

Abstract
Cell-cell interactions and communication are crucial to the proper function of complex mammalian physiology including neurocognitive and immune system functions. While many tools are available for observing and perturbing intracellular processes, relatively few exist to probe intercellular processes. Current techniques for studying interactions often rely on direct protein contact and few can manipulate diverse, functional outputs with tunable protein expression. To address these limitations, we have developed a small-molecule approach based on a trimethoprim prodrug-enzyme pair capable of reporting the presence of two different engineered cell populations with programable protein outputs. The approach relies on bacterial nitroreductase enzyme catalysis, which is orthogonal to normal mammalian biology, and diffusion of trimethoprim from “activator” cells to “receiver” cells. We test this strategy, which can theoretically regulate many different types of proteins, using biochemical and in vitro culture assays with optical and cytokine protein readouts. This describes the first small-molecule approach capable to detecting and controlling engineered cell-cell outputs, and we anticipate future applications especially relevant to the field of immuno-oncology.
Graphical Abstract
Introduction:
Cell-cell interactions underpin many physiological processes such as neuronal signaling1, immune responses to infection2, and tissue patterning during development3. Several tools have been developed to report and characterize cell interactions, often using fluorescent proteins. An example is complementary split-GFP fragments to delineate synaptic partners in the central nervous system4. Another example, LIPSTIC5, is used in an antigen presentation model where a membrane bound bacterial enzyme tags an interacting cell with a fluorophore. Finally, Synthetic Notch or SynNotch6 are chimeric receptors that rely on peptide-based recognition of epitope binding on cognate cells which drive a programmable genetic/protein output. However, most of these newer technologies rely on direct protein contact to elicit signal. A small-molecule based platform that uses passive diffusion from one cell to another may allow signaling over greater distance and over a longer period of time. Other groups have developed platforms where a small molecule’s native functionality is hindered by chemical appendages such that they conditionally report cell interactions. For example, quenched fluorescein esters could not fluoresce unless an orthogonal esterase removed the ester7 in specific cells of interest. In another instance, D-luciferin was modified with a galactose sugar molecule, preventing binding to firefly luciferase enzyme, unless activated by a beta-galactosidase in a nearby cell8. Yet these small-molecule technologies only report interactions optically and using fluorescence and bioluminescence respectively, and thus are not geared to drive programmable protein outputs.
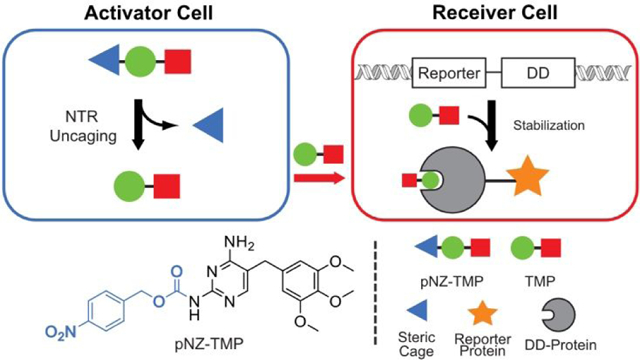
Here we report a strategy using the small-molecule antibiotic trimethoprim to control protein expression when two cell populations of interest are present in co-culture, thus creating a cellular “AND gate”. Our system uses the synthetic antibiotic trimethoprim (TMP), which is biologically inert in mammalian systems as it has over 10,000-fold selectivity for the E. coli dihydrofolate reductase (eDHFR) over the mammalian homologue9. Furthermore, the structure activity relationship of TMP is well characterized, as several studies have shown that the primary amines on the pyrimidine ring are crucial for high affinity binding10–12. To abrogate binding to eDHFR and develop a TMP prodrug, we derivatized the 2-position amine with a protecting group labile to enzymatic cleavage by orthogonal E. coli nitroreductase (NTR) expressed in “activator” cells. Moreover, we fused reporters, thermostable luciferase (tsLuc)13, yellow fluorescent protein (YFP), and interleukin-2 (IL-2) to a destabilized-domain (DD) eDHFR14 that is constitutively degraded unless bound to TMP ligand in a population of “receiver” cells. Therefore, our system conditionally returns signal only when activator- and receiver-cells are both present (Figure 1A-B).
Figure 1.
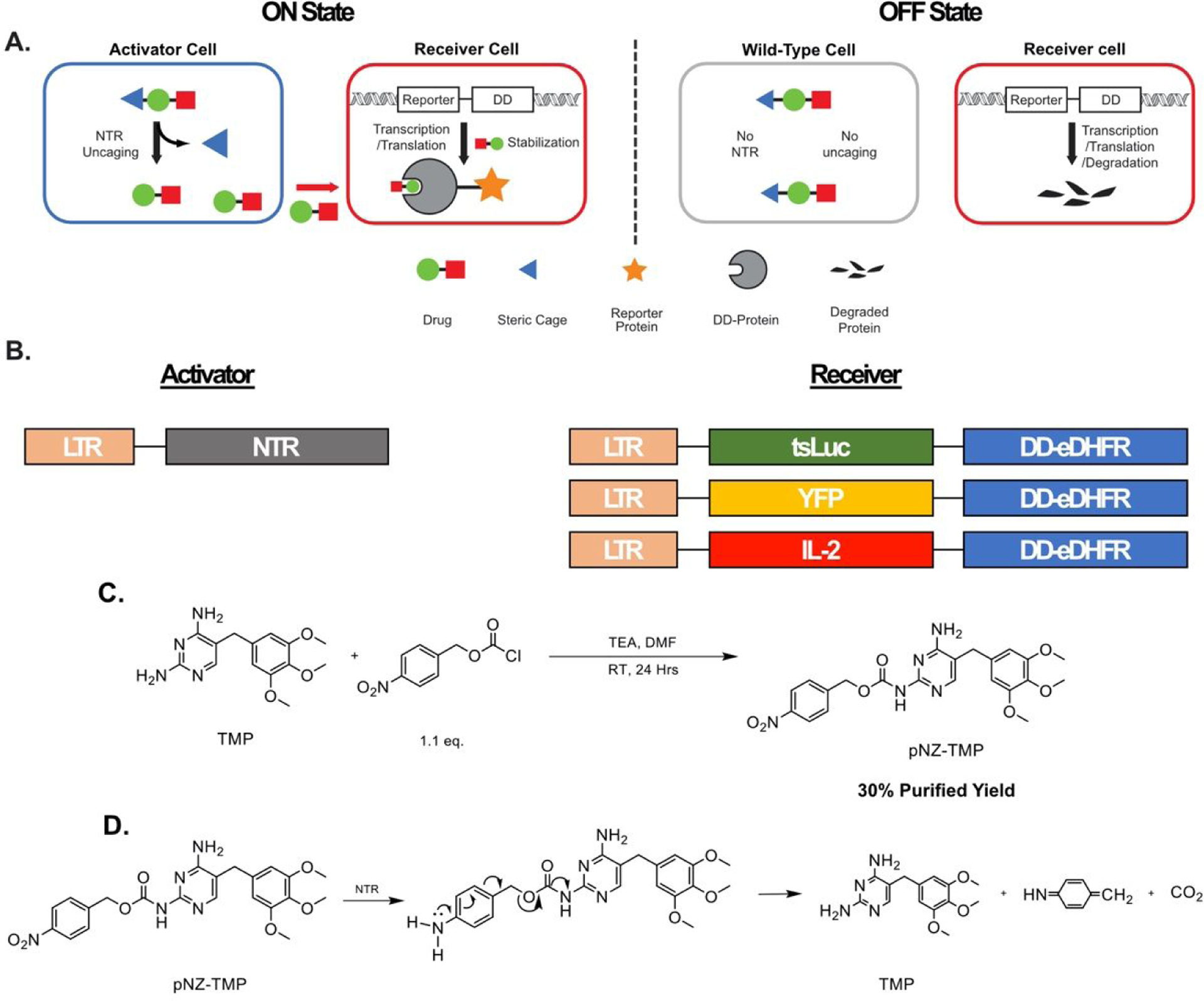
Schematic and synthesis of a chemical approach for programmable protein outputs based on engineered cells. (A) The system is composed of two cell types: an activator-cell population, bearing the bacterial nitroreductase enzyme, and a receiver-cell population, bearing a reporter-destabilizing domain fusion. In the ON-state, sterically “caged” prodrug, pNZ-TMP, is un-caged in activator-cells and diffuses out creating a local pool of free TMP. The free TMP diffuses into receiver-cells, binds to the destabilized domain, and prevents degradation of the entire Reporter-DD fusion. In the OFF-state, a receiver-cell population not in the presence of nitroreductase positive cells, is not exposed to free TMP resulting in constitutive degradation of the Reporter-DD. (B) Genetic constructs used for creation of activator- and receiver-cells. (C) One step synthesis of pNZ-TMP (D) Proposed uncaging mechanism of pNZ-TMP requiring complete reduction of the nitro substituent to the primary amine. The amine can then donate electrons into the aromatic ring, resulting in the cleavage of the carbamate linker, yielding TMP.
Results and Discussion:
The common peptide protecting group, para-nitrobenzyl carbamate15 (pNZ), was selected to derivatize TMP using a facile, one step synthesis to produce pNZ-TMP (30% purified yield, Figure 1C and S1–3). The prodrug was confirmed by LC-MS to release native TMP by chemical reduction using known conditions15 (Figure S4). Three types of receiver-cells were generated. One expressed a luminescent reporter using a thermostable firefly luciferase (tsLuc) construct, the second expressed a YFP14, and the third expressed IL-2. All were fused to a C-terminal destabilizing domain (DD), eDHFR, resulting in tsLuc-DD-eDHFR, YFP-DD-eDHFR, and IL-2-DD-eDHFR, respectively. The genetic constructs were transduced into HEK-293 or HCT-116 cells using retro or lentivirus and sorted by FACS. Activator-cells were prepared in a similar fashion using hemagglutinin (HA) tagged E. coli nitroreductase (nfsB).
The first objective was to determine if the steric pNZ cage prevented binding of the prodrug to eDHFR. For a dose response assay, luminescent receiver-cells were administered pNZ-TMP or TMP for 24-hour incubation. The results showed that TMP has a characteristic induction profile with ~5 nM EC50, which is similar to the known KD with E. coli DHFR16. Conversely, pNZ-TMP showed a right-shifted, flattened luminescent-induction curve with ~27-fold worse EC50 (EC50 139 nM, Figure 2A) that did not reach saturation. Dose response studies showed with YFP and IL-2 receiver cells also exhibited a flattened, right shifted profile when incubated with pNZ-TMP (Figure 2B–C). Time course assays with the fluorescent receiver-cells show differences, between TMP and pNZ-TMP abilities to bind and stabilize the DD-DHFR, in as little as 3 hours and suggest the overall serum stability of the pNZ caging group in complete media with cells (Figure 2D). Thus, derivatizing the 2-position amine with a pNZ moiety significantly hampers the ability of TMP to stabilize the DD.
Figure 2.
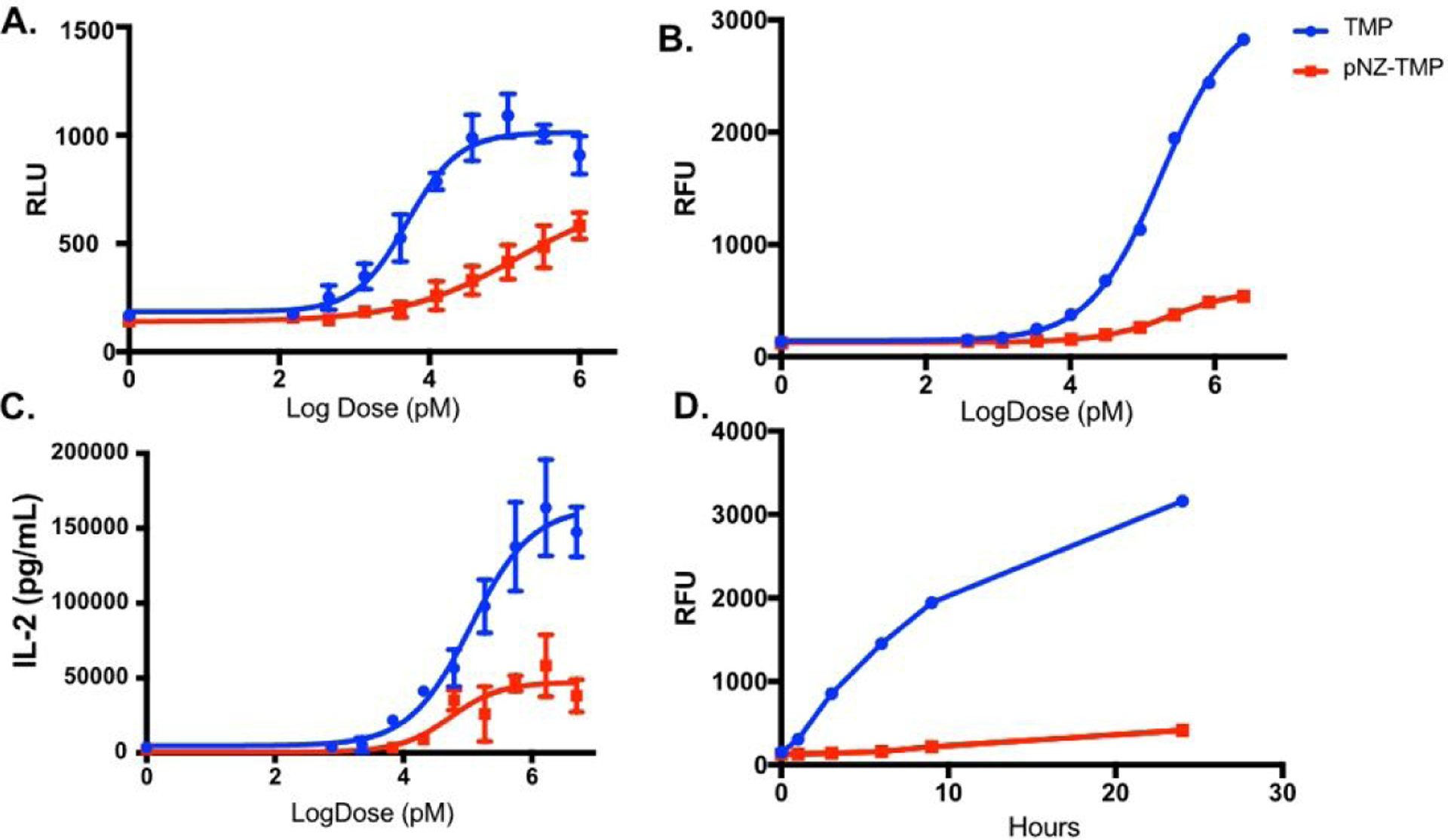
Prodrug binding assays. (A) Luminescent output of HCT-116 receiver-cells with varying doses of either TMP or pNZ-TMP. TMP shows an EC50 of 5 nM while pNZ has significantly reduced affinity. (B) Dose response assay with TMP and pNZ-TMP using destabilized YFP as reporter protein. (C) Dose response assay with TMP and pNZ-TMP using destabilized IL-2 as reporter protein. (D) Time course assay of reporter protein stabilization in fluorescent receiver-cells, with either TMP or pNZ-TMP, shows differences in fluorescence as early as 3 hours. Each experiment was repeated 3 times. Data is from a representative experiment; error bars represent S.D. for n = 6, 3, 4, and 4 technical replicates, respectively (A, B, C, & D).
Activator-cell protein expression of NTR was confirmed by western blot (Figure 3A) and function was assessed by testing their sensitivity to CB1954, a nitroreductase-activatable cytotoxic prodrug17. Comparison of HCT-116 wild-type (WT) and HCT-116 activator-cells incubated in the presence of CB1954 showed that activator-cells were significantly more sensitive to CB1954 cytotoxicity (Figure S6). To determine if pNZ-TMP could be uncaged by NTR, we performed a media transfer experiment, where pNZ-TMP was incubated with HCT-116 activator-cells or WT cells. After various incubation lengths (0–7 hours), the media was carefully removed, ensuring no cells were detached, and transferred to a plate containing HCT-116 receiver-cells. The receiver-cells were incubated with transferred media for 24 hours, at which point their luminescent intensity was measured (Figure 3B). Increasing exposure time of pNZ-TMP to NTR expressing activator-cells resulted in increasing luminescence (blue). Conversely, increasing incubation time of pNZ-TMP with WT cells that do not express NTR had no effect on luminescence (green). The media transfer experiment was then performed with HCT-116 IL-2-DD-eDHFR receiver cells (Figure 3C). Here, drugs were incubated for 48 hours with activator cells. Similar to luminescence experiments, high IL-2 expression was observed when pNZ-TMP was incubated with activator cells but not with WT cells.
Figure 3.
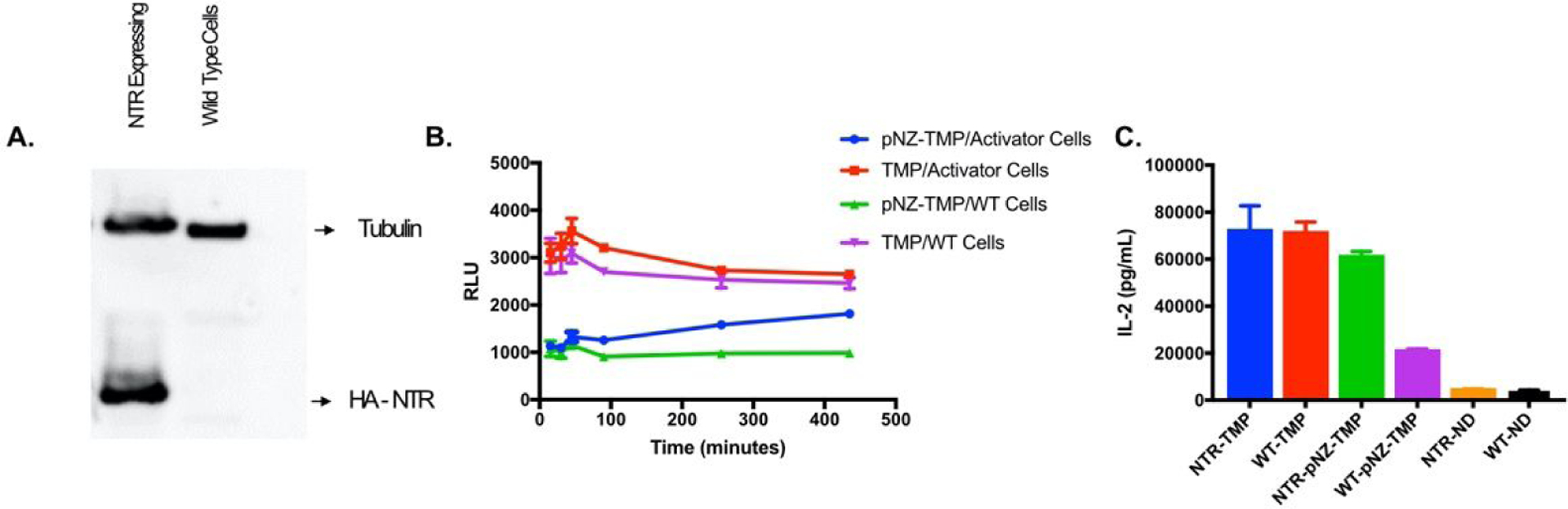
Selective activation of pNZ-TMP. (A) Western blot for HA tag on NTR in activator and WT control cells. Tubulin used as loading control. (B) Media transfer experiment where pNZ-TMP or TMP was incubated in the media of either activator or WT control cells for varying lengths of time before media was transferred to receiver-cells and incubated for another 24 hours. Receiver-cells cultured with pNZ-TMP containing media from activator-cells showed a time dependent increase in luminescent stabilization (blue). Conversely, receiver-cells cultured in pNZ-TMP containing media from WT cells showed consistently low luminescent stabilization (green). (C) Media Transfer experiment where pNZ-TMP or TMP was incubated in the media of either activator or WT control cells for 48 hours. The media was then transferred to IL-2-DD receiver cells and incubated for 24 hours before IL-2 was measured via ELISA. The experiments were repeated a total of three 3 times. Data are from a representative experiment; error bars represent S.D. for n = 4 technical replicates (B & C).
Lastly, a co-culture experiment was performed where activator or WT cells were plated together with receiver-cells (Figure 4A&C) to determine if the system could report interaction of two cell populations of interest in real-time. The ratio of activator- to receiver-cells was varied by changing the number of activator or WT cells while keeping the number of receiver-cells constant. Co-culture of WT with receiver-cells resulted in low-level background luminescent stabilization when pNZ-TMP was administered, but the ratio between the two cell types had no effect (purple curve) on luminescence. However, increasing the number of activator-cells in the co-culture resulted in increased luminescent output when pNZ-TMP was administered (blue curve) to the point of matching native TMP positive controls. Luminescence was not affected by WT or activator-cells when TMP was administered. Similar results were observed with IL-2 receiver cells, though the activated pNZ-TMP condition did not match the TMP positive control. A time course experiment with co-culture of WT/receiver-cells or activator/receiver-cells (Figure 4B) showed similar results. pNZ-TMP incubated with activator- and receiver-cells showed increasing luminescence over time, matching native TMP controls; WT and receiver-cell co-culture with pNZ-TMP administration showed low luminescence over multiple time points.
Figure 4.
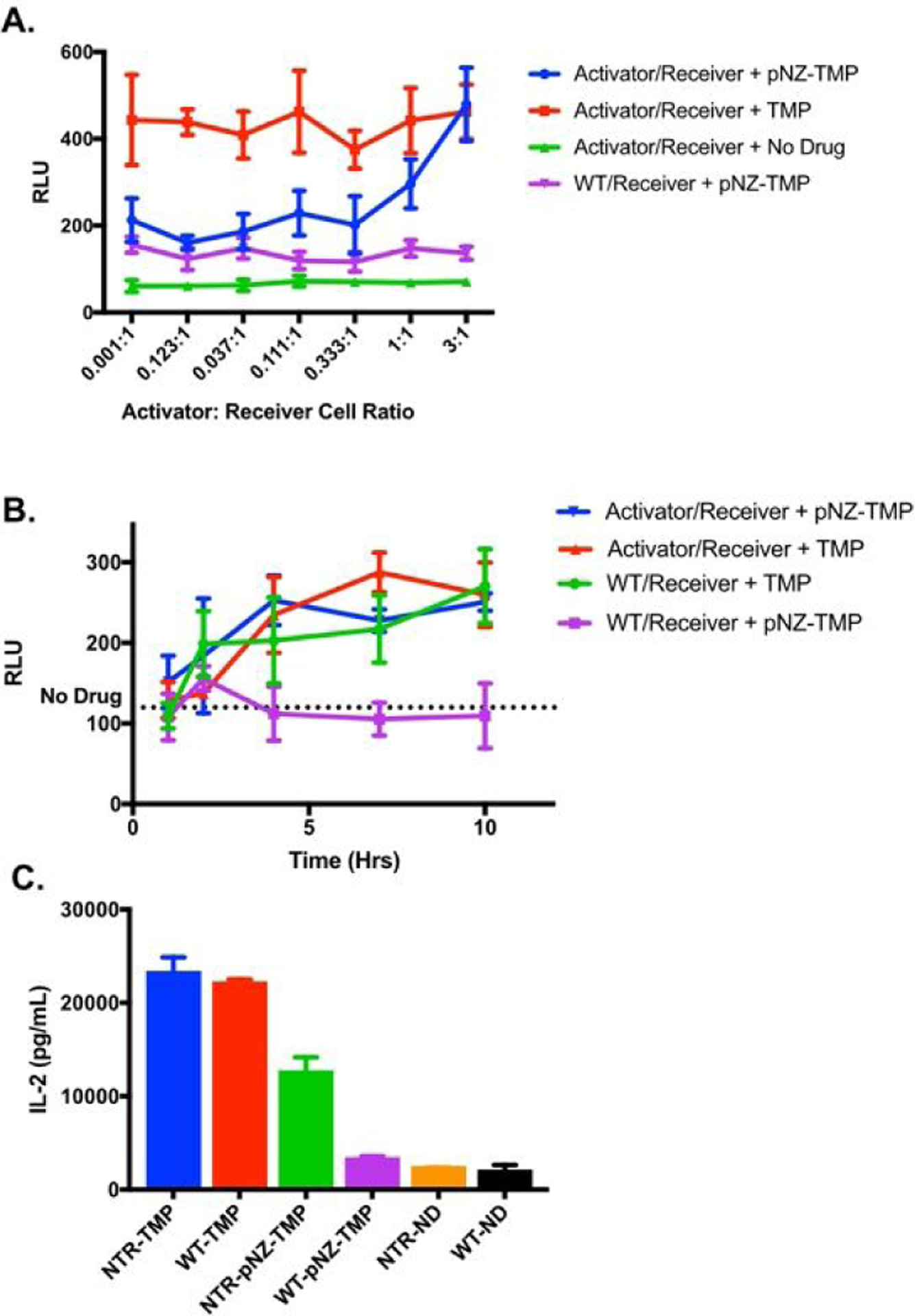
Co-culture experiments show specific uncaging and output stabilization over time and activator cell concentration. (A) Co-culture of WT and receiver-cells with pNZ-TMP shows low background luminescent stabilization (purple). Co-culture of activator- and receiver-cells with pNZ TMP shows increasing luminescence (blue) as the number of activator-cells increases until parity with TMP positive control (red). (B) Co-culture of activator- and receiver-cells with either pNZ-TMP or TMP (blue & red) shows increasing luminescent stabilization over time, while WT and receiver co-culture with pNZ-TMP (purple) remains near drug-free control levels. (C) Co-culture of activator or WT cells with IL-2-DD receiver cells with pNZ-TMP shows higher IL-2 concentrations when the activator and receiver cell pair are cultured together. Each experiment was repeated a total of 3 times. Data is from a representative experiment; error bars represent S.D. for n = 4 (A & C) and n = 5 (B) technical replicates.
Through genomic, proteomic, single cell analyses and other technical advances, a broad spectra of cell types with incredible diversity have been uncovered within tissues that were thought to be relatively homogeneous18. Untangling these network level interactions between cells will facilitate understanding of fundamental biology as well as disease pathophysiology. Of particular value are tools capable of tracking and manipulating specific cell populations of interest. To that end, we have developed a small-molecule strategy capable of discerning presence of two cell populations of interest. We have synthesized a sterically “caged” TMP with 27-fold reduced binding affinity for reporter-DD protein fusions in dose response assays. The “caged” prodrug is enzymatically liberated by an activator-cell, restoring high affinity for the protein target, and can diffuse into the surrounding medium as shown by a media transfer experiment. If the liberated drug reaches a nearby receiver-cell in sufficient concentration, it can stabilize DD fusion proteins eliciting a protein signal output, thereby reporting an interaction. This process was demonstrated in our co-culture assays, as high luminescent or cytokine signal required an activator- and receiver-cell to interact.
We used NTR as an uncaging enzyme orthogonal to mammalian nitroreductases. This rendered the TMP prodrug silent in a native mammalian system. However, other orthogonal enzymes such as beta-galactosidase8 or beta-lactamase19 have been used to conditionally unmask reporter systems. Moreover, a larger beta-lactam moiety or galactose sugar molecule might reduce the background DD stabilization we observed with pNZ-TMP in our dose-response and co-culture assays. Beta-lactamases and beta-galactosidases also have the added benefit of typically higher Kcat values for their substrates than bacterial nitroreductase20–22.
While we used optical proteins and a cytokine as our readout, the destabilizing system is capable of conditionally regulating many therapeutically relevant proteins including membrane or other secreted proteins. As cell-based therapies, such as CAR-T cells, mature and become more sophisticated, protein regulatory and gated control systems are likely to be routinely considered as adjunct, supporting technologies with great potential impact. With our system, researchers could use a gated approach targeting two antigens, which is a growing area of interest23,24. For example, one CAR-T cell bearing an activating enzyme could turn on protein expression in a second CAR-T cell targeting a less specific expressed antigen. This gated process would only occur in the presence of both CAR T populations, thus reducing the potential for individual CAR T cell “on-target/off-tumor” toxicity. Furthermore, use of activating enzyme that is tumor intrinsic and cognate small molecule cage could add another layer of regulation and/or safety. Many solid tumors use matrix metalloproteinases (MMPs)-2 and −9 in their metastatic processes. Previously, these enzymes have been leveraged in a prodrug strategy by conjugating their known peptide substrates to cytotoxic drugs such as doxorubcin25,26. With this system, it may be possible to use these or other natively expressed MMPs as activating enzymes to regulate therapeutic cell systems.
Supplementary Material
Acknowledgements
We thank R. Mach, J. Prescher, and R. Kohli for helpful discussions.
Funding Sources
No competing financial interests have been declared.
Research reported in this publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number UL1TR001878. MAS and this research is supported by the Burroughs Wellcome Fund Career Award for Medical Scientists, NIH Office of the Director Early Independence Award (DP5-OD26386). DAJ is supported by the NIH Pharmacological Sciences Predoctoral Research Training Program (NIHT32 GM008076), JDN was supported by the University of Pennsylvania Institute for Translational Medicine and Therapeutics (ITMAT) and NIH 5TL1TR001880-04.
Footnotes
Publisher's Disclaimer: This document is confidential and is proprietary to the American Chemical Society and its authors. Do not copy or disclose without written permission. If you have received this item in error, notify the sender and delete all copies.
Supporting Information. A PDF including methods all supplemental figures referenced in the text. This material is available free of charge via the Internet.
References:
- (1).Chao DL; Ma L; Shen K Transient Cell-Cell Interactions in Neural Circuit Formation. Nat. Rev. Neurosci 2009, 10 (4), 262–271. 10.1038/nrn2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Daneshpour H; Youk H Modeling Cell–Cell Communication for Immune Systems across Space and Time. Curr. Opin. Syst. Biol 2019, 18, 44–52. 10.1016/j.coisb.2019.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Mattes B; Scholpp S Emerging Role of Contact-Mediated Cell Communication in Tissue Development and Diseases. Histochem. Cell Biol 2018, 150 (5), 431–442. 10.1007/s00418-018-1732-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Wang G; Feinberg EH; Shen K; Bargmann CI; Fetter RD; VanHoven MK; Bendesky A GFP Reconstitution Across Synaptic Partners (GRASP) Defines Cell Contacts and Synapses in Living Nervous Systems. Neuron 2008, 57 (3), 353–363. 10.1016/j.neuron.2007.11.030. [DOI] [PubMed] [Google Scholar]
- (5).Pasqual G; Chudnovskiy A; Tas JMJ; Agudelo M; Schweitzer LD; Cui A; Hacohen N; Victora GD Monitoring T Cell-Dendritic Cell Interactions in Vivo by Intercellular Enzymatic Labelling. Nature 2018, 553 (7689), 496–500. 10.1038/nature25442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Morsut L; Roybal KT; Xiong X; Gordley RM; Coyle SM; Thomson M; Lim WA Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell 2016, 164 (4), 780–791. 10.1016/j.cell.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Tian L; Yang Y; Wysocki LM; Arnold AC; Hu A; Ravichandran B; Sternson SM; Looger LL; Lavis LD Selective Esterase-Ester Pair for Targeting Small Molecules with Cellular Specificity. Proc. Natl. Acad. Sci 2012, 109 (13), 4756–4761. 10.1073/pnas.1111943109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sellmyer MA; Bronsart L; Imoto H; Contag CH; Wandless TJ; Prescher JA Visualizing Cellular Interactions with a Generalized Proximity Reporter. Proc. Natl. Acad. Sci 2013, 110 (21). 10.1073/pnas.1218336110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Baccanari DP; Kuyper LF Basis of Selectivity of Antibacterial Diaminopyrimidines. J. Chemother 1993, 5 (6), 393–399. 10.1080/1120009x.1993.11741086. [DOI] [PubMed] [Google Scholar]
- (10).Rashid U; Ahmad W; Hassan SF; Qureshi NA; Niaz B; Muhammad B; Imdad S; Sajid M Design, Synthesis, Antibacterial Activity and Docking Study of Some New Trimethoprim Derivatives. Bioorganic Med. Chem. Lett 2016, 26 (23), 5749–5753. 10.1016/j.bmcl.2016.10.051. [DOI] [PubMed] [Google Scholar]
- (11).Ballister ER; Aonbangkhen C; Mayo AM; Lampson MA; Chenoweth DM Localized Light-Induced Protein Dimerization in Living Cells Using a Photocaged Dimerizer. Nat. Commun 2014, 5, 1–9. 10.1038/ncomms6475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zhang H; Aonbangkhen C; Tarasovetc EV; Ballister ER; Chenoweth DM; Lampson MA Optogenetic Control of Kinetochore Function. Nat. Chem. Biol 2017, 13 (10), 1096–1101. 10.1038/nchembio.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Tisi LC; White PJ; Squirrell DJ; Murphy MJ; Lowe CR; Murray JAH Development of a Thermostable Firefly Luciferase. Anal. Chim. Acta 2002, 457 (1), 115–123. 10.1016/S0003-2670(01)01496-9. [DOI] [Google Scholar]
- (14).Iwamoto M; Björklund T; Lundberg C; Kirik D; Wandless TJ A General Chemical Method to Regulate Protein Stability in the Mammalian Central Nervous System. Chem. Biol 2010, 17 (9), 981–988. 10.1016/j.chembiol.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Isidro-Llobet A; Guasch-Camell J; Álvarez M; Albericio F P-Nitrobenzyloxycarbonyl (PNZ) as a Temporary Na-Protecting Group in Orthogonal Solid-Phase Peptide Synthesis - Avoiding Diketopiperazine and Aspartimide Formation. European J. Org. Chem 2005, No. 14, 3031–3039. 10.1002/ejoc.200500167. [DOI] [Google Scholar]
- (16).Sellmyer MA; Lee I; Hou C; Weng C; Li S; Lieberman BP; Zeng C; Mankoff DA; Mach RH Bacterial Infection Imaging with [ 18 F]Fluoropropyl-Trimethoprim 2017, 114 (31). 10.1073/pnas.1703109114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Vass SO; Jarrom D; Wilson WR; Hyde EI; Searle PFE. Coli NfsA : An Alternative Nitroreductase for Prodrug Activation Gene Therapy in Combination with CB1954 2009, 100 (12), 1903–1911. 10.1038/sj.bjc.6605094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Gartner ZJ; Prescher JA; Lavis LD Unraveling Cell-to-Cell Signaling Networks with Chemical Biology. Nat. Chem. Biol 2017, 13 (6), 564–568. 10.1038/nchembio.2391. [DOI] [PubMed] [Google Scholar]
- (19).Yao H; So MK; Rao J A Bioluminogenic Substrate for in Vivo Imaging of β-Lactamase Activity. Angew. Chemie - Int. Ed 2007, 46 (37), 7031–7034. 10.1002/anie.200701931. [DOI] [PubMed] [Google Scholar]
- (20).Pitsawong W; Hoben JP; Miller AF Understanding the Broad Substrate Repertoire of Nitroreductase Based on Its Kinetic Mechanism. J. Biol. Chem 2014, 289 (22), 15203–15214. 10.1074/jbc.M113.547117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zygmunt DJ; Stratton CW; Kernodle DS Characterization of Four β-Lactamases Produced by Staphylococcus Aureus. Antimicrob. Agents Chemother 1992, 36 (2), 440–445. 10.1128/AAC.36.2.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Golan R; Zehavi U; Naim M; Patchornik A; Smirnoff P Inhibition of Escherichia Coli β-Galactosidase by 2-Nitro-1-(4,5-Dimethoxy-2-Nitrophenyl) Ethyl, a Photoreversible Thiol Label. Biochim. Biophys. Acta -Protein Struct. Mol. Enzymol 1996, 1293 (2), 238–242. 10.1016/0167-4838(95)00254-5. [DOI] [PubMed] [Google Scholar]
- (23).Dannenfelser R; Allen GM; VanderSluis B; Koegel AK; Levinson S; Stark SR; Yao V; Tadych A; Troyanskaya OG; Lim WA Discriminatory Power of Combinatorial Antigen Recognition in Cancer T Cell Therapies. Cell Syst 2020, 11 (3), 215–228.e5. 10.1016/j.cels.2020.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Han X; Wang Y; Wei J; Han W Multi-Antigen-Targeted Chimeric Antigen Receptor T Cells for Cancer Therapy. J. Hematol. Oncol 2019, 12 (1), 1–10. 10.1186/s13045-019-0813-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kline T; Torgov MY; Mendelsohn BA; Cerveny CG; Senter PD Novel Antitumor Prodrugs Designed for Activation by Matrix Metalloproteinases-2 and −9. Mol. Pharm 2004, 1 (1), 9–22. 10.1021/mp0340183. [DOI] [PubMed] [Google Scholar]
- (26).Albright CF; Graciani N; Han W; Yue E; Stein R; Lai Z; Diamond M; Dowling R; Grimminger L; Zhang SY; Behrens D; Musselman A; Bruckner R; Zhang M; Jiang X; Hu D; Higley A; DiMeo S; Rafalski M; Mandlekar S; Car B; Yeleswaram S; Stern A; Copeland RA; Combs A; Seitz SP; Trainor GL; Taub R; Huang P; Oliff A Matrix Metalloproteinase-Activated Doxorubicin Prodrugs Inhibit HT1080 Xenograft Growth Better than Doxorubicin with Less Toxicity. Mol. Cancer Ther 2005, 4 (5), 751–760. 10.1158/1535-7163.MCT-05-0006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.