

Abstract
DYT1 dystonia is a movement disorder mainly caused by a trinucleotide deletion (ΔGAG) in DYT1 (TOR1A), coding for torsinA. DYT1 dystonia patients show trends of decreased striatal ligand-binding activities to dopamine receptors 1 (D1R) and 2 (D2R). Dyt1 ΔGAG knock-in (KI) mice, which have the corresponding ΔGAG deletion, similarly exhibit reduced striatal D1R and D2R-binding activities and their expression levels. While the consequences of D2R reduction have been well characterized, relatively little is known about the effect of D1R reduction. Here, locomotor responses to D1R and D2R antagonists were examined in Dyt1 KI mice. Dyt1 KI mice showed significantly less responsiveness to both D1R antagonist SCH 23390 and D2R antagonist raclopride. The electrophysiological recording indicated that Dyt1 KI mice showed a significantly increased paired-pulse ratio of the striatal D1R-expressing medium spiny neurons and altered miniature excitatory postsynaptic currents. To analyze the in vivo torsinA function in the D1R-expressing neurons further, Dyt1 conditional knockout (Dyt1 d1KO) mice in these neurons were generated. Dyt1 d1KO mice had decreased spontaneous locomotor activity and reduced numbers of slips in the beam-walking test. Dyt1 d1KO male mice showed abnormal gait. Dyt1 d1KO mice showed defective striatal D1R maturation. Moreover, the mutant striatal D1R-expressing medium spiny neurons had increased capacitance, decreased sEPSC frequency, and reduced intrinsic excitability. The results suggest that torsinA in the D1R-expressing cells plays an important role in the electrophysiological function and motor performance. Medical interventions to the direct pathway may affect the onset and symptoms of this disorder.
Keywords: direct pathway, dopamine receptor, dystonia, paired-pulse facilitation, raclopride, SCH 23390
1. Introduction
Dystonia is a movement disorder characterized by sustained or intermittent muscle contractions causing abnormal postures such as twisting, repetitive movements, or both. Dystonia is often initiated or worsened by voluntary action and associated with overflow muscle activation [1]. DYT1 dystonia is an early-onset autosomal dominant inherited movement disorder caused by mutations in DYT1/TOR1A, coding for torsinA [Oppenheim’s dystonia; Online Mendelian Inheritance in Man (OMIM) identifier #128100; Dystonia 1]. The dystonic symptoms commonly start from the legs and often gradually expand to the whole body. Most patients have a heterozygous trinucleotide inframe deletion (ΔGAG), which causes a loss of a glutamic acid residue in the C-terminal region [2]. Other mutations in the gene, such as 18-bp deletion (p. F323_Y328del) [3, 4], 4-bp deletion (c.934_937delAGAG) [5], and a missense mutation (c.613T→A, p. F205I) [6], were also reported in rare cases. Moreover, a two-month-old male with a homozygous nonsense mutation showed severe arthrogryposis, developmental delay, and dystonic movements [7]. The existence of these various mutations suggests that partial loss of torsinA function contributes to exhibit the symptoms. Consistently, torsinA levels are reduced in the fibroblasts from DYT1 dystonia patients with the heterozygous ΔGAG mutation [8].
Functional alteration of the basal ganglia circuits appears to have a vital role in the pathogenesis of this disease [9, 10]. DYT1 dystonia patients exhibit abnormally high midbrain, cerebellar, and thalamic activity during movement measured by positron emission tomography [11]. Deep brain stimulation of globus pallidus internus is an effective treatment for the dystonic symptom [12]. Postmortem study of DYT1 dystonia patients shows reduced dopamine in rostral portions of the putamen and caudate nucleus [13], and increased striatal dopamine metabolism, and a trend toward a reduction in dopamine receptor 1 (D1R) and 2 (D2R) binding [14]. A positron emission tomography with [11C]-raclopride study suggested that striatal D2R availability is reduced in both manifesting and non-manifesting DYT1 mutation carriers [15]. While the hypothesis that D2R deficiency leading to an alteration of the indirect pathway causes the hyperkinesia seen in dystonia appears to be well supported, the exclusion of the involvement of the direct pathway has been made not by evidence but rather by a lack of substantial studies on D1R activity in dystonic patients.
Animal model studies also suggested functional alterations of the basal ganglia circuits in DYT1 dystonia. Consistent with the reductions of D1R and D2R binding activities in DYT1 dystonia patients, Dyt1 ΔGAG heterozygous knock-in (KI) mice show reductions of striatal D1R- and D2R-binding activities [16, 17]. Dyt1 KI mice exhibit long-term depression (LTD) deficits in the corticostriatal pathway [17], sustained contraction and co-contraction of agonist and antagonist muscles of the hind limbs [18], and motor deficits of the hindlimbs in the beam-walking test [19]. The motor deficits of hindlimbs are reproduced in another line of KI mice by the beam-walking test [20, 21]. The abnormal muscle contractions and motor deficits are attenuated by trihexyphenidyl [17, 18], commonly used for dystonia patients, suggesting that the same mechanism causes these symptoms in Dyt1 KI mice as humans. Consistent with DYT1 dystonia patients, torsinA levels are reduced in two independent lines of Dyt1 KI mice [8, 22, 23] and the cell culture models [24, 25], suggesting that the ΔGAG mutation causes a partial loss of torsinA function. Dyt1 knockdown (KD) mice [26] and Dyt1 heterozygous knockout (KO) mice [27] showed similar motor deficits to Dyt1 KI mice, suggesting that partial loss of torsinA contributes to the motor deficits. Moreover, cerebral cortex-specific Dyt1 conditional KO (cKO) mice and striatum-specific Dyt1 conditional knockout (sKO) mice show motor deficits similarly to Dyt1 KI mice, suggesting that loss of torsinA function in the corticostriatal pathway contributes to the motor deficits [28, 29]. Since Dyt1 sKO mice were generated by using Rgs9-Cre mice [30], torsinA is knocked out in both direct and indirect pathway medium spiny neurons (MSNs) [31]. Therefore, it was not clear whether direct pathway, indirect pathway or both contribute to the motor deficits. Recently, dopamine receptor 2-expressing-cell-specific Dyt1 conditional knockout (Dyt1 d2KO) mice were generated, which showed motor deficits, suggesting that loss of torsinA function in the indirect pathway contributes to the motor symptoms [32].
On the other hand, few studies focused on characterizing the direct pathway in DYT1 dystonia animal models. Although striatal D1R-binding and D1R protein levels are decreased in Dyt1 KI mice [16, 33], the functional alteration of the direct pathway in Dyt1 KI mice has not been assessed. The D1R-expressing MSNs of the direct pathway in the basal ganglia circuits is known to contribute to motor coordination. Here, the locomotor response to the D1R and D2R antagonists and the basic electrophysiological property of the striatal direct pathway MSNs were analyzed in Dyt1 KI mice. Moreover, dopamine receptor 1-expressing cell-specific Dyt1 conditional knockout (Dyt1 d1KO) mice were generated. The motor behavior, electrophysiological property of the D1R-expressing MSNs, and striatal dopamine receptor levels were measured in Dyt1 d1KO mice.
2. Material and methods
2.1. Mice
All animal experiments were conducted in compliance with the USPHS Guide for Care and Use of Laboratory Animals and approved by the Institutional Animal Use and Care Committees at the University of Florida and the University of Illinois at Urbana-Champaign. Dyt1 KI mice were prepared as previously described [19]. Drd1-EGFP and Drd1-Cre mice were purchased from MMRRC [Tg(Drd1-EGFP)X60Gsat/Mmmh, stock number: 000297; B6.FVB(Cg)-Tg(Drd1-cre)EY266Gsat/Mmucd, stock number: 034259] [34, 35]. Ai6 Cre reporter mice were purchased from Jackson Laboratory (stock number: 007906) [36]. Dyt1 loxP mice, which have exons 3–4 floxed in Dyt1, were prepared as previously described [28]. Mice were housed with ad libitum access to food and water under the condition of 12 hours of light and 12 hours of dark circles.
This study followed the recommended heterogenization of study samples with various conditions [37]. Adult mice of different ages were used for all behavioral tests. The data were analyzed with age as a covariate.
2.2. Pharmacological study of D1R and D2R antagonists
Since a previous study suggests that Dyt1 KI male mice show abnormal horizontal locomotion [19], male mice were used in this pharmacological study during the light phase. For the D1R antagonist experiment, eight wild-type control (CT) and seven Dyt1 KI male mice of 205–248 days old were acclimated in an open-field apparatus which consists of a 41×41×31 cm acryl case directly illuminated by a 60W white bulb. Spontaneous locomotor activities of the mice were recorded individually by infrared light beam sensors and DigiPro software (AccuScan Instruments) for 2 days, 1 h each day. The locomotor activity recorded for 1 h on the third day was used as the animal’s baseline. On the fourth day, all mice were injected with 50 μg/kg (10ml/kg in saline) of SCH 23390 and placed in the open-field apparatus for 1 h. Seven days later, mice were injected with 100 μg/kg (10ml/kg in saline) of SCH 23390, and motor activity was again measured for 1 h. The percentage reduction of total distance traveled was determined concerning base activity level and compared across animals.
Nine CT and eight Dyt1 KI male mice of 73–81 days old were used in the D2R antagonist experiment. On the first day, all mice were given an intraperitoneal injection of saline (5ml/kg). Twenty minutes after the injection, mice were placed in the open-field apparatus, and their spontaneous locomotor activity was monitored. A week later, the mice were injected with raclopride (0.1mg/kg, 5ml/kg in saline) 20 min before placement in the open-field monitor. After 35 days, the mice were injected with a larger quantity of raclopride (0.3 mg/kg, 5ml/kg in saline), and their activity level was measured in the same way. The percentage reductions of total distance traveled were determined concerning activity level after saline injection and compared across animals.
2.3. Generation of Drd1-EGFP Dyt1 KI double mutant mice
D1R-expressing cells in Dyt1 KI mice and their littermates were labeled with the enhanced green fluorescent protein (EGFP) by crossing Drd1-EGFP mice and Dyt1 KI mice. Drd1-EGFP Dyt1 KI double mutant mice and Drd1-EGFP littermate control (CT) mice were used to analyze the electrophysiological property of the striatal D1R-expressing MSNs.
2.4. Electrophysiological recording of the striatal D1R-expressing MSNs
The mice were anesthetized by inhalation of isoflurane, decapitated, and the brains were rapidly removed. 350 μm-thick coronal brain slices were cut with a Vibratome (Technical Products International, St. Louis, MO) as previously described [32]. Slices were first incubated in artificial cerebrospinal fluid (aCSF) containing (in mM) 124 NaCl, 26 NaHCO2, 1.25 NaH2PO4, 2.5 KCl, 1 CaCl2, 6 MgCl2, 10 D-glucose gassed with 95% O2 and 5% CO2 at 35°C for 30 min, then incubated at room temperature (22 °C). After at least 1 hr incubation, a slice was transferred to a submerged recording chamber with the continuous flow (2 ml/min) of aCSF as described above except for 2 mM CaCl2 and 2 mM MgCl2 and gassed with 95% O2-5% CO2 to have a pH 7.4. All recordings were carried out at 32°C to 33°C.
Whole-cell recordings were made from seven Drd1-EGFP Dyt1 KI double mutant (male, n = 4; female, n = 3) and six Drd1-EGFP littermate mice (male, n = 4; female, n = 2; 31–43 days old) using infrared differential interference contrast microscopy with a fluorescent optical unit and an Axopatch 1Damplifier (Axon Instruments, Foster City, CA). Striatal D1R-expressing MSNs were identified as EGFP-positive cells. The glass recording electrodes were prepared by pulling capillary glass tubes using a horizontal electrode puller (Sutter). Patch electrodes had a resistance of 3–5 MΩ when filled with intracellular solution containing (in mM): 125 K-gluconate, 8 NaCl, 10 HEPES, 2 MgATP, 0.3 Na3GTP, 0.2 EGTA, and 0.1% biocytin (pH 7.3 with KOH, osmolarity 290–300 mOsM). The mEPSCs were recorded at a holding potential of −70 mV and at the presence of 50 μM picrotoxin, which blocked GABAergic synaptic activity, and 1 μM TTX, which blocked the transmitter release driven by action potentials, in the recording bath solution. Series resistance was 15–20 MΩ, and cells were rejected if it changed more than 20% throughout the recording. All drugs were purchased from Sigma-Aldrich. The recording data were acquired using pClamp 10 software. The recordings were started 5 min after accessing cells to allow for stabilization of spontaneous synaptic activity. Analysis of mEPSCs was based on 5 min continuous recordings from each cell (CT, 16 cells/6 mice; KI, 15 cells/7 mice). Synaptic events were analyzed by using the Mini Analysis Program (Synaptosoft) with parameters optimized for each cell and then visually confirmed before analysis. The peak amplitude, 10–90% rise time and the decay time constant were measured based on the average of all events aligned by the rising phase.
To analyze the ratio of NMDA and AMPA receptor-dependent EPSCs, EPSCs were recorded first from individual neurons in the brain slices with a holding potential of +50 mV and the presence of GABA receptors blocker bicuculline (20 μM). Since EPSCs are known to be derived from both NMDA- and AMPA receptors, the AMPA component of EPSCs was isolated next by adding NMDA receptor blocker AP5 (50 μM) in the recording bath. The NMDA receptor-dependent EPECs were calculated by subtraction of the AMPA-dependent EPSCs from the total EPSCs. The NMDA/AMPA ratio was calculated from two Drd1-EGFP Dyt1 KI double mutant and three Drd1-EGFP littermate male mice (39 – 117 days old).
The paired-pulse ratio (PPR) was measured in the acute brain slices from four Drd1-EGFP Dyt1 KI double mutant and six Drd1-EGFP littermate male mice (39 – 117 days old) as previously described [27] with minor modification for MSN recording. The glass recording electrodes were filled with aCSF. The input resistance of each recording electrode was tested by applying a current pulse, and the tip was cut until the resistance of 1–3 MΩ was obtained. The recording electrodes were placed into the EGFP-positive MSNs for the whole-cell recording. Two different test stimuli with inter-stimulus intervals 20 ms and 50 ms were delivered from the surrounding area of the EGFP-positive MSNs by using a bipolar Teflon coated platinum stimulating electrode. Responses were recorded by using AxoClamp pClamp8 data acquisition software on a personal computer. All experimental stimuli were set to an intensity that evoked 50% of the maximum EPSP slope. All electrophysiological recordings were performed by an investigator blind to the genotypes.
2.5. Generation of Dyt1 d1KO mice
Drd1-cre+/− Dyt1 loxP+/− (double heterozygote: DHet) was generated by crossing the Drd1-cre+/− mice and either heterozygous Dyt1 loxP+/− or homozygous Dyt1 loxP−/− mice. Dyt1 d1KO mice were generated by crossing the DHet mice and either Dyt1 loxP+/− or Dyt1 loxP−/− mice (Supplementary Fig. 1). Genotyping was performed using PCR with tail DNA at two to three weeks old and specific primers for Cre and Dyt1 loxP [28]. The number of pups in each genotype was compared to analyze the neonatal lethality of the Dyt1 d1KO mice.
2.6. Fluorescence immunohistochemistry
Dyt1 d1KO and Dyt1 loxP−/− mice were anesthetized and perfused with 0.1M phosphate buffer (PB; pH 7.4) followed by 4% paraformaldehyde in 0.1M PB. The brains were dissected and stored in 4% paraformaldehyde in 0.1M PB at 4 °C overnight. The brains were put into 30% sucrose 0.1M PB until the brain sank. The brains were frozen with dry-ice powder and sliced with a sliding microtome at 35 μm thickness. The slices were stored in PBS and washed with 10 mM glycine/PBS for 5 min three times. The slices were then blocked with 2% gelatin in PBS for 15 min and washed with the glycine/PBS for 5 min. The slices were incubated with rabbit anti-torsinA (Abcam; ab34540; 1:714 dilution) and goat anti-Cre (Santa Cruz; sc-83398; 1:50 dilution) antibodies in 1% BSA/PBS at room temperature for 2 h. Slices were washed with 0.1% BSA/PBS for 5 min six times. The slices were treated with Alexa Flour 546 donkey anti-rabbit IgG (H+L; Invitrogen; A10040; 1:50 dilution) and Alexa Flour 488 donkey anti-goat IgG (H+L; Life Technologies; A11055; 1:50 dilution) secondary antibodies in 1% BSA/PBS for 2 h. The slices were washed with 0.1% BSA/PBS for 5 min six times and PBS for 5 min twice. The slices were incubated with TO-PRO-3 Iodide (Invitrogen; T3605; 1:1000 dilution) in PBS for 20 min to stain nuclei. The slices were washed in PBS for 5 min three times and mounted on Superfrost/plus microscope slides (Fisher scientific; 12-550-15) and dried overnight. The slices were covered with Vectashield Hardset antifade mounting medium (Vector Laboratories; H-1400) and cover glass No. 1 ½ (Corning; 2940–225). The confocal images were captured with Nikon A1R MP Laser-Scanning Confocal Microscope.
2.7. Motor behavioral characterization of Dyt1 d1KO mice
Motor performance of the Dyt1 d1KO (male, n = 9; female, n = 8) and CT littermate mice [male, n = 9 (Dyt1 loxP−/−, n = 2; Dyt1 loxP+/−, n = 6; Drd1-cre+/−, n = 1); female, n = 11 (Dyt1 loxP−/−, n = 4; Dyt1 loxP+/−, n = 6; Drd1-cre+/−, n = 1)] was evaluated during the light phase by the behavioral semi-quantitative assessments of motor disorders, open-field, accelerated-rotarod, beam-walking, and paw-print gait analysis tests in this order. The behavioral semi-quantitative assessments of motor disorders were performed as previously described [19, 38]. Each mouse at 165–277 days old was placed on a table, and assessments of hind paw clasping, hind paw dystonia, truncal dystonia, and balance adjustments to a postural challenge were made. The hind paw clasping was assessed as hind paw movements for postural adjustment and an attempt to straighten up while the mouse was suspended by the mid-tail.
An open-field test was performed during the light period essentially as previously described [39]. In brief, spontaneous locomotor activities of the mice at 173–285 days old were recorded individually by infrared light beam sensors in a 41 × 41 × 31 cm acryl case directly illuminated by a 60 W white bulb for 30 min at 1 min intervals using DigiPro software (AccuScan Instruments).
The accelerated rotarod test assesses the ability of mice to maintain balance and coordination. The motor performance of the mice at 182–294 days old was examined as previously described [19] with minor modification. An accelerating rotarod apparatus (Ugo Basile) was started at an initial speed of 4 rpm, and then each mouse was put on the same slot one by one. The rod speed was gradually accelerated at a rate of 0.2 rpm/s. The latency to fall was measured with a cutoff time of 5 min at a final rate of 64 rpm. Mice were tested for three trials on each day for 2 days. The trials within the same day were performed at about 1 h intervals.
The beam-walking test was performed as described earlier [19, 40–42]. The mice at 190–302 days old were trained to transverse a medium square beam (14 mm wide) in three consecutive trials each day for 2 days. The trained mice were tested twice on the medium square beam and medium round beam (17 mm diameter) on the third day, and small round beam (10 mm diameter) and small square beam (7 mm wide) on the fourth day. The hind paw slips on each side were recorded.
The paw-print test is an analysis of the animal’s gait. A runway with a dark goal box at the end was lined with a sheet of white paper [43]. Fore and hind paws of the mice at 196–308 days old were painted with water-soluble, non-toxic paint of different colors. Mice walked across the runway and into the goal box. One set of prints was collected for each animal after it walked continuously across the runway. The four center pairs of hind and forepaw prints of each set were analyzed for stride length, fore and hind base lengths, and distance of overlap of the paws.
2.8. Western blot analysis
Striata were dissected from Dyt1 d1KO (n = 3) and CT (n = 7) mice. Proteins were extracted in the lysis buffer containing 1% Triton X100 and quantified as previously described [22]. The proteins (30 μg) were separated on SDS-PAGE gels and transferred to PROTRAN nitrocellulose transfer membranes (Whatman). Precision Plus Protein All Blue Prestained Protein Standards (Bio-rad, #1610373) were also loaded as molecular weight markers. After blocking with 5% Non-fat milk (Bio-rad) in TBS-T buffer [20 mM Tris-Cl (pH7.6), 137 mM NaCl, 0.1% Tween 20], the membranes were cut between 50 and 37 kDa position. The high molecular weight part membranes were incubated with mouse monoclonal D1R (Santa Cruz, sc-33660, 1:800 dilution) or mouse monoclonal D2R antibodies (Santa Cruz, sc-5303, 1:200 dilution) at 4 °C overnight. After washing with TBS-T, the membranes were incubated with bovine anti-mouse IgG-HRP (Santa Cruz, sc-2371, 1:1000 dilution) for 1 h, and washed with TBS-T. The low molecular weight part membranes were incubated with Anti-GAPDH antibody HRP (Santa Cruz, sc-25778HRP, 1:500 dilution) overnight and washed with TBS-T. The chemiluminescence signals were produced with SuperSignal substrate (Pierce, #34096) and captured by an Alpha Innotech Innotech FluorChem FC2 camera, and the density of the bands was quantified by UN-SCAN-IT gel (Silk Scientific) or Image Studio Light (LI-COR). The experiments were performed in duplicate.
2.9. Electrophysiological recording of the striatal D1R-expressing MSNs in Dyt1 d1KO Ai6 mice
The D1R-expressing cells were genetically labeled with ZsGreen by crossing Ai6 mice and Drd1-Cre mice with or without Dyt1 loxP−/− allele [28]. Incorporations of the transgenic Ai6 and Drd1-Cre were confirmed by PCR-based genotyping with the specific primer set (Jackson Laboratory; protocol 28544; oIMR9020, oIMR9021, oIMR9103, oIMR9104) for Ai6 and another primer set (MMRRC; 034259-UCD; Dr1a F1, CreGS R1) for Drd1-Cre, respectively. Two Dyt1 d1KO Ai6 and three Drd1-cre+/− Ai6 mice at 58–61 days old were used for the electrophysiological recording of the striatal D1R-expressing MSNs.
The mice were anesthetized by inhalation of isoflurane, decapitated, and the brains were rapidly removed. Coronal brain slices were cut at 300 μm-thick in ice-cold, oxygenated cutting saline (in mM): 180 sucrose, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 10 D-glucose, 1 CaCl2, 10 MgCl2, and 10 glucose with a Vibratome (Leica VT 1000s) as previously described [31]. Slices were recovered in a holding chamber for 60 min at 35 °C with artificial cerebrospinal fluid (ACSF; in mM: 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 1 MgCl2, 2 CaCl2, 10 glucose, and bubbled with 95% O2-5% CO2 to have a pH 7.4. The slices were then incubated at room temperature for recording.
The slices were placed in a recording chamber and continuously perfused by ACSF that was bubbled via 5% CO2 and 95% O2 at a rate of 1.5 ml/min while being visualized with an upright microscope (Zeiss, Germany) using a 40× water-immersion objective with fluorescence optics. D1R-expressing striatal MSNs were identified as ZsGreen positive cells. All experiments were recorded at 32 °C by a dual automatic temperature controller (TC-344B). Recording patch pipette (6–10 MΩ) contained the following solutions (in mM): 125 K-gluconate, 8 NaCl, 10 HEPES, 2 MgATP, 0.3 NaGTP, 0.2 EGTA (pH 7.25–7.3 with KOH, osmolality, 290–300 mOsm) was used for both current and voltage-clamp recordings. Access resistances were <25 MΩ.
Spontaneous postsynaptic currents (sEPSCs) were recorded in ACSF in voltage-clamp mode After breaking through the cell membrane. Action potentials were evoked by injection of depolarizing 50 pA current pulse of 300 ms duration under current-clamp configuration in a brain slice. This process was repeated at ten increasingly depolarized potentials with 50pA current steps.
Events were detected using the Mini Analysis Program (Synaptosoft) with parameters optimized for each cell and then visually confirmed before analysis. The peak amplitude, 10–90% rise time, and the decay time constant were measured based on the average of all events aligned by the rising phase.
2.10. Statistics
The data were analyzed by the R program (ver. 4.0.4; R Foundation for Statistical Computing, Vienna, Austria) or SAS GENMOD procedure. The effects of D1R and D2R antagonists on the total horizontal movement were analyzed by a generalized linear mixed model (glm.nb) with a negative binomial distribution and glm with normal distribution, respectively, and hierarchically concerning genotype, weight, and age by Akaike Information Criterion (AIC). AIC is defined by AIC = (−2) log (maximum likelihood) + 2 (number of independently adjusted parameters within the model) [44]. When there are several competing models, the one with the minimum AIC value was used. Electrophysiological recording mEPSC data were analyzed by a generalized linear mixed model (lme) with the nested data. The genotype ratio of Dyt1 d1KO and CT littermate mice was analyzed with Fisher’s exact test. Data from the open-field, accelerated rotarod, and paw-print were analyzed by glm concerning genotype, sex, weight, and age by AIC. The slip numbers in the beam-walking test for all four beams were analyzed together by glm.nb and concerning genotype, sex, weight, and age by AIC. The densities of D1R (80kDa, 65kDa) and D2R (109 kDa, 96 kDa) bands standardized with that of GAPDH were analyzed by R glm for the normal distribution data with genotype and trial as variables. The densities of D1R (48 kDa) and D2R (70kDa) bands standardized with GAPDH were analyzed by R gln.nb with genotype and trial as variables. The sEPSC and evoked action potential data were analyzed by SAS GENMOD procedure with a negative binomial distribution (AP), gamma distribution (frequency, rise, decay, Cm, Rm, Tau, MP), and normal distribution (amplitude). Correlation between the resting membrane potential and the sEPSC or the evoked action potential was calculated by Pearson’s product-moment correlation program (R cor.test). Significance was assigned at p < 0.05.
3. Results
3.1. Reduced locomotor responses to D1R and D2R antagonists
Dyt1 KI mice show hyperactive locomotion and motor deficits [19]. Dyt1 KI mice also show alteration of dopamine metabolism and decreased striatal D1R and D2R levels [16, 17]. Since locomotor activity is tightly linked to dopaminergic alterations, the locomotor activity levels of Dyt1 KI mice in response to the D1R and D2R antagonists were measured in the open field apparatus, respectively.
Total horizontal distances traveled in each mouse without drug and that after administration of D1R antagonist SCH23390 at 50 μg/kg (Fig. 1A) and 100 μg/kg (Fig. 1B) were measured. Dyt1 KI mice showed significantly smaller reduction of total horizontal distance in comparison to CT mice with SCH 23390 at 50 μg/kg [mean ± standard errors (SE); CT, 42 ± 6 %, n = 8; Dyt1 KI, 55 ± 17 %, n = 7; z (11) = 2.193; p = 0.028] and a trend of the reduction at 100 μg/kg dose [CT, 18 ± 3 %, n = 8; Dyt1 KI, 29 ± 6 %, n = 7; z (13) = 1.942; p = 0.052], suggesting that Dyt1 KI mice showed a less response to the D1R antagonist (Fig. 1C). The results suggest a faster saturation of receptors due to the reduction of D1R numbers in Dyt1 KI mice.
Fig. 1.
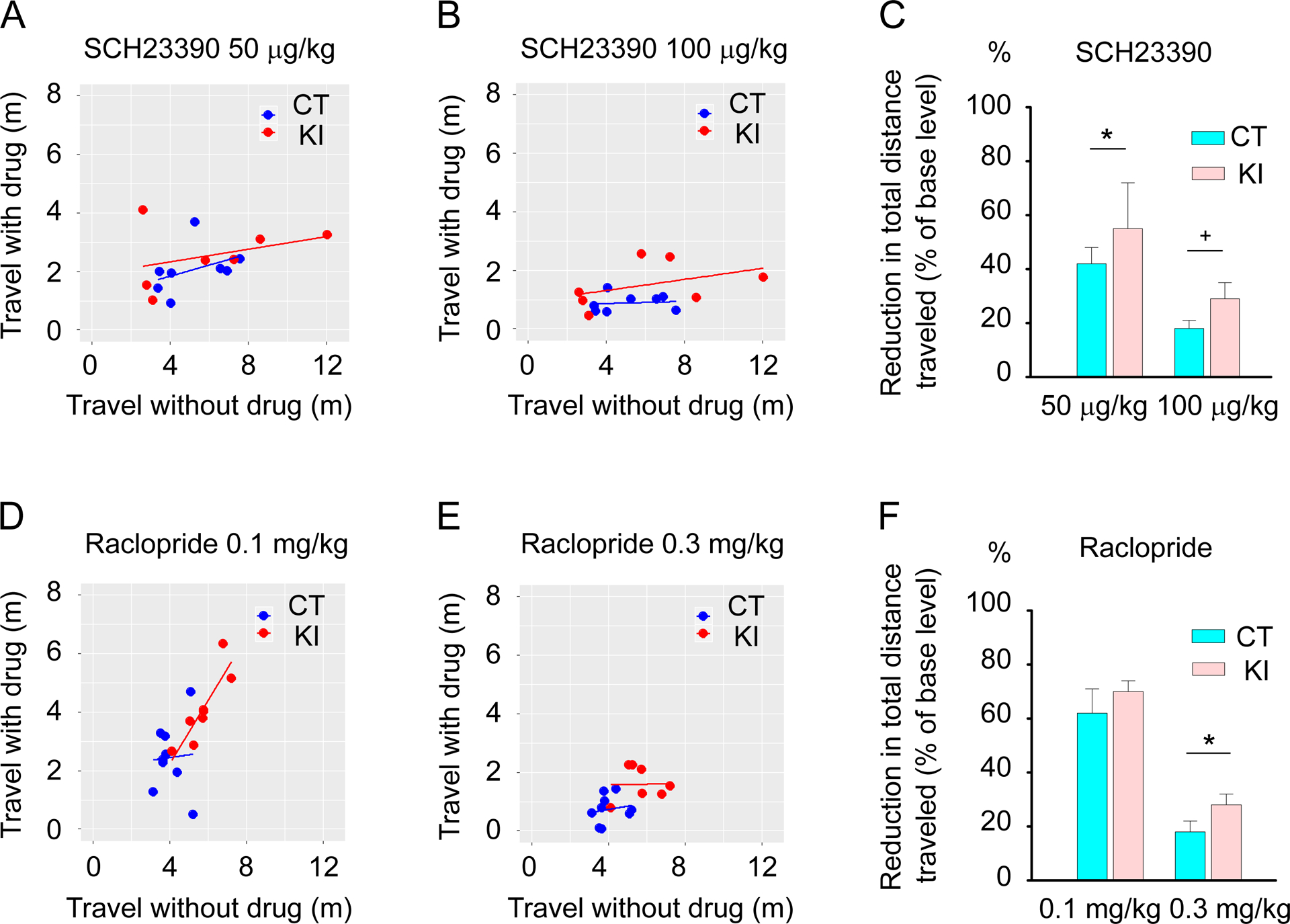
The in vivo effects of dopamine receptor antagonists on locomotion in Dyt1 KI and CT mice. Total horizontal distance traveled of each mouse without drug and that after administration of D1R antagonist (SCH23390) at 50 μg/kg (A) and 100 μg/kg (B). (C) Dyt1 KI mice showed significantly less response in the total horizontal distance to SCH23390 at 50 μg/kg (p = 0.028) and a trend of less response at 100 μg/kg (p = 0.052). Total horizontal distance traveled of each mouse without drug and that after administration of D2R antagonist (raclopride) at 0.1 mg/kg (D) and 0.3 mg/kg (E). (F) Dyt1 KI mice showed a significant less response of drug-induced attenuation of locomotion in the total horizontal distance to raclopride at 0.3 mg/kg (p = 0.026), but not at 0.1 mg/kg (p = 0.53). +p < 0.1, *p < 0.05.
Total horizontal distances traveled in each mouse without drug and that after administration of D2R antagonist raclopride at 0.1 mg/kg (Fig. 1D) and 0.3 mg/kg (Fig. 1E) were measured. At a starting dose of 0.1 mg/kg of raclopride, Dyt1 KI and CT mice showed similar levels of locomotor activity reduction [CT, 62 ± 9 %, n = 9; Dyt1 KI, 70 ± 4 %, n = 8; t (14) = 0.637; p = 0.53; Fig. 1F]. On the other hand, Dyt1 KI mice showed a significantly smaller reduction of total horizontal distance in comparison to CT mice at a higher dose of 0.3 mg/kg [CT, 18 ± 4 %, n = 9; Dyt1 KI, 28 ± 4 %, n = 8; t (14) = 2.495; p = 0.026; Fig. 1F], suggesting that Dyt1 KI mice showed less response to the D2R antagonist. The result suggests a faster saturation of the receptors due to the reduction of D2R numbers in Dyt1 KI mice.
3.2. Decreased rise and decay times of the D1R-expressing MSNs mEPSCs in Drd1-EGFP Dyt1 KI mice
Electrophysiological properties of the EGFP-labeled D1R-expressing striatal MSNs were characterized by using acute brain slices from control Drd1-EGFP mice (CT, 16 cells/6 mice) and Drd1-EGFP Dyt1 KI double-mutant mice (KI, 15 cells/7 mice). There was no significant alteration in the frequency of mEPSCs between Drd1-EGFP and Drd1-EGFP Dyt1 KI mice [mean ± SE; Hz; CT, 3.2 ± 0.3; KI, 3.6 ± 0.4; t (29) = 0.868; p = 0.39; Fig. 2A–C]. As shown in the representative averaged peak traces (Fig. 2D), there was no significant alteration in the amplitude of the mEPSCs between Drd1-EGFP and Drd1-EGFP Dyt1 KI mice [pA; CT, 11.7 ± 1.4; KI, 12.7 ± 2.3; t (28) = 1.4333; p = 0.16; Fig. 2E, F]. On the other hand, Drd1-EGFP Dyt1 KI mice showed significantly decreased rise [ms; CT, 2.3 ± 0.2; KI, 2.1 ± 0.2; t (28) = −2.249; p = 0.033; Fig. 2G, H] and decay times [ms; CT, 3.5 ± 0.4; KI, 3.2 ± 0.2; t (28) = −2.068; p = 0.048; Fig. 2I, J] of the mEPSCs in comparison to Drd1-EGFP control mice. The results suggest a functional alteration of the postsynaptic elements of the D1R-expressing MSNs in Drd1-EGFP Dyt1 KI mice.
Fig. 2.
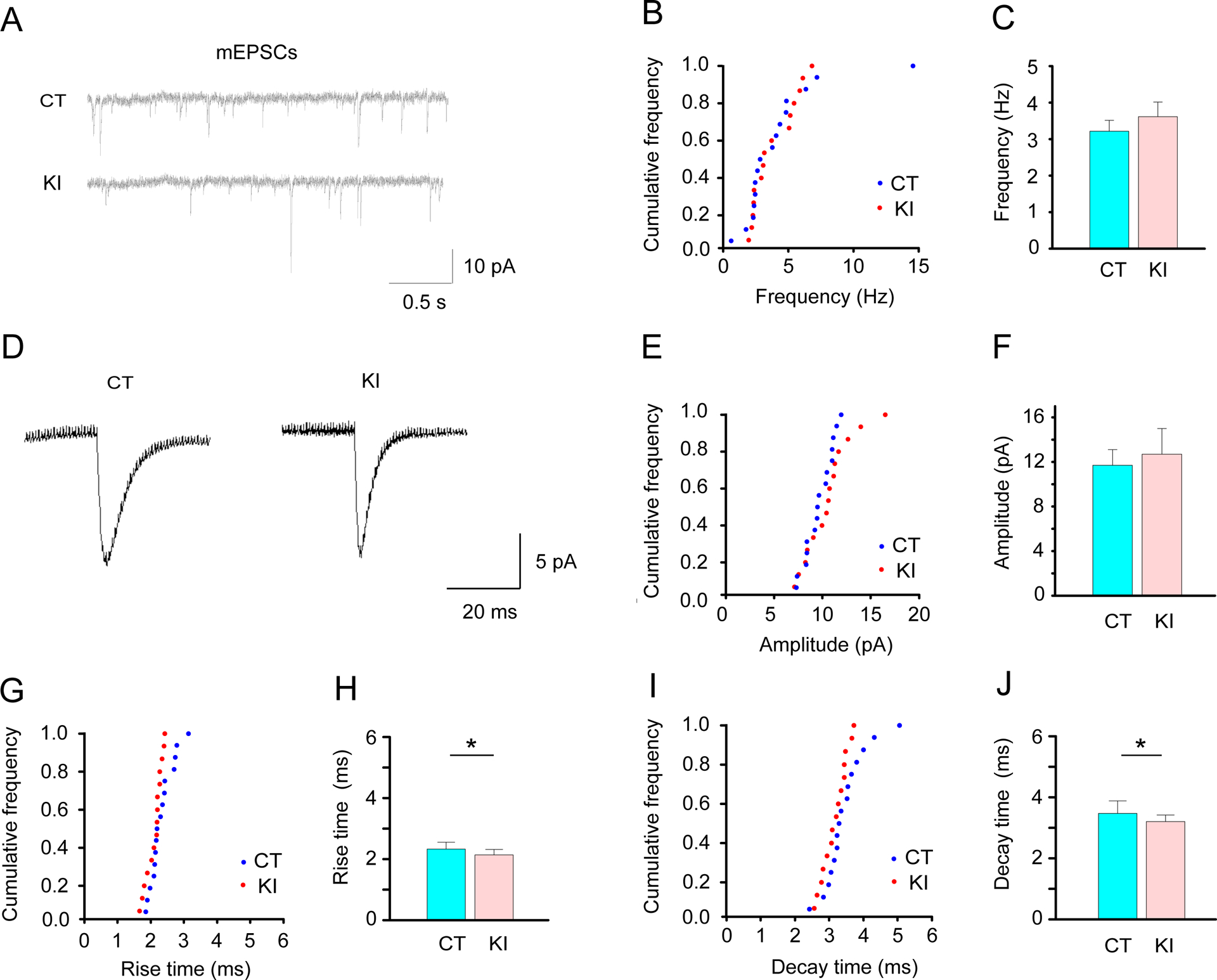
Characterization of mEPSC of the striatal D1R-expressing MSNs in Drd1-EGFP Dyt1 KI mice. (A) Representative mEPSC traces of the D1R-expressing MSNs in Drd1-EGFP (CT) and Drd1-EGFP Dyt1 KI (KI) mice. (B) The cumulative frequency of the mEPSC frequency (Hz) is shown as previously described [76]. (C) There was no significant difference in the frequency between CT and KI mice. (D) Representative averaged peak traces of the mEPSCs in CT and KI mice. (E) Cumulative frequency of the mEPSC amplitude (pA). (F) There was no significant difference in the amplitude between CT and KI mice. (G) Cumulative frequency of the mEPSC rise time (ms). (H) Drd1-EGFP Dyt1 KI mice showed significantly decreased rise time (ms) of the mEPSCs. (I) Cumulative frequency of the mEPSC decay time (ms). (J) Drd1-EGFP Dyt1 KI mice showed significantly decreased decay time (ms) of the mEPSCs. The vertical bars represent means ± SE in C, F, H, and J. *p < 0.05.
3.3. No significant alteration of the AMPA and NMDA ratio of the D1R-expressing MSNs in Drd1-EGFP Dyt1 KI mice
Since the rise and the decay times of EPSCs are known to be affected by postsynaptic elements, the NMDA/AMPA ratio was compared between Drd1-EGFP control mice (CT, 9 cells/3 mice) and Drd1-EGFP Dyt1 KI double-mutant mice (KI, 5 cells/2 mice). As shown in the representative traces (Fig. 3A), there was no significant alteration of the NMDA/AMPA ratio of the D1R-expressing MSNs between Drd1-EGFP and Drd1-EGFP Dyt1 KI mice [mean ± SE; CT, 0.58 ± 0.05; KI, 0.57 ± 0.05; t (12) = −0.067; p = 0.95; Fig. 3B]. The result suggests that the altered rise and decay times of mEPSCs in the Drd1-EGFP Dyt1 KI mice were not caused by the alteration of the NMDA/AMPA ratio of the D1R-expressing MSNs.
Fig. 3.
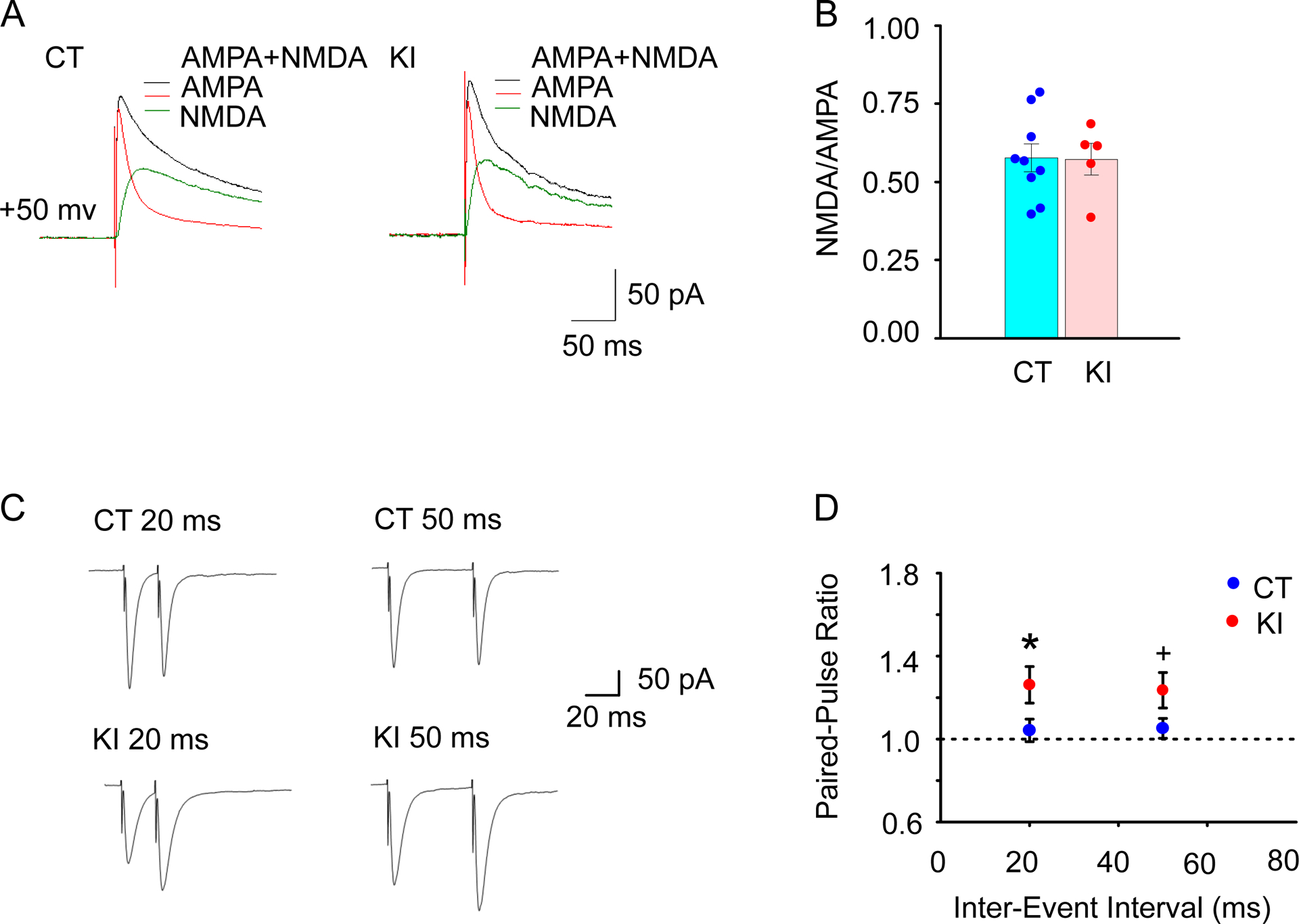
Electrophysiological characterization of the postsynaptic components and the presynaptic property. (A) The representative EPSC traces from individual neurons recorded in Drd1-EGFP (CT) and Drd1-EGFP Dyt1 KI (KI) mice with a holding potential of +50 mV and the presence of GABA receptors blocker Bicuculline (20 μM). Black and red traces were obtained before and after adding AP5 (50 μM), respectively. NMDA component (green line) was obtained after the subtraction of red traces from the black traces. (B) Ratios of NMDA to AMPA components of individual neurons were not significantly different between CT and KI mice. The vertical bars represent means ± SE. Dots indicate the data derived from each cell. (C) Representative traces of paired-pulse stimulation in the D1R-expressing MSNs, which were evoked with inter-event intervals of 20 ms and 50 ms in CT and KI mouse brain slices. (D) Paired-pulse ratio (2nd/1st response) of the D1R-expressing MSNs in CT and KI mice. Drd1-EGFP Dyt1 KI mice showed significantly enhanced paired-pulse facilitation with 20 ms inter-stimulus interval stimuli comparing to CT mice and a trend of enhanced paired-pulse facilitation with 50 ms inter-stimulus interval stimuli. The circles and bars represent means ± SE in D. Blue and red circles represent CT and KI mice, respectively. +p < 0.1, *p < 0.05.
3.4. Enhanced paired-pulse facilitation of the D1R-expressing MSNs in Drd1-EGFP Dyt1 KI mice
Since the spontaneous and evoked neurotransmitter releases may use distinct mechanisms, the evoked glutamate release from the presynaptic neurons was further measured in whole-cell recording mode by stimulating the surrounding area of the EGFP-labeled D1R-expressing MSNs in Drd1-EGFP control mice (CT, 20 cells/6 mice) and Drd1-EGFP Dyt1 KI double-mutant mice (KI, 10 cells/4 mice). Two different test stimuli with 20 ms and 50 ms inter-stimulus intervals were delivered. These stimuli mimic the action potentials from the presynaptic neurons to the EGFP-labeled D1R-expressing MSNs. As shown in the representative traces (Fig. 3C), Drd1-EGFP Dyt1 KI showed paired-pulse facilitation in both conditions. Drd1-EGFP Dyt1 KI mice showed significantly increased PPR with 20 ms stimulation in comparison to Drd1-EGFP mice [CT, 1.04 ± 0.05; KI, 1.26 ± 0.09; t (28) = 2.083; p = 0.047; Fig. 3D], and a trend of increased PPR with 50 ms stimulation [CT, 1.05 ± 0.05; KI, 1.24 ± 0.09; ; t (28) = 1.835; p = 0.077; Fig. 3D]. When all data were analyzed together, regardless of the inter-stimulus intervals, Drd1-EGFP Dyt1 KI mice showed significantly increased PPR in comparison to Drd1-EGFP mice [PPR ± SE; CT, 1.05 ± 0.04; KI, 1.25 ± 0.06; t (28) = 2.146; p = 0.041]. Since PPR is inversely proportional to the probability of synaptic vesicle release [45], the results suggest that glutamate release deficits from the presynaptic neurons to the striatal D1R-expressing MSNs in Dyt1 KI mice.
3.5. Generation of Dyt1 d1KO mice
To examine the in vivo effect of torsinA loss in D1R-expressing cells, we generated Dyt1 d1KO mice and characterized their motor phenotypes. The genotype ratio of Dyt1 d1KO and CT littermate mice did not deviate from Mendel’s rule (Supplementary table 1), suggesting that Dyt1 d1KO mice are neither embryonic nor neonatal lethal. Dyt1 d1KO mice grew up to adulthood without apparent developmental delay. The loss of torsinA in the striatal D1R-expressing MSNs in Dyt1 d1KO mice was confirmed by fluorescence immunohistochemistry (Supplementary Fig.2). The representative confocal image of the striatum from a Dyt1 d1KO mouse showed a loss of torsinA in Cre-expressing MSNs. Since Cre is expressed from the D1R promoter in Dyt1 d1KO mice, the results suggested that Dyt1 was removed from the striatal D1R-expressing MSNs.
3.6. No overt dystonic symptoms in Dyt1 d1KO mice
Motor performance of Dyt1 d1KO mice was characterized in behavioral tests, i.e., behavioral semi-quantitative assessments of motor disorders, open field, accelerated rotarod, beam-walking, and paw-print gait analysis, in this order. The behavioral semi-quantitative assessments of motor disorders were performed as previously described [19, 38]. There were no overt behavioral alterations between Dyt1 d1KO mice and CT mice, suggesting that Dyt1 d1KO mice did not exhibit obvious dystonic symptoms.
3.7. Decreased spontaneous locomotion of Dyt1 d1KO mice in the open field test
The locomotion of Dyt1 d1KO mice was examined in an open field apparatus. Dyt1 d1KO mice showed significantly decreased movement number (Fig. 4A) and time (Fig. 4B) and increased rest time of spontaneous locomotor activities comparing to CT mice (Supplementary table 2). Dyt1 d1KO mice also showed a trend of reduced horizontal activity (Fig. 4C), total distance (Fig. 4D), and anticlockwise revolution (Supplementary table 2). The data were stratified by sex and analyzed further in each sex. Dyt1 d1KO male mice showed significantly decreased movement time (Fig. 4B), horizontal activity (Fig. 4C), total distance (Fig. 4D), vertical movement number, marginal movement distance, central distance, and increased rest time comparing to CT mice (Supplementary table 2). Although the central distance was also decreased, there was no significant difference in the central distance ratio between Dyt1 d1KO male mice and CT mice. Therefore, the decreased central distance in Dyt1 d1KO male mice was derived simply from the decreased locomotion rather than an anxiety-like phenotype. Moreover, Dyt1 d1KO male mice showed a trend of decreased movement number (Fig. 4A), clockwise revolution, and anticlockwise revolution (Supplementary table 2). Furthermore, Dyt1 d1KO female mice showed significantly decreased movement number of spontaneous locomotor activities comparing to CT mice (Fig. 4A; Supplementary table 2). Overall, the open-field test results suggest that loss of torsinA in D1R-expressing cells decreased locomotion. Since D1R-expressing direct pathway MSNs generally promote movement, knockout of torsinA in D1R-expressing cells may cause malfunction of the direct pathway and exhibit decreased locomotion.
Fig. 4.
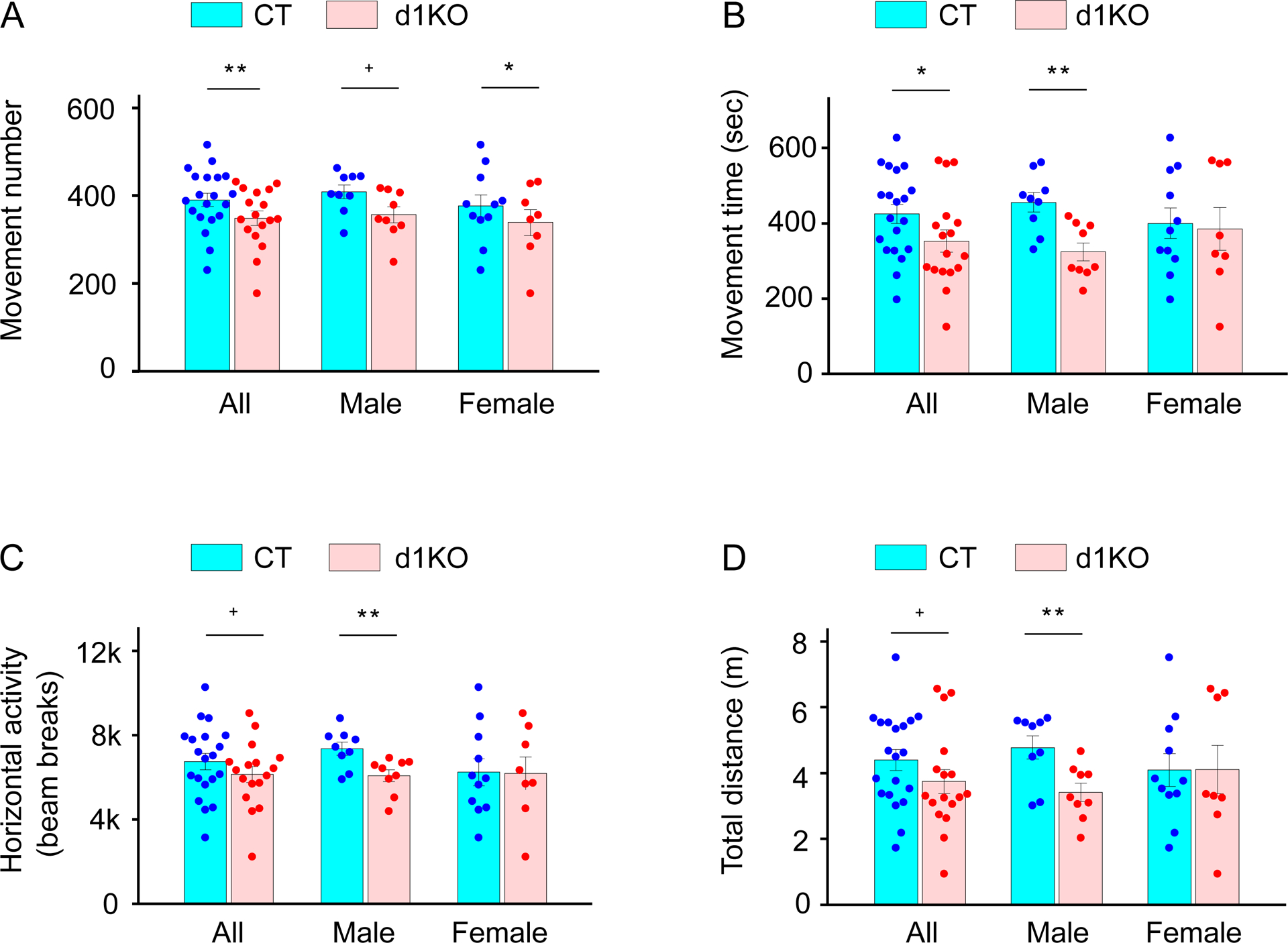
Open-field test. (A) Movement number. (B) Movement time (sec). (C) Horizontal activity (beam breaks). (D) The total horizontal distance (m). The vertical bars indicate means ± SE. The black dots indicate data points from individual mice. +p < 0.1, *p < 0.05, **p < 0.01. The detailed open-field data are shown in Supplementary Table 2.
3.8. Motor coordination and balance of Dyt1 d1KO mice
Motor coordination and balance were analyzed by an accelerated rotarod test. Since motor coordination and balance were influenced by body weight, the effect was incorporated in the results of the statistical analysis. Dyt1 d1KO mice did not show significant alteration in the rotarod performance [latency to fall (s; means ± SE) of all trials; CT, 113 ± 7; n = 20; d1KO, 116 ± 7; n = 17; t (32) = 0.512; p = 0.61; Fig. 5A]. Comparison of the latency to fall in each trial did not show significant alteration between Dyt1 d1KO and CT mice, either (trial 1, t (34) = −0.583; p = 0.56; trail 2, t (34) = −0.506; p = 0.62; trial 3, t (34) = 1.009; p = 0.32; trail 4, t (34) = 0.670; p = 0.51; trial 5, t (34) = 0.141; p = 0.89; trial 6, t (34) = 1.405; p = 0.17). The latency to fall data were stratified by sex and analyzed further. Dyt1 d1KO male and female mice did not show significant alteration in latency to fall comparing to CT male [CT male, 107 ± 13, n = 9; d1KO male, 113 ± 9, n = 9; t (14) = 0.966; p = 0.35; Fig. 5A] and female mice [CT female, 117 ± 9; n = 11; d1KO female, 121 ± 10, n = 8; t (17) = 0.244; p = 0.81; Fig. 5A], respectively. The results suggest that Dyt1 d1KO mice did not show abnormal motor performance with four limbs, which was similarly to Dyt1 KI mice [19].
Fig. 5.
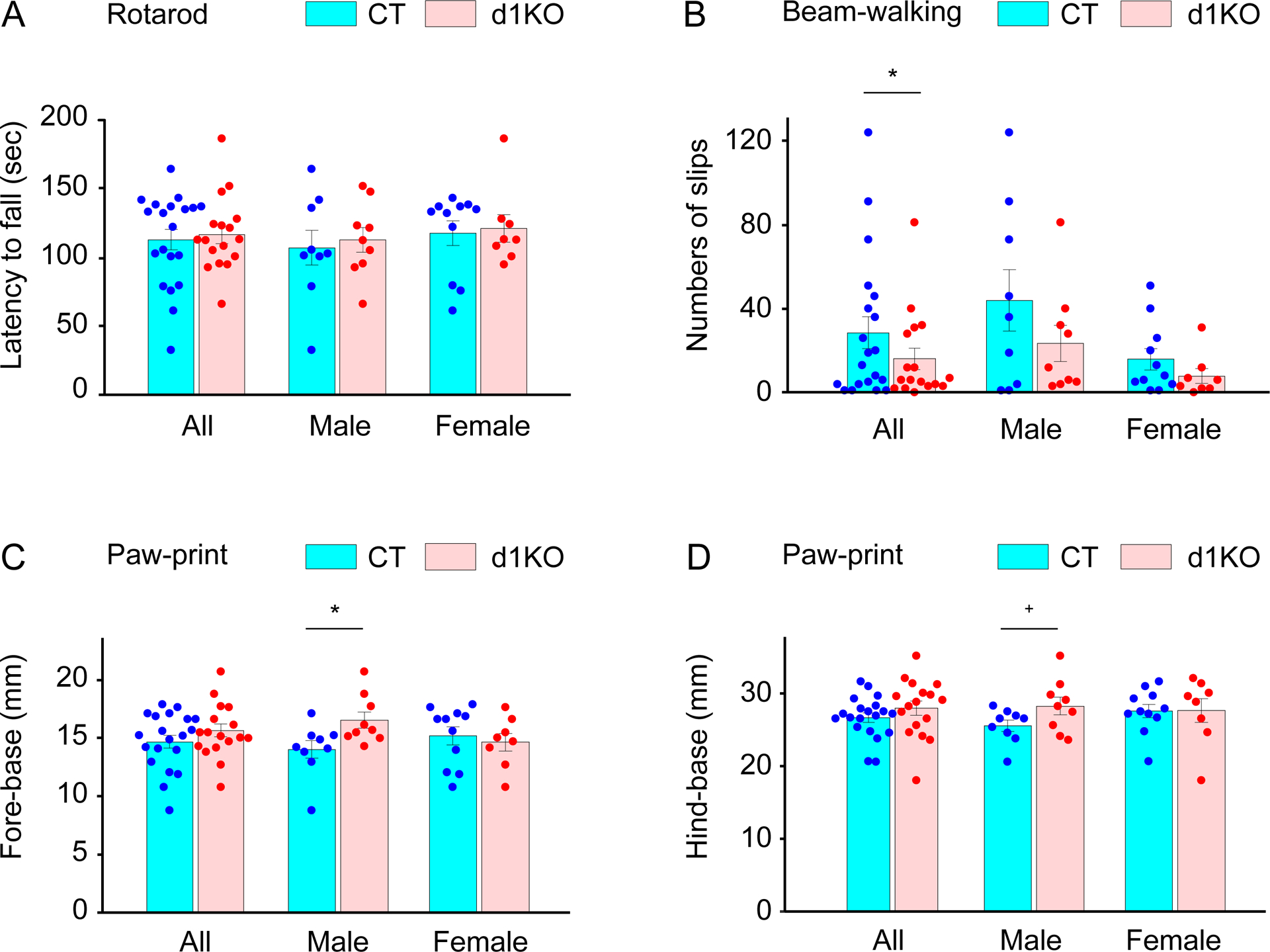
Motor behaviors of Dyt1 d1KO mice in the accelerated rotarod, beam-walking, and paw-print tests. (A) Latency to fall (s) in the accelerated rotarod test. (B) Slip numbers in the beam-walking test. Fore-base (mm; C) and hind-base (mm; D) in paw-print gait analysis. The vertical bars indicate means ± SE. +p < 0.1, *p < 0.05. The detailed paw-print data are shown in Supplementary Table 3.
The motor performance of hindlimbs was further analyzed by the beam-walking test and slip numbers of the hindlimbs were compared between Dyt1 d1KO mice and CT mice. Dyt1 d1KO mice showed significantly reduced slip numbers comparing to CT mice in the beam-walking test [slip numbers in all trials (means ± SE); CT, 28.5 ± 7.6, n = 20; d1KO, 16.1 ± 5.1, n = 17; z (33) = −2.074; p = 0.038; Fig. 5B]. Since sex significantly contributed to the slip numbers [z (33) = −2.954; p = 0.0031], the data were stratified by sex and analyzed further. Dyt1 d1KO male and female mice show the same trend of fewer slip numbers comparing to CT male and female mice, respectively [CT male, 43.9 ± 14.6, n = 9; d1KO male, 23.4 ± 8.5, n = 9; z (15) = −1.360; p = 0.17; CT female, 15.9 ± 5.0, n = 11; d1KO female, 7.9 ± 3.6, n = 8; z (16) = −1.582; p = 0.11; Fig. 5B]. Overall, Dyt1 d1KO mice showed better motor performance of hindlimbs in the beam-walking test, which was similarly to the cerebellar Purkinje-cell specific Dyt1 conditional KO (pKO) mice [46].
3.9. Alteration of the gait of Dyt1 d1KO male mice in paw-print analysis
The gait was further analyzed by the paw-print test. When both male and female mice were analyzed together, Dyt1 d1KO mice did not show significant alteration in all parameters of the paw-print test (Fig. 5C, D; Supplementary table 3). The data were stratified by sex and analyzed further. Dyt1 d1KO male mice showed significantly wider fore-base [t (16) = 2.437; p = 0.027; Fig. 5C] and a trend of wider hind-base [t (16) = 1.861; p = 0.081; Fig. 5D]. On the other hand, Dyt1 d1KO female mice did not show significant alteration in all parameters. It should be noted that both Dyt1 cKO and Dyt1 KD male mice show a significantly narrower hind base [28]. Contrastingly, Dyt1 d1KO male mice showed the opposite direction of abnormal gait in the present paw-print analysis.
3.10. Defected striatal D1R maturation process in Dyt1 d1KO mice
The D1R and D2R levels in the striatal tissue protein extract were compared between CT (n = 7) and Dyt1 d1KO (n = 3) mice by Western blot analysis. Multiple bands corresponding to the mature (80 kDa) and two premature (65 kDa and 48 kDa) forms of D1R were detected (Fig.6A). There was a trend of reduction of the D1R mature form in Dyt1 d1KO mice (80kDa; mean ± SE; CT, 100 ± 4%; d1KO, 88 ± 5%; t (19) = −1.90; p = 0.074; Fig.6B). On the other hand, there were significant increases of the D1R premature forms (65 kDa: CT, 100 ± 13%; d1KO, 190 ± 36%; t (19) = 3.49, p = 0.0028, Fig.6C; 48 kDa: CT, 100 ± 21%; d1KO, 297 ± 120%; t (19) = 4.10, p = 0.000041; Fig.6D) in Dyt1 d1KO mice. The results suggest that the loss of torsinA in the D1R-expressing cells affects striatal D1R maturation.
Fig.6.
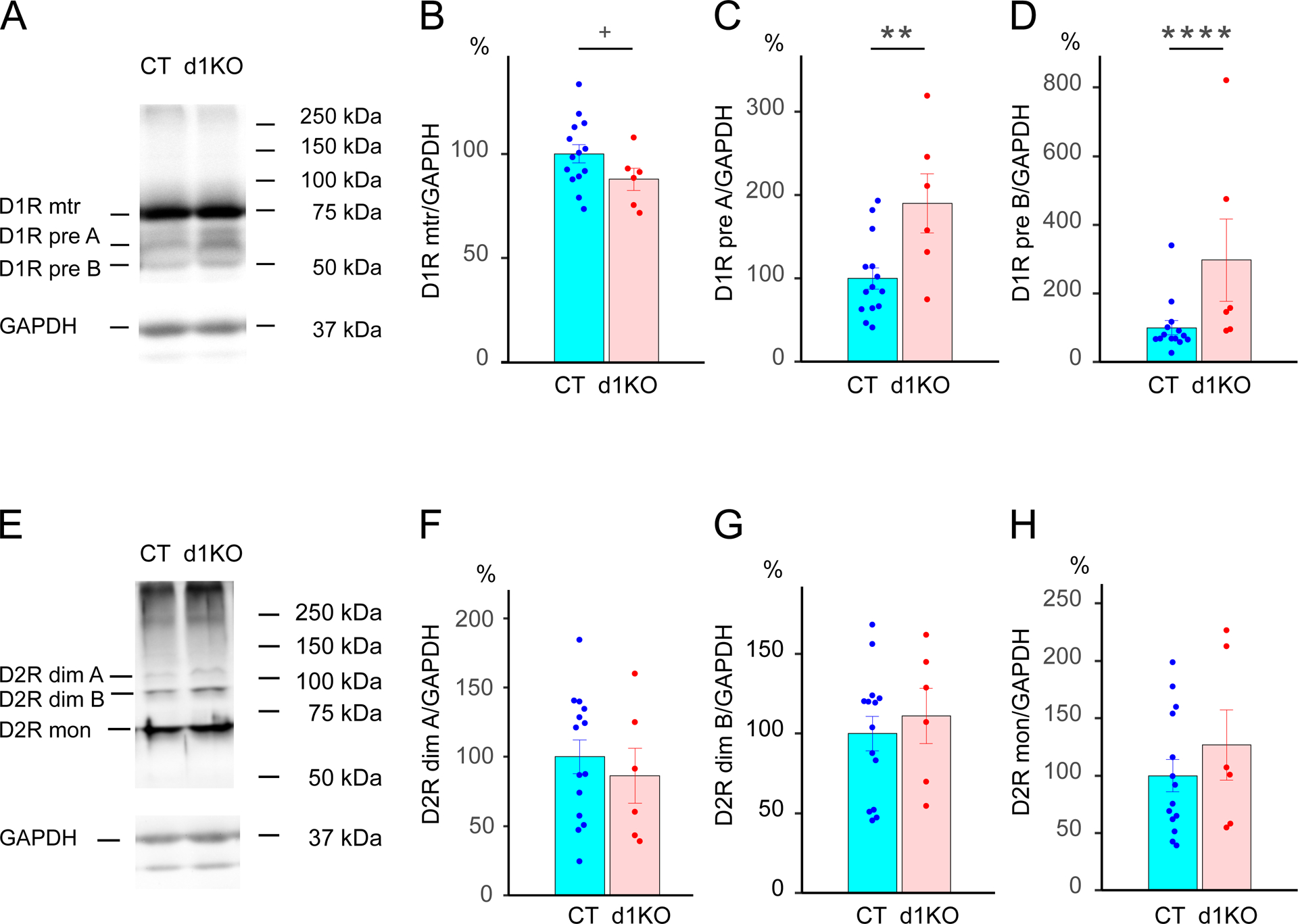
Western blot analysis of the striatal D1R and D2R in CT and Dyt1 d1KO mice. (A) Representative images of D1R and GAPDH bands. (B) The trend of reduction of the quantified mature form of D1R in Dyt1 d1KO mice. Significant increase of premature form A (C) and B (D) of D1R in Dyt1 d1KO mice. (E) Representative images of D2R and GAPDH bands. There was no significant alteration in dimers band A (F), dimers band B (G), and monomer form of D2R (H). +p < 0.1. **p < 0.01. ****p < 0.0001.
Multiple bands corresponding to D2R dimers migrated at 109 kDa and 96 kDa, and monomers migrated at 70kDa [47, 48] were detected (Fig.6E). There was no significant alteration in D2R dimers band A (109 kDa: CT, 100 ± 12%; d1KO, 86 ± 20%; t (19) = −0.656, p = 0.52; Fig.6F), D2R dimers band B (96 kDa: CT, 100 ± 11%; d1KO, 111 ± 17%; t (19) = 0.546, p = 0.59; Fig.6G), or D2R monomers (70 kDa: CT, 100 ± 14%; d1KO, 127 ± 31%; t (19) = 1.208, p = 0.23; Fig.6H). The results suggest that the loss of torsinA in the D1R-expressing cells does not affect striatal D2R maturation.
3.11. Decreased sEPSC frequency of the striatal D1R-expressing MSNs in Dyt1 d1KO Ai6 mice
The D1R-expressing cells were genetically labeled with ZsGreen. Electrophysiological properties of the striatal D1R-expressing MSNs were characterized by whole-cell patch clump in acute brain slices from Dyt1 d1KO Ai6 (11 cells/2 mice) and control Drd1-Cre Ai6 mice (15 cells/3 mice). The representative sEPSC traces of the striatal D1R-expressing MSNs were obtained as shown in Fig.7A. There was a significant reduction of sEPSC frequencies in Dyt1 d1KO Ai6 mice (Hz; CT, 5.7 ± 0.3; d1KO, 4.3 ± 0.4; Z = −2.16, p = 0.031; Fig.7B, C). The representative averaged peak traces were obtained as shown in Fig. 7D. There was no significant alteration in amplitude (pA; CT, 6.8 ± 0.5; d1KO, 6.4 ± 1.0; Z = −0.32, p = 0.75; Fig.7E, F), rise time (ms; CT, 2.2 ± 0.1; d1KO, 2.4 ± 0.1; Z = 1.29, p = 0.20; Fig.7G, H), or decay time (ms; CT, 4.1 ± 0.3; d1KO, 3.8 ± 0.2; Z = −0.88, p = 0.38; Fig.7I, J) of the sEPSCs between Dyt1 d1KO Ai6 and Drd1-Cre+/− Ai6 mice. The results suggest that the loss of torsinA in the D1R-expressing cells decreases presynaptic transmitter release.
Fig.7.
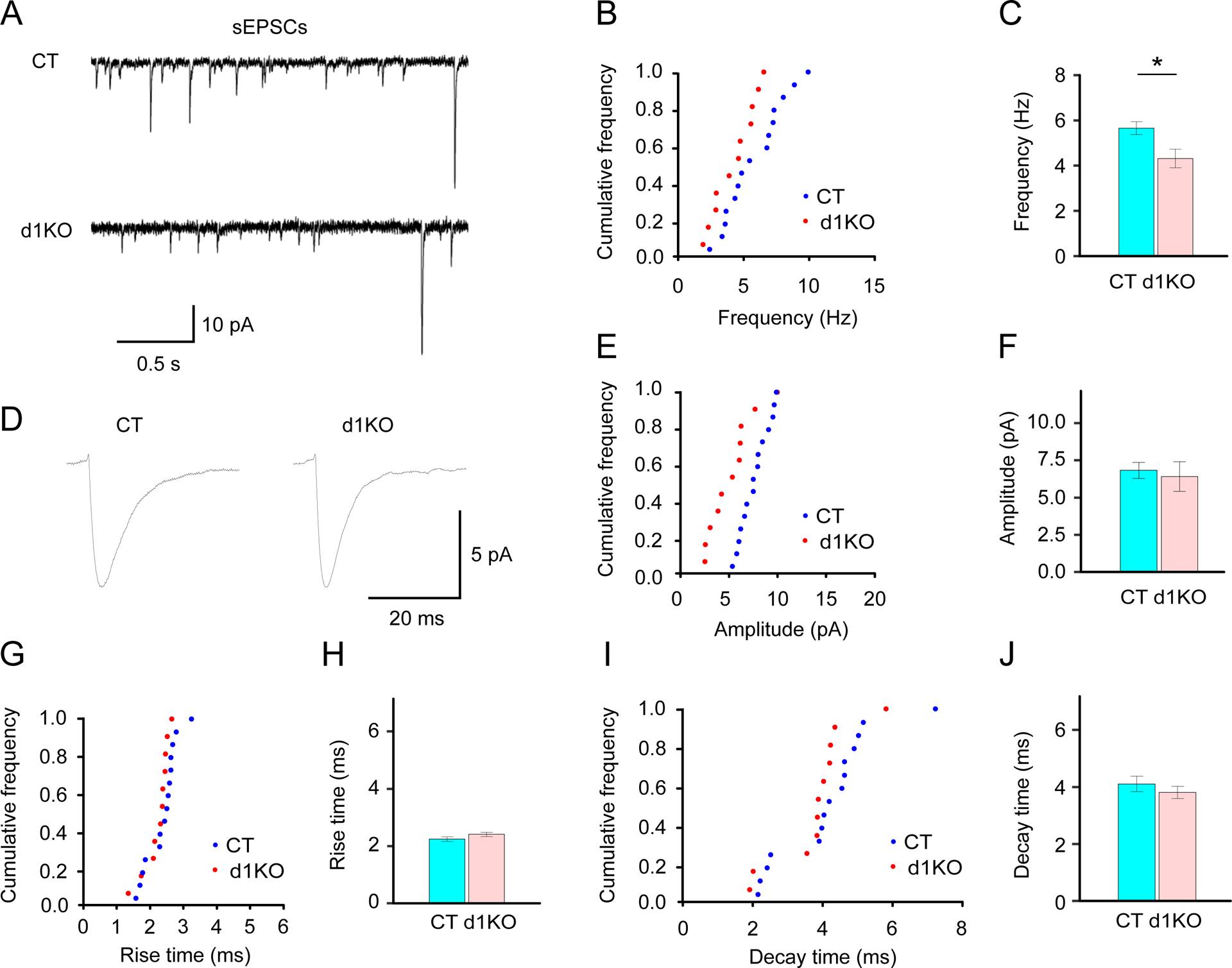
Characterization of sEPSC of the striatal D1R-expressing MSNs in Dyt1 d1KO Ai6 mice. Representative individual (A) and averaged peak (D) traces of sEPSCs in Drd1-cre+/− Ai6 (CT) and Dyt1 d1KO Ai6 (d1KO) mice. Cumulative frequency of the frequency (B), amplitude (E), rise time (G), and decay time (I) of sEPSCs. (C) There was a significant reduction of the sEPSC frequency in the Dyt1 d1KO Ai6 mice. There was no significant difference in the amplitude (F), rise time (H), and decay time (J) of the sEPSCs. The vertical bars represent means ± SE. *p < 0.05.
3.12. Increased membrane capacitance of the striatal D1R-expressing MSNs in Dyt1 d1KO Ai6 mice
Electrophysiological membrane properties were compared (Table 1). The striatal D1R-expressing MSNs in Dyt1 d1KO Ai6 mice showed significantly increased membrane capacitance (p = 0.012) and a trend of increase in the resting membrane potential (p = 0.080). On the other hand, there was no significant alteration in membrane resistance (p = 0.75) or time constant (p = 0.27) between Dyt1 d1KO Ai6 and Drd1-Cre Ai6 mice. Since membrane capacitance is proportional to the membrane surface area [49], the result suggests that D1R-expressing MSNs are bigger than those in control mice.
Table 1.
Electrophysiological membrane properties of the striatal direct pathway MSNs in Dyt1 d1KO mice
| Properties | CT (17 cells/3 mice) | d1KO (17 cells/2 mice) | Z | p |
|---|---|---|---|---|
| Cm (pF) | 40.01 ± 5.62 | 69.69 ± 7.74 | 2.50 | 0.012 * |
| Rm (MΩ) | 125.74 ± 14.70 | 118.77 ± 9.96 | −0.32 | 0.75 |
| Tau (ms) | 1.19 ± 0.05 | 1.11 ± 0.03 | −1.10 | 0.27 |
| MP (mV) | −91.19 ± 0.27 | −89.92 ± 0.58 | −1.75 | 0.080+ |
Cm: Capacitance; Rm: membrane resistance; Tau: Time constant; MP: resting membrane potential; SAS GENMOD procedure was used to calculate p-values with log link and gamma distribution. Nested data in each mouse were used for the analysis. Means ± standard errors are shown.
p<0.1,
p<0.05.
3.13. Decreased frequency of the evoked action potentials of the striatal D1R-expressing MSNs in Dyt1 d1KO Ai6 mice
Action potentials were evoked by current steps to characterize the intrinsic property of the striatal D1R-expressing MSNs in Dyt1 d1KO Ai6 mice (d1KO) and Drd1-cre+/− Ai6 (CT) mice (Fig.8). The average action potential numbers evoked by all sweeps were significantly reduced in d1KO mice comparing to those in CT mice (CT, 8.2 ± 1.8; d1KO, 4.0 ± 0.9; Z = −2.22, p = 0.027). The results suggest that the loss of torsinA in the D1R-expressing cells decreases their intrinsic excitability.
Fig.8.
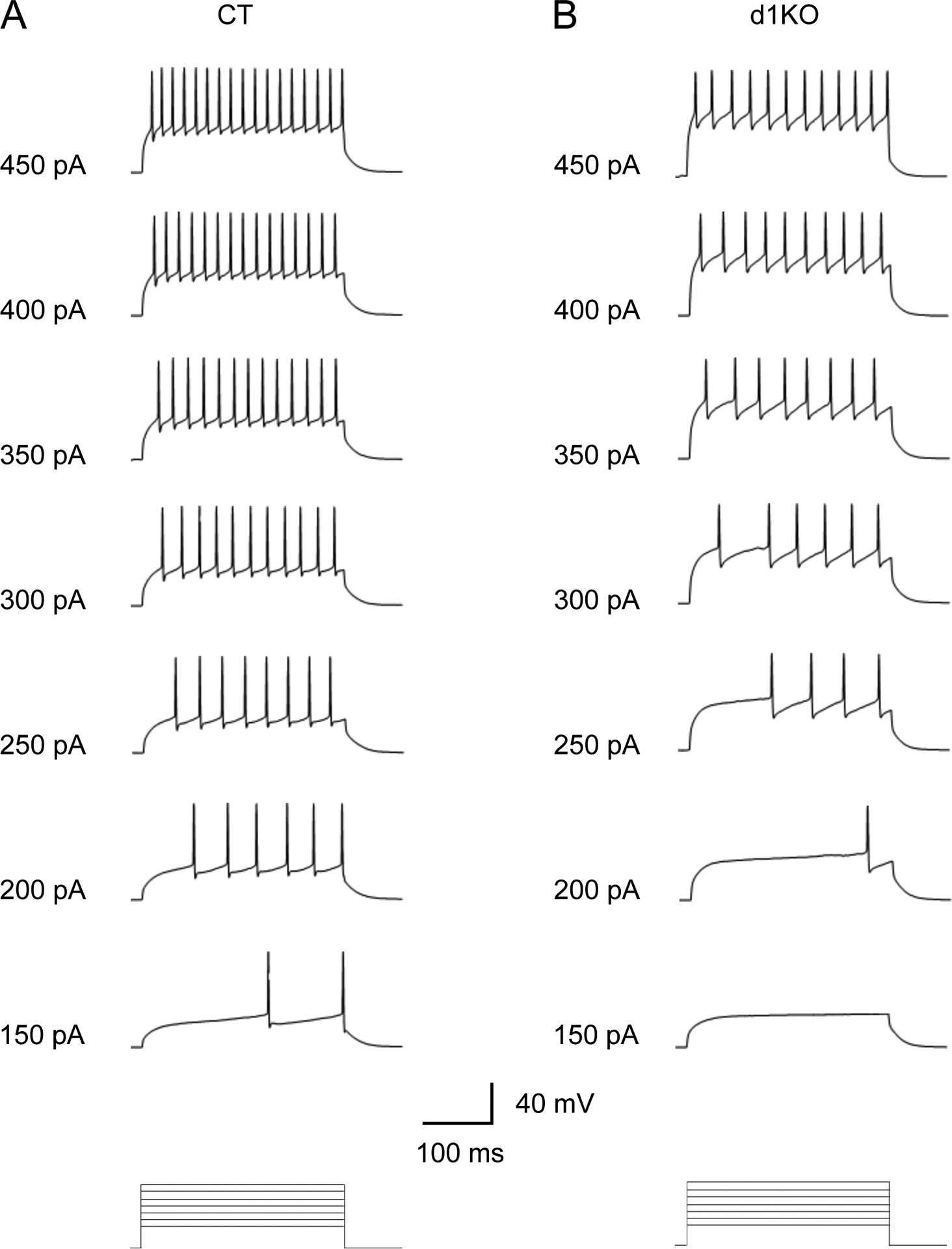
Representative traces of the evoked firing of the striatal D1R-expressing MSNs in Drd1-cre+/− Ai6 (CT) and Dyt1 d1KO Ai6 (d1KO) mice. The evoked action potential numbers were significantly reduced in Dyt1 d1KO Ai6 mice.
3.14. Correlations between the resting membrane potential and sEPSC frequency or evoked action potential of the striatal D1R-expressing MSNs
Correlation between the resting membrane potential and sEPSC frequency in the same MSNs was calculated in both genotypes together; there was no significant correlation between them [t(24) = −0.236, p = 0.82]. The data were then stratified by genotype and analyzed separately. There was a significant positive correlation between the resting membrane potential and sEPSC frequency in Dyt1 d1KO Ai6 mice [t(9) = 2.66, p = 0.026], whereas there was no significant correlation in CT mice [t(13) = −0.764, p = 0.46]. Similarly, the correlation between the resting membrane potential and the evoked action potentials in the same MSNs was calculated, and there was no significant correlation [t(30) = 1.64, p = 0.11]. When they were analyzed separately in each genotype, there was a significant positive correlation in Dyt1 d1KO Ai6 mice [t(13) = 2.95, p = 0.011], whereas there was no significant correlation in CT mice [t(15) = 0.511, p = 0.62]. These correlations suggest that the activity of the striatal D1R-expressing MSNs in Dyt1 d1KO Ai6 mice profoundly depends on the resting membrane potential, whereas that in CT mice is controlled by more complex mechanism.
4. Discussion
Here, behavioral and electrophysiological properties of the direct pathway were characterized in Dyt1 KI mice. Moreover, a new line of D1R-expressing cell-specific conditional knockout mice of torsinA was generated and characterized. Dopamine receptor antagonists induced reductions of spontaneous locomotion in both Dyt1 KI mice and CT mice. Consistent with the reduced striatal D1R and D2R levels [16, 17], Dyt1 KI mice were significantly less responsive to D1R and D2R antagonists. Dyt1 KI mice showed significantly reduced rise and decay times of mEPSCs and increased PPR of D1R-expressing MSNs. Dyt1 d1KO mice were generated and showed decreased locomotor activity in the open-field test and reduced slips in the beam-walking test. Dyt1 d1KO male mice showed abnormal gait. Biochemically, Dyt1 d1KO mice showed defective striatal D1R maturation. D1R-expressing MSNs without torsinA showed increased capacitance, decreased sEPSC frequency and intrinsic excitability. The results suggest that torsinA in the direct pathway plays an important role in electrophysiological and motor function.
A decrease in striatal D1R generally should lead to hypoactivity, while a reduction in striatal D2R can impair the indirect pathway and leads to hyperactivity. On the other hand, a recent hypothesis suggests that all MSNs might either facilitate or inhibit movement depending on the form of synaptic plasticity expressed at a specific moment [50]. Consistently, both D1R antagonist SCH 23390 [51] and D2R antagonist raclopride [52] reduce locomotor activity in rodents. Since Dyt1 KI mice have reduced binding to both striatal D1R and D2R [16, 17], SCH 23390 and raclopride were used to examine their pharmacological profile further. As expected, both D1R and D2R antagonists reduced spontaneous locomotor activity in the CT mice. However, Dyt1 KI mice were significantly less responsive to SCH 23390 and raclopride, consistent with their reduced striatal D1R and D2R levels. The response reductions suggest that the heterozygous ΔGAG mutation affects both direct and indirect pathways in vivo.
Although dystonia is different from an increase in locomotion as measured by the open-field apparatus, both are thought to involve the dopaminergic system. Acute treatment of D1R or D2R antagonists leads to an inhibition of movement. Homozygous D1R KO mice show hyperactivity [53], whereas homozygous long-form D2R KO mice show hypoactivity in the open field [54]. On the other hand, the heterozygous knockout of both D1R and D2R does not impact open field activity [55]. Dyt1 KI mice have an increased locomotor activity [19], which may be originated from complex changes elsewhere [17, 56, 57].
Dyt1 KI mice have reduced rise and decay time and increased PPR. The reduced rise and decay time may affect the neurotransmission in the direct pathway. The shorter decay time of mEPSCs kinetics was also reported in the cultured hippocampal neurons from another line of heterozygous ΔE-torsinA mouse, whereas they show normal rise time [58]. The difference in the rise time between the previous study and the current data may depend on differences in recording conditions, types of neurons, genetic background, or their combinations. On the other hand, the shorter decay time in the different neurons from different lines suggests that the heterozygous Dyt1 ΔGAG mutation potentially affects glutamatergic neurotransmission. Moreover, the increased PPR suggests that a deficit of presynaptic glutamate release. It should be noted that Dyt1 KI mice also show increased PPR in hippocampal slices [59] while d1KO mice had decreased frequency of sEPSCs. These results suggest that both ΔGAG mutation and lack of torsinA induce presynaptic release deficits in various neurons in different brain regions. Although Dyt1 KI mice show LTD deficits, they do not show long-term potentiation (LTP) deficits in the corticostriatal pathway [17]. Dyt1 KI mice also show normal LTP in the hippocampus [60], suggesting that this mutation also induces the same long-term plasticity in these distinct brain regions as well. TorsinA is known to interact with snapin, which is implicated in presynaptic neurotransmitter release [61]. Defective snapin function may underly the synaptic deficits in both brain regions.
TorsinA functions as a molecular chaperon. The loss of torsinA in D1R-expressing MSNs may affect the D1R maturation. The striatal D1R-expressing MSNs in Dyt1 d1KO mice showed a trend of reduced D1R mature form and significantly increased D1R immature forms in the striatal tissues. D1R is a stimulatory G-protein coupled metabotropic receptor. A partial loss of D1R function is supposed to reduce the activity of D1R-expressing MSNs. Consistently, Dyt1 d1KO Ai6 mice showed a decrease of evoked action potentials, suggesting decreased intrinsic excitability. Furthermore, the D1R-expressing MSNs in Dyt1 d1KO Ai6 mice showed a significant increase in membrane capacitance. After both Dyt1 alleles are knocked out in the D1R-expressing MSNs in Dyt1 d1KO Ai6 mice, these neurons do not produce torsinA. On the other hand, these electrophysiological properties were not observed in Dyt1 KI mice, which express a reduced amount of torsinA in the striatum [22]. Therefore, the results suggest that the residual torsinA function in Dyt1 KI mice may act to avoid these electrophysiological alterations observed in Dyt1 d1KO Ai6 mice.
Various genetic dystonia animal models have been developed to investigate the mechanism of this disorder [62–65]. Consistent with Dyt1 KI mice, cell type-specific Dyt1 conditional KO mice do not show overt dystonic symptoms. For example, Dyt1 cKO and Dyt1 sKO mice show motor deficits similarly to Dyt1 KI mice, suggesting that loss of torsinA function in the corticostriatal pathway contributes to exhibit the symptoms [28, 29]. Moreover, cholinergic neuron-specific Dyt1 conditional KO mice show motor deficits in accelerated rotarod test [66] and beam walking [67]. On the other hand, Dyt1 pKO mice show morphological alteration of Purkinje cells [68] and improvement of motor performance in the beam-walking test [46], suggesting loss of torsinA function has different effects on neuronal circuits and motor behavior. Motor deficits but not overt dystonic symptoms, motor learning deficits, and alteration of striatal monoamine metabolism have been reported in other genetic dystonia mouse models. These models contain a growing number of other targeted and transgenic rodent models of DYT1 dystonia [69–71], DYT11 myoclonus-dystonia [40, 42, 72–74], and DYT12 dystonia-parkinsonism [41, 75]. These studies and the present study suggest that a loss of torsinA function in different cells affects different parts of neuronal circuits and induces different motor behavioral phenotypes. Dyt1 d1KO mice showed hypoactivity in the open field test and decreased slips in the beam-walking test. Since these phenotypes were in the opposite direction of those in Dyt1 KI male mice [19], intervening in the direct pathway may attenuate motor symptoms in DYT1 dystonia.
Supplementary Material
Highlights.
Dyt1 KI mice were less responsive to both D1R and D2R antagonists in open field test.
Dyt1 KI mice showed an increased paired-pulse ratio of the D1R medium spiny neurons.
Dyt1 d1KO mice had significantly decreased spontaneous locomotor activity.
Dyt1 d1KO mice had fewer slip numbers in beam-walking and abnormal gait.
Dyt1 d1KO mice showed decreased excitability and increased capacitance of D1R MSNs.
Acknowledgments
We thank the animal colony staff at the University of Florida and the University of Illinois for animal care, Kelly Kwon, Jason R. Gandre, Alyka Glor P. Fernandez, Robert Yuen, Douglas E. Smith, Pallavi Girdhar, and other undergraduate students for their technical assistance.
Funding
This work was supported by Tyler’s Hope for a Dystonia Cure, Inc., “Mini-Moonshot” Fixel-MBI Pilot Grant Mechanism for Dystonia and Related Disorders, National Institutes of Health (NS047692, NS054246, NS065273, NS072872, NS074423, NS075012, NS082244, NS 111498, and NS118397), startup funds from the Lucille P. Markey Charitable Trust and Beckman Institute (UIUC), Department of Neurology (UF), Dystonia Medical Research Foundation, and Bachmann-Strauss Dystonia and Parkinson Foundation, Inc., and the Office of the Assistant Secretary of Defense for Health Affairs through the Peer-Reviewed Medical Research Program (W81XWH1810099 and W81XWH2110198). Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the Department of Defense. Nikon A1RMPsi-STORM4.0 multiphoton/super-resolution imaging system was acquired by the MBI, Office of Research, College of Medicine, and an NIH Shared Instrumentation Grant (1S10OD020026).
Abbreviation
- aCSF
artificial cerebrospinal fluid
- AIC
Akaike Information Criterion
- CT
control
- DA
dopamine
- DF
degrees of freedom
- DHet
double heterozygote
- Dyt1 d1KO mice
D1R-expressing cell-specific Dyt1 conditional knockout mice
- Dyt1 d2KO mice
D2R-expressing cell-specific Dyt1 conditional knockout mice
- D1R
dopamine receptor 1
- D2R
dopamine receptor 2
- Dyt1 KI mice
Dyt1 ΔGAG heterozygous knock-in mice
- Dyt1 pKO mice
cerebellar Purkinje-cell specific Dyt1 conditional KO mice
- EGFP
enhanced green fluorescent protein
- KD
knockdown
- KO
knockout
- LTD
long-term depression
- LTP
long-term potentiation
- mEPSC
miniature excitatory postsynaptic current
- MSNs
medium spiny neurons
- PPR
paired-pulse ratio
- SE
standard errors
- sEPSC
spontaneous excitatory postsynaptic current
- TTX
tetrodotoxin
Footnotes
Conflict of interest
The authors declare no competing financial interests.
References
- [1].Albanese A, Bhatia K, Bressman SB, Delong MR, Fahn S, Fung VS, Hallett M, Jankovic J, Jinnah HA, Klein C, Lang AE, Mink JW, Teller JK, Phenomenology and classification of dystonia: a consensus update, Mov Disord 28(7) (2013) 863–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, de Leon D, Brin MF, Raymond D, Corey DP, Fahn S, Risch NJ, Buckler AJ, Gusella JF, Breakefield XO, The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein, Nat Genet 17(1) (1997) 40–8. [DOI] [PubMed] [Google Scholar]
- [3].Leung JC, Klein C, Friedman J, Vieregge P, Jacobs H, Doheny D, Kamm C, DeLeon D, Pramstaller PP, Penney JB, Eisengart M, Jankovic J, Gasser T, Bressman SB, Corey DP, Kramer P, Brin MF, Ozelius LJ, Breakefield XO, Novel mutation in the TOR1A (DYT1) gene in atypical early onset dystonia and polymorphisms in dystonia and early onset parkinsonism, Neurogenetics 3(3) (2001) 133–43. [DOI] [PubMed] [Google Scholar]
- [4].Doheny D, Danisi F, Smith C, Morrison C, Velickovic M, De Leon D, Bressman SB, Leung J, Ozelius L, Klein C, Breakefield XO, Brin MF, Silverman JM, Clinical findings of a myoclonus-dystonia family with two distinct mutations, Neurology 59(8) (2002) 1244–6. [DOI] [PubMed] [Google Scholar]
- [5].Ritz K, Gerrits MC, Foncke EM, van Ruissen F, van der Linden C, Vergouwen MD, Bloem BR, Vandenberghe W, Crols R, Speelman JD, Baas F, Tijssen MA, Myoclonus-dystonia: clinical and genetic evaluation of a large cohort, J Neurol Neurosurg Psychiatry 80(6) (2009) 653–658. [DOI] [PubMed] [Google Scholar]
- [6].Calakos N, Patel VD, Gottron M, Wang G, Tran-Viet KN, Brewington D, Beyer JL, Steffens DC, Krishnan RR, Zuchner S, Functional evidence implicating a novel TOR1A mutation in idiopathic, late-onset focal dystonia, J Med Genet 47(9) (2010) 646–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Isik E, Aykut A, Atik T, Cogulu O, Ozkinay F, Biallelic TOR1A mutations cause severe arthrogryposis: A case requiring reverse phenotyping, Eur J Med Genet 62(9) (2019) 103544. [DOI] [PubMed] [Google Scholar]
- [8].Goodchild RE, Kim CE, Dauer WT, Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope, Neuron 48(6) (2005) 923–32. [DOI] [PubMed] [Google Scholar]
- [9].Breakefield XO, Blood AJ, Li Y, Hallett M, Hanson PI, Standaert DG, The pathophysiological basis of dystonias, Nat Rev Neurosci 9(3) (2008) 222–34. [DOI] [PubMed] [Google Scholar]
- [10].Balint B, Mencacci NE, Valente EM, Pisani A, Rothwell J, Jankovic J, Vidailhet M, Bhatia KP, Dystonia, Nat Rev Dis Primers 4(1) (2018) 25. [DOI] [PubMed] [Google Scholar]
- [11].Eidelberg D, Moeller JR, Antonini A, Kazumata K, Nakamura T, Dhawan V, Spetsieris P, deLeon D, Bressman SB, Fahn S, Functional brain networks in DYT1 dystonia, Ann Neurol 44(3) (1998) 303–12. [DOI] [PubMed] [Google Scholar]
- [12].Fox MD, Alterman RL, Brain Stimulation for Torsion Dystonia, JAMA Neurol 72(6) (2015) 713–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Furukawa Y, Hornykiewicz O, Fahn S, Kish SJ, Striatal dopamine in early-onset primary torsion dystonia with the DYT1 mutation, Neurology 54(5) (2000) 1193–5. [DOI] [PubMed] [Google Scholar]
- [14].Augood SJ, Hollingsworth Z, Albers DS, Yang L, Leung JC, Muller B, Klein C, Breakefield XO, Standaert DG, Dopamine transmission in DYT1 dystonia: a biochemical and autoradiographical study, Neurology 59(3) (2002) 445–8. [DOI] [PubMed] [Google Scholar]
- [15].Asanuma K, Ma Y, Okulski J, Dhawan V, Chaly T, Carbon M, Bressman SB, Eidelberg D, Decreased striatal D2 receptor binding in non-manifesting carriers of the DYT1 dystonia mutation, Neurology 64(2) (2005) 347–9. [DOI] [PubMed] [Google Scholar]
- [16].Yokoi F, Dang MT, Liu J, Gandre JR, Kwon K, Yuen R, Li Y, Decreased dopamine receptor 1 activity and impaired motor-skill transfer in Dyt1 DeltaGAG heterozygous knock-in mice, Behav Brain Res 279 (2015) 202–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dang MT, Yokoi F, Cheetham CC, Lu J, Vo V, Lovinger DM, Li Y, An anticholinergic reverses motor control and corticostriatal LTD deficits in Dyt1 DeltaGAG knock-in mice, Behav Brain Res 226(2) (2012) 465–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].DeAndrade MP, Trongnetrpunya A, Yokoi F, Cheetham CC, Peng N, Wyss JM, Ding M, Li Y, Electromyographic evidence in support of a knock-in mouse model of DYT1 Dystonia, Mov Disord 31(11) (2016) 1633–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dang MT, Yokoi F, McNaught KS, Jengelley TA, Jackson T, Li J, Li Y, Generation and characterization of Dyt1 DeltaGAG knock-in mouse as a model for early-onset dystonia, Exp Neurol 196(2) (2005) 452–63. [DOI] [PubMed] [Google Scholar]
- [20].Song CH, Fan X, Exeter CJ, Hess EJ, Jinnah HA, Functional analysis of dopaminergic systems in a DYT1 knock-in mouse model of dystonia, Neurobiol Dis 48(1) (2012) 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liu Y, Xing H, Yokoi F, Vaillancourt DE, Li Y, Investigating the role of striatal dopamine receptor 2 in motor coordination and balance: insights into the pathogenesis of DYT1 dystonia, Behav Brain Res (2021) 113137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yokoi F, Yang G, Li J, DeAndrade MP, Zhou T, Li Y, Earlier onset of motor deficits in mice with double mutations in Dyt1 and Sgce, J Biochem 148(4) (2010) 459–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cao S, Hewett JW, Yokoi F, Lu J, Buckley AC, Burdette AJ, Chen P, Nery FC, Li Y, Breakefield XO, Caldwell GA, Caldwell KA, Chemical enhancement of torsinA function in cell and animal models of torsion dystonia, Dis Model Mech 3(5–6) (2010) 386–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Giles LM, Chen J, Li L, Chin LS, Dystonia-associated mutations cause premature degradation of torsinA protein and cell-type-specific mislocalization to the nuclear envelope, Hum Mol Genet 17(17) (2008) 2712–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gordon KL, Gonzalez-Alegre P, Consequences of the DYT1 mutation on torsinA oligomerization and degradation, Neuroscience 157(3) (2008) 588–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dang MT, Yokoi F, Pence MA, Li Y, Motor deficits and hyperactivity in Dyt1 knockdown mice, Neurosci Res 56(4) (2006) 470–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yokoi F, Chen HX, Dang MT, Cheetham CC, Campbell SL, Roper SN, Sweatt JD, Li Y, Behavioral and electrophysiological characterization of Dyt1 heterozygous knockout mice, PLoS One 10(3) (2015) e0120916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yokoi F, Dang MT, Mitsui S, Li J, Li Y, Motor deficits and hyperactivity in cerebral cortex-specific Dyt1 conditional knockout mice, J Biochem 143(1) (2008) 39–47. [DOI] [PubMed] [Google Scholar]
- [29].Yokoi F, Dang MT, Li J, Standaert DG, Li Y, Motor deficits and decreased striatal dopamine receptor 2 binding activity in the striatum-specific Dyt1 conditional knockout mice, PLoS One 6(9) (2011) e24539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Dang MT, Yokoi F, Yin HH, Lovinger DM, Wang Y, Li Y, Disrupted motor learning and long-term synaptic plasticity in mice lacking NMDAR1 in the striatum, Proc Natl Acad Sci U S A 103(41) (2006) 15254–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lyu S, Xing H, DeAndrade MP, Liu Y, Perez PD, Yokoi F, Febo M, Walters AS, Li Y, The Role of BTBD9 in Striatum and Restless Legs Syndrome, eNeuro 6(5) (2019) ENEURO.0277–19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yokoi F, Oleas J, Xing H, Liu Y, Dexter KM, Misztal C, Gerard M, Efimenko I, Lynch P, Villanueva M, Alsina R, Krishnaswamy S, Vaillancourt DE, Li Y, Decreased number of striatal cholinergic interneurons and motor deficits in dopamine receptor 2-expressing-cell-specific Dyt1 conditional knockout mice, Neurobiol Dis 134 (2020) 104638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhang L, McCarthy DM, Sharma N, Bhide PG, Dopamine receptor and Galpha(olf) expression in DYT1 dystonia mouse models during postnatal development, PLoS One 10(4) (2015) e0123104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, Heintz N, A gene expression atlas of the central nervous system based on bacterial artificial chromosomes, Nature 425(6961) (2003) 917–25. [DOI] [PubMed] [Google Scholar]
- [35].Gong S, Doughty M, Harbaugh CR, Cummins A, Hatten ME, Heintz N, Gerfen CR, Targeting Cre recombinase to specific neuron populations with bacterial artificial chromosome constructs, J Neurosci 27(37) (2007) 9817–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H, A robust and high-throughput Cre reporting and characterization system for the whole mouse brain, Nat Neurosci 13(1) (2010) 133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Voelkl B, Altman NS, Forsman A, Forstmeier W, Gurevitch J, Jaric I, Karp NA, Kas MJ, Schielzeth H, Van de Casteele T, Würbel H, Reproducibility of animal research in light of biological variation, Nat Rev Neurosci 21(7) (2020) 384–393. [DOI] [PubMed] [Google Scholar]
- [38].Fernagut PO, Diguet E, Stefanova N, Biran M, Wenning GK, Canioni P, Bioulac B, Tison F, Subacute systemic 3-nitropropionic acid intoxication induces a distinct motor disorder in adult C57Bl/6 mice: behavioural and histopathological characterisation, Neuroscience 114(4) (2002) 1005–17. [DOI] [PubMed] [Google Scholar]
- [39].Cao BJ, Li Y, Reduced anxiety- and depression-like behaviors in Emx1 homozygous mutant mice, Brain Res 937(1–2) (2002) 32–40. [DOI] [PubMed] [Google Scholar]
- [40].Yokoi F, Dang MT, Li J, Li Y, Myoclonus, motor deficits, alterations in emotional responses and monoamine metabolism in epsilon-sarcoglycan deficient mice, J Biochem 140(1) (2006) 141–6. [DOI] [PubMed] [Google Scholar]
- [41].DeAndrade MP, Yokoi F, van Groen T, Lingrel JB, Li Y, Characterization of Atp1a3 mutant mice as a model of rapid-onset dystonia with parkinsonism, Behav Brain Res 216(2) (2011) 659–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yokoi F, Dang MT, Zhou T, Li Y, Abnormal nuclear envelopes in the striatum and motor deficits in DYT11 myoclonus-dystonia mouse models, Hum Mol Genet 21(4) (2012) 916–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Carter RJ, Morton AJ, Dunnett SB, Motor Coordination and Balance in Rodents, in: Crawley J (Ed.), Current Protocols in Neuroscience, John Wiley & Sons, Inc. 2001, pp. 8.12.1–8.12.14. [DOI] [PubMed] [Google Scholar]
- [44].Akaike H, A new look at the statistical model identification, IEEE Transactions on Automatic Control 19(6) (1974) 716–723. [Google Scholar]
- [45].Debanne D, Guerineau NC, Gahwiler BH, Thompson SM, Paired-pulse facilitation and depression at unitary synapses in rat hippocampus: quantal fluctuation affects subsequent release, J Physiol 491 (Pt 1) (1996) 163–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Yokoi F, Dang MT, Li Y, Improved motor performance in Dyt1 DeltaGAG heterozygous knock-in mice by cerebellar Purkinje-cell specific Dyt1 conditional knocking-out, Behav Brain Res 230(2) (2012) 389–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ng GY, O’Dowd BF, Lee SP, Chung HT, Brann MR, Seeman P, George SR, Dopamine D2 receptor dimers and receptor-blocking peptides, Biochem Biophys Res Commun 227(1) (1996) 200–4. [DOI] [PubMed] [Google Scholar]
- [48].Zawarynski P, Tallerico T, Seeman P, Lee SP, O’Dowd BF, George SR, Dopamine D2 receptor dimers in human and rat brain, FEBS Lett 441(3) (1998) 383–6. [DOI] [PubMed] [Google Scholar]
- [49].Golowasch J, Thomas G, Taylor AL, Patel A, Pineda A, Khalil C, Nadim F, Membrane capacitance measurements revisited: dependence of capacitance value on measurement method in nonisopotential neurons, J Neurophysiol 102(4) (2009) 2161–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Calabresi P, Picconi B, Tozzi A, Ghiglieri V, Di Filippo M, Direct and indirect pathways of basal ganglia: a critical reappraisal, Nat Neurosci 17(8) (2014) 1022–30. [DOI] [PubMed] [Google Scholar]
- [51].Hoffman DC, Beninger RJ, The D1 dopamine receptor antagonist, SCH 23390 reduces locomotor activity and rearing in rats, Pharmacol Biochem Behav 22(2) (1985) 341–2. [DOI] [PubMed] [Google Scholar]
- [52].Simon VM, Parra A, Minarro J, Arenas MC, Vinader-Caerols C, Aguilar MA, Predicting how equipotent doses of chlorpromazine, haloperidol, sulpiride, raclopride and clozapine reduce locomotor activity in mice, Eur Neuropsychopharmacol 10(3) (2000) 159–64. [DOI] [PubMed] [Google Scholar]
- [53].Xu M, Moratalla R, Gold LH, Hiroi N, Koob GF, Graybiel AM, Tonegawa S, Dopamine D1 receptor mutant mice are deficient in striatal expression of dynorphin and in dopamine-mediated behavioral responses, Cell 79(4) (1994) 729–42. [DOI] [PubMed] [Google Scholar]
- [54].Wang Y, Xu R, Sasaoka T, Tonegawa S, Kung MP, Sankoorikal EB, Dopamine D2 long receptor-deficient mice display alterations in striatum-dependent functions, J Neurosci 20(22) (2000) 8305–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kobayashi M, Iaccarino C, Saiardi A, Heidt V, Bozzi Y, Picetti R, Vitale C, Westphal H, Drago J, Borrelli E, Simultaneous absence of dopamine D1 and D2 receptor-mediated signaling is lethal in mice, Proc Natl Acad Sci U S A 101(31) (2004) 11465–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Scarduzio M, Zimmerman CN, Jaunarajs KL, Wang Q, Standaert DG, McMahon LL, Strength of cholinergic tone dictates the polarity of dopamine D2 receptor modulation of striatal cholinergic interneuron excitability in DYT1 dystonia, Exp Neurol 295 (2017) 162–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Downs AM, Fan X, Donsante C, Jinnah HA, Hess EJ, Trihexyphenidyl rescues the deficit in dopamine neurotransmission in a mouse model of DYT1 dystonia, Neurobiol Dis 125 (2019) 115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kakazu Y, Koh JY, Iwabuchi S, Gonzalez-Alegre P, Harata NC, Miniature release events of glutamate from hippocampal neurons are influenced by the dystonia-associated protein torsinA, Synapse 66(9) (2012) 807–22. [DOI] [PubMed] [Google Scholar]
- [59].Yokoi F, Cheetham CC, Campbell SL, Sweatt JD, Li Y, Pre-synaptic release deficits in a DYT1 dystonia mouse model, PLoS One 8(8) (2013) e72491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Yokoi F, Dang MT, Miller CA, Marshall AG, Campbell SL, Sweatt JD, Li Y, Increased c-fos expression in the central nucleus of the amygdala and enhancement of cued fear memory in Dyt1 DeltaGAG knock-in mice, Neurosci Res 65(3) (2009) 228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Granata A, Watson R, Collinson LM, Schiavo G, Warner TT, The dystonia-associated protein torsinA modulates synaptic vesicle recycling, J Biol Chem 283(12) (2008) 7568–79. [DOI] [PubMed] [Google Scholar]
- [62].Oleas J, Yokoi F, DeAndrade MP, Pisani A, Li Y, Engineering animal models of dystonia, Mov Disord 28(7) (2013) 990–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Oleas J, Yokoi F, DeAndrade MP, Li Y, Rodent Models of Autosomal Dominant Primary Dystonia. In: Movement Disorders: Genetics and Models (LeDoux MS, ed), pp483–505, Second ed., Academic Press Elsevier, New York, 2015. [Google Scholar]
- [64].Richter F, Richter A, Genetic animal models of dystonia: common features and diversities, Prog Neurobiol 121 (2014) 91–113. [DOI] [PubMed] [Google Scholar]
- [65].Imbriani P, Ponterio G, Tassone A, Sciamanna G, El Atiallah I, Bonsi P, Pisani A, Models of dystonia: an update, J Neurosci Methods 339 (2020) 108728. [DOI] [PubMed] [Google Scholar]
- [66].Sciamanna G, Hollis R, Ball C, Martella G, Tassone A, Marshall A, Parsons D, Li X, Yokoi F, Zhang L, Li Y, Pisani A, Standaert DG, Cholinergic dysregulation produced by selective inactivation of the dystonia-associated protein torsinA, Neurobiol Dis 47(3) (2012) 416–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Liu Y, Xing H, Sheng W, Singh KN, Korkmaz AG, Comeau C, Anika M, Ernst A, Yokoi F, Vaillancourt DE, Frazier CJ, Li Y, Alteration of the cholinergic system and motor deficits in cholinergic neuron-specific Dyt1 knockout mice, Neurobiol Dis 154 (2021) 105342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Zhang L, Yokoi F, Jin YH, Deandrade MP, Hashimoto K, Standaert DG, Li Y, Altered Dendritic Morphology of Purkinje cells in Dyt1 DeltaGAG Knock-In and Purkinje Cell-Specific Dyt1 Conditional Knockout Mice, PLoS One 6(3) (2011) e18357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Zhao Y, DeCuypere M, LeDoux MS, Abnormal motor function and dopamine neurotransmission in DYT1 DeltaGAG transgenic mice, Exp Neurol 210(2) (2008) 719–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Page ME, Bao L, Andre P, Pelta-Heller J, Sluzas E, Gonzalez-Alegre P, Bogush A, Khan LE, Iacovitti L, Rice ME, Ehrlich ME, Cell-autonomous alteration of dopaminergic transmission by wild type and mutant (DeltaE) TorsinA in transgenic mice, Neurobiol Dis 39(3) (2010) 318–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Alvarez-Fischer D, Grundmann M, Lu L, Samans B, Fritsch B, Moller JC, Schaefer MK, Hartmann A, Oertel WH, Bandmann O, Prolonged generalized dystonia after chronic cerebellar application of kainic acid, Brain Res 1464 (2012) 82–8. [DOI] [PubMed] [Google Scholar]
- [72].Yokoi F, Dang MT, Yang G, Li J, Doroodchi A, Zhou T, Li Y, Abnormal nuclear envelope in the cerebellar Purkinje cells and impaired motor learning in DYT11 myoclonus-dystonia mouse models, Behav Brain Res 227(1) (2012) 12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Zhang L, Yokoi F, Parsons DS, Standaert DG, Li Y, Alteration of Striatal Dopaminergic Neurotransmission in a Mouse Model of DYT11 Myoclonus-Dystonia, PLoS One 7(3) (2012) e33669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Xiao J, Vemula SR, Xue Y, Khan MM, Carlisle FA, Waite AJ, Blake DJ, Dragatsis I, Zhao Y, LeDoux MS, Role of major and brain-specific Sgce isoforms in the pathogenesis of myoclonus-dystonia syndrome, Neurobiol Dis 98 (2017) 52–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Moseley AE, Williams MT, Schaefer TL, Bohanan CS, Neumann JC, Behbehani MM, Vorhees CV, Lingrel JB, Deficiency in Na,K-ATPase alpha isoform genes alters spatial learning, motor activity, and anxiety in mice, J Neurosci 27(3) (2007) 616–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Dorris DM, Cao J, Willett JA, Hauser CA, Meitzen J, Intrinsic excitability varies by sex in prepubertal striatal medium spiny neurons, J Neurophysiol 113(3) (2015) 720–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.