

Abstract
Burkholderia cepacia complex (Bcc) poses a serious health threat to people with cystic fibrosis or compromised immune systems. Infections often arise from Bcc strains, which are highly resistant to many classes of antibiotics, including β-lactams. β-Lactam resistance in Bcc is conferred largely via PenA-like β-lactamases. Avibactam was previously shown to be a potent inactivator of PenA1. Here, we examined the inactivation mechanism of PenA1, a class A serine carbapenemase from Burkholderia multivorans using β-lactamase inhibitors (β-lactam-, diazabicyclooctane-, and boronate-based) with diverse mechanisms of action. In whole cell based assays, avibactam, relebactam, enmetazobactam, and vaborbactam restored susceptibility to piperacillin against PenA1 expressed in Escherichia coli. The rank order of potency of inactivation in vitro based on kinact/KI or k2/K values (range: 3.4 × 102 to 2 × 106 M−1 s−1) against PenA1 was avibactam > enmetazobactam > tazobactam > relebactam > clavulanic acid > vaborbactam. The contribution of selected amino acids (S70, K73, S130, E166, N170, R220, K234, T237, and D276) in PenA1 toward inactivation was evaluated using site-directed mutagenesis. The S130A, R220A, and K234A variants of PenA1 were less susceptible to inactivation by avibactam. The R220A variant was purified and assessed via steady-state inhibition kinetics and found to possess increased Ki-app values and decreased kinact/KI or k2/K values against all tested inhibitors compared to PenA1. Avibactam was the most affected by the alanine replacement at 220 with a nearly 400-fold decreased acylation rate. The X-ray crystal structure of the R220A variant was solved and revealed loss of the hydrogen bonding network between residues 237 and 276 leaving a void in the active site that was occupied instead by water molecules. Michaelis−Menten complexes were generated to elucidate the molecular contributions of the poorer in vitro inhibition profile of vaborbactam against PenA1 (k2/K, 3.4 × 102 M−1 s−1) and was compared to KPC-2, a class A carbapenemase that is robustly inhibited by vaborbactam. The active site of PenA1 is larger than that of KPC-2, which impacted the ability of vaborbactam to form favorable interactions, and as a result the carboxylate of vaborbactam was drawn toward K234/T235 in PenA1 displacing the boronic acid from approaching the nucleophilic S70. Moreover, in PenA1, the tyrosine at position 105 compared to tryptophan in KPC-2, was more flexible rotating more than 90°, and as a result PenA1’s Y105 competed for binding with the cyclic boronate vs the thiophene moiety of vaborbactam, further precluding inhibition of PenA1 by vaborbactam. Given the 400-fold decreased k2/K for the R220A variant compared to PenA1, acyl−enzyme complexes were generated via molecular modeling and compared to the PenA1-avibactam crystal structure. The water molecules occupying the active site of the R220A variant are unable to stabilize the T237 and D276 region of the active site altering the ability of avibactam to form favorable interactions compared to PenA1. The former likely impacts the ability of all inhibitors to effectively acylate this variant enzyme. Based on the summation of all evidence herein, the utility of these newer β-lactamase inhibitors (i.e., relebactam, enmetazobactam, avibactam, and vaborbactam) in combination with a β-lactam against B. multivorans producing PenA1 and the R220A variant is promising.
Keywords: β-lactamases, Burkholderia, β-lactam, PenA, carbapenemase, β-lactamase inhibitor, relebactam, avibactam, vaborbactam, enmetazobactam
Graphical Abstract
Alarmingly, the Centers for Disease Control and Prevention (CDC) reported in 2019 that every 15 min a person dies from an antibiotic resistant infection in the United States.1 Intrinsically resistant to antibiotics (e.g., polymyxins, β-lactams), the Burkholderia cepacia complex (Bcc) is a group of Gram-negative bacteria that are opportunistic pathogens and pose a serious threat to those with cystic fibrosis or compromised immune systems. All Bcc carry an inducible chromosomal class A serine β-lactamase of the PenA-family; the spectra of hydrolytic activity for each PenA family member varies by species as well as within species due to sequence heterogeneity.2,3
One strategy to combat β-lactamase-mediated antibiotic resistance is to combine a β-lactam with a β-lactamase inhibitor. β-Lactamase inhibitors possess the potential to be of great importance for treating infections as they rescue β-lactams from hydrolysis by β-lactamases. There are three major classes of β-lactamase inhibitors that are active against class A serine enzymes: β-lactam-based (e.g., clavulanic acid, tazobactam, enmetazobactam), diazabicyclooctanes (DBOs) (e.g., avibactam and relebactam), and boronates (e.g., vaborbactam) (Figure 1). The inhibitors’ mechanisms of action with β-lactamases vary by type (Figure 2).
Figure 1.
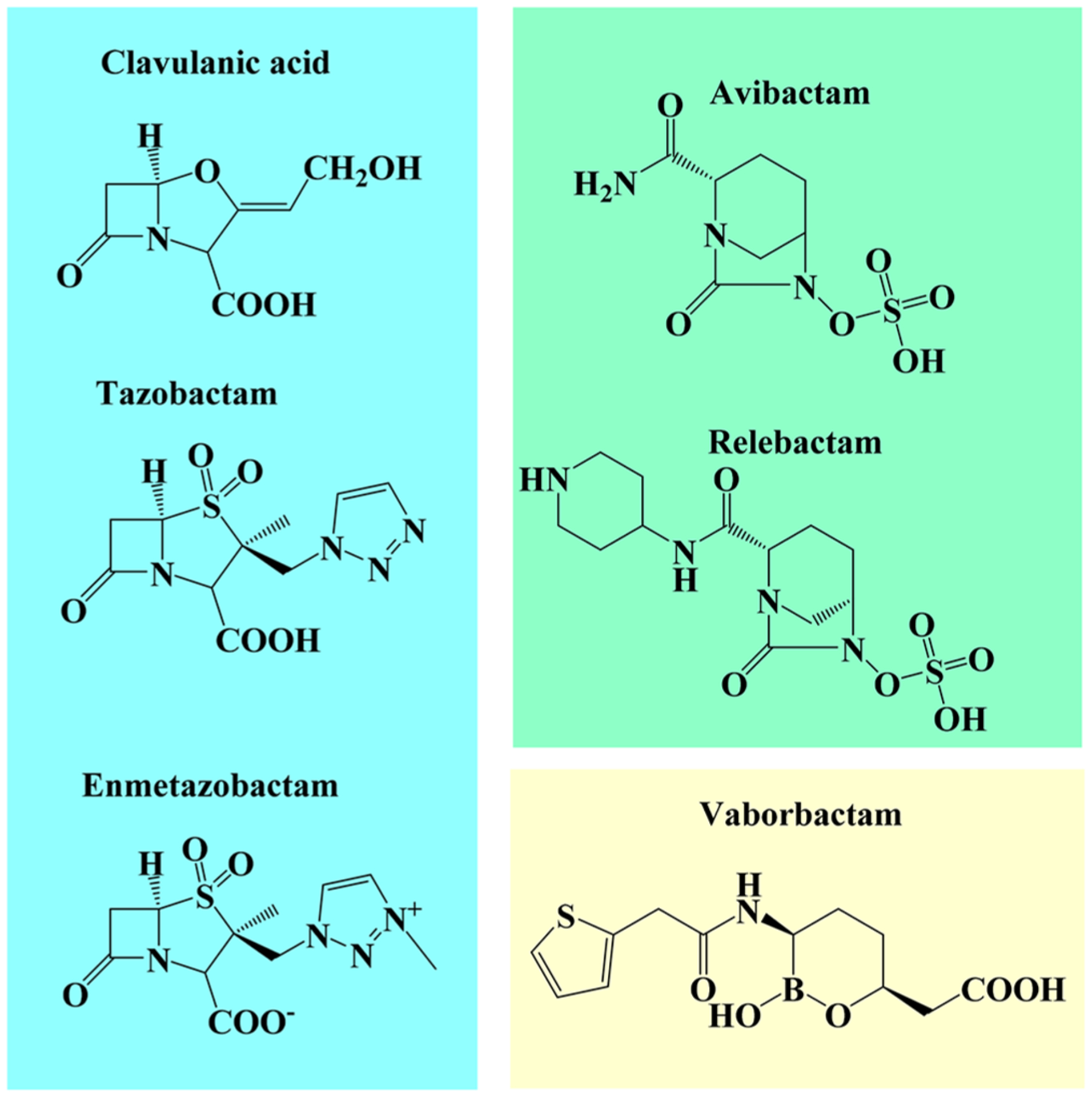
Chemical structures of β-lactamase inhibitors used in this study.
Figure 2.
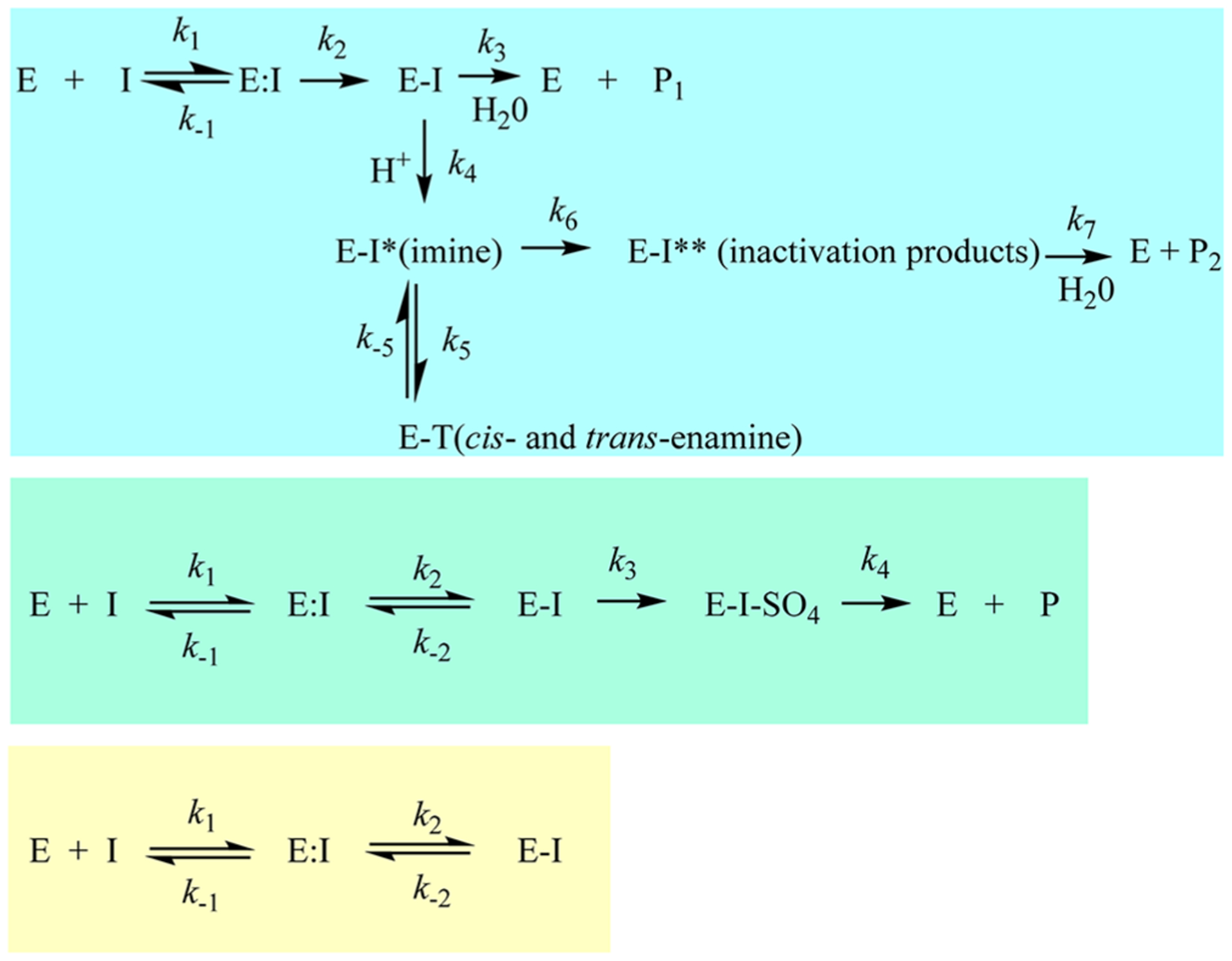
Reaction schemes for the three different β-lactamase inhibitor types (I) with a β-lactamase (E).
β-Lactam-based inhibitors form acyl-complexes with the β-lactamase active site but are rapidly converted to various intermediates (imine, cis- and trans-enamine), can fragment, and are eventually hydrolyzed to inactive products (Figure 2, top).4 The β-lactam-based inhibitors, clavulanic acid, sulbactam, and tazobactam, were the first to reach clinical use. Interest in this class has continued as a novel sulfone, enmetazobactam (formerly AAI101), partnered with cefepime is currently being evaluated in clinical trials. Enmetazobactam is a zwitterion, which results in enhanced bacteria cell penetration as well as potent β-lactamase inactivation rates.5
DBOs also form an acyl-complex with the active site; however, this process is reversible and active inhibitor can be released upon recyclization of the DBO scaffold (Figure 2, middle).1,6 As with β-lactam-based inhibitors, DBOs are also hydrolyzed, albeit the rate is slow, and with serine β-lactamases, typically after desulfation of the inhibitor.1,7–9 Two DBOs, avibactam and relebactam, are approved by the U.S. Food and Drug Administration (FDA) paired with ceftazidime and imipenemcilastatin, respectively. Ceftazidime-avibactam is the first β-lactam-β-lactamase inhibitor combination approved to treat Enterobacterales-producing KPC-type carbapenemases, thus targeting a CDC urgent heath threat.1 Several novel “dualaction” DBO β-lactamase inhibitors (e.g., durlobactam, zidebactam, and nacubactam) that also inactivate penicillin binding proteins are in development.10
Boronate inhibitors form a dative bond between the boron atom of the inhibitor and the active site serine hydroxyl, which is reversible and results in the release of an active inhibitor (Figure 2, bottom). To date, hydrolysis of boronate inhibitors by β-lactamases is not described, likely due to the unique chemistry of these compounds that precludes hydrolysis. One boronate, vaborbactam, is approved for use with meropenem by the FDA in the United States. Novel boronates (e.g., taniborbactam and QPX7728) that possess the most extensive β-lactamase inhibition profiles (classes A, B, C, and D) are in the pipeline.10
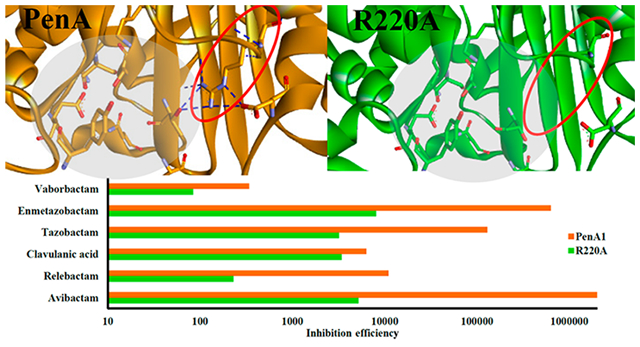
Inhibiting the PenA β-lactamase with β-lactams that are poor substrates for the β-lactamase and/or the addition of a β-lactamase inhibitor is an essential criterion for antimicrobial activity against Bcc. Previously, avibactam was found to be a potent inactivator of the PenA1 β-lactamase from Burkholderia multivorans, a member of Bcc.11 Here, the activity of three newer β-lactamase inhibitors from all three classes, enmetazobactam, relebactam, and vaborbactam, are tested against PenA1 and compared to avibactam, clavulanic acid, and tazobactam. Moreover, the contribution of selected amino acids in various active site motifs (S70XXK73, S130DN132, R164-Ω-loop-D179, K234TG) toward inhibition is evaluated by making single amino acid substitutions (S70A, K73A, S130A, E166A, N170A, R220A, K234A, T237A, and D276A) in PenA1 (Figure 3). The roles of these motifs in PenA1 were previously described in detail.3 Briefly, S70 is the nucleophile, K73, S130, E166, and K234 participate in acylation and deacylation, K234, R220, and D276 are involved in binding and stabilization, and E166 and N170 anchor the deacylation water molecule.
Figure 3.
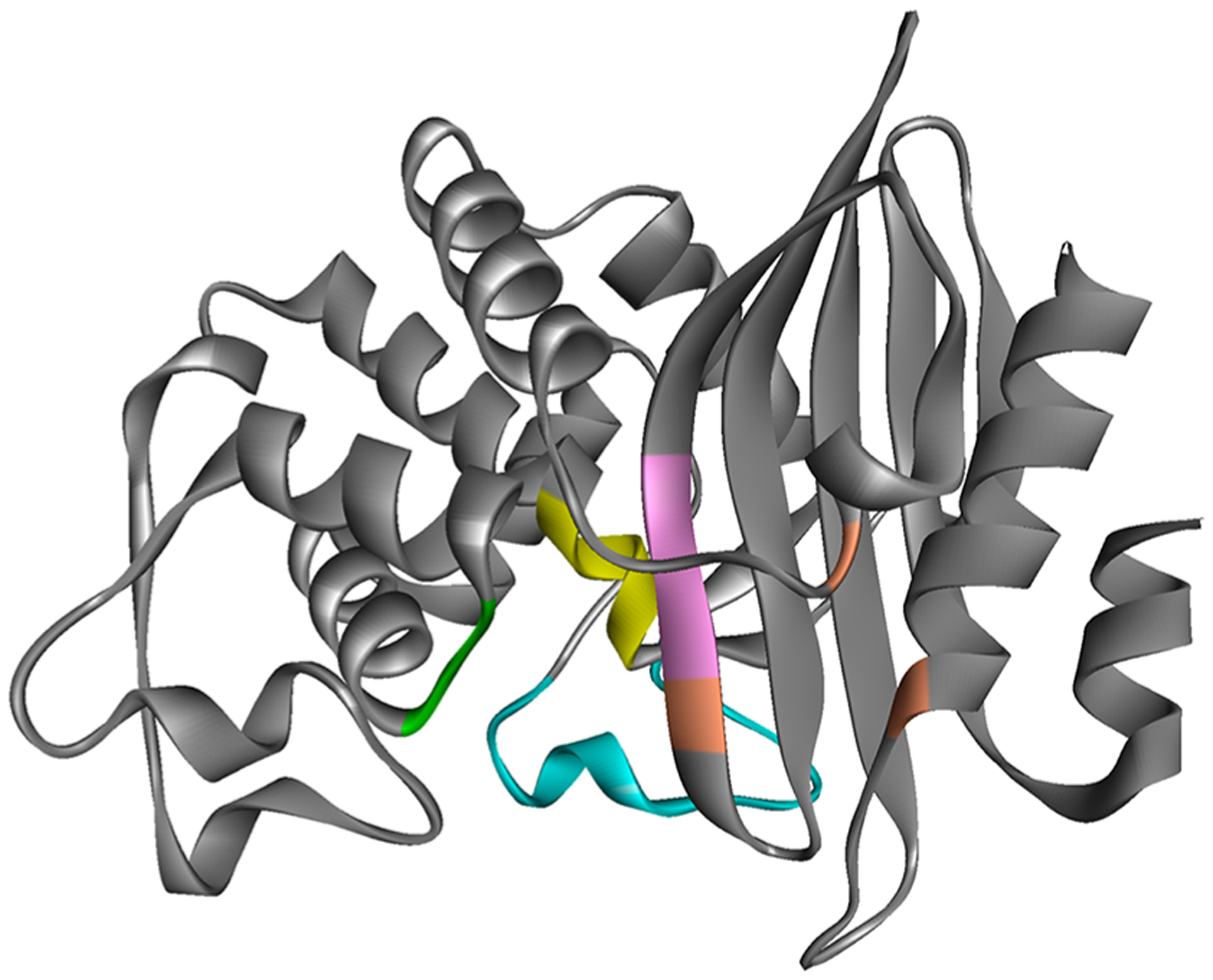
PenA1 structure (PDB 3W4Q) showing the location of SXXK motif (yellow), SDN loop (green), Ω-loop (cyan), KTG motif (pink), and residue T237, R220, and D276 (positioned left to right in orange).
RESULTS
Enmetazobactam, Relebactam, and Vaborbactam Are as Effective as Avibactam against an Isogenic Strain Producing PenA1.
To evaluate the efficacy of the various β-lactamase inhibitors against PenA1, the blaPenA1 gene was cloned and expressed in Escherichia coli DH10B for susceptibility testing. Piperacillin was selected as the partner β-lactam, as previously piperacillin was found to be a potent partner against clinical isolates of Bcc.12 The newer β-lactamase inhibitors, avibactam, relebactam, enmetazobactam, and vaborbactam reduced piperacillin minimum inhibitory concentrations (MICs) to the susceptible range (MICs: 2−8 μg/mL) for E. coli with blaPenA1, while clavulanic acid and tazobactam did not (MICs: 128−256 μg/mL) (Table 1).
Table 1.
MIC Profiles for E. coli DH10B Carrying blaPenA1 and the blaPenA1 Mutants against Select β-Lactams and β-Lactam−β-Lactamase Inhibitor Combinationss
| Strain | Piperacillin | Piperacillin-Avibactam | Piperacillin-Relebactam | Piperacillin-Clavulanate | Piperacillin-Enmetazobactam | Piperacillin-Tazobactam | Piperacillin-Vaborbactam |
|---|---|---|---|---|---|---|---|
| pBC SK(+) empty | 2 | 2 | 2 | 2 | 4 | 4 | 2 |
| pBC SK(+) blaPenA1 | 256 | 2 | 8 | 128 | 4 | 256 | 4 |
| pBC SK(+) blaPenA1 S70A | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| pBC SK(+) blaPenA1 K73A | 16 | 8 | 32 | 8 | 16 | 16 | 32 |
| pBC SK(+) blaPenA1 S130A | 32 | 16 | 32 | 16 | 8 | 8 | 4 |
| pBC SK(+) blaPenA1 E166A | 16 | 2 | 2 | 2 | 2 | 4 | 2 |
| pBC SK(+) blaPenA1 N170A | 128 | 2 | 2 | 2 | 4 | 2 | 4 |
| pBC SK(+) blaPenA1 R220A | 32 | 4 | 8 | 8 | 4 | 8 | 32 |
| pBC SK(+) blaPenA1 K234A | 4 | 4 | 4 | 4 | 4 | 8 | 4 |
| pBC SK(+) blaPenA1 T237A | 128 | 2 | 4 | 4 | 2 | 4 | 8 |
| pBC SK(+) blaPenA1 D276A | 64 | 2 | 2 | 2 | 2 | 2 | 4 |
The concentration of piperacillin was varied (1−256 μg/mL) and β-lactamase inhibitors were held at a constant concentration of 4 μg/mL. CLSI interpretative indices for Enterobacterales were used for piperacillin MICs: ≥128 μg/mL = resistant, ≥32−64 μg/mL = intermediate, and ≤16 μg/mL = susceptible. MICs are highlighted from white, light blue, medium blue to dark blue based on the lowest value to the highest value, respectively.
Residues K73, S130, and R220 Play a Role in Lowering Piperacillin-β-Lactamase Inhibitor MICs.
To gain insights into the inactivation mechanism of PenA1 by β-lactamase inhibitors, single amino acid substitutions at positions S70A, K73A, S130A, E166A, N170A, R220A, K234A, T237A, and D276A were generated in PenA1. With the exception of the S70A variant, the PenA1 variants were produced at detectable levels via immunoblot (Figure 4). Lack of detectable expression of the S70A variant correspondingly resulted in MICs of 2 μg/mL against all tested antibiotics for the strain carrying the S70A variant similar to E. coli with the empty vector (Table 1). All other variants were detected; however, the production of the S130A, N170A, T237A, and D276A variants was less than that of wild-type PenA1 (Figure 4). Yet, E. coli producing the S130A, N170A, T237A, and D276A variants remained nonsusceptible (32−128 μg/mL) to piperacillin (Table 1). Thus, the lower expression levels of S130A, N170A, T237A, and D276A variants did not meaningfully alter piperacillin MICs. Cells with the K73A and E166A variants, which were produced at levels similar to that of wild type, tested in the susceptible range (16 μg/mL) (Table 1). The amino acid substitution of K234A abrogated piperacillin MICs (256 → 4 μg/mL) when expressed in E. coli; protein expression levels were similar to that of wild type (Table 1 and Figure 4). The R220A variant was expressed at wild-type levels and its MIC to piperacillin was reduced (256 → 32 μg/mL) but remained in the resistant range.
Figure 4.
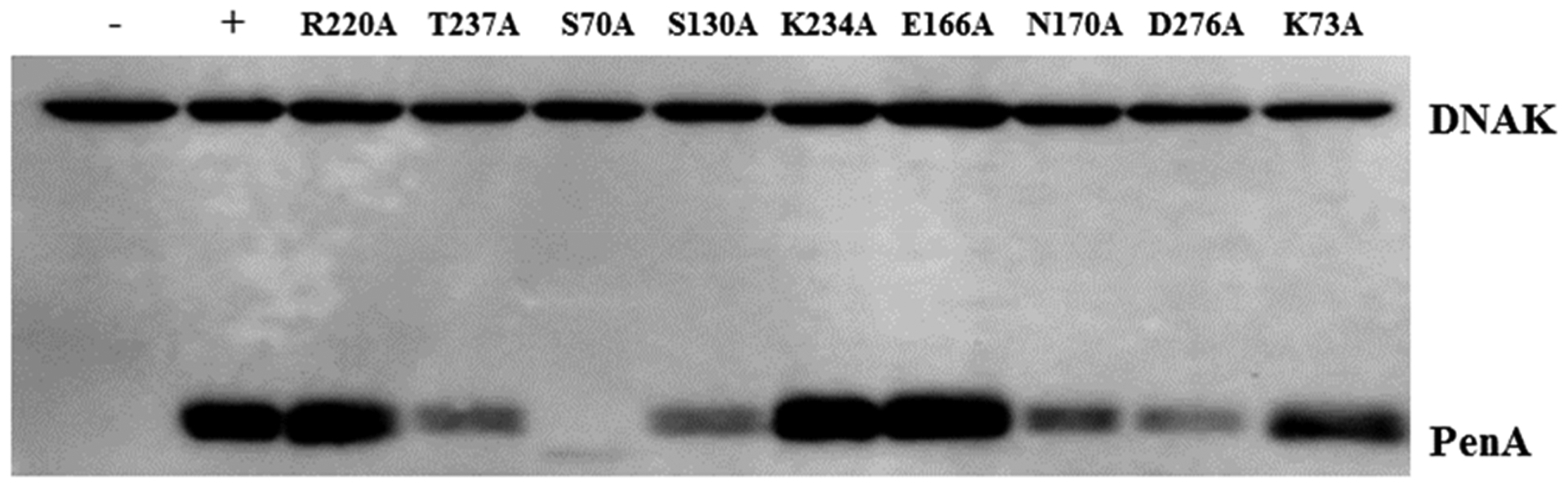
Immunoblotting of crude extracts using anti-PenA1 and anti-DNAK antibodies to reveal the expression levels of PenA1 and the variants; negative (−), empty vector control; positive (+), PenA1 wild-type. PenA1 and variants were expressed from pBC SK(+) in E. coli DH10B.
When the different classes of β-lactamase inhibitors were combined with piperacillin, various patterns emerged. The addition of all β-lactamase inhibitors to E. coli producing the K73A and S130A variants resulted in only minor effects on piperacillin MICs (16−32 → 8−32 μg/mL) (Table 1). The only exception was when vaborbactam was combined with piperacillin with the S130A variant as the MIC decreased by 8-fold. The β-lactamase inhibitors remained largely effective against E. coli with the E166A, N170A, T237A, and D276A variants (Table 1). The variant possessing an intermediate effect to the combinations was R220A.
Avibactam Is a Poor Inhibitor of the S130A, R220A, and K234A Variants.
Crude extracts of E. coli producing the variants were screened for activity against a chromogenic reporter β-lactam substrate, nitrocefin and further experimentation with the S70A, E166A and K73A variants was discontinued due to very low hydrolytic activity. To kinetically screen the remaining variants, avibactam was used. Determining the IC50 values for avibactam using crude extracts revealed that the S130A and K234A variants were poorly inhibited by avibactam, while the T237A, N170A, and D276A variants’ inhibition was largely unaffected (Table 2). As variants at positions S13013,14 and K23415,16 were previously examined in other class A β-lactamases with the two newer classes of β-lactamase inhibitors (i.e., DBOs and boronates), the R220A variant, which possessed a 31-fold higher IC50 toward avibactam, was selected for further analysis.
Table 2.
Avibactam Inhibitory Concentrations (IC50) Using Crude Protein Extracts from E. coli Producing the Different Variants and Wild-Type PenA1a
| β-lactamase | avibactam IC50 (μM) |
|---|---|
| PenA1 | 1 |
| R220A | 31 |
| T237A | 0.12 |
| S130A | >500 |
| K234A | >800 |
| N170A | 2.6 |
| D276A | 1 |
100 μM nitrocefin was used for all trials as the substrate.
Avibactam Is the Most Potent Inactivator of the PenA1 Carbapenemase.
To assess the efficiency of the various classes of β-lactamase inhibitors toward inactivation of PenA1, steady-state kinetic inhibition parameters were obtained with avibactam, relebactam, clavulanic acid, tazobactam, enmetazobactam, and vaborbactam using the purified wild-type PenA1 β-lactamase. The kinetic analysis of the β-lactamase inhibitors reveals that avibactam was the most potent inhibitor of PenA1 with the lowest Ki-app value of 0.5 μM, highest acylation rate, k2/K of 2 × 106 M−1 s−1, and least amount of drug turned over in 15 min at five molecules (Table 3). Enmetazobactam and tazobactam are the subsequent most potent agents against PenA1 with low Ki-app values (0.6−1.1 μM) and high acylation rates in the 105 M−1 s−1 range (Table 3). Conversely, vaborbactam was the worst inhibitor of PenA1 with a Ki-app of 38 μM and k2/K of 240 M−1 s−1 (Table 3). Relebactam was more effective then clavulanic acid as 5000 molecules of clavulanic acid were hydrolyzed in 15 min by PenA1 compared to 10 molecules of relebactam in 15 min (Table 3).
Table 3.
Steady-State Kinetic Parameters with Purified PenA1 and R220A β-Lactamases
| Ki-app (μM) | PenA1 | R220A | fold increase |
|---|---|---|---|
| avibactam | 0.5 ± 0.1 | 13 ± 3 | 26 |
| relebactam | 9 ± 1 | 110 ± 17 | 12 |
| clavulanic acid | 5 ± 1 | 26 ± 3 | 5 |
| tazobactam | 0.6 ± 0.1 | 36 ± 10 | 60 |
| enmetazobactam | 1.1 ± 0.3 | 24 ± 5 | 22 |
| vaborbactam | 38 ± 4 | 108 ± 10 | 3 |
| k2/K (M−1 s−1) | PenA1 | R220A | fold decrease |
| avibactam | 2 ± 1 × 106 | 5.2 ± 0.1 × 103 | 385 |
| relebactam | 1.1 ± 0.2 × 104 | 2.3 ± 0.8 × 102 | 48 |
| clavulanic acid | 6.3 ± 0.1 × 103 | 3.4 ± 0.2 × 103 | 1.9 |
| tazobactam | 1.3 ± 1.0 × 105 | 3.2 ± 0.4 × 103 | 41 |
| enmetazobactam | 6.3 ± 2.0 × 105 | 8.1 ± 1.5 × 103 | 77 |
| vaborbactam | 3.4 ± 0.1 × 102 | 8.4 ± 2.0 × 101 | 4 |
| tn (15 min) | PenA1 | R220A | fold decrease |
| avibactam | 5 | 10 | 2b |
| relebactam | 10 | 50 | 5b |
| clavulanic acid | 5000 | 2000 | 2.5 |
| tazobactam | 50 | 10 | 5 |
| enmetazobactam | 100 | 10 | 10 |
| vaborbactam | 200a | 100a | 2 |
Represents the stoichiometry of inactivation, as vaborbactam does not undergo side reactions or hydrolysis.
The represented fold change values are increased not decreased for the DBOs.
Position R220 Is Critical for β-Lactamase Inhibition.
To investigate the role of position R220 in β-lactamase inhibition, the R220A variant of PenA1 was purified for kinetic analysis and compared to wild-type PenA1. The R220A substitution decreased the potency of all inhibitors regardless of mechanism (Table 3). The Ki-app values increased, while the k2/K values decreased for all agents when tested against the R220A variant. Ironically, the inhibitor most affected by the R220A substitution was avibactam with the acylation rate decreasing nearly 400-fold (Table 3). Vaborbactam was the inhibitor least affected by the R220A substitution with a 2-fold increase in Ki-app (Table 3). The only evidence of mechanism-based effects on inhibition were observed with the tn at 15 min, as the R220A substitution resulted in increased turnover of DBOs but not β-lactam-based inhibitors or the boronate (Table 3).
Lack of Arginine at 220 Abrogates the 237−220−276 Hydrogen Bonding Network.
To understand the mechanistic contribution of R220 toward β-lactamase inhibition, the R220A variant of PenA1 was crystallized (Table 4). The R220A crystal structure (PDB 7D5J) was obtained at 1.5 Å resolution at pH 6.5, while the previously published PenA1 crystal structure (PDB 3W4Q) was at a 1.2 Å resolution at pH 4.2.3 The R220A and PenA1 β-lactamase crystals were in the space group C2 with three molecules per asymmetric unit and in a monomeric state with protein B-factors of 15.43 Å2 and 16.39 Å2, respectively.3
Table 4.
X-ray Data Collection and Results from Phenix Refinement for PDB 7D5J
| parameter | value |
|---|---|
| X-ray data collection (XDS) | |
| source | Photon Factory AR-NW12a |
| detector | Pilatus3 S2M |
| wavelength (Å) | 1.000 |
| space group | C2 |
| cell dimensions | |
| a, b, c (Å) | 123.3, 71.2, 84.5 |
| α, β, γ (deg) | 90, 90.04, 90 |
| resolution (highestres.shell) (Å) | 42.27−1.51 (1.53−1.51)a |
| observations | 44 234 990 (19 251) |
| unique reflections | 114 986 (5243) |
| completeness (%) | 98.2 (93.0) |
| redundancy | 3.8 (3.7) |
| Iav/σ(I) | 12.5 (2.5) |
| CC1/2 | 0.988 (0.640) |
| Rmerge(I) | 0.056 (0.365) |
| refinement results (Phenix) | |
| resolution range (Å) | 34.88−1.51 (1.56−1.51)a |
| no. of reflections used [F > 0σ(F)] | 113 426 (10 858) |
| Rwork/Rfreeb (%) | 17.19 (23.77)/19.10 (27.37) |
| Rtotal (%) | 17.27 |
| residues in Ramachandran zones (%) | |
| favored/allowed/disallowed | 98.7/1.3/0 |
| RMSD values from ideality | |
| bond lengths (Å) | 0.005 |
| bond angles (deg) | 0.83 |
| mean B-factors (number of atoms) | |
| protein (no.) | 15.43 (5730) |
| MES (no.) | 27.05 (36) |
| water molecules (no.) | 24.00 (732) |
| all atoms (no.) | 16.45 (6498) |
Data for highest resolution shell are in parentheses.
Rfree was calculated from 5% of reflections
The overall structures of PenA1 and the R220A variant were very similar (rms deviation is 0.29 Å for 35−285 Cα atoms) with protein folds consisting of an α-helical region followed by five antiparallel β-sheet strands and additional α-helices. Active site residues (S70, K73, S130, N132, E166, N170, K234, T235, G236, and T237) were well modeled in the 2Fo−Fc map (Figure 5a) and were superimposable (Figure 5b) with a RMSD of 0.39 Å for all the atoms present in these amino acids. A higher RMSD was mainly caused by a different rotamer of residue T237. The R220A substitution did not affect the overall active site architecture as evidenced by 0.39 Å rms deviation for active site residues.
Figure 5.
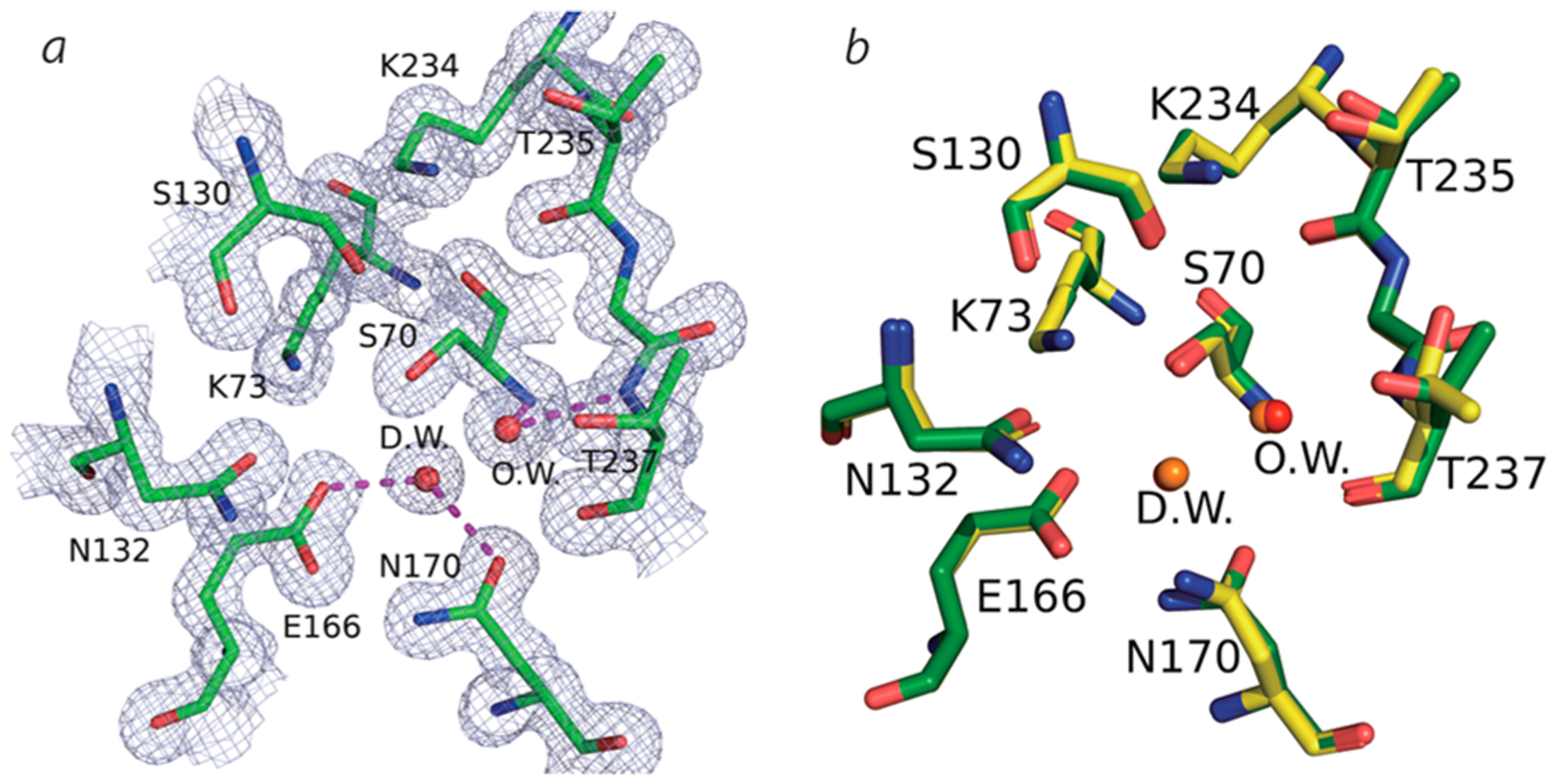
(a) 2Fo−Fc electron density map (1.5σ) around the active site of R220A variant enzyme. O.W., oxyanion water; D.W., deacylation water. (b) Superposition of PenA1 (yellow, PDB 3W4Q) and the R220A variant (green, PDB 7D5J).
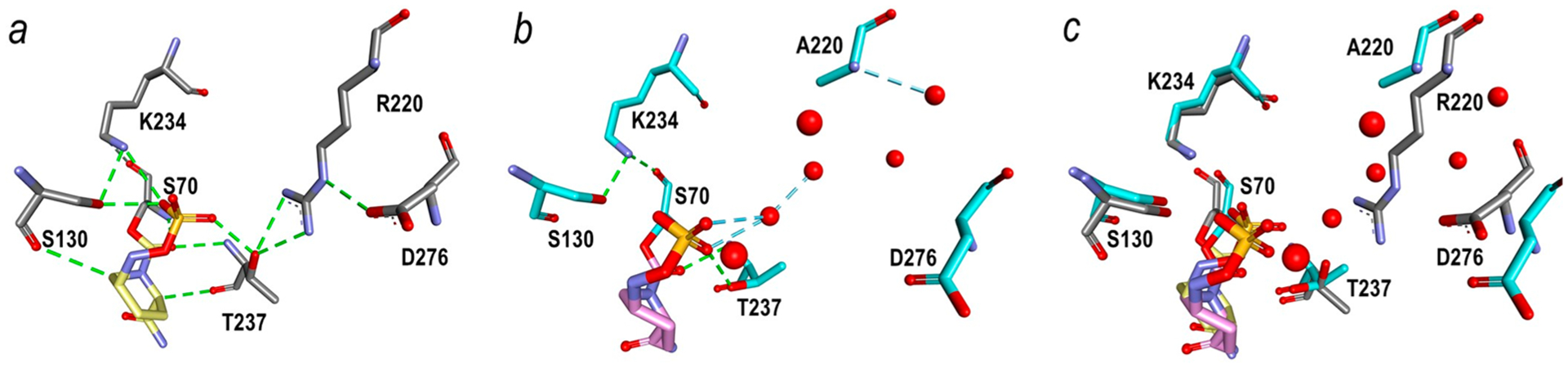
In the wild-type PenA1 structure, the guanidium group of R220 formed a four membered intramolecule hydrogen bonding network, R220:NH1-D245:O (3.1 Å distance), R220:NH1-G236:0 (2.9 Å), R220:NH2-T237:OG (3.0 Å), and NH2:NED276:OD1(2.7 Å) (Figure 6a). In the R220A variant, four water molecules, W3, W4, W5, W6 of ChainA, now occupied the space normally occupied by the guanidium side chain of arginine and the major hydrogen bonds between 237 and 220−276 were lost (Figure 6b). As a result, the positioning of D276:OD1 was also moved by 2.7 Å and the side chain of D276 lacked electron density (Figure 6c). The B-factor of Cα in the A270-D276 region increased with the loss of the hydrogen bonds and migration of D276.
Figure 6.
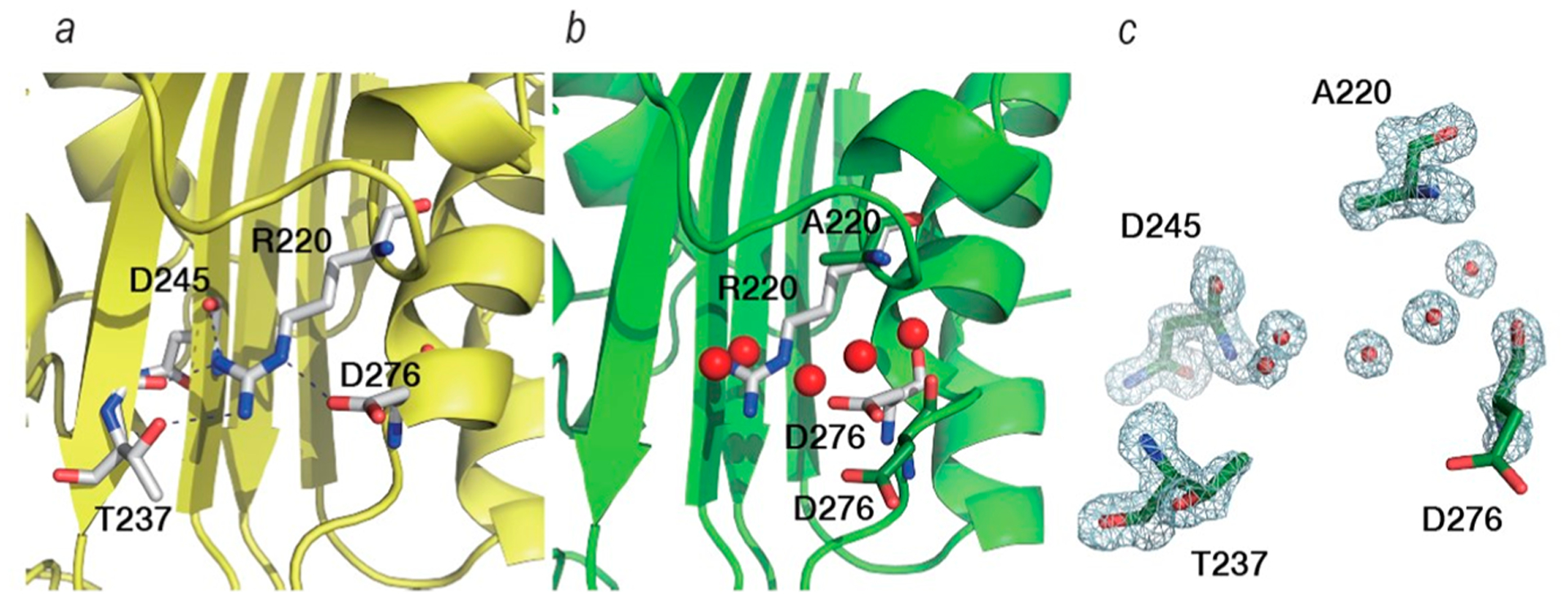
Residues around position R220 of PenA1 (PDB 3W4Q) (a) and A220 of the R220A variant (PDB 7D5J) (b). White sticks show R220 of PenA1 and its hydrogen-bonding network (dashed back lines). Green sticks show A220 and D276 of the R220A variant. Red spheres in (b) are water molecules observed near the A220 of the R220A variant. Electron density around A220 (PDB 7D5J) reveals the occupancy by water molecules due to loss of the guanidium and unresolved density for the carboxylate of D276 (c).
The B-factor of Cα in the A270-D276 region increased with the loss of the hydrogen bonds and migration of D276 (Figure 7). Whereas average B-factors for PenA1 and the R220A variant were 16.39/15.43 for the whole molecule, they were 20.87/25.01 for the Cα atoms of the A270-D276 region, and 17.62/24.26 for the A274-D276 region, respectively. Moreover, the B-factor of Cα in the V225-G228 region of the R220A variant was also increased as well as the area around P254. Although two different traces were modeled in this region in wild-type PenA1, the second trace could not be built in 2Fo−Fc map of the R220A variant. Overall, the three regions in α/β domain of the R220A variant possessed higher B-factors than PenA1.
Figure 7.
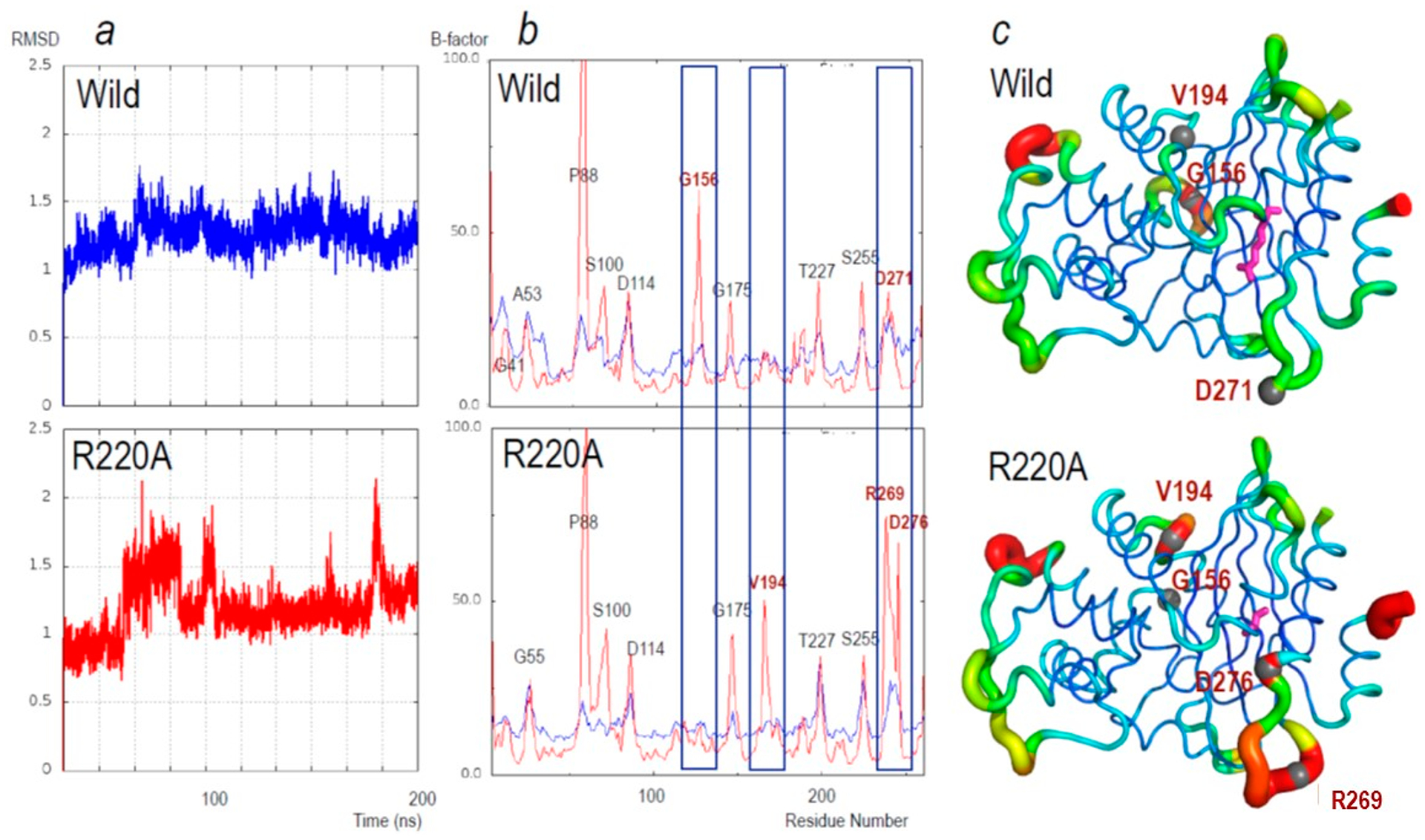
(a) Changes in the root-mean-square deviation (RMSD) for main-chain atoms during the 200 ns simulations relative to the starting structure. Upper, PenA1 (wild, blue lines) and lower, R220A (R220A, red lines). (b) Average B-factors of main-chain atoms of the individual amino acid residues during the 200 ns MD simulations (red); the average B-factors of the crystal structure (blue). Upper: PenA1 (Wild) and Lower: R220A (R220A). Labels on the peak indicate the residue using standard class A β-lactamase numbering. Residues with the largest changes in B-factor are in red font and highlighted by blue rectangles. (c) B-factors from MD simulation putty representation on Pymol of (upper) PenA1 and (lower) R220A. The color changes from blue (B-factors below 5.0), while cyan, green, yellow, and red (B factors of more than 50.0). The thickness of the lines reflects the height of the B-factor. The stick model (magenta) indicates the position of R220 and A220, respectively. Sphere atom models and red font indicate the peak position found in panel (b).
To further analyze the movement of the β-lactamases, molecular dynamics (MD) simulations were performed using the crystal structures of PenA1 (PDB 3W4Q) and the R220A variant (PDB 7D5J). RMSD value of the R220A variant for main chain atoms from the initial structure was relatively unstable compared to PenA1 (Figure 7a). This observation suggests that the movement of main chain amino acids of the R220A variant is larger than for PenA1. The average B-factors for main chain atoms calculated from MD simulation trajectories are shown in Figure 7b and c. The B-factor peaks were clearly different in three regions between PenA1 and the R220A variant. In the R269-D276 region of the R220A variant, B-factors were clearly larger than PenA1. This may be the result of loss of the NH2:R220-OD1:D276 hydrogen bond. Other differences were observed in the region of G156 and V194. The former was disappearance of the peak at G156 and the appearance of a new peak at V194 for the R220A variant.
The Active Site Architecture of PenA1 Impedes Inactivation by Vaborbactam.
To gain insight into why vaborbactam is the worst inhibitor of PenA1 (k2/K, 3.4 ± 0.1 ×102 M−1 s−1) in vitro, Michaelis−Menten complexes were generated with PenA1 and vaborbactam and compared to Michaelis−Menten complexes of KPC-2 and vaborbactam, as vaborbactam is a potent inactivator of KPC-2.17 The KPC-2 active site is narrower than that of PenA1; thus, during docking into KPC-2, vaborbactam was restrained into a conformation, which allowed for productive interactions (Figure 8a). In KPC-2, the boronic acid moiety was “guided” toward the S70 within ≈2 Å, due in a major part to W105 maintaining steric interactions with the cyclic boronate region of vaborbactam to promote favorable complex formation. Conversely, the active site of PenA1 is larger allowing vaborbactam to form different conformations and interactions within the active site during docking. Besides the larger active site of PenA1, another factor that influenced vaborbactam binding was that PenA1 has a tyrosine at position 105 as opposed to a tryptophan in KPC-2 (Figure 8b). Position Y105 in PenA1 was very flexible, flipping more than 90°. The movement of Y105 further restricted the access of vaborbactam into the PenA1 active site by competing for binding between the cyclic boronate and thiophene rings on vaborbactam. With PenA1, the boronic acid moiety was prevented from making productive interactions with S70, due to multiple conformations that vaborbactam adopted. Furthermore, the molecular docking suggests that the interactions of the carboxylate group of vaborbactam with K234 and T235 was preventing the boronate from reaching S70 as it was more than 4.0 Å away.
Figure 8.
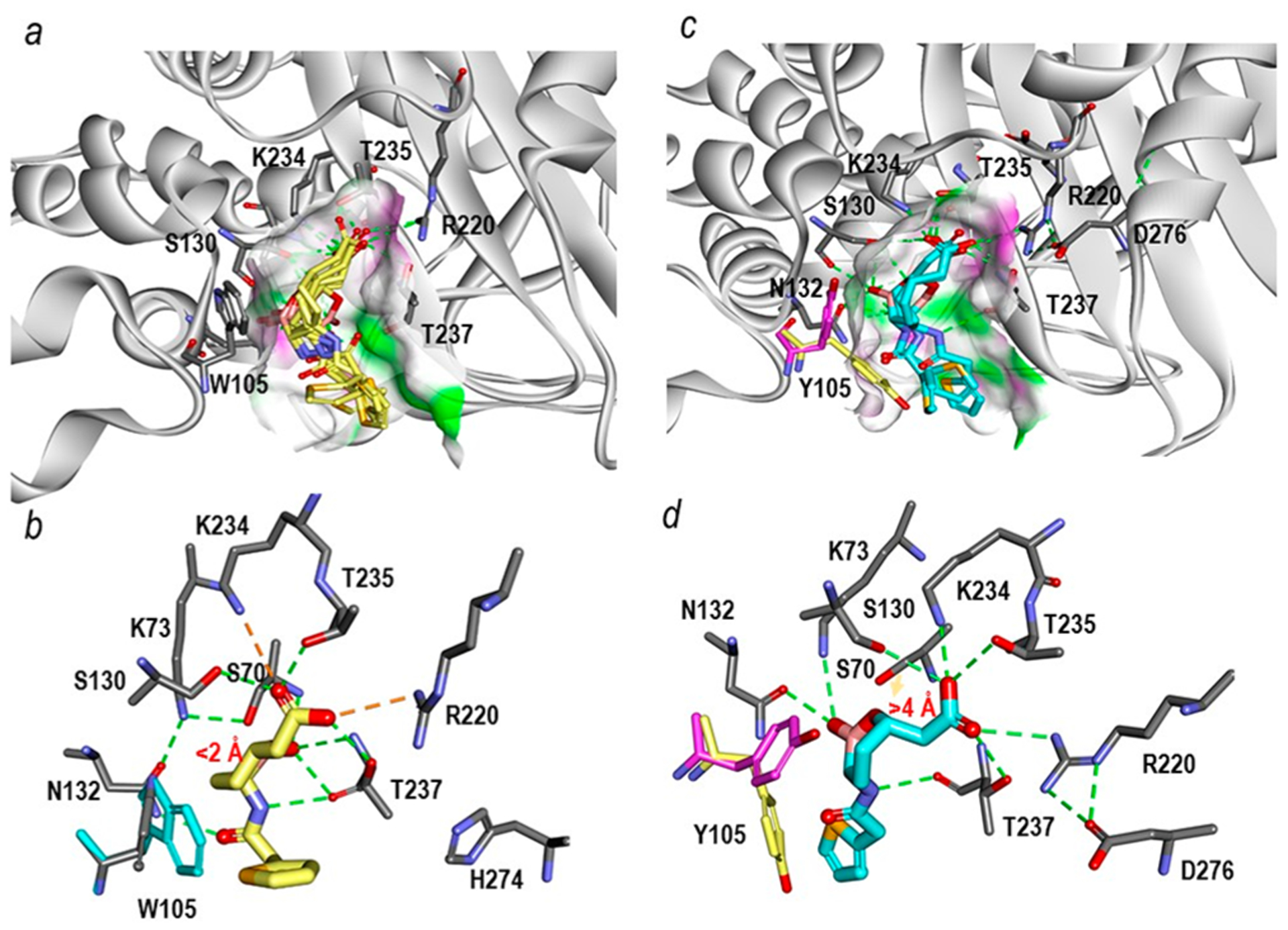
Michaelis−Menten complexes of vaborbactam in the sp2 configuration shown in multiple different poses obtained upon docking into the active sites of KPC-2 (PDB 2OV5) and PenA1 (PDB 3W4Q). (a) Within KPC-2, several docked vaborbactam molecules (yellow) formed favorable complexes. The surface of the active site was colored by hydrogen-bonding type with hydrogen donors colored in magenta and hydrogen acceptors in green. The surface further reveals the difference in the active site sizes between the two enzymes with the KPC-2 active site being narrower than that of PenA1. (b) A close up of the active site interactions in KPC-2 with a single selected vaborbactam pose obtained from docking reveals favorable interactions with the boronate near S70 (<2 Å) and stacking interactions between W105 (cyan) and cyclic boronate moiety. Hydrogen bonds are represented by dashed green lines; stronger hydrogen bonding interactions (e.g., ionic interactions) are in orange dashed lines. (c) Conversely, with PenA1, the multiple poses of vaborbactam (cyan) obtained from the docking formed mostly unfavorable interactions that were driven by larger active site of PenA1. Surface representations are the same as described in panel (a) for KPC-2. (d) A close up of the PenA1 active site with a single selected vaborbactam pose obtained from docking reveals that the carboxylate moiety of vaborbactam competes for binding to K234 and T235, thus moving the boronate away from S70 (>4 Å); the faint orange arrow points out the distance between hydroxide side chain of S70 and the boronate moiety. Moreover, during docking two conformations of Y105 (pink/yellow) were observed that competed for interactions with cyclic boronate moiety and the thiophene ring of vaborbactam. The hydrogen bonding interactions are the same as described in panel (b) for KPC-2.
R220 Aides in Productive β-Lactamase Inhibitor Binding.
To evaluate the impact of the R220A substitution on β-lactamase inhibitor binding, an acyl−enzyme complex was constructed using the inhibitor most effected by the R220A substitution, avibactam (~400-fold decrease in k2/K) with the R220A variant and compared to the PenA1-avibactam crystal structure (PDB 7DOO). In PenA1, avibactam formed favorable interactions with the active site residues S130, K234, and T237 (Figure 9a). The interactions with T237 were aided via hydrogen bonds to R220 and D276. Not surprisingly, in the R220A variant, the loss of the arginine side chain resulted in a void that was filled with water molecules and the 237−220−276 hydrogen bonding network was disrupted. The positions of T237 and D276 were moved and avibactam was contorted and less productively positioned (Figure 9b and c).
Figure 9.
Crystal structure of PenA1 (gray) with avibactam (yellow) (PDB 7DOO) (a) was compared to a docked acyl−enzyme molecular model of avibactam (pink) in the R220A variant (cyan) (b), and an overlay of panels (a) and (b) is found in panel (c). Only one docking pose is presented for avibactam in the PenA1 and R220A variant active sites in the acyl-complex as avibactam is a more potent inhibitor of the PenA1 and R220A variants compared to vaborbactam. Hydrogen bonds are denoted by dashed lines (green, residue-based hydrogen bonds; blue, water-based hydrogen bonds); red spheres are water molecules.
DISCUSSION
Herein, the abilities of β-lactamase inhibitors with diverse inactivation mechanisms were evaluated against PenA1 from B. multivorans. The recently approved β-lactamase inhibitors, relebactam and vaborbactam, and a β-lactamase inhibitor in development, enmetazobactam, were found to be equally effective at lowering piperacillin MICs as avibactam against E. coli producing wild-type PenA1. Not surprisingly, the older β-lactamase inhibitors were not effective, as clavulanic acid and tazobactam were previously shown to not lower MICs when partnered with a β-lactam against strains producing PenA1, as PenA1 is able to hydrolyze these inhibitors.3 Interestingly, enmetazobactam with similar in vitro kinetic constants against PenA1 outperformed tazobactam when partnered with piperacillin against E. coli producing PenA1 with MICs of 4 μg/mL compared to 256 μg/mL, respectively. This discrepancy is likely the direct result of the increased penetration of enmetazobactam into the bacterial cell due to its zwitterion chemistry.5 Comparable observations were previously reported with another class A carbapenemase, KPC-2.5
To further evaluate inactivation mechanisms, single amino acid substitutions (S70A, K73A, S130A, E166A, N170A, R220A, K234A, T237A, and D276A) were generated in PenA1. E. coli producing the S70A variant possessed piperacillin MICs equivalent to E. coli with the empty vector and the protein was not detected using immunoblotting; thus, this substitution was not further assessed. In other class A β-lactamases (e.g., TEM-1), S70G variants possess a gain in protein stability, while the S70A variants do not, suggesting that the removal of the Cβ atom is needed for the gain in stability.18 Here, the substitution of S70A in PenA likely destabilized the protein, leading to degradation as inferred from lack of detection via immunoblot. The K73A, S130A, R220A, and K234A variants became less susceptible to inhibition by β-lactamase inhibitors, while E. coli carrying the E166A variant was susceptible to piperacillin and piperacillin-β-lactamase inhibitor combinations. The role of N170, T237, and D276 in inhibition is likely ancillary, as substitutions at these positions only slightly impacted piperacillin−β-lactamase inhibitor combination MICs and avibactam’s IC50. Of the variants less susceptible to inhibition, the R220A variant was chosen for further characterization as the detailed biochemical analysis of S130 and K234 variants has previously been studied with select DBOs and boronates.13–16 Understanding the impact of a substitution at position 220 in class A carbapenemases on the inhibition by the newer β-lactamase inhibitors, enmetazobactam, avibactam, relebactam, and vaborbactam has not been explored or only explored in limited detail (i.e., susceptibility testing)14 and never before in PenA1.
In other class A β-lactamases (e.g., TEM-1 and SHV-1, narrow-spectrum penicillinases; PER-2, extended-spectrum β-lactamase (ESBL) and KPC-2, carbapenemase), position R220 or its equivalent (R244 in TEM and SHV) was shown to be important for binding the C3/C4 carboxylate or sulfonate of β-lactams and β-lactamase inhibitors.19–23 Substitutions at position R244 and R220 in narrow-spectrum penicillinases and ESBLs lead to decreased inhibition by the older β-lactamase inhibitors (i.e., clavulanic acid, sulbactam, and tazobactam). Conversely, strains expressing class A carbapenemases (e.g., KPC-2 and PenA1 with R220) are naturally resistant to inhibition by these older β-lactamase inhibitors. Bacteria producing PER-2 or KPC-2 with substitutions at position R220 also lead to resistance toward β-lactam-avibactam combinations.14,24 Herein, we find that substituting an alanine for arginine at position R220 in PenA1 resulted in dramatic effects on the inhibitory kinetics with Ki-app values increasing from 2-fold up to 98-fold and acylation rates decreasing by 2− 385-fold against all three classes of β-lactamases inhibitors: β-lactam-based, DBO-based, and boronic acid−based. Thus, substitutions at R220 can further extend the inhibitor resistance profile in vitro toward newer β-lactamase inhibitors; implications toward susceptibility are discussed below.
The X-ray crystal structure of the R220A variant revealed the loss of a major hydrogen bonding network, 237−220−276, that is important for substrate binding in class A β-lactamases.3,23,25 The void left by the missing side chain of arginine leaves a pocket that was occupied by water molecules, and the positioning of 237 and 276 was migrated. These modifications led to decreased piperacillin resistance when the R220A variant is expressed in E. coli (256 → 32 μg/mL). These observations were supported by previous studies with KPC-2 and PER-2, as substitutions at R220 decrease the hydrolytic activity of the enzyme toward β-lactams.23,25
Vaborbactam was the β-lactamase inhibitor with the lowest k2/K value and highest Ki-app against PenA1. By generating Michaelis−Menten complexes of PenA1 with vaborbactam using molecular modeling and comparing this data to KPC-2, a carbapenemase that is inhibited well by vaborbactam, three major differences were observed. The larger active site of PenA1 led to conformational flexibility in vaborbactam that was further exacerbated by the movement of Y105, and the hydrogen bonding of the carboxylate of vaborbactam to K234 and T235. These unproductive interactions distanced the boronate moiety from S70 and delayed acylation. An important role for W105 vs Y105 in binding of vaborbactam was previously observed between KPC-2 and CTX-M-15, a class A ESBL that supports our observations.13 Interestingly, despite the lower acylation rate of vaborbactam against PenA1 in vitro, whole-cell assays revealed that when vaborbactam was combined with piperacillin, MICs were reduced (256 → 4 μg/mL). Thus, despite these “rocky” in vitro interactions, vaborbactam exhibited potency against cells producing PenA1, suggesting that vaborbactam was eventually able to form a stable inhibitory complex with PenA1.
The inhibitor that was impacted the most by the R220A substitution was avibactam with an increased Ki-app of 13 μM compared to 0.5 μM for the R220A variant vs PenA1 and decreased acylation rates by ~400-fold. Molecular modeling of the acyl−enzyme complex of R220A with avibactam was compared to the PenA1-avibactam crystal structure and not surprisingly the loss of the 237−220−276 hydrogen bonding network was found to help guide the avibactam into position. Thus, without the guanidium to bridge 237 to 276, the inhibitor had difficulty binding to the enzyme.
The R220A substitution in PenA1 gave rise to a β-lactamase that was not inhibited as well as PenA1 vs all classes of β-lactamase inhibitors as revealed by the in vitro kinetic analysis. Yet, the R220A variant expressed in E. coli remained susceptible in whole cell assays likely due to decreased β-lactamase activity of the R220A variant compared to wild-type (piperacillin MIC 32 vs 256 μg/mL, respectively). Thus, despite this “inhibitor-resistance” in vitro, strains producing the R220A variant will likely be susceptible to newer generation β-lactam−β-lactamase combinations (e.g., ceftazidime−avibactam, meropenem−vaborbactam, and imipenem−cilastatin−relebactam). The evolution of the R220A variant of PenA1 in B. multivorans is likely to be suppressed due to its corresponding loss in β-lactamase activity. The only likely clinical impact of variants at position R220 in PenA1 would be if they were observed in combination with additional amino acid substitutions that lead to enhanced β-lactamase activity (e.g., complex mutants).26,27 The complex mutant of TEM-2 is an example of such a case; the TEM-121 enzyme possessed an R244S substitution leading to decreased potency of clavulanic acid but also carried substitutions (E104 K, R164S, A237T, and E240 K) that lead to an ESBL phenotype with enhanced activity against ceftazidime and aztreonam.27 Complex mutants of PenA1 may hamper the activity of β-lactam−β-lactamase inhibitor combinations against B. multivorans but fortunately have not been observed to date. In summary, combinations of the newer β-lactamase inhibitors (i.e., enmetazobactam, avibactam, relebactam, and vaborbactam) with β-lactams will likely remain favorable therapies for B. multivorans carrying PenA1 and the R220A variant.
MATERIAL AND METHODS
Strains and Plasmids.
The construction of plasmids pBC SK(+) blaPenA1 and pGEX-6p-2 blaPenA1 in E. coli was previously described.3 Using the Quikchange XL Kit (Agilent), site-directed mutagenesis was performed on pBC SK(+) blaPenA1 to generate mutations encoding the S70A, K73A S130A, E166A, N170A, R220A, K234A, T237A, and D276A variants of PenA1, as described previously.28 In addition, mutations leading to the R220A variant of PenA1 were introduced into pGEX-6p-2 blaPenA1. The pBC SK(+) constructs were expressed in E. coli DH10B cells and used for susceptibility testing, immunoblotting, and IC50 determinations, while pGEX-6p-2 blaPenA1 was introduced into E. coli Origami 2 DE3 cells and used for PenA1 protein expression.
Antibiotics.
Piperacillin, clavulanic acid, and tazobactam were purchased from Sigma-Aldrich. Avibactam was obtained from Advanced Chemblocks. Enmetazobactam, vaborbactam, and relebactam were acquired from MedChemExpress.
Susceptibility Testing.
MICs were determined with a Steers replicator using the guidelines of the Clinical Laboratory Standards Institute on E. coli DH10B cells expressing the pBC SK(+) blaPenA1 and the PenA1 variants as well as an empty pBC SK(+) vector control.29 The β-lactamase inhibitors (clavulanic acid, tazobactam, enmetazobactam, avibactam, vaborbactam, and relebactam) were held at a concentration of 4 μg/mL, while the piperacillin concentration was varied. MICs were performed in triplicate with reported values being the mode.
Immunoblotting.
E. coli DH10B expressing PenA1 and the variants were grown to log phase (optical density at λ = 600 nm (OD600 nm) between 0.6 and 0.7) in lysogeny broth, pelleted, and frozen. The cells were lysed to prepare crude extracts in 50 mM Tris-chloride (Tris-Cl) at pH 7.4 with 1 mM magnesium sulfate, lysozyme (40 mg/Liter), and benzonase nuclease (Novagen) (1.0 U/mL). Ethylenediaminetetraacetic acid pH 7.8 (2.0 mM) was added to complete the extraction. The lysed cells were centrifuged at 12 000 rpm for 10 min to remove the cellular debris, as previously described.23 The extracts were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride membranes. The membranes were blocked in 5% nonfat dry milk in 20 mM Tris-Cl with 150 mM NaCl pH 7.4 (TBS) for 1 h and probed in 5% nonfat dry milk in TBS with 1:30 000 dilution of polyclonal anti-PenA1 antibody and a loading control, 1:5000 dilution of monoclonal anti-DNAK antibody (Stressgen), for 1 h. DNAK was used as the loading control. Membranes were washed five times for 10 min with TBS with 0.05% Tween 20 (TBST). Blots were incubated for 1 h in 1:10 000 dilutions of goat anti-rabbit-HRP and goat anti-mouse-HRP antibodies in 5% nonfat dry milk in TBS. Blots were washed five times for 10 min with TBST, developed using the ECL-Plus kit (GE Healthcare Life Sciences) according to the manufacturers’ instructions, and imaged on a Fotodyne Luminary/FX system.
Protein Purification for Steady-State Kinetics.
E. coli Origami 2 DE3 cells with the pGEX-6p-2 plasmid expressing the PenA1 or the R220A variant were grown in superoptimal broth (SOB) medium at 37 °C until the OD600 was about 0.6. Cultures were induced with 0.1 mM isopropyl β-D-1-thiogalactopyrano-side (IPTG) and incubated for 2 h. Cells were pelleted and frozen at −20 °C. Crude protein extracts were obtained from these pellets as described above for immunoblotting. Crude extracts were run over GSTrap FF HiTrap affinity columns and eluted with 50 mM Tris-HCl with 20 mM reduced glutathione and 1 mM dithiothreitol, pH 8.0 as instructed by the manufacturer (GE Healthcare). The protein was further purified by ÄKTA fast protein liquid chromatography (FPLC) using a Sephadex 16/60 gel filtration chromatography column (GE Life Sciences) using 10 mM phosphate-buffered saline (PBS) at pH 7.4. The proteins were maintained in 10 mM PBS pH 7.4, and the GST tags were cleaved with PreScission Protease (GE Healthcare) at 4 °C. The PenA1 and R220 variant β-lactamases were separated from the GST tag by running the digests over a GSTrap FF HiTrap affinity column. At every step, the presence and purities of the proteins were checked using SDS-PAGE gel electrophoresis and stained with Coomassie blue. Protein was then concentrated on a polyethersulfone membrane using nitrogen gas, and the concentration was measured by comparing its absorbance at 280 nm and the protein’s extinction coefficient (Δϵ, 32 555 m−1 cm−1), determined by the ExPASy’s ProtParam tool.30 Bovine serum albumin (BSA) at a concentration of 20 mg/mL was added to the R220A pure protein for stabilization.
Kinetics.
All kinetic parameters were obtained using an Agilent 8453 Diode Array spectrophotometer with the reactions in 10 mM PBS at pH 7.4 at room temperature.3,11 For determining the amount of inhibitor required to reduce activity by 50%, or IC50, E. coli DH10B expressing the PenA1 and the variants were grown and lysed as described above for immunoblotting. Using the crude extracts, IC50 values were determined using a direct competition assay under steady-state conditions.31 Nitrocefin was used as the reporter substrate at a fixed concentration of 100 μM. Crude extracts, avibactam, and nitrocefin were mixed manually, and the initial velocity was monitored. Inverse initial steady-state velocities (1/v0) versus inhibitor concentration (I) were plotted, and the IC50 values were determined by dividing the value for the y-intercept by the slope of line.32
The apparent Ki (Ki-app) values were determined as described above for IC50 values except using purified PenA1 or the R220A variant. Also, the data were corrected to account for the use of nitrocefin as a reporter substrate (PenA1 Km = 147 μM and R220A variant Km = 193 μM) via eq 1.
| (1) |
To obtain the acylation rate k2/K, PenA1 or the R220A variant was mixed with increasing concentrations of avibactam using nitrocefin at 100 μM as a reporter substrate and reactions were monitored over time.6 Progress curves were fit to eq 2 to obtain kobs values.
| (2) |
Here, Vf is the final velocity, V0 is the initial velocity, and A0 is the initial absorbance at λ = 482 nm. The data were plotted as kobs vs [inhibitor]. If a saturation curve was obtained from the kobs vs [inhibitor] graph, then a modified Michaelis−Menten equation was used to determine k2/Kobs, previously described as kinact/KI.31 Conversely, if a line was obtained, then the slope of the line was determined to be k2/Kobs.6 The k2/Kobs values were adjusted for the use of nitrocefin as an indicator substrate to obtain the k2/K value (eq 3).
| (3) |
Partition ratios or turnover number (tn) at 15 min for PenA1 and the R220A variant with all β-lactamase inhibitors except vaborbactam (where this value is stoichiometry of inactivation, as it does not undergo side reactions or hydrolysis) were obtained by incubating the enzymes with increasing concentrations of inhibitor at room temperature in 10 mM PBS, pH 7.4. The ratio of inhibitor to enzyme (I:E) necessary to inhibit the hydrolysis of nitrocefin by greater than 95% was determined.
Crystallography.
For crystallography, another protein expression plasmid was constructed by having the blaPenA1 synthesized with codons optimized for expression in E. coli (Hokkaido System Science) and cloned into pET48b(+) expression vector with an human rhinovirus (HRV) 3C cleavage site before its multiple-cloning site. The N-terminal Ala-Arg of PenA was replaced by Gly-Pro-Leu-Gly-Ser (3 amino acids addition) to allow for cleavage by the HRV 3C protease. The R220A substitution was constructed by conducted site-directed mutagenesis of the nucleotides corresponding to position 220 using the Quikchange XL mutagenesis kit as described above. The R220A variant was purified by a HisTrap FF crude column (GE Healthcare) equilibrated with 20 mM sodium phosphate buffer pH 7.4, 20 mM imidazole, 0.5 M NaCl, followed by the cleavage of His-tag by HRV 3C protease. The cleaved protein was again purified by using the HisTrap HP column (GE Healthcare). HiTrap SP HP (GE Healthcare) equilibrated with 20 mM MES buffer, pH 6.3 was used for the next step and eluted with a 0−0.5 M NaCl gradient. The protein was further purified by gel filtration using a HiLoad 16/60 Superdex 200 pg (GE Healthcare) system with a running buffer of 20 mM HEPES pH 7.5 and 100 mM NaCl. The protein was concentrated to 15 mg/mL. The R220A variant β-lactamase was crystallized at room temperature by the vapor diffusion method using a 250 μL reservoir (25% polyethylene glycol 8 kDa [PEG8K], 0.1 M MES at pH 6.5)) with a 4 μL hanging drop (7.5 mg/mL protein, 12.5% PEG8K, 0.05 M MES at pH 6.5). Adding acetone to the drop (final 4%) improved the crystal size and shape.
The R220A variant crystal was cryoprotected by the addition of 20% glycerol to the PEG holding solution. Loop-mounted crystals were flash-cooled and kept at 100 K with a nitrogen gas stream. The 1.0° oscillation images were collected on a Pilatus3 S2M detector (DECTRIS AG, Baden, Switzerland) with synchrotron radiation at beamline NW12A of the Advanced Ring of the Photon Factory, Tsukuba, Japan. Only one crystal was used. The XDS programs were used to reduce and scale X-ray intensities (Table 4).33 Molecular replacement using the PenA1 structure (PDB 3W4Q) as a search model and model refinement were done using the PHENIX program.34 Coot was used for manual model fitting.35 The resolution limit was determined so that the I/σ would be higher than 2.0. Authors will release the atomic coordinates (PDB 7D5J) and experimental data upon article publication.
MD Simulation and Analysis of Simulation Results.
The initial coordinates of the atoms in PenA1 and R220A β-lactamases were respective crystal structures. The crystal waters except for the deacylation and oxyanion waters were removed, and then water molecules were generated around the substrate and enzyme by the 3D-RISM atom placement algorithm.36 The β-lactamases were placed in a rectangular box filled with TIP3P water molecules. The model also contains sodium and chloride ions to neutralize the model system. The final model size was ca. 80 Å × 70 Å × 87 Å and the total number of atoms was about 48 000 for both models. The ff14SB force field was applied to the enzyme.37 Minimization, heating, and pre-equilibration were carried out using the sander module of AMBER18.38 Production run of MD simulation was carried out using the pmemd module.39 Calculation procedure is similar to previous works.40,41 The production runs of MD simulation were carried out for 200 ns. The cutoff distance for the electrostatic and van der Waals energy terms was set to 12.0 Å. The particle mesh Ewald method was applied to calculate the long-distance electrostatic force. The integration time step was 1 fs. The ptraj module was utilized to obtain the snapshot structures from the simulation trajectory, the root-mean-square deviation (RMSD), and the B-factor. In the calculation of the B-factor, only main chain atoms (N, Cα, and C) were taken into account.
Molecular Modeling.
Molecular representations were obtained as previously described using the BIOVIA Discovery Studio 2017 (DS2017; Accelrys, Inc., San Diego, CA) molecular modeling software.42 To obtain the Michaelis−Menten complexes of KPC-2 and PenA1 with vaborbactam, the apo structures of KPC-2 (PDB 2OV5) and PenA1 (PDB 3W4Q) were used. Vaborbactam with its boronate in an sp2 configuration was constructed using the Fragment Builder tools and minimized using the Standard Dynamics Cascade protocol of DS2017, and it was only successfully docked into PenA1 and KPC-2 using the LibDock module of DS2017.43–45 To generate the acyl−enzyme complex of the R220A variant with avibactam, the crystal coordinates of the R220A variant (PDB 7D5J) were used and the acyl−avibactam from the PenA1−avibactam crystal structure (PDB 7DOO) was minimized using the Standard Dynamics Cascade protocol of DS2017 and automatically docked in the R220A variant using the the Flexible docking module of DS2017. In all cases, the crystallographic water molecules were maintained during modeling or added after molecular docking. The β-lactamases were solvated and minimized to a root-mean-square deviation of 0.03 Å using the conjugate gradient method. The generated conformations were further analyzed. The best conformations (e.g., avibactam carbonyl/vaborbactam boronate oriented toward the oxyanion hole) were evaluated, and the best scoring poses were reported. The scoring was based on LibDock scores and the energy values (i.e., the internal ligand energy and the receptor−ligand interaction energy), which are reported as negative values (lower values indicate more favorable binding. This enables the energy to be used as a scoring function.
ACKNOWLEDGMENTS
Research reported in this publication was supported by funds and/or facilities provided by the Cleveland Department of Veterans Affairs, the Veterans Affairs Merit Review Program BX002872 to K.M.P.-W. from the U.S. Department of Veterans Affairs Biomedical Laboratory Research and Development Service. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government. Funds from the Cystic Fibrosis Foundation #691309 also support the laboratory of K.M.P.-W. These studies were partly supported by the Grant-in-Aid for Scientific Research (C) Grant Number JP 20K07484 to M.N. This work has been performed under the approval of the Photon Factory Program Advisory Committee (Proposal No. 2018G613).
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acsinfecdis.0c00682
The authors declare no competing financial interest.
Contributor Information
Michiyoshi Nukaga, Department of Pharmaceutical Sciences, Josai International University, Togane City, Chiba 283-8555, Japan.
Michael J. Yoon, Research Service, Louis Stokes Cleveland VAMC, Cleveland, Ohio 44106, United States; Department of Biochemistry, Case Western Reserve University, Cleveland, Ohio 44106, United States
Magdalena A. Taracilia, Research Service, Louis Stokes Cleveland VAMC, Cleveland, Ohio 44106, United States; Department of Medicine, Case Western Reserve University, Cleveland, Ohio 44106, United States
Tyuji Hoshino, Graduate School of Pharmaceutical Sciences, Chiba University, Chuo-ku, Chiba 263-8522, Japan.
Krisztina M. Papp-Wallace, Research Service, Louis Stokes Cleveland VAMC, Cleveland, Ohio 44106, United States; Department of Biochemistry and Department of Medicine, Case Western Reserve University, Cleveland, Ohio 44106, United States;.
REFERENCES
- (1).Ehmann DE, Jahic H, Ross PL, Gu RF, Hu J, Durand-Reville TF, Lahiri S, Thresher J, Livchak S, Gao N, Palmer T, Walkup GK, and Fisher SL (2013) Kinetics of avibactam inhibition against class A, C, and D β-lactamases. J. Biol. Chem 288 (39), 27960–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Becka SA, Zeiser ET, Marshall SH, Gatta JA, Nguyen K, Singh I, Greco C, Sutton GG, Fouts DE, LiPuma JJ, and Papp-Wallace KM (2018) Sequence heterogeneity of the PenA carbapenemase in clinical isolates of Burkholderia multivorans. Diagn. Microbiol. Infect. Dis 92 (3), 253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Papp-Wallace KM, Taracila MA, Gatta JA, Ohuchi N, Bonomo RA, and Nukaga M (2013) Insights into β-lactamases from Burkholderia species, two phylogenetically related yet distinct resistance determinants. J. Biol. Chem 288 (26), 19090–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Drawz SM, and Bonomo RA (2010) Three decades of β-lactamase inhibitors. Clin. Microbiol. Rev 23 (1), 160–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Papp-Wallace KM, Bethel CR, Caillon J, Barnes MD, Potel G, Bajaksouzian S, Rutter JD, Reghal A, Shapiro S, Taracila MA, Jacobs MR, Bonomo RA, and Jacqueline C (2019) Beyond piperacillin-tazobactam: cefepime and AAI101 as a potent β-lactam-β-lactamase inhibitor combination. Antimicrob. Agents Chemother 63 (5), e00105–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ehmann DE, Jahic H, Ross PL, Gu RF, Hu J, Kern G, Walkup GK, and Fisher SL (2012) Avibactam is a covalent, reversible, non-β-lactam β-lactamase inhibitor. Proc. Natl. Acad. Sci. U.S. A 109 (29), 11663–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lohans CT, Brem J, and Schofield CJ (2017) New Delhi metallo-β-lactamase 1 catalyzes avibactam and aztreonam hydrolysis. Antimicrob. Agents Chemother 61 (12), e01224–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Choi H, Paton RS, Park H, and Schofield CJ (2016) Investigations on recyclisation and hydrolysis in avibactam mediated serine β-lactamase inhibition. Org. Biomol. Chem 14 (17), 4116–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Abboud MI, Damblon C, Brem J, Smargiasso N, Mercuri P, Gilbert B, Rydzik AM, Claridge TD, Schofield CJ, and Frere JM (2016) Interaction of avibactam with class B metallo-β-lactamases. Antimicrob. Agents Chemother 60 (10), 5655–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Papp-Wallace KM (2019) The latest advances in β-lactam/β-lactamase inhibitor combinations for the treatment of Gram-negative bacterial infections. Expert Opin. Pharmacother 20 (17), 2169–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Papp-Wallace KM, Becka SA, Zeiser ET, Ohuchi N, Mojica MF, Gatta JA, Falleni M, Tosi D, Borghi E, Winkler ML, Wilson BM, LiPuma JJ, Nukaga M, and Bonomo RA (2017) Overcoming an extremely drug resistant (XDR) pathogen: avibactam restores susceptibility to ceftazidime for Burkholderia cepacia complex isolates from cystic fibrosis patients. ACS Infect. Dis 3 (7), 502–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zeiser ET, Becka SA, Wilson BM, Barnes MD, LiPuma JJ, and Papp-Wallace KM (2019) ″Switching partners″: piperacillinavibactam is a highly potent combination against multidrug-resistant Burkholderia cepacia complex and Burkholderia gladioli cystic fibrosis isolates. J. Clin. Microbiol 57 (8), e00181–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Pemberton OA, Tsivkovski R, Totrov M, Lomovskaya O, and Chen Y (2020) Structural basis and binding kinetics of vaborbactam in class A β-lactamase inhibition. Antimicrob. Agents Chemother 64 (10), e00398–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Papp-Wallace KM, Winkler ML, Taracila MA, and Bonomo RA (2015) Variants of β-lactamase KPC-2 that are resistant to inhibition by avibactam. Antimicrob. Agents Chemother 59 (7), 3710–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Barnes MD, Taracila MA, Good CE, Bajaksouzian S, Rojas LJ, van Duin D, Kreiswirth BN, Jacobs MR, Haldimann A, Papp-Wallace KM, and Bonomo RA (2019) Nacubactam Enhances Meropenem Activity against Carbapenem-Resistant Klebsiella pneumoniae Producing KPC. Antimicrob. Agents Chemother 63 (8), e00432–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Winkler ML, Rodkey EA, Taracila MA, Drawz SM, Bethel CR, Papp-Wallace KM, Smith KM, Xu Y, Dwulit-Smith JR, Romagnoli C, Caselli E, Prati F, van den Akker F, and Bonomo RA (2013) Design and exploration of novel boronic acid inhibitors reveals important interactions with a clavulanic acid-resistant sulfhydryl-variable (SHV) β-lactamase. J. Med. Chem 56 (3), 1084–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tsivkovski R, and Lomovskaya O (2020) Biochemical activity of vaborbactam. Antimicrob. Agents Chemother 64 (2), e01935–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Stojanoski V, Adamski CJ, Hu L, Mehta SC, Sankaran B, Zwart P, Prasad BV, and Palzkill T (2016) Removal of the side chain at the active-site serine by a glycine substitution increases the stability of a wide range of serine β-lactamases by relieving steric strain. Biochemistry 55 (17), 2479–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Thomson JM, Distler AM, Prati F, and Bonomo RA (2006) Probing active site chemistry in SHV β-lactamase variants at Ambler position 244. Understanding unique properties of inhibitor resistance. J. Biol. Chem 281 (36), 26734–44. [DOI] [PubMed] [Google Scholar]
- (20).Lin S, Thomas M, Mark S, Anderson V, and Bonomo RA (1999) OHIO-1 β-lactamase mutants: the Arg244Ser mutant and resistance to β-lactams and β-lactamase inhibitors. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol 1432 (1), 125–36. [DOI] [PubMed] [Google Scholar]
- (21).Giakkoupi P, Tzelepi E, Legakis NJ, and Tzouvelekis LS (1998) Substitution of Arg-244 by Cys or Ser in SHV-1 and SHV-5 β- lactamases confers resistance to mechanism-based inhibitors and reduces catalytic efficiency of the enzymes. FEMS Microbiol. Lett 160 (1), 49–54. [DOI] [PubMed] [Google Scholar]
- (22).Zafaralla G, Manavathu EK, Lerner SA, and Mobashery S (1992) Elucidation of the role of arginine-244 in the turnover processes of class A β-lactamases. Biochemistry 31 (15), 3847–52. [DOI] [PubMed] [Google Scholar]
- (23).Papp-Wallace KM, Taracila MA, Smith KM, Xu Y, and Bonomo RA (2012) Understanding the molecular determinants of substrate and inhibitor specificities in the carbapenemase KPC-2: exploring the roles of Arg220 and Glu276. Antimicrob. Agents Chemother 56 (8), 4428–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ruggiero M, Papp-Wallace KM, Brunetti F, Barnes MD, Bonomo RA, Gutkind G, Klinke S, and Power P (2019) Structural insights into the inhibition of the extended-spectrum β-lactamase PER-2 by avibactam. Antimicrob. Agents Chemother 63 (9), e00487–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Ruggiero M, Curto L, Brunetti F, Sauvage E, Galleni M, Power P, and Gutkind G (2017) Impact of mutations at Arg220 and Thr237 in PER-2 β-lactamase on conformation, activity, and susceptibility to inhibitors. Antimicrob. Agents Chemother 61 (6), e02193–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Canton R, Morosini MI, Martin O, de la Maza S, and de la Pedrosa EGG (2008) IRT and CMT beta-lactamases and inhibitor resistance. Clin. Microbiol. Infect 14 (Suppl 1), 53–62. [DOI] [PubMed] [Google Scholar]
- (27).Poirel L, Mammeri H, and Nordmann P (2004) TEM-121, a novel complex mutant of TEM-type β-lactamase from Enterobacter aerogenes. Antimicrob. Agents Chemother 48 (12), 4528–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Papp-Wallace KM, Becka SA, Taracila MA, Zeiser ET, Gatta JA, LiPuma JJ, and Bonomo RA (2017) Exploring the role of the Ω-loop in the evolution of ceftazidime resistance in the PenA β-lactamase from Burkholderia multivorans, an important cystic fibrosis pathogen. Antimicrob. Agents Chemother 61 (2), e01941–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Clinical and Laboratory Standards Institute (CLSI) (2020) Performance Standards for Antimicrobial Susceptibility Testing, 30th ed., CLSI supplement, Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- (30).Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, and Bairoch A (2005) The Proteomics Protocols Handbook; Humana Press. [Google Scholar]
- (31).Papp-Wallace KM, Bethel CR, Distler AM, Kasuboski C, Taracila M, and Bonomo RA (2010) Inhibitor resistance in the KPC-2 β-lactamase, a preeminent property of this class A β-lactamase. Antimicrob. Agents Chemother 54 (2), 890–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kopelovich L, Sweetman L, and Nisselbaum JS (1971) Kinetics of the inhibition of aspartate aminotransferase isozymes by DL-glyceraldehyde 3-phosphate. Eur. J. Biochem 20 (3), 351–62. [DOI] [PubMed] [Google Scholar]
- (33).Kabsch W (2010) Xds. Acta Crystallogr., Sect. D: Biol. Crystallogr 66 (Pt 2), 125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Liebschner D, Afonine PV, Baker ML, Bunkoczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJ, Moriarty NW, Oeffner RD, Poon BK, Prisant MG, Read RJ, Richardson JS, Richardson DC, Sammito MD, Sobolev OV, Stockwell DH, Terwilliger TC, Urzhumtsev AG, Videau LL, Williams CJ, and Adams PD (2019) Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct Biol 75 (Pt 10), 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010) Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 66 (Pt 4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Sindhikara DJ, Yoshida N, and Hirata F (2012) Placevent: an algorithm for prediction of explicit solvent atom distribution-application to HIV-1 protease and F-ATP synthase. J. Comput. Chem 33 (18), 1536–43. [DOI] [PubMed] [Google Scholar]
- (37).Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, and Simmerling C (2015) ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput 11 (8), 3696–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Case D, Betz R, Cerutti DS, Cheatham T, Darden T, Duke R, Giese TJ, Gohlke H, Götz A, Homeyer N, Izadi S, Janowski P, Kaus J, Kovalenko A, Lee T, LeGrand S, Li P, Lin C, Luchko T, and Kollman P (2016) Amber16, University of California, San Francisco. [Google Scholar]
- (39).Salomon-Ferrer R, Gotz AW, Poole D, Le Grand S, and Walker RC (2013) Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. explicit solvent Particle Mesh Ewald. J. Chem. Theory Comput 9 (9), 3878–88. [DOI] [PubMed] [Google Scholar]
- (40).Fudo S, Yamamoto N, Nukaga M, Odagiri T, Tashiro M, and Hoshino T (2016) Two distinctive binding modes of endonuclease inhibitors to the N-terminal region of influenza virus polymerase acidic subunit. Biochemistry 55 (18), 2646–60. [DOI] [PubMed] [Google Scholar]
- (41).Fudo S, Qi F, Nukaga M, and Hoshino T (2017) Influence of precipitants on molecular arrangement and space group of protein crystals. Cryst. Growth Des 17, 534–542. [Google Scholar]
- (42).Papp-Wallace KM, Taracila M, Wallace CJ, Hujer KM, Bethel CR, Hornick JM, and Bonomo RA (2010) Elucidating the role of Trp105 in the KPC-2 β-lactamase. Protein Sci. 19 (9), 1714–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Diller DJ, and Merz KM Jr. (2001) High throughput docking for library design and library prioritization. Proteins: Struct., Funct., Genet 43 (2), 113–24. [DOI] [PubMed] [Google Scholar]
- (44).Diller DJ, and Li R (2003) Kinases, homology models, and high throughput docking. J. Med. Chem 46 (22), 4638–47. [DOI] [PubMed] [Google Scholar]
- (45).Rao SN, Head MS, Kulkarni A, and LaLonde JM (2007) Validation studies of the site-directed docking program LibDock. J. Chem. Inf. Model 47 (6), 2159–71. [DOI] [PubMed] [Google Scholar]