 150 mM NaCl, 1% NP40, 0.1% SDS, 0.5% Na deoxycholate,1mM EDTA, 1mM DTT) with the addition of PhosSTOP and Complete Mini proteasome inhibitors (Roche). Protein lysates were denatured using a protein sample buffer and separated on an SDS-PAGE gel. The samples were transferred to a nitrocellulose membrane using a wet-transfer system from BioRad. The membrane was then blocked with PBS-Tween-20 (0.1%) containing 5% nonfat dry milk for at least 30 min and incubated with the primary antibody overnight. The membrane was washed three times with PBS-Tween and incubated with the secondary antibodies conjugated to horseradish peroxidase (HRP). Super Signal West Pico Plus Chemiluminescent substrate (Thermo Scientific) was used to develop the membranes on a Chemidoc imaging system (BioRad).
Western blot analysis for lysates from the nuclear protein fraction was performed by utilizing the NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific) with the addition of PhosSTOP and Complete Mini proteasome inhibitors (Roche). Western blot analysis for tumor tissues from in vivo mice experiments was performed by lysing powdered tumor tissue (obtainer with a tissue pulverizer) in RIPA buffer (50 mM Tris pH 8, 150 mM NaCl, 1% NP40, 0.1% SDS, 0.5% Na deoxycholate, 1mM DTT) with the addition of PhosSTOP and Complete Mini proteasome inhibitors (Roche). The antibodies used in the study was listed in the key resources table. Where mentioned, protein levels were quantified using Image Lab software (BioRad) to analyze optical density of western blots normalized to loading control.
Quantitative polymerase chain reaction (qPCR)
RNAs from cell lysates were isolated using TRIzol (Invitrogen) purification on Direct-zol RNA Microprep columns (Zymo Research, CA, USA) according to manufacturer’s instructions with DNase treatment. Following the RNA isolation, the quality of the RNA samples was checked by running them on an agarose gel. Single-stranded cDNA was synthesized by using 0.5–1μg RNA as a template with High-Capacity cDNA Reverse transcription kit (Applied Biosystems). cDNA samples were diluted 1:10 and 1 μL of template was used in a PowerUP SYBR Green master mix reaction run on an QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems). qPCR primer sequences are listed in Table S3.
Polysome fractionation and isolation of polysome-associated RNA for qPCR
MEFs expressing control or Pmpcb sRNAs were induced for 2 days with Dox. Prior to harvesting, the medium of the cells was changed with fresh medium for at least 3 hours and incubated with 100 μg/ml cycloheximide (Sigma) for 3min in the culture. Cells were then washed with PBS containing cycloheximide and resuspended in lysis buffer containing 10mM Tris-HCl (pH 8.0), 150mM NaCl, 1.5mM MgCl2, 0.25% NP-40, 0.1% Triton X-100, 50mM DTT, 150 μg/ml cycloheximide, and 640U/ml Rnasin for 30–45min on ice, vortexing every 10min. Lysates were centrifuged at 10,000rpm at 4°C for 10 min. The supernatants were adjusted by OD260 concentration and loaded onto a 30%–60% sucrose gradient before centrifugation at 37,500 rpm for 5 hr at 4°C in a Beckman L8-70M ultracentrifuge. Samples were separated on a Biocomp fractionation system to evaluate polysome profiles and collect polysome fractions. 13 fractions were collected for each sample in total and RNA was isolated from each fraction, except the first one. 750ul of Trizol LS (Invitrogen) and 0.4ul of 1 mg/ml glycogen (Roche) was added to 250μl of sucrose lysate and incubated for 15min at room temperature. 200ul of chloroform was added to the mix before spinning down the samples for 15 min at 12,000 g at 4°C. RNA was precipitated by adding 500ul of isopropanol to the supernatant overnight at −80°C. The next day the samples were spun down for 20min at 12,000 g at 4°C. The pellet obtained was washed with 70% ethanol and resuspended in dH2O. Any trace DNA was removed with the Invitrogen TurboDNA-free kit. cDNA synthesis and qPCR analysis were performed as described previously.
CellTiter Glo assay
Cells were seeded on 96-well plate, incubated overnight to attach and then treated with A-1331852 (MedChemExpress) at concentrations indicated in Figures 1D and 1E. In the case of combinational treatment, 0.2μM eFT-508 (Effector) were also added to the cell medium. 24- or 72-hours post-treatment, Cell-titer Glo Assay (Promega) was performed following the manufacturer’s protocol and the absorbance at 490nm was measured on Glomax plate reader (Promega).
Analysis of global protein synthesis
MEFs expressing control or Pmpcb sgRNAs induced with Dox for 48h were incubated in methionine-free media supplemented with 10% FBS and 30 μCi of 35S labeled methionine/cysteine for 1 hour. Cells were washed and lysed as described for standard western blot procedures. Equal amounts of total proteins were separated out on a 10% SDS polyacrylamide gel and transferred to nitrocellulose membrane. Membranes were exposed to autoradiography film for 12–48 hours and developed 35S methionine/cysteine incorporation was quantified using ImageJ software to analyze optical density and normalized to β-actin levels.
Luciferase reporter assay
5′UTRs were amplified from cDNA synthesized from MEFs and cloned into the pGL3 luciferase reporter vector (Promega) upstream of the Firefly luciferase open reading frame. MEFs expressing control or Pmpcb sgRNAs were seeded in a 6-well plate and induced with Dox for 24 h. After that, cells were transfected using Lipofectamine LTX (Invitrogen) with 900ng of pGL3-FLuc-Sv40 and 100ng of RLuc. Dox was not removed from the medium to allow 48h induction in total. 24h post-transfection cells were harvested: a fraction was lysed in Trizol for RNA extraction and the rest were lysed in a passive lysis buffer for 20 min. The Rluc/Fluc activity was assessed using the Dual-luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions, using a Glomax microplate luminometer (Promega). RNA extraction was performed using the Zymogen DirectZol kit following manufacturer’s instructions TurboDNA-free kit (Thermofisher) was used to remove transfected DNA completely. cDNA synthesis was performed by the SuperScript III first-strand synthesis kit (Invitrogen) using specific oligos for FLuc, RLuc and actin. qPCR analyses were performed as described previously.
UPRmt transcription factors profiling
eIF4E+/+ and eIF4E+/− MEFs expressing control or Pmpcb sgRNAs were induced with Dox for 48h and nuclear fraction of lysates were isolated using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific) with the addition of PhosSTOP and Complete Mini proteasome inhibitors (Roche). The same protein amount of nuclear fractions were used for each sample and the mitochondrial UPR transcription factors profiling plate array was used by following manufacturer’s protocol (Signosis, FA-1010).
Annexin staining
eIF4E+/+ and eIF4E+/− MEFs expressing control or Pmpcb sgRNAs induced with Dox for 48h harvested and resuspended in binding buffer containing 50 mM HEPES, 700 mM NaCl, 12.5 mM CaCl2, pH 7.4, stained with FITC-Annexin V (Thermo) for 15 min on ice and analyzed immediately on Attune NxT flow cytometer (Invitrogen).
Transwell assay
eIF4E+/+ and eIF4E+/− MEFs expressing control or Pmpcb sgRNAs induced with Dox for 48h. Cells were washed and dissociated from the plate using dissociation buffer containing EDTA. Cells were washed twice with serum-free medium. Equal numbers of cells were resuspended in 100 μl serum-free medium and seeded on a polycarbonate insert with 8μm pore size (VWR, Cat No 3422) in a 24-well plate where each well contains 750 μl DMEM with serum. After 4 hours, each insert was washed twice in PBS and fixed in 750 μl 4% paraformaldehyde for 10 min at room temperature. After washing with PBS, the mash parts of the inserts were cut and mounted on a slide with Prolong Diamond Antifade mountant with DAPI (Invitrogen). Slides were analyzed on Zeiss spinning disk confocal microscopy. The images were quantified by Fiji software. For each condition, three inserts were prepared and the experiment is repeated at least 2 times.
Immunofluorescence
eIF4E+/+ and eIF4E+/− MEFs expressing control or Pmpcb sgRNAs were seeded on a coverslip coated with poly-Lysine (Sigma) and induced with Dox for 48h. The cells were washed with PBS and fixed with ice cold methanol for 15 min at −20. After washing with PBS three times, cells were blocked in PBS containing 0.3% Triton X-100 and 5% goat-serum for 30 min. The primary antibody was diluted in PBS containing 0.3% Triton X-100 and 1%BSA (LC3I/II, Cst #4108, 1:200 dilution) and incubated overnight at 4°C. The cells were then washed again and incubated with fluorochrome-conjugated secondary antibody diluted in an antibody dilution buffer for 1 hour at room temperature in the dark. Finally, the coverslips were washed with PBS and mounted on a slide with Prolong Diamond Antifade mountant with DAPI (Invitrogen). Slides were analyzed on Zeiss spinning disk confocal microscopy. The images were quantified by Fiji software.
RNA-seq
eIF4E+/+ and eIF4E+/− MEFs expressing scramble, Magoh, eIF4A3 and Rbm8a sgRNAs were induced with Dox for 48 hours in triplicates, total RNA was isolated by Trizol and ribosomal RNA was removed by Ribo-Zero (Illumina). RNA seq libraries were prepared by ScriptSeq v2 RNA-seq library preparation kit (Epicenter) following the manufacturer’s protocol and sequenced as paired-end 100 bp on HiSeq 4000 by the Center for Advance Technology core at UCSF. RNA-seq reads were aligned to mouse GRCm38 genome by using HISAT v2.0.5 (Kim et al., 2015) with default settings. Gene read counts and expression values (measured by Transcripts Per Kilobase Million, TPM) were further calculated by RSEM v1.3.0 (Li and Dewey, 2011) with default settings. Differential expression analyses were conducted with DESeq2 (Love et al., 2014) R package, where only genes with an average TPM over 0.5 were involved. Genes with p value < 0.05 and fold change > 2 were considered as differentially expressed genes. GO and KEGG enrichment analyses were carried out with DAVID v6.8 (Huang et al., 2009), where differentially expressed genes were compared against a background gene set containing all of the expressed genes (average TPM > 0.5). TCGA data (gene expression values and progression data) were downloaded from cBioPortal. The interaction effect between Magoh and eIF4E on progression were tested by ‘coxph’ function in ‘survival’ (Therneau and Grambsch, 2000) R package. Survival curves were generated by ggsurvplot function in ‘survminer’ R package (https://github.com/kassambara/survminer).
QUANTIFICATION AND STATISTICAL ANALYSIS
Graphpad Prism 9.0 was used for statistical analysis. The statistical details of each experiment can be found in the corresponding figure legend. The replicate number of each experiment was indicated as “n” in the figure legends. For in vivo experiments, “n” corresponds to number of animals used in the study. All the graphs in this study represents the mean value + SEM. Asterisks represent p values as *p < 0.01, **p < 0.001, ***p < 0.0001.
Supplementary Material
Highlights.
Genome-wide CRISPRi screen reveals more than 600 synthetic lethal partners of eIF4E
Functional interaction between eIF4E and Bcl-xL is important for tumor growth
Mitochondrial dysfunction triggers an eIF4E-dependent adaptive stress response
Interaction between eIF4E and EJC controls the migratory capacity of cancer cells
ACKNOWLEDGMENTS
We thank members of the Ruggero laboratory for discussions, especially Dr. Joanna Kovalski and Dr. Yichen Xu for critical inputs on the manuscript. We also acknowledge Preclinical Therapeutics and the Laboratory for Cell Analysis core facilities at the University of California, San Francisco (UCSF) for assistance in our study. D.K.-O. was funded by a EMBO long-term fellowship (ALTF 1005-2015) and a Human Frontier Science Program long-term fellowship (LT000711/2016-L). Z.H. and S.E.B. are supported by a research agreement between the University of California, Berkeley and Tata Consultancy Services. L.A.G. is supported by NIH DP2 CA239597 and a Goldberg-Benioff Endowed Professorship. This work was supported by NIH grant no. R35CA242986 (to D.R.) and by the American Cancer Society (RP-19-181-01-RMC; American Cancer Society Research Professor Award) (to D.R.).
Footnotes
DECLARATION OF INTERESTS
G.G.C. is an employee and shareholder of eFFECTOR Therapeutics, Inc. L.A.G. has filed patent applications related to CRISPRi/a screening. L.A.G. consults for and holds equity in Chroma Medicine. S.T.W. is a founder and President and CEO of eFFECTOR Therapeutics, Inc. D.R. is a shareholder of eFFECTOR Therapeutics, Inc., and a member of its scientific advisory board. The remaining authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109321.
REFERENCES
- Anastas JN, and Moon RT (2013). WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 13, 11–26. [DOI] [PubMed] [Google Scholar]
- Anderson GR, Wardell SE, Cakir M, Crawford L, Leeds JC, Nussbaum DP, Shankar PS, Soderquist RS, Stein EM, Tingley JP, et al. (2016). PIK3CA mutations enable targeting of a breast tumor dependency through mTOR-mediated MCL-1 translation. Sci. Transl. Med 8, 369ra175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barna M, Pusic A, Zollo O, Costa M, Kondrashov N, Rego E, Rao PH, and Ruggero D (2008). Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 456, 971–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassik MC, Kampmann M, Lebbink RJ, Wang S, Hein MY, Poser I, Weibezahn J, Horlbeck MA, Chen S, Mann M, et al. (2013). A systematic mammalian genetic interaction map reveals pathways underlying ricin susceptibility. Cell 152, 909–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, and Mardis ER (2018). The emerging clinical relevance of genomics in cancer medicine. Nat. Rev. Clin. Oncol 15, 353–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm V, and Gehring NH (2016). Exon junction complexes: Supervising the gene expression assembly line. Trends Genet. 32, 724–735. [DOI] [PubMed] [Google Scholar]
- Boehm JS, Hession MT, Bulmer SE, and Hahn WC (2005). Transformation of human and murine fibroblasts without viral oncoproteins. Mol. Cell. Biol 25, 6464–6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Aktas BH, Wang Y, He X, Sahoo R, Zhang N, Denoyelle S, Kabha E, Yang H, Freedman RY, et al. (2012). Tumor suppression by small molecule inhibitors of translation initiation. Oncotarget 3, 869–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F-M, Huang L-J, Ou-Yang F, Kan J-Y, Kao L-C, and Hou M-F (2020). Activation of mitochondrial unfolded protein response is associated with Her2-overexpression breast cancer. Breast Cancer Res. Treat 183, 61–70. [DOI] [PubMed] [Google Scholar]
- Durieux J, Wolff S, and Dillin A (2011). The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144, 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faller WJ, Jackson TJ, Knight JRP, Ridgway RA, Jamieson T, Karim SA, Jones C, Radulescu S, Huels DJ, Myant KB, et al. (2015). mTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature 517, 497–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii K, Shi Z, Zhulyn O, Denans N, and Barna M (2017). Pervasive translational regulation of the cell signalling circuitry underlies mammalian development. Nat. Commun 8, 14443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furic L, Rong L, Larsson O, Koumakpayi IH, Yoshida K, Brueschke A, Petroulakis E, Robichaud N, Pollak M, Gaboury LA, et al. (2010). eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proc. Natl. Acad. Sci. USA 107, 14134–14139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gakh O, Cavadini P, and Isaya G (2002). Mitochondrial processing peptidases. Biochim. Biophys. Acta 1592, 63–77. [DOI] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. (2014). Genome-scale CRISPR-mediated control of gene repression and activation. Cell 159, 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guntuku L, Naidu VGM, and Yerra VG (2016). Mitochondrial dysfunction in gliomas: pharmacotherapeutic potential of natural compounds. Curr. Neuropharmacol 14, 567–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighat A, Mader S, Pause A, and Sonenberg N (1995). Repression of cap-dependent translation by 4E-binding protein 1: Competition with p220 for binding to eukaryotic initiation factor-4E. EMBO J. 14, 5701–5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn WC, Dessain SK, Brooks MW, King JE, Elenbaas B, Sabatini DM, DeCaprio JA, and Weinberg RA (2002). Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol. Cell. Biol 22, 2111–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: The next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Horlbeck MA, Gilbert LA, Villalta JE, Adamson B, Pak RA, Chen Y, Fields AP, Park CY, Corn JE, Kampmann M, and Weissman JS (2016). Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. eLife 5, 914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, and Auwerx J (2013). Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497, 451–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh AC, Costa M, Zollo O, Davis C, Feldman ME, Testa JR, Meyuhas O, Shokat KM, and Ruggero D (2010). Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell 17, 249–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, et al. (2012). The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485, 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009). Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RM, Branda J, Johnston KA, Polymenis M, Gadd M, Rustgi A, Callanan L, and Schmidt EV (1996). An essential E box in the promoter of the gene encoding the mRNA cap-binding protein (eukaryotic initiation factor 4E) is a target for activation by c-myc. Mol. Cell. Biol 16, 4754–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny TC, Hart P, Ragazzi M, Sersinghe M, Chipuk J, Sagar MAK, Eliceiri KW, LaFramboise T, Grandhi S, Santos J, et al. (2017). Selected mitochondrial DNA landscapes activate the SIRT3 axis of the UPRmt to promote metastasis. Oncogene 36, 4393–4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konicek BW, Stephens JR, McNulty AM, Robichaud N, Peery RB, Dumstorf CA, Dowless MS, Iversen PW, Parsons S, Ellis KE, et al. (2011). Therapeutic inhibition of MAP kinase interacting kinase blocks eukaryotic initiation factor 4E phosphorylation and suppresses outgrowth of experimental lung metastases. Cancer Res. 71, 1849–1857. [DOI] [PubMed] [Google Scholar]
- Le Hir H, Saulière J, and Wang Z (2016). The exon junction complex as a node of post-transcriptional networks. Nat. Rev. Mol. Cell Biol 17, 41–54. [DOI] [PubMed] [Google Scholar]
- Lessene G, Czabotar PE, and Colman PM (2008). BCL-2 family antagonists for cancer therapy. Nat. Rev. Drug Discov 7, 989–1000. [DOI] [PubMed] [Google Scholar]
- Leverson JD, Phillips DC, Mitten MJ, Boghaert ER, Diaz D, Tahir SK, Belmont LD, Nimmer P, Xiao Y, Ma XM, et al. (2015). Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci. Transl. Med 7, 279ra40. [DOI] [PubMed] [Google Scholar]
- Levy JMM, Towers CG, and Thorburn A (2017). Targeting autophagy in cancer. Nat. Rev. Cancer 17, 528–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang R, Lin Y, Ye J-Z, Yan X-X, Liu Z-H, Li Y-Q, Luo X-L, and Ye H-H (2017). High expression of RBM8A predicts poor patient prognosis and promotes tumor progression in hepatocellular carcinoma. Oncol. Rep 37, 2167–2176. [DOI] [PubMed] [Google Scholar]
- Liang J, Jia X, Wang K, and Zhao N (2018). High expression of TFEB is associated with aggressive clinical features in colorectal cancer. OncoTargets Ther. 11, 8089–8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little AS, Smith PD, and Cook SJ (2013). Mechanisms of acquired resistance to ERK1/2 pathway inhibitors. Oncogene 32, 1207–1215. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinus RD, Garth GP, Webster TL, Cartwright P, Naylor DJ, Høj PB, and Hoogenraad NJ (1996). Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur. J. Biochem 240, 98–103. [DOI] [PubMed] [Google Scholar]
- Melber A, and Haynes CM (2018). UPRmt regulation and output: A stress response mediated by mitochondrial-nuclear communication. Cell Res. 28, 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naviaux RK, Costanzi E, Haas M, and Verma IM (1996). The pCL vector system: Rapid production of helper-free, high-titer, recombinant retroviruses. J. Virol 70, 5701–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa L, and Germain D (2011). Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J. Cell Sci 124, 1396–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK, Ferrone CR, et al. (2015). Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 524, 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourdehnad M, Truitt ML, Siddiqi IN, Ducker GS, Shokat KM, and Ruggero D (2013). Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc. Natl. Acad. Sci. USA 110, 11988–11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poveda-Huertes D, Matic S, Marada A, Habernig L, Licheva M, Myketin L, Gilsbach R, Tosal-Castano S, Papinski D, Mulica P, et al. (2020). An early mtUPR: Redistribution of the nuclear transcription factor Rox1 to mitochondria protects against intramitochondrial proteotoxic aggregates. Mol. Cell 17, 180–188.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirós PM, Langer T, and López-Otín C (2015). New roles for mitochondrial proteases in health, ageing and disease. Nat. Rev. Mol. Cell Biol 16, 345–359. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenwald IB, Rhoads DB, Callanan LD, Isselbacher KJ, and Schmidt EV (1993). Increased expression of eukaryotic translation initiation factors eIF-4E and eIF-2 alpha in response to growth induction by c-myc. Proc. Natl. Acad. Sci. USA 90, 6175–6178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskoski R Jr. (2012). ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Res 66, 105–143. [DOI] [PubMed] [Google Scholar]
- Senft D, and Ronai ZA (2016). Regulators of mitochondrial dynamics in cancer. Curr. Opin. Cell Biol 39, 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, and Lowe SW (1997). Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602. [DOI] [PubMed] [Google Scholar]
- Shpilka T, and Haynes CM (2018). The mitochondrial UPR: Mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol 19, 109–120. 10.1038/nrm.2017.110. [DOI] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen ISY, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therneau TM, and Grambsch PM (2000). Modeling Survival Data: Extending the Cox Model chapter 8 (Springer; ), section 8.2. [Google Scholar]
- Truitt ML, and Ruggero D (2016). New frontiers in translational control of the cancer genome. Nat. Rev. Cancer 16, 288–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truitt ML, Conn CS, Shi Z, Pang X, Tokuyasu T, Coady AM, Seo Y, Barna M, and Ruggero D (2015). Differential requirements for eIF4E dose in normal development and cancer. Cell 162, 59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasan N, Baselga J, and Hyman DM (2019). A view on drug resistance in cancer. Nature 575, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas S, Zaganjor E, and Haigis MC (2016). Mitochondria and cancer. Cell 166, 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC (2012). Mitochondria and cancer. Nat. Rev. Cancer 12, 685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waskiewicz AJ, Flynn A, Proud CG, and Cooper JA (1997). Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 16, 1909–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E (2012). Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 12, 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe AL, Singh K, Zhong Y, Drewe P, Rajasekhar VK, Sanghvi VR, Mavrakis KJ, Jiang M, Roderick JE, Van der Meulen J, et al. (2014). RNA G-quadruplexes cause eIF4A-dependent oncogene translation in cancer. Nature 513, 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, and Ruggero D (2020). The role of translation control in tumorigenesis and its therapeutic implications. Annu. Rev. Cancer Biol 4, 437–457. [Google Scholar]
- Xu Y, Poggio M, Jin HY, Shi Z, Forester CM, Wang Y, Stumpf CR, Xue L, Devericks E, So L, et al. (2019). Translation control of the immune checkpoint in cancer and its therapeutic targeting. Nat. Med 25, 301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, and Hoogenraad NJ (2002). A mitochondrial specific stress response in mammalian cells. EMBO J. 21, 4411–4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Zhou Q, He L, and Chen L (2021). Mitochondrial unfolded protein response: An emerging pathway in human diseases. Free Radic. Biol. Med 163, 125–134. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original data for Figures 6C and 6D (RNA Seq) in the paper is available at GEO with accession number GEO: GSE175417.