

FAN1-MLH1 interaction governs the processing of DNA interstrand crosslinks and disease-associated repeats.
Abstract
FAN1, a DNA structure-specific nuclease, interacts with MLH1, but the repair pathways in which this complex acts are unknown. FAN1 processes DNA interstrand crosslinks (ICLs) and FAN1 variants are modifiers of the neurodegenerative Huntington’s disease (HD), presumably by regulating HD-causing CAG repeat expansions. Here, we identify specific amino acid residues in two adjacent FAN1 motifs that are critical for MLH1 binding. Disruption of the FAN1-MLH1 interaction confers cellular hypersensitivity to ICL damage and defective repair of CAG/CTG slip-outs, intermediates of repeat expansion mutations. FAN1-S126 phosphorylation, which hinders FAN1-MLH1 association, is cell cycle–regulated by cyclin-dependent kinase activity and attenuated upon ICL induction. Our data highlight the FAN1-MLH1 complex as a phosphorylation-regulated determinant of ICL response and repeat stability, opening novel paths to modify cancer and neurodegeneration.
INTRODUCTION
In 2010, four different research groups independently reported the identification of human FAN1 as a DNA repair nuclease required for interstrand cross-link (ICL) repair (1–4). Further studies revealed interactions between FAN1 and mismatch repair (MMR) proteins of the MutL family (MLH1, PMS2, and MLH3) (2, 4, 5). However, the relevance and functional implications of the FAN1-MutL complexes for genome stability remains to be determined.
MMR proteins and, more recently, FAN1 have been implicated in disease-associated trinucleotide repeat instability. Expansions of repeats are associated with a growing number of neurological disorders, including Huntington’s disease (HD), a dominantly inherited condition caused by an expanded CAG repeat in the huntingtin (HTT) gene (6, 7). Both MLH1 and FAN1 were identified as HD age-of-onset modifier genes in genome-wide association studies of HD individuals (8–10). Previous findings demonstrated that Mlh1 and Mlh3 are essential for somatic CAG expansions in HD knock-in mice, implicating MutLγ (MLH1-MLH3) as one of the drivers of HD pathogenesis (11, 12). In contrast, FAN1 was proposed to have a protective role in HD progression by minimizing ongoing HTT CAG expansion mutations in cultured mitotic and postmitotic neuronally differentiated HD patient cells (10, 13). Notably, the enhanced somatic CAG expansions of the Htt CAG tract in tissues (brain and liver) of Fan1 knockout mice were blocked by simultaneous Mlh1 inactivation, suggesting that FAN1 acts to suppress the MLH1-dependent CAG expansions in HD patient tissues (14). Fan1 similarly suppresses somatic CGG repeat expansions in Fragile X mice (15). However, it remains to be demonstrated whether a direct physical interaction between FAN1 and MLH1 influences processing of damaged DNA or expanded CAG repeats.
Here, we uncover the molecular details of human FAN1-MutL complexes and show that FAN1 interacts with MLH1 via two adjacent MLH1-binding sites, referred to as MLH1-interacting protein (MIP) box and MLH1-interacting motif (MIM). We further propose that the FAN1-MLH1 interaction is regulated during the cell cycle and inhibited in G2/M upon cyclin-dependent kinase 1/2 (CDK1/2)–mediated phosphorylation of FAN1-S126 within the MIP box. We provide evidence that the same site can also be targeted by CDK5, an atypical and neuron-specific member of the CDK family. Functionally, we demonstrate that the FAN1-MLH1 interaction promotes the assembly of both proteins into DNA damage foci in response to mitomycin C (MMC) treatment and is required for ICL and repeat slip-out processing but largely dispensable for MMR. Disruption of the FAN1-MLH1 interaction leads to hypersensitivity to DNA cross-linking agents and defective repair of slipped-CAG/CTG repeats. Both phenotypes are rescued when the MLH1 interaction–defective FAN1 variant is additionally impaired in its endonuclease activity or dimerization. Together, our data provide a first mechanistic rationale for the role of the FAN1-MLH1 complex in ICL repair and CAT/CTG repeat stability.
RESULTS
FAN1-MLH1 interaction is regulated by phosphorylation of the MIP box in FAN1
To address these challenges, we first had to define the molecular details of the FAN1-MLH1 interaction. EXO1 and BLM bind to the C-terminal domain (CTD) of MLH1 via a tetrapeptide motif called the MIP box, with the consensus sequence Sx(Y/F)F (16–18). Sequence analysis revealed that FAN1 homologs contain a putative MIP box, 126SPYF129 (Fig. 1A and fig. S1A). Isothermal titration calorimetry (ITC) measurements revealed that recombinant MLH1-CTD (fig. S1B) binds 8-mer peptides encompassing MIP motifs of human FAN1, EXO1, and BLM with similar affinities (Fig. 1B, fig. S1C, and table S3). FAN1 residues S126, Y128, and F129 (but not P127) are critical for MLH1 interaction, as their substitution with alanines eliminated detectable binding to MLH1-CTD (Fig. 1B, fig. S1C, and table S3). Coimmunoprecipitation (co-IP) experiments in HEK293 cells transfected with FLAG-tagged FAN1 variants confirmed that S126A, Y128A, and Y128A/F129A (denoted as MIP*) impaired the interaction with endogenous full-length MLH1, while P127A had no effect (Fig. 1C). Residue E669 of MLH1 is known to be critical for its interaction with MIP box proteins (16, 19). FAN1 and BLM were pulled down from HeLa nuclear extracts with MLH1-CTD wild-type (wt) but not with the E669A variant (fig. S2A), further confirming that FAN1 126SPYF129 represents a bona fide MIP box.
Fig. 1. FAN1 contains a MIP box that is negatively regulated by CDK-mediated phosphorylation at S126.
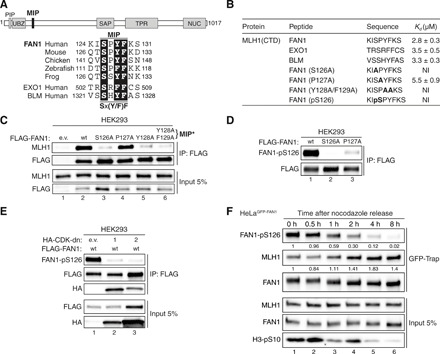
(A) Top: Protein domain architecture of human FAN1 indicating the location of the MIP box. PIP, proliferating cell nuclear antigen–interacting peptide box; UBZ, ubiquitin-binding zinc finger; SAP, SAF-A/B, Acinus, and PIAS; TPR, tetratricopeptide repeat; NUC, nuclease. Bottom: Sequence alignment of the FAN1 orthologs with the MIP box of human EXO1 and BLM. (B) Dissociation constants (Kd) indicating the binding affinity of MLH1 CTD for the indicated peptides from FAN1, EXO1, and BLM as determined by ITC. NI, no interaction (weak and constant signal under the condition used). (C) Immunoblots of FLAG-M2 affinity resin immunoprecipitations (IPs) of extracts from HEK293 cells transfected either with empty vector (e.v.), FLAG-FAN1 wild type (wt), or the variant constructs. The Y128A/F129A mutation is denoted as MIP*. (D) Immunoblots of FLAG-M2 affinity resin IPs of extracts from HEK293 cells transfected with the indicated FLAG-FAN1 constructs. (E) Immunoblots of FLAG-M2 affinity resin IPs of extracts from HEK293 cells cotransfected with FLAG-FAN1 and vectors expressing hemagglutinin (HA)–tagged dominant-negative (dn) forms of CDK1 or CDK2. (F) Immunoblots of green fluorescent protein (GFP)–Trap IPs of extracts from HeLaGFP-FAN1 cells synchronized in mitosis by thymidine-nocodazole block. Mitotic cells were replated in fresh medium for the indicated time periods. The relative ratios of pS126 to FAN1 and MLH1 to FAN1 in GFP-Trap samples were quantified by densitometry using ImageJ. (C to F) The antibodies used are shown on the left.
Phosphorylation at S126 in the 8-mer FAN1 MIP peptide abolished interaction with MLH1-CTD (Fig. 1B, fig. S1B, and table S3). Analogously, a FAN1 phospho-mimetic mutant (S126E) failed to stably interact with MLH1 (fig. S2B). To confirm that FAN1 is S126-phosphorylated, we generated a phospho-specific antibody against FAN1-pS126 and found that it recognized FAN1 wt but not its S126A or P127A variants (Fig. 1D). The latter suggested that S126 within the FAN1 MIP is likely to be phosphorylated by proline-directed kinases such as CDKs. Consistent with this idea, we found FAN1-pS126 levels to be substantially lower in cells expressing catalytically inactive, dominant-negative mutants of CDK1 or CDK2 (Fig. 1E and fig. S2C). Intriguingly, FAN1-S126 phosphorylation was also stimulated by CDK5 in the presence of its neuronal-specific activator p35 (fig. S2D).
To learn about the effect of key residues in FAN1, we used the “protein replacement” system to generate stable U2OS cell lines, in which endogenous FAN1 was knocked down and substituted by its inducibly expressed enhanced green fluorescent protein (eGFP)–tagged variants (U2OSGFP-FAN1) (20). Treatment of U2OSGFP-FAN1 cells with roscovitine, a pan-CDK inhibitor, or RO-3306, a selective CDK1 inhibitor, decreased FAN1-pS126 levels (fig. S2E), suggesting that FAN1-S126 phosphorylation is cell cycle–regulated. Synchronization of HeLaGFP-FAN1 cells with nocodazole revealed that FAN1-pS126 levels peak in G2/M are markedly reduced during G1 and remain low through S phase before rising again in G2 (fig. S2F). FAN1-pS126 levels inversely correlated with the amounts of MLH1 interacting with FAN1 (Fig. 1F). Notably, MMC treatment reduced FAN1-S126 phosphorylation (fig. S2G), likely as a consequence of DNA damage–induced cell cycle checkpoint activation. Together, these findings suggested that the FAN1-MLH1 interaction is negatively regulated through the cell cycle by CDK-mediated phosphorylation of S126 in the FAN1 MIP box.
FAN1 contains a second conserved MLH1-binding site
Three human MLH1 partner proteins, MLH3, PMS2, and PMS1, share a homologous 36–amino acid domain implicated in MLH1 interaction (21). Just downstream of the FAN1 MIP box, we noticed a highly conserved 12–amino acid sequence with extensive homology to a part of that domain, which we therefore designated as MIM (Fig. 2A and fig. S3, A and B). ITC analyses revealed that MLH1-CTD binds 12-mer peptides encompassing MIMs of FAN1 and MLH3 with similar affinities (Fig. 2B, fig. S3C, and table S3), suggesting that they may compete for the same binding site in MLH1. In contrast, the putative MIM peptide of PMS2 displayed lower binding affinity, while that of PMS1 was not stably interacting with MLH1-CTD (Fig. 2B, fig. S3C, and table S3). We then mapped the critical residues within the FAN1 MIM for interaction with MLH1 through site-directed mutagenesis. Co-IP experiments with different FLAG-FAN1 constructs revealed two highly conserved leucine residues, L155 and L159, to be primarily responsible for MLH1 interaction, with the corresponding double mutant L155A/L159A (denoted as MIM*) exhibiting a cumulative effect when compared to the single mutants (Fig. 2C and fig. S3D). ITC analysis of FAN1 and MLH3 mutant MIM peptides further confirmed the critical role of the two leucine residues within their MIMs for MLH-CTD binding (fig. S3E).
Fig. 2. An adjacent MLH1-interacting motif (MIM) in FAN1 synergizes with the MIP box to promote stable MLH1 binding.
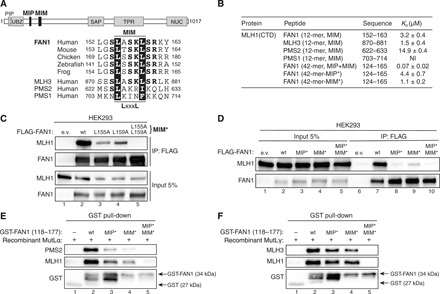
(A) Top: Protein domain architecture of human FAN1 indicating the location of the MIP box and MIM. Bottom: Sequence alignment of FAN1 orthologs with the putative MIM of human MLH3, PMS2, and PMS1. (B) ITC was used to determine the Kd indicating the binding affinity of MLH1-CTD for the indicated peptides. (C) Immunoblots of FLAG-M2 affinity resin IPs of extracts from HEK293 cells transfected either with e.v. or indicated FLAG-FAN1 expression constructs. The L155A/L159A mutation is denoted as MIM*. (D) Immunoblots of FLAG-M2 affinity resin IPs of extracts from HEK293 cells transfected either with e.v. or indicated FLAG-FAN1 expression constructs. (E and F) Recombinant MutLα (E) or MutLγ (F) (200 nM) was subjected to pull-down reactions using the indicated recombinant GST-FAN1 (amino acids 118 to 177) variants. (C to F) The antibodies used are shown on the left.
To define the individual impact of MIP and MIM on MLH1 binding, we subjected FAN1-derived 42-mer peptides containing both motifs (fig. S3A) to ITC analysis. Notably, having both MIP and MIM sites on a contiguous peptide resulted in ~40-fold increased binding affinity to MLH1-CTD [dissociation constant (Kd) = 0.07 μM] compared to the MIP- or MIM-only peptides (Kd = 2.8 and 3.2 μM, respectively) (Fig. 2B, fig. S3F, and table S3). This synergistic effect was not observed with 42-mer peptides carrying defective MIP* or MIM* variants (Fig. 2B, fig. S3F, and table S3). Independent contributions of each motif were corroborated for the full-length proteins by co-IP, showing that FLAG-FAN1 single MIP* or MIM* mutants exhibited residual interaction with MLH1, whereas the compound MIP*/MIM* mutant was fully deficient in binding to MLH1 (Fig. 2D). Last, GST-FAN1 pull-down assays with purified recombinant MutLα (MLH1-PMS2) or MutLγ (MLH1-MLH3) confirmed the cooperativity between both motifs and demonstrated that FAN1 is unlikely to compete with PMS2 or MLH3 for MLH1 binding (Fig. 2, E and F). We also observed specific binding of MutLα to GST-FAN1, using lysates from HEK293T cells transfected with MLH1- and PMS2-expressing vectors (fig. S3G). Together, our findings suggested that direct and specific docking of FAN1 to MLH1-CTD promotes the formation of stable complexes between FAN1 and MutLα or MutLγ.
This prompted us to evaluate whether FAN1-MLH1 interaction functions in MMR, although currently available data do not indicate a prominent role for FAN1 in this repair pathway (4). Acquired resistance to DNA alkylating agents generating O6-methylguanine–containing mismatches, such as the chemotherapeutic temozolomide (TMZ), is a common feature of MMR-deficient tumor cells (22–24). Consistent with this, depletion of MLH1 from U2OS cells results in TMZ resistance (fig. S4, A to C). In contrast, cells depleted of FAN1, or cell lines inducibly expressing FAN1 mutants either defective in nuclease activity (D960A, nd*) or MLH1 binding (MIP*/MIM*), were efficiently killed by TMZ (fig. S4, A to C), indicating that the FAN1-MLH1 interaction is unlikely to act in canonical MMR.
FAN1-MLH1 interaction confers resistance to cross-link damage
To confirm the existence of FAN1-MLH1 complexes in cells, we performed in situ proximity ligation assays (PLA). Nuclear PLA signals were readily detected in U2OS cells inducibly expressing GFP-FAN1 wt but not the MIP*/MIM* variant and significantly increased in number upon MMC treatment, indicating that the FAN1-MLH1 interaction might operate in ICL repair (Fig. 3A). To corroborate this finding, we generated a mouse monoclonal antibody against the nuclease domain of human FAN1 (amino acids 871 to 1017), specifically detecting endogenous FAN1 expression in U2OS nuclear extracts and HeLa whole-cell lysates by Western blotting (fig. S5, A and B). Using this new tool, we could confirm the MMC-dependent increase in the number of nuclear FAN1-MLH1 PLA foci in HeLa cells (fig. S5C).
Fig. 3. FAN1-MLH1 interaction promotes resistance to cross-link damage.
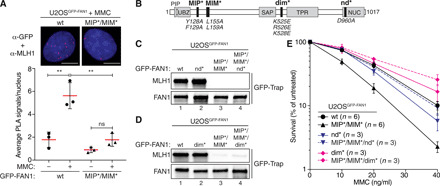
(A) PLA was used to evaluate FAN1-MLH1 association in U2OSGFP-FAN1 wt and U2OSGFP-FAN1 MIP*/MIM* cells mock-treated or treated with MMC (150 ng/ml) for 24 hours. Representative images are shown. Scale bars, 10 μm. Scatterplot displays quantification of the PLA signals per nucleus from at least 100 cells. Data display the means ± SD from three independent experiments. Statistical significance was calculated by unpaired t test. **P < 0.01; ns, not significant. (B) Schematic representation of human FAN1, highlighting positions of mutations used, including MIP*, MIM*, dimerization-defective (dim*), and nuclease-defective (nd*) FAN1 variants. (C and D) Immunoblots of GFP-Trap IPs of extracts from indicated U2OSGFP-FAN1 cells. The antibodies used are shown on the left. (E) Clonogenic survival assay of the indicated U2OSGFP-FAN1 cells exposed to increasing doses of MMC. Viability of untreated cells was defined as 100%. Data are presented as the means ± SEM.
Increased MMC hypersensitivity was evident in cells expressing the MLH1 interaction–defective FAN1 MIP*/MIM* variant compared to cells expressing FAN1 wt (Fig. 3, B to E, and fig. S5, D and E). Simultaneous loss of MLH1 binding and DNA nuclease activity (=MIP*/MIM*/nd*) rescued clonogenic survival after MMC treatment to near wt levels (Fig. 3, C and E, and fig. S5D). We observed nearly identical chemosensitivity profiles upon treatment of these cells with the ICL-inducing agent cisplatin (fig. S5, F and G). The FAN1 D960A (nd*) mutant is completely deficient in both endo- and exonuclease activities (25). In contrast, the FAN1 triple mutant (K525E/R526/K528E, here referred to as dim*; Fig. 3B) incapacitates both FAN1 head-to-tail dimerization and endonucleolytic activity while retaining exonucleolytic activity (25). Notably, expression of the compound MIP*/MIM*/dim* FAN1 variant also fully restored MMC resistance (Fig. 3, D and E, and fig. S5E).
Loss of FAN1-MLH1 interaction results in persistent ssDNA signaling
Quantitative image-based cytometry (QIBC) analysis established that homodimerization is required for FAN1 foci formation in response to MMC treatment, whereas MLH1 binding is largely dispensable (Fig. 4A and fig. S6A). Conversely, MLH1 localization to ICL damage was significantly impaired in both FAN1 dim* and MIP*/MIM* variants, indicating that accumulation of MLH1 at ICLs relies on its physical interaction with FAN1 (fig. S6B). Moreover, we found that cells expressing the MIP*/MIM* variant displayed a prolonged G2/M arrest after MMC treatment, which was not observed in cells expressing the MIP*/MIM*/dim* (Fig. 4B and fig. S6C). Single-stranded DNA (ssDNA) accumulated in MMC-treated MIP*/MIM*-expressing G2/M cells compared to FAN1 wt cells, as detected by native 5-bromo-2′-deoxyuridine (BrdU) staining (fig. S6D). Accumulation of ssDNA was not observed in MIP*/MIM*/dim* cells (fig. S6D). Replication protein A (RPA), a heterotrimeric complex consisting of RPA1, RPA2, and RPA3 subunits, is the major ssDNA binding factor in eukaryotic cells and an indispensable player during DNA replication and repair (26). Consistently, we found increased RPA1 and RPA2 loading on MMC-damaged chromatin in cells expressing the MIP*/MIM* variant but not in cells expressing MIP*/MIM*/nd* (nuclease-dead) or MIP*/MIM*/dim* (defective in endo- but not exonuclease) forms of FAN1 (Fig. 4C and fig. S6, E and F). Last, both CHK1 and RPA2 phosphorylation were elevated in MIP*/MIM* cells upon 24-hour treatment with a high dose of MMC (Fig. 4D), indicative of activation of the DNA damage response. This effect was somewhat diminished for the MIP*/MIM*/nd* or MIP*/MIM*/dim* relative to the MIP*/MIM* (Fig. 4D). Together, these findings are consistent with the idea that correct MLH1 binding is critical to prevent cytotoxic endonucleolytic DNA cleavage by MLH1-free FAN1 in response to ICL damage.
Fig. 4. FAN1-MLH1 interaction prevents extensive ssDNA formation following cross-link damage.
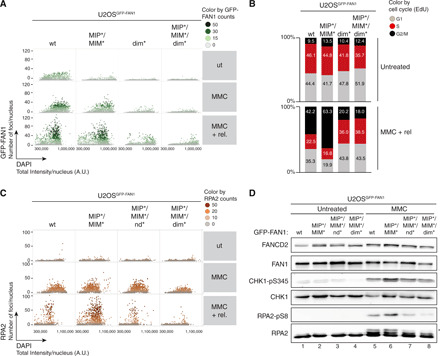
(A) QIBC analysis of GFP-FAN1 foci in U2OSGFP-FAN1 cells mock-treated, treated with MMC (20 ng/ml) for 24 hours, or MMC-treated and then released for 72 hours. Color-coded scatterplots indicate the number of GFP-FAN1 foci per nucleus. ut, untreated. (B) Same cells as in (A) were pulse-labeled with ethynyl deoxyuridine (EdU) during the last 30 min before harvesting and subjected to the Click-IT reaction. Cell cycle distribution was evaluated by QIBC using the 4′,6-diamidino-2-phenylindole (DAPI) and EdU signals (fig. S5C). (C) QIBC of RPA2 foci in U2OSGFP-FAN1 cells mock-treated, treated with MMC (20 ng/ml) for 24 hours, or MMC-treated and then released for 72 hours. Color-coded scatterplots indicate the number of RPA2 foci per nucleus. A.U., arbitrary units. (D) Same cells as in (C) were treated with MMC (300 ng/ml) for 24 hours, and lysates were analyzed by immunoblotting using the indicated antibodies. Asterisk indicates hyperphosphorylated form of RPA2.
FAN1-MLH1 interaction promotes DNA repair of CAG slip-outs
Several studies have implicated FAN1 in repeat diseases (8–10, 15). Repeat expansions occur because of aberrant or escaped repair of unusual slipped-DNA structures that form upon out-of-register annealing of complementary repeat strands during DNA repair, replication or transcription (27, 28). Slipped-DNAs occur at the expanded CAG/CTG repeat disease locus in myotonic dystrophy type 1 patient tissues, where their levels directly correlated with the levels of somatic repeat expansions, providing support for the involvement of slipped-DNAs in expansions (29). We therefore set out to investigate the involvement of the FAN1-MLH1 complex in the processing of slipped-strand DNAs formed by CAG/CTG repeats, using an established in vitro slipped-DNA repair assay (30–32). Modulation of CAG instability with a ligand that binds to slipped-DNAs in brains of HD mice, where the ligand also blocked in vitro CAG slip-out repair by cell extracts, supports this assay as an impression of the in vivo maintenance of CAG repeats (30). We previously demonstrated the requirement of a DNA nick for strand-specific slip-out repair, as in base-base MMR, the nick determines the strand that will be repaired and the continuous strand serves as the template for repair (31, 32). The absence of a nick led to poor correction of base-base mismatches and heteroduplexes with limited strand bias. The cell extract preparations used for slip-out repair are competent in a broad range of DNA repair processes, including MMR, and are capable of inducing replication-mediated CAG/CTG repeat expansions and contractions (33). We and others revealed that MLH1 is required for the repair of short slip-outs of a single excess repeat (34, 35). Thus, before interrogating the role of the FAN1-MLH1 interaction in slip-out repair, we first established the sensitivity of long slip-out (excess of 20 repeats) repair to MLH1 levels. The repair-proficient human cell line TK6 and its MLH1-null CRISPR-Cas9–produced derivative have been described (36). Using TK6 and TK6-MLH1−/− cell extracts, we reveal that MLH1 is involved in slip-out repair, and repair efficiency depended specifically on the titrated levels of MLH1 (fig. S7A). A parallel experiment using extracts of the MLH1-null HCT116 and its corrected derivative HCT116 + chromosome 3 (37) revealed identical results (fig. S7B). Thus, the repair of both short and long slip-outs is sensitive to MLH1.
The involvement of the FAN1-MLH1 interaction was next assessed. Specifically, extracts from U2OS clones inducibly expressing GFP-FAN1 variants were tested for their capacity to repair slipped-DNA substrates that model repeat contraction or expansion intermediates, containing a nick on the opposite or on the same strand of the slip-out, respectively (Fig. 5A). We established that the MLH1 interaction–defective FAN1 MIP*/MIM* variant was significantly less efficient in mediating the repair of a substrate with 20 excess CAG repeats than the FAN1 wt protein (fig. S8A). Thus, efficient repair of slipped-CAG/CTG repeats requires the FAN1-MLH1 interaction. Next, we found that slip-out repair was restored when MLH1 interaction–defective FAN1 protein was also made defective in either its DNA nuclease activity (MIP*/MIM*/nd*) or in dimer formation (MIP*/MIM*/dim*), a scenario which is analogous to the MMC-induced cellular phenotypes (Fig. 5B and Fig. 3E). Identical results were observed with all long slip-out substrates tested, regardless of the slip-out repeat motif (CAG or CTG) or location of the nick (fig. S8, B to D). Conversely, the FAN1-MLH1 interaction was dispensable for the processing of short, isolated single repeat slip-outs (fig. S9, A and B). To further validate the importance of the FAN1-MLH1 interaction in the repair of repeat slip-outs, we synthesized a FAN1 peptide containing both MLH1-binding motifs that efficiently inhibited complex formation between endogenous FAN1 and MLH1 in HeLa extracts (Fig. 5C). Supplementing the extracts with the FAN1 wt peptide caused a concentration-dependent decrease in CAG slip-out repair, which was not observed with the compound MIP*/MIM* mutant peptide (Fig. 5D). The peptide selectively inhibited the FAN1-MLH1 interaction, as it did not cause a further reduction in slip-out repair when added to extracts derived from MIP*/MIM* mutant U2OS cells (Fig. 5E).
Fig. 5. FAN1-MLH1 interaction promotes repair of CAG/CTG DNA slip-outs.
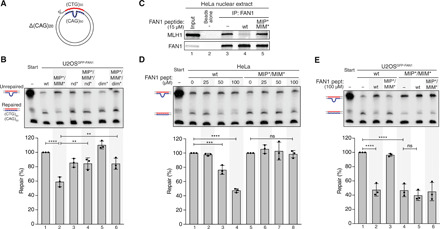
(A) Scheme of the slipped (CTG)30/(CAG)50 substrate. (B) The (CAG)20 slip-out substrate was incubated with extracts from U2OSGFP-FAN1 cells expressing indicated FAN1 variants. Repair efficiency was quantified by densitometric analysis of Southern blots. Values represent the mean of three independent experiments. Error bars represent ±SD. Statistical significance was calculated by ordinary one-way analysis of variance (ANOVA) test followed by Tukey’s multiple comparisons. **P < 0.005, ***P < 0.0005, ****P < 0.00005. (C) HeLa nuclear extract was incubated with FAN1-derived 60-mer peptides (amino acids 118 to 177) containing wt or mutant MIP-MIM, immunoprecipitated using anti-FAN1 antibody, and immunoblotted with the indicated antibodies. (D) Extracts from HeLa cells were supplemented with increasing concentrations of FAN1 peptides and incubated with the (CAG)20 slip-out substrate. Repair efficiency was calculated as described in (B). (E) Extracts of U2OSGFP-FAN1 wt and U2OSGFP-FAN1 MIP*/MIM* cells were supplemented with indicated FAN1 peptides and incubated with the (CAG)20 slip-out substrate. Repair efficiency was calculated as described in (B).
DISCUSSION
Here, we revealed the molecular basis of the interaction between FAN1 and MLH1, its negative regulation by CDK-mediated phosphorylation of FAN1-S126, and its functional relevance for processing ICLs and slipped-DNA intermediates of repeat expansion mutations (as detailed in Fig. 6). Our data suggest that disrupting FAN1-MLH1 association, either by MIP-MIM mutations or by a MIP-MIM mimetic peptide, can lead to less efficient ICL processing leading to enhanced cytotoxicity and diminished repair of slipped-DNAs. When FAN1 is unable to bind MLH1, additional loss of FAN1 endo- but not exonuclease activity can restore both deleterious traits associated with disruption of the FAN1-MLH1 interaction. Given that the FAN1-MLH1 interaction protects cells from ICL-induced cell death, its modulation might be harnessed to enhance MMC chemosensitivity for cytoreductive surgical debulking in cases of persistent MMC chemoresistance (38). Likewise, the rescue of reduced slip-out repair in extracts expressing the MLH1 interaction–defective FAN1 occurs when FAN1 is also endonuclease-defective but exonuclease-proficient, suggesting that endonucleolytic cleavage of the slip-out substrates by MLH1-free FAN1 diminishes slip-out repair. Moreover, peptide-mediated suppression of MLH1 binding to FAN1 also inhibits slip-out repair in vitro. Thus, modulation of the FAN1-MLH1 interaction might represent a novel opportunity to alter the metabolism of disease-associated repeats.
Fig. 6. FAN1-MLH1 interaction affects the repair of ICLs and slipped-DNA repeats.
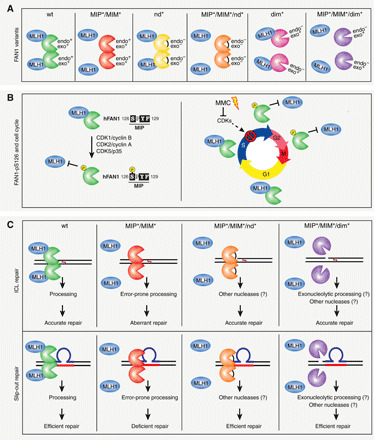
Summary of the FAN1-MLH1 complex interaction and a plausible model for its involvement in ICL and slip-out repair. (A) Scheme of the FAN1-MLH1 interaction in different states of wt or mutated FAN1. Protein orientation is unknown and arbitrarily presented for ease. (B) Left: FAN1 is subjected to CDK-mediated phosphorylation at S126 located within the MIP box. Right: Regulation of FAN1-MLH1 interaction through the cell cycle and in response to ICL damage. (C) Top: The FAN1-MLH1 complex localizes to ICL damaged chromatin to preserve genome stability and ensure cell viability. In cells, devoid of the FAN1-MLH1 interaction aberrant ICL repair and cell death ensues. Inactivation of FAN1’s endo- and exonuclease activities by mutating D960A or inhibition of FAN1 dimerization, affecting FAN1’s endo- but not exonucleolytic activity, would alleviate the toxicity of the FAN1 MIP/MIM-mutated variant and promote cell proliferation and survival. Bottom: Inhibition of the FAN1-MLH1 interaction (MIP*/MIM*) blocks repair of slipped-DNAs. Repair defects were rescued when the MLH1-interacting–defective FAN1 was also defective either in both its endonuclease activities or in dimer formation (endo- but not exonuclease defective). Therefore, the regulatory aspects of the FAN1 binding to MLH1 aligns the pathway of ICL repair with that of slipped-DNA processing. Mutations: MIP*, Y128A/F129A; MIM*, L155A/L159A; nd*, D960A; dim*, K525E/R526E/K528E.
The MutLα (MLH1-PMS2) endonuclease, activated by MutSβ (MSH2-MSH3), PCNA, and RFC, can weakly incise both strands of short CAG or CTG slip-out containing DNAs (1 to 4 excess repeat units), without displaying a strand or polarity bias (35). In contrast, the MutLγ (MLH1-MLH3) endonuclease, activated by MutSβ, preferentially incises the strand opposite of CAG or CTG slip-outs, often cleaving upstream or downstream of the slip-out DNA (12). FAN1 and the MMR genes MSH3, MLH1, MLH3, and PMS2 were identified as genetic modifiers of the age-of-onset of HD families and at least six other CAG repeat expansion disorders (39). Specifically, noncoding and coding variants of these DNA repair genes are associated with an earlier or later age of disease onset (40). MMR proteins are required for somatic CAG and CGG expansions in tissues of HD, Myotonic dystrophy type 1 (DM1), and Fragile X syndrome (FXS) transgenic mice, as their deficiency leads to stabilization of CAG/CTG repeat tracts (41). Specifically, in the absence of Mlh1 and Mlh3, the spontaneous ongoing ultrafrequent somatic CAG expansions arising in wt mice were completely stabilized, while in the absence of Pms2, somatic expansions were partially stabilized and very rare large contractions (~1.6 to 14 contractions per 1000 cells; >20 U per event) were increased by 1.5- to 3.5-fold (42). This contrasts with the increased hyperexpansions of repeats in tissues that arise in the absence of Fan1 (14). Curiously, HD mice deficient for both Mlh1 and Fan1 showed no somatic CAG expansions, indicating that MLH1 is critical for driving repeat instability in FAN1-deficient mice (14). Interpretations of this are that MLH1 is strictly required (i) for FAN1’s apparent ability to suppress hyperexpansion or (ii) to drive the hyperexpansions, which are otherwise suppressed by FAN1. It is nonetheless perplexing that FAN1 and MLH1, which form a complex, have completely opposing effects on the spontaneous ongoing CAG expansions in tissues of HD mice, which reflect somatic expansions in patients. Mlh1 is absolutely required to drive CAG expansions, as Mlh1 deficiency abolishes expansions (11). In contrast, Fan1 deficiency leads to enhanced somatic CAG expansion rates in cultured patient cells and HD mice, suggesting that the role of FAN1 in HD patients is to attenuate the levels of CAG expansion (10, 13, 14). It is tempting to speculate that the rate of repeat expansion and thus of disease onset might be controlled by the FAN1-MLH1 interaction status. Usdin and colleagues (43) revealed that Exo1, which interacts with Mlh1 through its MIP box, could suppress against Mlh1-dependent CGG expansions in Fragile X mice. Further studies are clearly needed to establish the determinants of how expanded repeats are processed by MutL complexes interacting mutually exclusive with FAN1 or EXO1.
Squitieri and colleagues (44) showed that MMC treatment of HD patient cells significantly diminished the rate of ongoing CAG expansions of the mutant HTT but did not affect the nonexpanded gene. This suggests that DNA lesions, whose repair involves the FAN1-MLH1 complex and its regulation by posttranslational modification, can indeed have an impact on CAG instability. Overall, our findings presented herein begin to dissect the critical interactions and reveal commonalities of the FAN1-MLH1 complex in ICL processing and repeat stability/instability.
MATERIALS AND METHODS
Cell culture
U2OS, HEK293, HEK293T (Invitrogen, Life Technologies), and HeLa Kyoto FAN1 N-FLAP (The Hyman Lab, MPI-CBG) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS), penicillin (100 U/ml), and streptomycin (100 μg/ml). HCT116 cells were grown in McCoy’s 5A medium supplemented with 10% FCS, penicillin (100 U/ml), and streptomycin (100 μg/ml). TK6 cells were cultivated in RPMI 1640 medium supplemented with 10% heat-inactivated horse serum (Gibco, Life Technologies), sodium pyruvate (200 μg/ml), penicillin (100 U/ml), and streptomycin (100 μg/ml). U2OS Flp-In T-REx and HeLa Flp-In T-REx (Invitrogen, Life Technologies) cells were maintained in medium supplemented with blasticidin (10 μg/ml) and hygromycin B (250 μg/ml). The Flp-In T-REx system (Thermo Fisher Scientific) was used to generate cell lines stably expressing different short hairpin RNA (shRNA)–resistant forms of GFP-FAN1 constructs under the control of a doxycycline (Dox)–inducible promoter (20). To learn about the effect of key residues in FAN1, as previously, we used this protein replacement system to generate stable U2OS cell lines, in which the endogenous FAN1 was knocked down by coexpression of an anti-FAN1 shRNA and substituted by its eGFP-tagged variants. This method makes use of a stably integrated vector containing two Dox-inducible cassettes: one expressing shRNA targeting mRNA that encodes an endogenous protein of choice and the second expressing cDNA encoding the desired protein variants. Dox treatment of these stable cell lines leads to degradation of the mRNA encoding the endogenous FAN1 protein and to expression of the desired variants. Briefly, expression vectors pAIO-TO/eGFP-FAN1 and the Flp recombinase expression plasmid, pOG44, were mixed in a 1:9 ratio and transfected into U2OS or HeLa Flp-In T-REx cells using Lipofectamine 3000 (Thermo Fisher Scientific). Forty-eight hours later, cells were plated at different dilutions and, 72 hours after transfection, the medium was supplemented with hygromycin B (250 μg/ml) and blasticidin S (12.5 μg/ml). The medium was replaced every 2 to 3 days, and the cells were maintained in selection medium for approximately 14 days. Resistant colonies were picked and single-cell clones analyzed for GFP expression by flow cytometry and immunoblotting. For G2/M synchronization, HeLaGFP-FAN1 Flp-In T-REx cells were cultured in the presence of 2 mM thymidine for 24 hours, released for 3 hours, and then incubated with nocodazole (100 ng/ml) for 18 hours, washed three times with 1× phosphate-buffered saline (PBS) and cultured for the indicated times in DMEM growth medium.
Antibodies and plasmids
A complete list of all primary antibodies and DNA plasmids used throughout this study can be found in tables S1 and S2, respectively. Secondary horseradish peroxidase (HRP)–conjugated anti-mouse and anti-rabbit antibodies were from GE Healthcare, and the HRP-conjugated anti-goat antibody was from Santa Cruz Biotechnology. Alexa Fluor 488, 594, and 647–conjugated secondary antibodies were purchased from Thermo Fisher Scientific. The phospho-specific antibody against FAN1-pS126 was produced by Creative Biolabs (NY, USA) by immunizing rabbits with a synthetic keyhole limpet hemocyanin–conjugated FAN1 phospho-peptide (119-REVKQKIpSPYFKSN-132) of human FAN1. The monoclonal antibody against FAN1 was produced by GenScript (NJ, USA) by immunizing mice with recombinant His-tagged FAN1 (amino acids 871 to 1017) purified from Escherichia coli.
Chemicals
BrdU, 4′,6-diamidino-2-phenylindole (DAPI), MMC, thymidine, nocodazole, cisplatin, O6-benzylguanine (O6-BG), and TMZ were purchased from Sigma-Aldrich. R-roscovitine and RO-3306 were purchased from Calbiochem. Dox was purchased from TaKaRa Clontech.
Isothermal titration calorimetry
Human MLH1 CTD (amino acids 486 to 756) was purified for ITC analyses as described previously (fig. S1B) (16, 17). The synthetic peptides used in this study were purchased from GeneCust (Boynes, France) at 95% purity. The binding between MLH1(CTD) and the different peptides was measured by using a VP-ITC (Cytiva). Before the measurements, all the solutions were degassed under vacuum. The reaction cell was loaded with 15 μM MLH1 protein, and the syringe contained 150 μM peptide solutions. Thirty injections of 10-μl volume were made at 210-s intervals. Control experiments were performed at the same concentration of peptides with buffer in the cell. Thermodynamic parameters ΔH° (enthalpy change), n (stoichiometry), and Ka (association constant) were obtained by nonlinear least-squares fitting of the experimental data by using the single set of independent binding sites model of the Origin software provided with the instrument. The free energy of binding (ΔG°) and entropy (ΔS°) were obtained by using the classical thermodynamic formulae: ΔG° = −RT × ln Ka and ΔG° = ΔH°–TΔS°, where R is the gas constant and T is the absolute temperature in kelvin. The titrations were carried out at 30°C in 50 mM Na2HPO4 (pH 8.0), 150 mM NaCl, and 10 mM β-mercaptoethanol. See table S3 for the summary of all ITC data.
His-MLH1 pull-down assays
His6-GST-MLH1-CTD constructs were expressed in E. coli strain BL21. Cultures were grown at 37°C with shaking at 250 rpm. Protein expression was induced at OD600 (optical density at 600 nm) 0.6 by the addition of 0.5 mM isopropyl-β-d-thiogalactopyranoside at 18°C for 20 hours. The cells were lysed in lysis buffer [1× PBS, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM benzamidine, and 1× complete protease inhibitor cocktail (Roche)] and sonicated for 5 min. The mixture was rotated at 4°C for 1 hour. The soluble fraction was retrieved by centrifugation at 13,000 rpm at 4°C for 30 min and incubated with nickel–nitrilotriacetic acid beads in pull-down buffer for 2 hours at 4°C. HeLa nuclear extract (CilBiotech) was incubated with MLH1-bound beads for 2 hours at 4°C. The beads were washed three times with NTEN300 [0.5% NP-40, 20 mM tris-HCl (pH 7.4), 0.1 mM EDTA, and 300 mM NaCl] and once with TEN100 [20 mM tris-HCl (pH 7.4), 0.1 mM EDTA, and 100 mM NaCl]. The recovered complexes were boiled in SDS sample buffer and analyzed by SDS–polyacrylamide gel electrophoresis (PAGE) followed by immunoblotting.
Coimmunoprecipitation
Following procedures previously described in (45), cells were lysed in NP-40 buffer [50 mM tris-HCl (pH 7.5), 120 mM NaCl, 1 mM EDTA, 6 mM EGTA, 15 mM sodium pyrophosphate, and 1% NP-40, supplemented with phosphatase inhibitors (20 mM NaF and 1 mM Na3VO4) and protease inhibitors (1 mM benzamidine and 0.1 mM PMSF)], incubated with Benzonase (Merck) for at least 30 min at 4°C, and clarified by centrifugation. A total of 1 to 3 mg of lysates was incubated with anti-FLAG M2 (Sigma-Aldrich) or GFP-Trap affinity resin (ChromoTek) for 2 hours or overnight at 4°C. The beads were then washed three times with NP-40 buffer and once with TEN100 [50 mM tris-HCl (pH 7.5), 140 mM NaCl, and 5 mM EDTA]. Retrieved protein complexes were boiled in SDS sample buffer and analyzed by SDS-PAGE followed by immunoblotting.
FAN1 60-mer peptides
Custom-designed FAN1 60-mer peptides (amino acids 118 to 177), wt, and MIP*/MIM* (Y128A/F129A/L155A/L159A) were purchased from GenScript and synthesized according to standard practice (N-terminal acetylation and C-terminal amidation). Lyophilized peptides with a purity >85% were dissolved in water at 5 mg/ml.
FAN1 immunoprecipitation
One milligram of HeLa nuclear extract was incubated with or without 15 μM FAN1 60-mer peptides in 0.5 ml of NP-40 buffer for 2 hours at 4°C with rotation. One microgram of anti-FAN1 rabbit polyclonal antibody was added to the samples and incubated overnight at 4°C with rotation. Protein A Sepharose beads (CL4B, Sigma-Aldrich) were equilibrated in NP-40 buffer, and 25 μl of bead slurry was then added to each sample and incubated for 2 hours at 4°C with rotation. The beads were then washed three times with NP-40 buffer and once with 1× TEN100 buffer, boiled in SDS sample buffer, and analyzed by SDS-PAGE followed by immunoblotting.
siRNA transfections
Small interfering RNA (siRNA) oligos were transfected at a final concentration of 20 nM using Lipofectamine RNAiMAX (Thermo Fisher Scientific). FAN1 (5′-GUAAGGCUCUUUCAACGUA-3′) and MLH1 (5′-GUGUUCUUCUUUCUCUGUA-3′) siRNAs were purchased from Microsynth. CNTL siRNA was purchased from Ambion (catalog no. 4390846).
Colony formation assays
For TMZ treatment, U2OS cells were transfected with siCNTL, siFAN1, or siMLH1 for 24 hours. The cells were then plated in six-well plates previously coated with poly-l-lysine (0.01%, Sigma-Aldrich). U2OSGFP-FAN1 cells were seeded in the presence of Dox (100 ng/ml) in six-well plates. Twenty-four hours after seeding, the cells were treated with 10 μM O6-BG for 2 hours. The medium was then replaced with serum-free medium supplemented or not with TMZ for 3 hours, and the cells were then washed twice with 2 ml of 1× PBS before adding add fresh growth medium. For MMC and cisplatin treatment, U2OSGFP-FAN1 cells were seeded in the presence of Dox (100 ng/ml) in six-well plates. Twenty-four hours after the seeding, the cells were treated with MMC or cisplatin for 24 hours. For MMC colony formation assays, plates were then washed twice with 2 ml of 1× PBS before adding fresh growth medium. For cisplatin colony formation assays, medium was left on the plates after 24 hours of treatment and topped up with fresh growth medium. In all colony formation assays, the cells were cultured for 13 days at 37°C before fixation with crystal violet solution [0.5% crystal violet and 20% ethanol (w/v)]. For analysis, plates were scanned and analyzed with the ImageJ Plugin ColonyArea using the parameter “intensity percent” (46).
Immunoblotting
If not stated otherwise, cells were lysed in NP-40 buffer supplemented with phosphatase inhibitors (20 mM NaF and 1 mM Na3VO4) and protease inhibitors (1 mM benzamidine and 0.1 mM PMSF), incubated with Benzonase (Merck) for at least 30 min at 4°C, and clarified by centrifugation. Nuclear and cytoplasmic fractions of U2OS cells were generated according to the nuclear extraction protocol of Thermo Fisher Scientific. Proteins were resolved by SDS-PAGE and transferred to nitrocellulose membranes. Immunoblots were performed with the indicated antibodies, and proteins were visualized using the Advansta WesternBright ECL reagent and the Fusion Solo S imaging system.
In situ PLA
U2OSGFP-FAN1 cells were seeded in the presence of Dox (100 ng/ml). Twenty-four hours after seeding, U2OSGFP-FAN1 or HeLa cells were grown in the absence or presence of MMC (150 ng/ml) for 24 hours. Following procedures previously described in (47), cells were preextracted for 5 min on ice and fixed in 4% formaldehyde in 1× PBS (w/v) for 15 min on ice. In situ PLA was performed using Duolink PLA technology (Sigma-Aldrich) according to the manufacturer’s instructions. Briefly, coverslips were blocked for 30 min at 37°C and incubated with the respective primary antibodies for 1 hour at room temperature (RT). Coverslips were washed twice in 1× PBS for 5 min. Then, anti-Mouse PLUS and anti-Rabbit MINUS PLA probes (Sigma-Aldrich) were coupled to the primary antibodies for 1 hour at 37°C. After three wash steps in wash buffer A (10 mM tris, 150 mM NaCl, and 0.05% Tween 20) for 5 min, the PLA probes were ligated for 30 min at 37°C. The coverslips were then washed three times for 5 min in wash buffer A. Amplification using the “Duolink In Situ Detection Reagents Red” (Sigma-Aldrich) was performed at 37°C for 100 min. After amplification, the coverslips were washed twice in wash buffer B (200 mM tris and 100 mM NaCl) for 10 min and once in 0.01× wash buffer B for 1 min. Last, the coverslips were mounted using Vectashield Mounting Media (Vector Laboratories) containing DAPI dihydrochloride, sealed and imaged on a Leica DMI 6000 fluorescence microscope at ×63 magnification.
Quantitative image–based cytometry
Following procedures previously described in (48), automated multichannel wide-field microscopy for QIBC was performed on an Olympus ScanR Screening system equipped with an inverted motorized Olympus IX83 microscope, a motorized stage, infrared-laser hardware autofocus and a Hamamatsu ORCA-FLASH 4.0 V2 cooled scientific complementary metal-oxide semiconductor camera (2048 × 2048 pixels; pixel size, 6.45 μm by 6.45 μm; 12-bit dynamics). Images were analyzed with the Olympus ScanR Image Analysis Software version 3.0.1, a dynamic background correction was applied, nuclei segmentation was performed using an integrated intensity–based object detection module using the DAPI signal, and foci segmentation was performed using an integrated spot detection module. All downstream analyses were focused on properly detected nuclei containing a 2C-4C DNA content as measured by total and mean DAPI intensities. Fluorescence intensities were quantified and are depicted as arbitrary units. Color-coded scatterplots of asynchronous cell populations were generated with Spotfire data visualization software version 7.0.1 (TIBCO). Within one experiment, similar cell numbers were compared for the different conditions. For visualizing discrete data in scatterplots, mild jittering (random displacement of data points along the discrete data axes) was applied to demerge overlapping data points. Representative scatterplots are shown.
Immunofluorescence microscopy
U2OSGFP-FAN1 cells were grown on 13-mm coverslips. The cells were treated with MMC (20 ng/ml) for 24 hours, followed by release into fresh DMEM growth media for 72 hours where indicated. Ethynyl deoxyuridine (EdU) labeling was performed for the last 30 min as previously described. The cells were incubated with 0.5% Triton X-100 in PBS for 2 min, washed one time in 1× PBS before fixation in 4% formaldehyde in 1× PBS for 15 min. The primary antibodies and secondary antibodies were diluted in filtered DMEM containing 10% FCS and 0.02% sodium azide. Antibody incubation was performed for 1 to 2 hours at RT. Following antibody incubation, the cells were washed once with 1× PBS and incubated for 10 min with 1× PBS containing DAPI (0.5 μg/ml). After three washing steps with 1× PBS, the coverslips were briefly washed with distilled water and mounted on glass slides with 6 μl of Mowiol-based mounting media [Mowiol 4.88 (Calbiochem) in glycerol/tris).
Native BrdU assay
U2OSGFP-FAN1 cells were seeded in the presence of Dox (100 ng/ml) and BrdU (25 μM). Twenty-four hours after seeding, the cells were treated or not with MMC (20 ng/ml) for 24 hours. Plates were then washed twice with 1× PBS before adding fresh growth medium containing Dox (100 ng/ml) and BrdU (25 μM). Forty-eight hours later, cells were harvested by trypsinization and preextracted in 0.2% Triton X-100/1× PBS for 10 min on ice. After fixation in 4% formaldehyde/1× PBS, cells were washed three times with 1% bovine serum albumin/PBS. Cells were incubated in 1× Perm/Wash buffer (BD Biosciences) containing the primary antibody against BrdU for 1 hour at RT, followed by a wash step with 1× Perm/Wash buffer. For secondary antibody staining, cells were incubated in 1× Perm/Wash buffer (BD Biosciences) containing Alexa Flour 647–conjugated secondary antibody for 1 hour at RT, followed by a wash step with 1× Perm/Wash buffer. Cells were then counterstained with DAPI (1 μg/ml)/ribonuclease (0.1 mg/ml), and fluorescence was measured on an Attune NxT Flow Cytometer (Thermo Fisher Scientific) and analyzed with FlowJo X (Tree Star).
Slipped-DNA repair assay
A set of previously characterized human DM1 mutagenic intermediates containing pure (CTG)x•(CAG)y repeats where x = 30 or 50 and y = 50 or 30 have been used to perform slipped-strand repair reactions. Circular slipped hybrid DNAs were prepared by hybridizing linearized double-stranded DNAs to circular ssDNAs containing different numbers of repeats. Circular slipped hybrids were electrophoretically resolved on agarose and then gel-purified. Circular substrates containing a one repeat slip-out were made by hybridizing single-stranded circular DNA with a (CAG)47 repeat tract with a double-stranded linearized plasmid containing (CTG)48•(CAG)48 repeats. Detailed procedures for making the substrates have been described previously (28). To prepare cell extracts for repair assay, U2OS Flp-In T-REx cells were induced to express GFP-FAN1-wt or FAN1 mutants using Dox (100 nM) for 72 hours. Cell extracts were prepared as described (33) from the U2OS Flp-In T-Rex, TK6, TK6 MLH1−/−, HCT116, HCT116 complemented with chromosome 3, and repair-proficient HeLa cells. Slipped-strand repair reactions were conducted as described (32). Briefly, 22 fmol of circular slipped-substrates was incubated in 30 mM Hepes-KOH (pH 7.8); creatine kinase (100 μg/ml); 40 mM creatine phosphate; 4 mM ATP; 200 μM each CTP, GTP, and UTP; 33 mM each cold dNTP; and 40 μg of extract at 37°C for 1 hour. Last, after releasing the repeat-containing fragment (EcoRI/HindIII digestion) and resolving on native 4% acrylamide gels, the products were electrotransferred to a membrane and hybridized with a radiolabeled EcoRI/HindIII fragment–containing 17 repeats. Repair efficiency is the proportion of radio intensity of the repair product relative to all repeat-containing fragments (ImageQuant Molecular Dynamics v1.2). To analyze the effect of synthetic 60-mer FAN1 peptide on repair efficiency, cell extracts were incubated in the absence or presence of either wt or mutant synthetic 60-mer FAN1 peptide (GenScript) for 30 min at RT before starting the repair reactions.
Acknowledgments
We thank the Hyman Lab for providing HeLa Kyoto cell lines stably expressing GFP-tagged FAN1. We thank J. Rouse for providing sheep anti-FAN1 antibodies (4). We thank P. Cejka for providing purified recombinant MLH1-MLH3 heterodimer (MutLγ) (49). We thank S. Takeda for providing the TK6 and its derivative MLH1−/− cell lines (36). We thank R. Boland for providing the HCT116 and its derivative HCT116 + chr3 cell lines (37). We thank G. B. Panigrahi and A. Deshmukh for helpful discussions and the Center for Microscopy and Image Analysis (University of Zurich, Switzerland), G. B. Panigrahi, P. Wang, and P. Patel (The Hospital for Sick Children) for technical support. Funding: This work was supported by grants from the Swiss National Science Foundation (31003A_176161 to A.A.S. and PP00P3_179057 to M.A.), the Swiss Cancer Research Foundation (KFS-4702-02-2019 to A.A.S.), the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation program (ERC-2016-STG 714326 to M.A.), the French Infrastructure for Integrated Structural Biology (FRISBI; ANR-10-INBS-0005 to J.B.C.), and the Fondation ARC pour la Recherche sur la Cancer (fellowship to J.D.). Work in the Pearson laboratory is supported by the Petroff Family Fund, the Kazman Family Fund, the Marigold Foundation, Canada Foundation for Innovation, Canadian Institutes for Health Research (CIHR FRN148910), and the Natural Sciences and Engineering Research Council (NSERC). C.E.P. holds a Tier 1 Canada Research Chair in Disease-Associated Genome Instability. Author contributions: A.A.S. conceived and designed the study with significant input from A.P., M.M., and C.E.P. A.A.S., C.E.P., A.P., and M.M. prepared the figures and wrote the manuscript. J.J. instigated the research upon discovering FAN1 as binding partner of MutL homologs and proofread the manuscript. J.D., F.I., V.R., and J.-B.C. conducted and analyzed ITC assays. R.G. performed sequence analyses and molecular modeling. V.S. and M.A. conducted and analyzed QIBC assays. C.Z. performed clonogenic assays. M.M. performed, analyzed, and prepared figures for all slip-out repair assays. A.P. established cell lines. All other experiments were performed and analyzed by A.P. with support from C.Z., G.C., K.M.F., and N.L.M. Competing interests: A.A.S. and C.E.P. plan to submit a patent based on parts of the results and inventions presented in this study. All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/31/eabf7906/DC1
REFERENCES AND NOTES
- 1.Liu T., Ghosal G., Yuan J., Chen J., Huang J., FAN1 acts with FANCI-FANCD2 to promote DNA interstrand cross-link repair. Science 329, 693–696 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Smogorzewska A., Desetty R., Saito T. T., Schlabach M., Lach F. P., Sowa M. E., Clark A. B., Kunkel T. A., Harper J. W., Colaiácovo M. P., Elledge S. P., A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol. Cell 39, 36–47 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kratz K., Schöpf B., Kaden S., Sendoel A., Eberhard R., Lademann C., Cannavo E., Sartori A. A., Hengartner M. O., Jiricny J., Deficiency of FANCD2-associated nuclease KIAA1018/FAN1 sensitizes cells to interstrand crosslinking agents. Cell 142, 77–88 (2010). [DOI] [PubMed] [Google Scholar]
- 4.MacKay C., Déclais A.-C., Lundin C., Agostinho A., Deans A. J., MacArtney T. J., Hofmann K., Gartner A., West S. C., Helleday T., Lilley D. M. J., Rouse J., Identification of KIAA1018/FAN1, a DNA repair nuclease recruited to DNA damage by monoubiquitinated FANCD2. Cell 142, 65–76 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cannavo E., Gerrits B., Marra G., Schlapbach R., Jiricny J., Characterization of the interactome of the human MutL homologues MLH1, PMS1, and PMS2. J. Biol. Chem. 282, 2976–2986 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Usdin K., House N. C. M., Freudenreich C. H., Repeat instability during DNA repair: Insights from model systems. Crit. Rev. Biochem. Mol. Biol. 50, 142–167 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Massey T. H., Jones L., The central role of DNA damage and repair in CAG repeat diseases. Dis. Model. Mech. 11, dmm031930 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium , Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell 162, 516–526 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium , CAG repeat not polyglutamine length determines timing of Huntington’s disease onset. Cell 178, 887–900.e14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim K.-H., Hong E. P., Shin J. W., Chao M. J., Loupe J., Gillis T., Mysore J. S., Holmans P., Jones L., Orth M., Monckton D. G., Long J. D., Kwak S., Lee R., Gusella J. F., MacDonald M. E., Lee J.-M., Genetic and functional analyses point to FAN1 as the source of multiple Huntington disease modifier effects. Am. J. Hum. Genet. 107, 96–110 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pinto R. M., Dragileva E., Kirby A., Lloret A., Lopez E., St Claire J., Panigrahi G. B., Hou C., Holloway K., Gillis T., Guide J. R., Cohen P. E., Li G.-M., Pearson C. E., Daly M. J., Wheeler C., Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington’s disease mice: Genome-wide and candidate approaches. PLOS Genet. 9, e1003930 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kadyrova L. Y., Gujar V., Burdett V., Modrich P. L., Kadyrov F. A., Human MutLγ, the MLH1-MLH3 heterodimer, is an endonuclease that promotes DNA expansion. Proc. Natl. Acad. Sci. U.S.A. 117, 3535–3542 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goold R., Flower M., Moss D. H., Medway C., Wood-Kaczmar A., Andre R., Farshim P., Bates G. P., Holmans P., Jones L., Tabrizi S. J., FAN1 modifies Huntington’s disease progression by stabilizing the expanded HTT CAG repeat. Hum. Mol. Genet. 28, 650–661 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loupe J. M., Pinto R. M., Kim K.-H., Gillis T., Mysore J. S., Andrew M. A., Kovalenko M., Murtha R., Seong I., Gusella J. F., Kwak S., Howland D., Lee R., Lee J.-M., Wheeler V. C., MacDonald M. E., Promotion of somatic CAG repeat expansion by Fan1 knock-out in Huntington’s disease knock-in mice is blocked by Mlh1 knock-out. Hum. Mol. Genet. 29, 3044–3053 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao X.-N., Usdin K., FAN1 protects against repeat expansions in a Fragile X mouse model. DNA Repair (Amst.). 69, 1–5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dherin C., Gueneau E., Francin M., Nunez M., Miron S., Liberti S. E., Rasmussen L. J., Zinn-Justin S., Gilquin B., Charbonnier J.-B., Boiteux S., Characterization of a highly conserved binding site of Mlh1 required for exonuclease I-dependent mismatch repair. Mol. Cell. Biol. 29, 907–918 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gueneau E., Dherin C., Legrand P., Tellier-Lebegue C., Gilquin B., Bonnesoeur P., Londino F., Quemener C., Le Du M.-H., Marquez J. A., Moutiez M., Gondry M., Boiteux S., Charbonnier J.-B., Structure of the MutLα C-terminal domain reveals how Mlh1 contributes to Pms1 endonuclease site. Nat. Struct. Mol. Biol. 20, 461–468 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Goellner E. M., Putnam C. D., Graham 5th W. J., Rahal C. M., Li B.-Z., Kolodner R. D., Identification of Exo1-Msh2 interaction motifs in DNA mismatch repair and new Msh2-binding partners. Nat. Struct. Mol. Biol. 25, 650–659 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amin N. S., Nguyen M. N., Oh S., Kolodner R. D., exo1-Dependent mutator mutations: Model system for studying functional interactions in mismatch repair. Mol. Cell. Biol. 21, 5142–5155 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Porro A., Berti M., Pizzolato J., Bologna S., Kaden S., Saxer A., Ma Y., Nagasawa K., Sartori A. A., Jiricny J., FAN1 interaction with ubiquitylated PCNA alleviates replication stress and preserves genomic integrity independently of BRCA2. Nat. Commun. 8, 1073 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kondo E., Horii A., Fukushige S., The interacting domains of three MutL heterodimers in man: hMLH1 interacts with 36 homologous amino acid residues within hMLH3, hPMS1 and hPMS2. Nucleic Acids Res. 29, 1695–1702 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Desai A., Gerson S., Exo1 independent DNA mismatch repair involves multiple compensatory nucleases. DNA Repair (Amst.). 21, 55–64 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin S. A., Lord C. J., Ashworth A., Therapeutic targeting of the DNA mismatch repair pathway. Clin. Cancer Res. 16, 5107–5113 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Shinsato Y., Furukawa T., Yunoue S., Yonezawa H., Minami K., Nishizawa Y., Ikeda R., Kawahara K., Yamamoto M., Hirano H., Tokimura H., Aria K., Reduction of MLH1 and PMS2 confers temozolomide resistance and is associated with recurrence of glioblastoma. Oncotarget 4, 2261–2270 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao Q., Xue X., Longerich S., Sung P., Xiong Y., Structural insights into 5′ flap DNA unwinding and incision by the human FAN1 dimer. Nat. Commun. 5, 5726 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iftode C., Daniely Y., Borowiec J. A., Replication protein A (RPA): The eukaryotic SSB. Crit. Rev. Biochem. Mol. Biol. 34, 141–180 (1999). [DOI] [PubMed] [Google Scholar]
- 27.Pearson C. E., Tam M., Wang Y.-H., Montgomery S. E., Dar A. C., Cleary J. D., Nichol K., Slipped-strand DNAs formed by long (CAG)*(CTG) repeats: Slipped-out repeats and slip-out junctions. Nucleic Acids Res. 30, 4534–4547 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakamori M., Pearson C. E., Thornton C. A., Bidirectional transcription stimulates expansion and contraction of expanded (CTG)*(CAG) repeats. Hum. Mol. Genet. 20, 580–588 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Axford M. M., Wang Y.-H., Nakamori M., Zannis-Hadjopoulos M., Thornton C. A., Pearson C. E., Detection of slipped-DNAs at the trinucleotide repeats of the myotonic dystrophy type I disease locus in patient tissues. PLOS Genet. 9, e1003866 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakamori M., Panigrahi G. B., Lanni S., Gall-Duncan T., Hayakawa H., Tanaka H., Luo J., Otabe T., Li J., Sakata A., Caron M.-C., Joshi N., Prasolava T., Chiang K., Masson J.-Y., Wold M. S., Wang X., Lee M. Y. W. T., Huddleston J., Munson K. M., Davidson S., Layeghifard M., Edward L.-M., Gallon R., Santibanez-Koref M., Murata A., Takahashi M. P., Eichler E. E., Shlien A., Nakatani K., Mochizuki H., Pearson C. E., A slipped-CAG DNA-binding small molecule induces trinucleotide-repeat contractions in vivo. Nat. Genet. 52, 146–159 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Panigrahi G. B., Lau R., Montgomery S. E., Leonard M. R., Pearson C. E., Slipped (CTG)*(CAG) repeats can be correctly repaired, escape repair or undergo error-prone repair. Nat. Struct. Mol. Biol. 12, 654–662 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Panigrahi G. B., Slean M. M., Simard J. P., Gileadi O., Pearson C. E., Isolated short CTG/CAG DNA slip-outs are repaired efficiently by hMutSβ, but clustered slip-outs are poorly repaired. Proc. Natl. Acad. Sci. U.S.A. 107, 12593–12598 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Panigrahi G. B., Cleary J. D., Pearson C. E., In vitro (CTG)*(CAG) expansions and deletions by human cell extracts. J. Biol. Chem. 277, 13926–13934 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Panigrahi G. B., Slean M. M., Simard J. P., Pearson C. E., Human mismatch repair protein hMutLα is required to repair short slipped-DNAs of trinucleotide repeats. J. Biol. Chem. 287, 41844–41850 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pluciennik A., Burdett V., Baitinger C., Iyer R. R., Shi K., Modrich P., Extrahelical (CAG)/(CTG) triplet repeat elements support proliferating cell nuclear antigen loading and MutLα endonuclease activation. Proc. Natl. Acad. Sci. U.S.A. 110, 12277–12282 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rahman M. M., Mohiuddin M., Keka I. S., Yamada K., Tsuda M., Sasanuma H., Andreani J., Guerois R., Borde V., Charbonnier J.-B., Takeda S., Genetic evidence for the involvement of mismatch repair proteins, PMS2 and MLH3, in a late step of homologous recombination. J. Biol. Chem. 295, 17460–17475 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koi M., Umar A., Chauhan D. P., Cherian S. P., Carethers J. M., Kunkel T. A., Boland C. R., Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces N-methyl-N’-nitro-N-nitrosoguanidine tolerance in colon tumor cells with homozygous hMLH1 mutation. Cancer Res. 54, 4308–4312 (1994). [PubMed] [Google Scholar]
- 38.Shannon N. B., Tan J. W., Tan H. L., Wang W., Chen Y., Lim H. J., Tan Q. X., Hendrikson J., Ng W. H., Loo L. Y., Skanthakumar T., Wasudevan S. D., Kon O. L., Lim T. K. H., Tan G. H. C., Chia C. S., Soo K. C., Ong C.-A. J., Teo M. C. C., A set of molecular markers predicts chemosensitivity to Mitomycin-C following cytoreductive surgery and hyperthermic intraperitoneal chemotherapy for colorectal peritoneal metastasis. Sci. Rep. 9, 10572 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deshmukh A. L., Porro A., Mohiuddin M., Lanni S., Panigrahi G. B., Caron M.-C., Masson J.-Y., Sartori A. A., Pearson C. E., FAN1, a DNA repair nuclease, as a modifier of repeat expansion disorders. J Huntingtons Dis. 10, 95–122 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wright G. E. B., Black H. F., Collins J. A., Gall-Duncan T., Caron N. S., Pearson C. E., Hayden M. R., Interrupting sequence variants and age of onset in Huntington’s disease: Clinical implications and emerging therapies. Lancet Neurol. 19, 930–939 (2020). [DOI] [PubMed] [Google Scholar]
- 41.Schmidt M. H. M., Pearson C. E., Disease-associated repeat instability and mismatch repair. DNA Repair (Amst.). 38, 117–126 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Gomes-Pereira M., Fortune M. T., Ingram L., McAbney J. P., Monckton D. G., Pms2 is a genetic enhancer of trinucleotide CAG.CTG repeat somatic mosaicism: Implications for the mechanism of triplet repeat expansion. Hum. Mol. Genet. 13, 1815–1825 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Zhao X., Zhang Y., Wilkins K., Edelmann W., Usdin K., MutLγ promotes repeat expansion in a Fragile X mouse model while EXO1 is protective. PLOS Genet. 14, e1007719 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cannella M., Maglione V., Martino T., Ragona G., Frati L., Li G.-M., Squitieri F., DNA instability in replicating Huntington’s disease lymphoblasts. BMC Med. Genet. 10, 11 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ferretti L. P., Himmels S.-F., Trenner A., Walker C., von Aesch C., Eggenschwiler A., Murina O., Enchev R. I., Peter M., Freire R., Porro A., Sartori A. A., Cullin3-KLHL15 ubiquitin ligase mediates CtIP protein turnover to fine-tune DNA-end resection. Nat. Commun. 7, 12628 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guzmán C., Bagga M., Kaur A., Westermarck J., Abankwa D., ColonyArea: An ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLOS ONE 9, e92444 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Przetocka S., Porro A., Bolck H. A., Walker C., Lezaja A., Trenner A., von Aesch C., Himmels S.-F., D’Andrea A. D., Ceccaldi R., Altmeyer M., Sartori A. A., CtIP-mediated fork protection synergizes with BRCA1 to suppress genomic instability upon DNA replication stress. Mol. Cell 72, 568–582.e6 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Kilic S., Lezaja A., Gatti M., Bianco E., Michelena J., Imhof R., Altmeyer M., Phase separation of 53BP1 determines liquid-like behavior of DNA repair compartments. EMBO J. 38, e101379 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cannavo E., Sanchez A., Anand R., Ranjha L., Hugener J., Adam C., Acharya A., Weyland N., Aran-Guiu X., Charbonnier J.-B., Hoffmann E. R., Borde V., Matos J., Cejka P., Regulation of the MLH1-MLH3 endonuclease in meiosis. Nature 586, 618–622 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/31/eabf7906/DC1