

Abstract
Aberrant activation of FGFR signaling occurs in many cancers, and ATP-competitive FGFR inhibitors have received regulatory approval. Despite demonstrating clinical efficacy, these inhibitors exhibit dose-limiting toxicity, potentially due to a lack of selectivity amongst the FGFR family and are poorly tolerated. Here, we report the discovery and characterization of DGY-09-192, a bivalent degrader that couples the pan-FGFR inhibitor BGJ398 to a CRL2VHL E3 ligase recruiting ligand, which preferentially induces FGFR1&2 degradation while largely sparing FGFR3&4. DGY-09-192 exhibited two-digit nanomolar DC50s for both wildtype FGFR2 and several FGFR2-fusions, resulting in degradation-dependent antiproliferative activity in representative gastric cancer and cholangiocarcinoma cells. Importantly, DGY-09-192 induced degradation of a clinically relevant FGFR2 fusion protein in a xenograft model. Taken together, we demonstrate that DGY-09-192 has potential as a prototype FGFR degrader.
Keywords: protein degradation, FGFR2, FGFR1, degrader, cholangiocarcinoma
Graphical Abstract
Current approved FGFR inhibitors lack selectivity within the FGFR family, which may contribute to their poor tolerability. Here, we describe DGY-09-192, a selective FGFR1&2 degrader that destabilized wildtype FGFR1&2 and FGFR2 fusion proteins, had potent anti-proliferative activity in FGFR2-dependent cells, and possessed pharmacokinetics properties suitable for in vivo degradation. Thus, FGFR degradation may be a promising therapeutic approach.
Introduction
The human genome encodes four FGFRs (FGFR1–4), with FGFR4 being the most divergent.[1][2] This family of receptor tyrosine kinases and their ligands exhibit highly specific temporal and spatial expression patterns and regulate diverse physiological processes including embryogenesis, angiogenesis, metabolism, fibrogenesis, proliferation, and differentiation.
Dysregulation of FGFR signaling is an oncogenic driver in many cancers, including breast, biliary tract, gastro-esophageal, and hepatocellular carcinomas[3], and can occur through receptor or ligand overexpression, gene amplification, activating mutations, or chromosomal fusions. Moreover, feedback activation of FGFR1 signaling has been identified as a mechanism of adaptive resistance to RTK/RAS/MEK pathway inhibition.
The oncogenic role of aberrant FGFR signaling has prompted extensive efforts to develop therapeutic agents targeting this pathway.[4] First-generation FGFR inhibitors, such as dovitinib,[5] nintedanib (BIBF1120),[6] and ponatinib (AP23534),[7] are multi-targeted agents that potently inhibit VEGFR and PDGFR. Although ponatinib[7b] and nintedanib[8] are approved for the treatment of myeloid leukemia and non-small-cell lung cancer, respectively, the toxicity profile of these agents limits their therapeutic doses.
To improve selectivity, a strategy of covalently targeting the conserved cysteine on the activation loop of FGFR paralogs has been achieved by FIIN1–3,[9] PRN1371,[10] BLU554,[11] and TAS-120.[12] Separately, second generation reversible FGFR inhibitors such as AZD4547,[13] BGJ398,[14] JNJ-42756493[15] and INCB054828[16] have improved selectivity for the FGFR family and have demonstrated efficacy against FGFR-dependent cancers in clinical trials, leading to the recent approval of JNJ-42756493 and INCB054828 for the treatment of advanced urothelial and biliary tract cancers, respectively (Figure 1). Nevertheless, despite significant progress in the development of potent and selective FGFR inhibitors over the last decade, the long-term efficacy of these agents in cancer treatment has been hampered by the rapid onset of acquired resistance, occurring commonly through the mutation of the gatekeeper residue. For example, while the covalent inhibitor TAS-120 is effective against some FGFR mutants, it cannot overcome the gatekeeper mutation.[17] Moreover, cancer cells may develop resistance to covalent inhibitors like TAS-120 by mutating the residue targeted by the inhibitor, a resistance mechanism previously demonstrated for BTK[18] and EGFR[19] covalent inhibitors.
Figure 1.
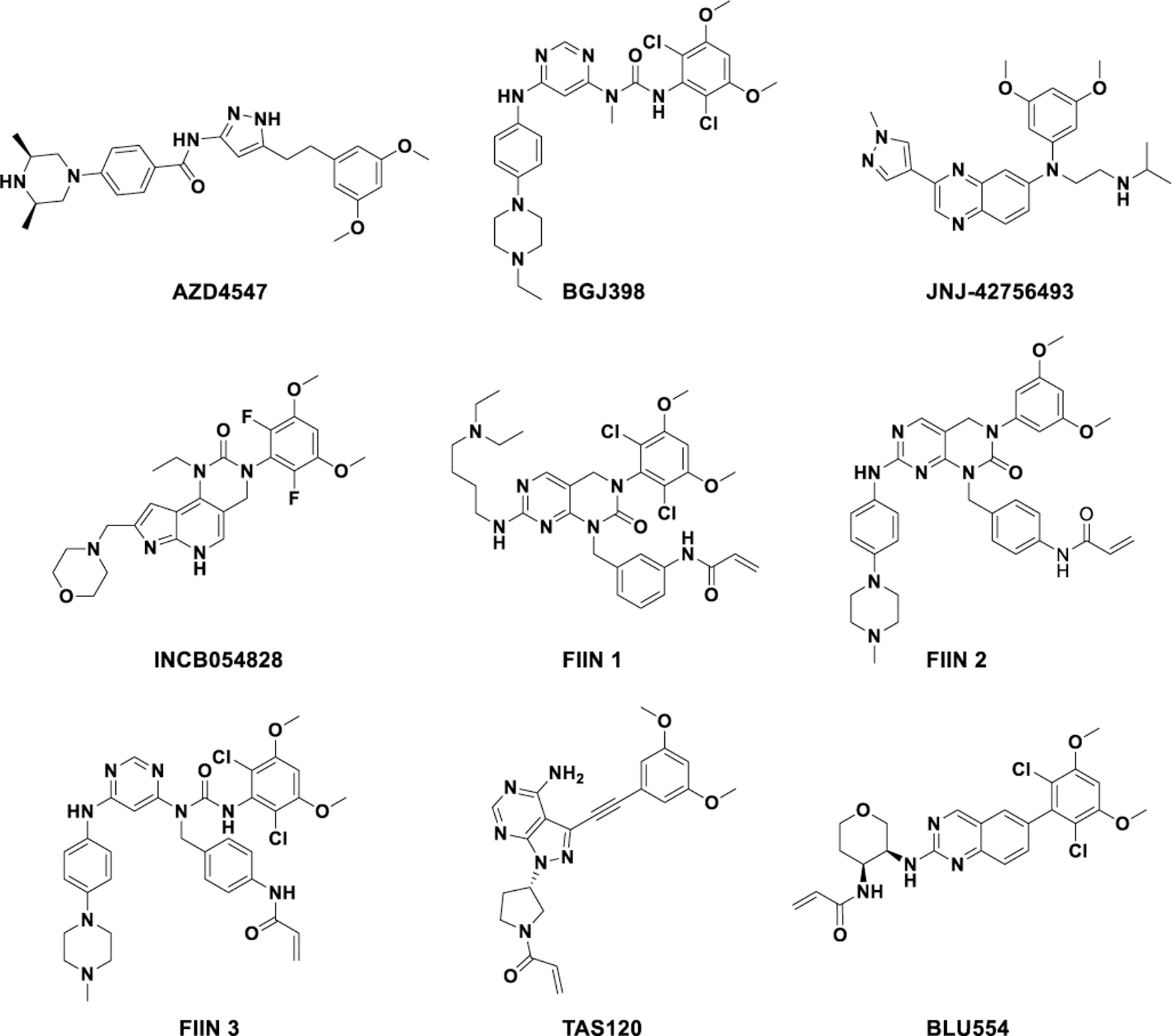
Structures of selective FGFR inhibitors.
Moreover, second generation FGFR inhibitors are pan-FGFR inhibitors with class-specific on-target toxicities.[15a, 20] Many patients require dose reduction, and thus drug exposure within tumors may be sub-optimal. Thus, inhibitors with selectivity within the FGFR family could be therapeutically beneficial. However, given the high structural similarity in the ATP-binding pocket between FGFR1–3, developing selective inhibitors is challenging and none have been reported to date. Currently, the only small molecule with selectivity within the FGFR family is BLU554, which targets a unique cysteine (Cys 554) located in the hinge region of FGFR4.[15]
Recently, degradation strategies that utilize heterobifunctional molecules to recruit an E3 ubiquitin ligase complex to a target protein for ubiquitination and subsequent proteasome-mediated degradation have been employed to increase target selectivity when compared to their parental inhibitors[21][22]. For example, a degrader that was constructed with a promiscuous kinase inhibitor TAE684 and CRBN ligand caused selective degradation of only a subset of the TAE684 binding kinase targets.[23] Furthermore, we have recently developed degraders that can differentiate among close paralogs within the same family, such as CDK4 versus CDK6[24] or CDK9 versus other CDKs[25]. Here, we describe the development, characterization and validation of DGY-09-192 as a dual degrader of FGFR1 and 2, with minimal activity towards FGFR3 and 4.
Results and Discussion
To design FGFR degraders, we first examined two high-resolution crystal structures of FGFR bound to ATP-competitive inhibitors BGJ398 and FIIN1–3 and identified the solvent-exposed piperazine as appropriate sites for linker installation.
We then synthesized several imide-based CRBN-targeting compounds with various linkers attached via the piperazine (Table 1&2). The prepared molecules remained potent biochemical inhibitors of FGFR2 as shown in Table 1&2. Next, we evaluated whether these compounds induced degradation of the TEL-FGFR2 fusion protein (which includes the FGFR2 kinase domain) in Ba/F3 cells. As shown in Figure 2A, 24h treatment with 1 μM of each degrader resulted in reduction of TEL-FGFR2 fusion protein levels in Ba/F3 cells. Encouraged by this result, we then tested these compounds in the KATO III gastric cancer cell line harboring amplification of full length FGFR2. Surprisingly, immunoblot analysis revealed no decrease of FGFR2 levels up to 24h treatment (Figure 2B). While the reasons for the differential degradation observed in the Ba/F3 versus the KATO III cells remains unclear, potential contributing factors include differences in protein expression, cellular localization of TEL-FGFR2 (cytoplasmic) compared to FGFR2 (membrane bound), CRBN availability, and/or ability to form a ternary complex with FGFR2.
Table 1.
Biochemical IC50s of BGJ398-IMiDs based degraders.
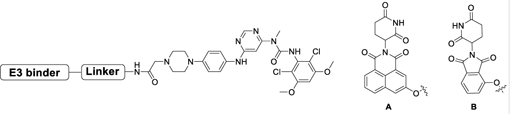 | |||
|---|---|---|---|
| Compound | Linker | E3 binder | FGFR2 IC50 (nM)[a] |
| DGY-09-037 | A | 51 | |
| DGY-09-073 | B | 33 | |
| DGY-09-038 | A | 63 | |
| DGY-09-036 | A | 54 | |
| DGY-09-041 | A | 23 | |
All the IC50s are measured by Z’-LYTE kinase assay by Invitrogen.
Table 2.
Biochemical IC50s of FIIN2- IMiDs based degraders.
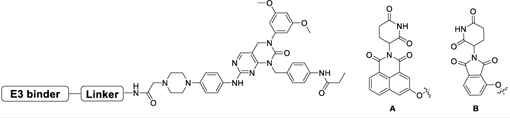 | |||
|---|---|---|---|
| Compound | Linker | E3 binder | FGFR2 IC50 (nM)[a] |
| DGY-09-068 | A | 93 | |
| DGY-09-074 | B | 42 | |
| DGY-09-077 | A | 72 | |
All the IC50s are measured by Z’-LYTE kinase assay by Invitrogen.
Figure 2.
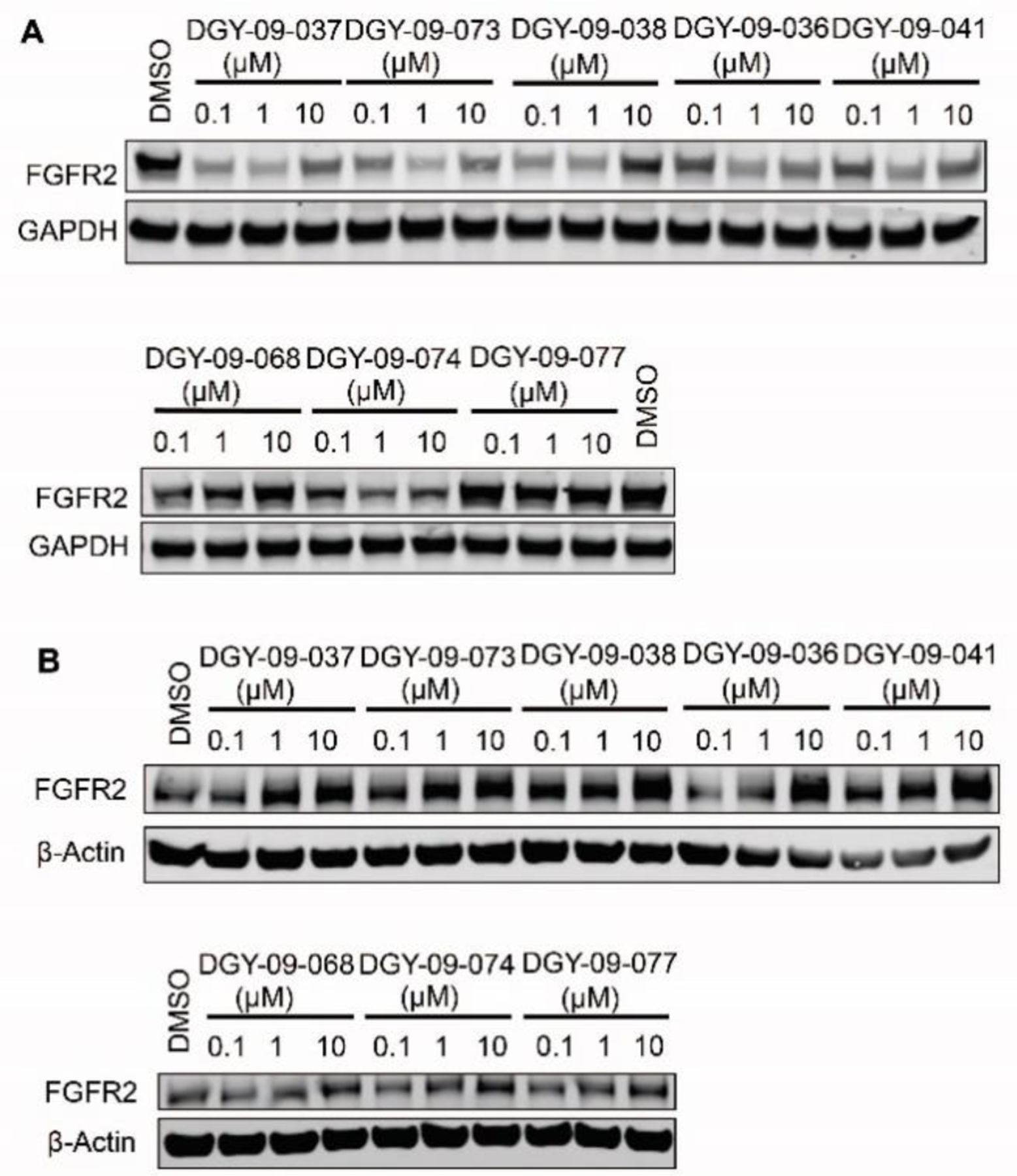
Screening of IMiDs based degraders. Immunoblot analysis of FGFR2 in A) TEL-FGFR2 Ba/F3 cells and B) Kato III cells treated with the indicated BGJ398-IMiDs or FIIN2-IMiDs based compounds for 24h.
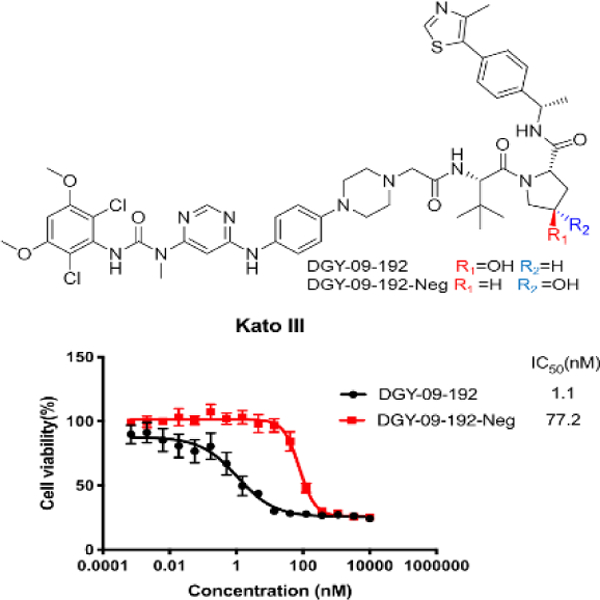
To examine whether the nature of the E3 ligase affects the potency of FGFR2 degraders, we synthesized several bivalent compounds in which BGJ398 was linked with a VHL recruiting ligand connected through a variety of exit vectors (Table 3). Overall, all VHL-based degrader molecules displayed a similar biochemical inhibition of FGFR2. However, DGY-09-192 showed the most potent degradation of full length FGFR2 protein in KATO III cells after 24h treatment at 1 µM (Figure 3B). A dose titration revealed a DC50 of 70 nM (Dmax=74%) after 6h treatment, while the hook effect was not observed until concentrations above 5 µM (Figure 3C, S4). A time course displayed signification FGFR2 degradation within 4h, which was sustained up to 16h (Figure 3D). To investigate whether DGY-09-192 could target different FGFR-fusion proteins, which might present distinct steric constraints that affect tertiary complex formation,[27] we tested the compound in CCLP-1-FP cells with ectopic expression of the FGFR2-PHGDH fusion and ICC13–7 cells that express the FGFR2-OPTN fusion endogenously. These fusions contain nearly the full coding sequence of FGFR2, including the extracellular, transmembrane, and kinase domains, fused in-frame to partners encoding dimerization domains. As shown in Figure 3E&F, 100 nM and 1 µM of DGY-09-192 resulted in the dose-dependent degradation of FGFR fusion proteins in both CCLP-1-FP and ICC13–7 cells.
Table 3.
Biochemical IC50s of FGFR2 for BGJ398-VHL based degraders.
 | |||
|---|---|---|---|
| Compound | Linker | E3 binder | FGFR2 IC50 (nM)[a] |
| DGY-09-192 |  |
C | 34 |
| NJH-05–166 |  |
C | 30 |
| NJH-05–132 | 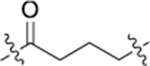 |
C | 10 |
| NJH-05-123 | C | 16 | |
| NJH-05-043 | 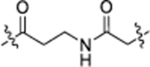 |
C | 30 |
| NJH-05-044 | 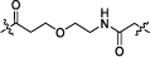 |
C | 33 |
| DGY-11-123 | D | 59 | |
| DGY-11-122 | E | 43 | |
| DGY-09-192-Neg | 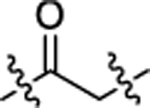 |
F | 28 |
All the IC50s are measured by Z’-LYTE kinase assay by Invitrogen.
Figure 3.
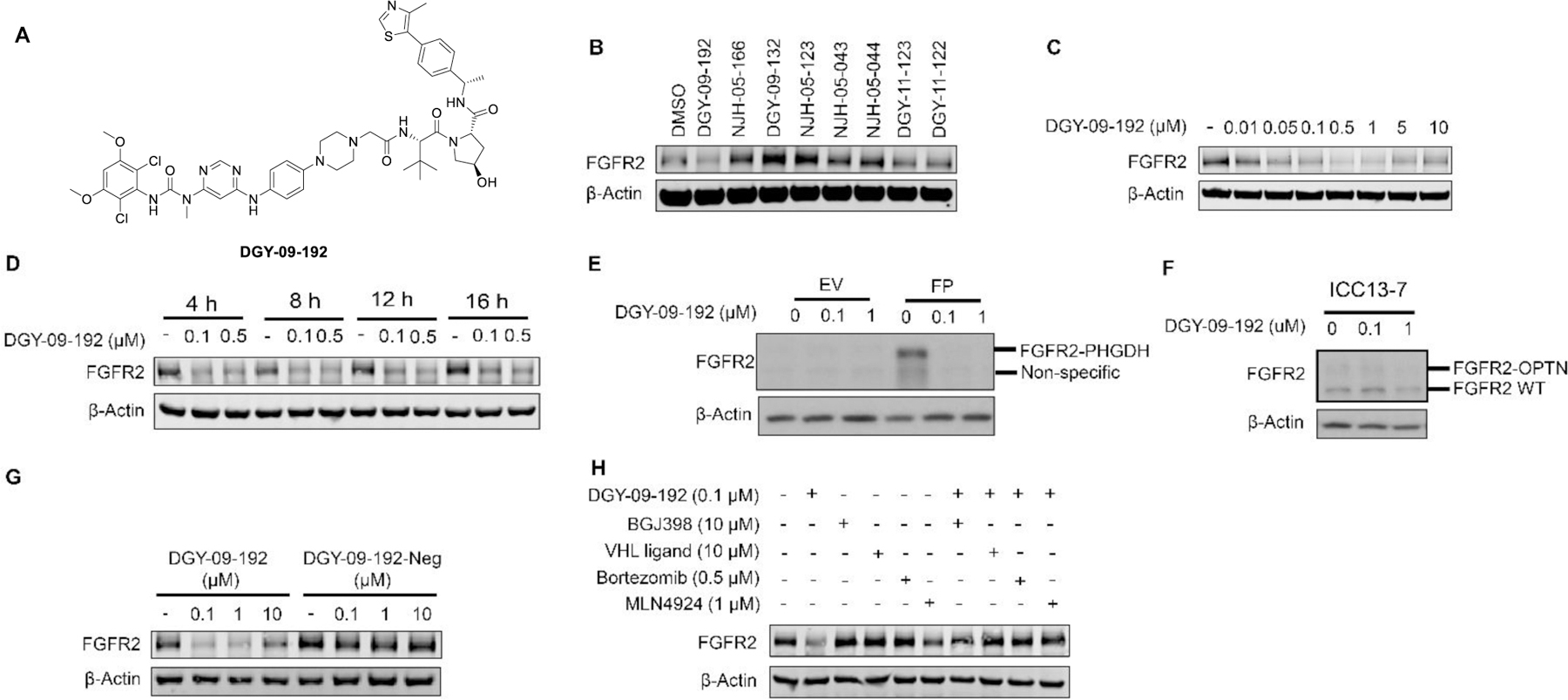
DGY-09-192 induced degradation of FGFR2 or FGFR2-fusion in a VHL dependent manner. A. Structure of DGY-09-192. B. Immunoblot analysis of FGFR2 in Kato III cells treated with 1 µM of the indicated compounds for 24h. C. Immunoblot analysis of FGFR2 in Kato III cells treated with indicated concentrations of DGY-09-192 for 6h. D. Immunoblot analysis of FGFR2 in Kato III cells treated with DGY-09-192 at indicated time points. E. Immunoblot analysis of FGFR2-PHGDH (FP) fusion in CCLP-1 cells and F) FGFR2-OPTN fusion in ICC13–7 cells treated with DGY-09-192 for 24h. G. Immunoblot analysis of FGFR2 in Kato III cells treated with DGY-09-192 or DGY-09-192-Neg for 4h. H. Immunoblot analysis of FGFR2 in Kato III cells pretreated for 2h with BGJ398, VHL ligand, Bortezomib or MLN4924, and then treated with DGY-09-192 for 4h. EV is short for empty vector.
To investigate whether the observed FGFR2 degradation induced by DGY-09-192 is VHL-dependent, we synthesized the negative control compound DGY-09-192-Neg, featuring a VHL ligand previously shown to exhibit a substantial reduction in VHL binding affinity[28] (Figure S2). As expected, DGY-09-192-Neg showed similar biochemical inhibition of FGFR kinase activity as DGY-09-192 (Table 3), but could not degrade FGFR2 (Figure 3G). In addition, pretreatment with either BGJ398 or VHL ligand rescued FGFR2 degradation induced by DGY-09-192. Similar rescue was observed with a proteasome inhibitor (bortezomib) and a NEDD8 activating E1 enzyme inhibitor (MLN4924), which inhibits the process of ubiquitination and proteasomal degradation (Figure 3H). Taken together, these data support a VHL-dependent mechanism for DGY-09-192 induced FGFR2 degradation.
We next inspected the degradation selectivity of DGY-09-192. We observed that DGY-09-192 is an equally potent inhibitor for all FGFR isoforms (Table S1). However, immunoblot analysis and quantitative proteomics indicated that DGY-09-192 is a highly selective degrader of FGFR1 and 2, while sparing FGFR3 and 4. As shown in Figure 4A, 100 nM of DGY-09-192 effectively degraded FGFR1 protein in CCLP1 cells (DC50 = 4.35 nM, Dmax=85%, Figure S4), but had minimal effects on FGFR3 and FGFR4 protein levels in JHH7 cells (Figure S5&S6). To further assess the proteome-wide degradation selectivity, we performed quantitative mass spectrometry-based proteomics following 5h treatment in Kelly cells which express both FGFR1 and FGFR2. As shown in Figure 4B, DGY-09-192 exhibited a high selectivity for FGFR1 and FGFR2 with PDE6D as an off-target (Figure S7). PDE6D has a large lipophilic binding pocket and has been previously reported as a degradable target for both IMiDs and VHL based degraders.[29]
Figure 4.
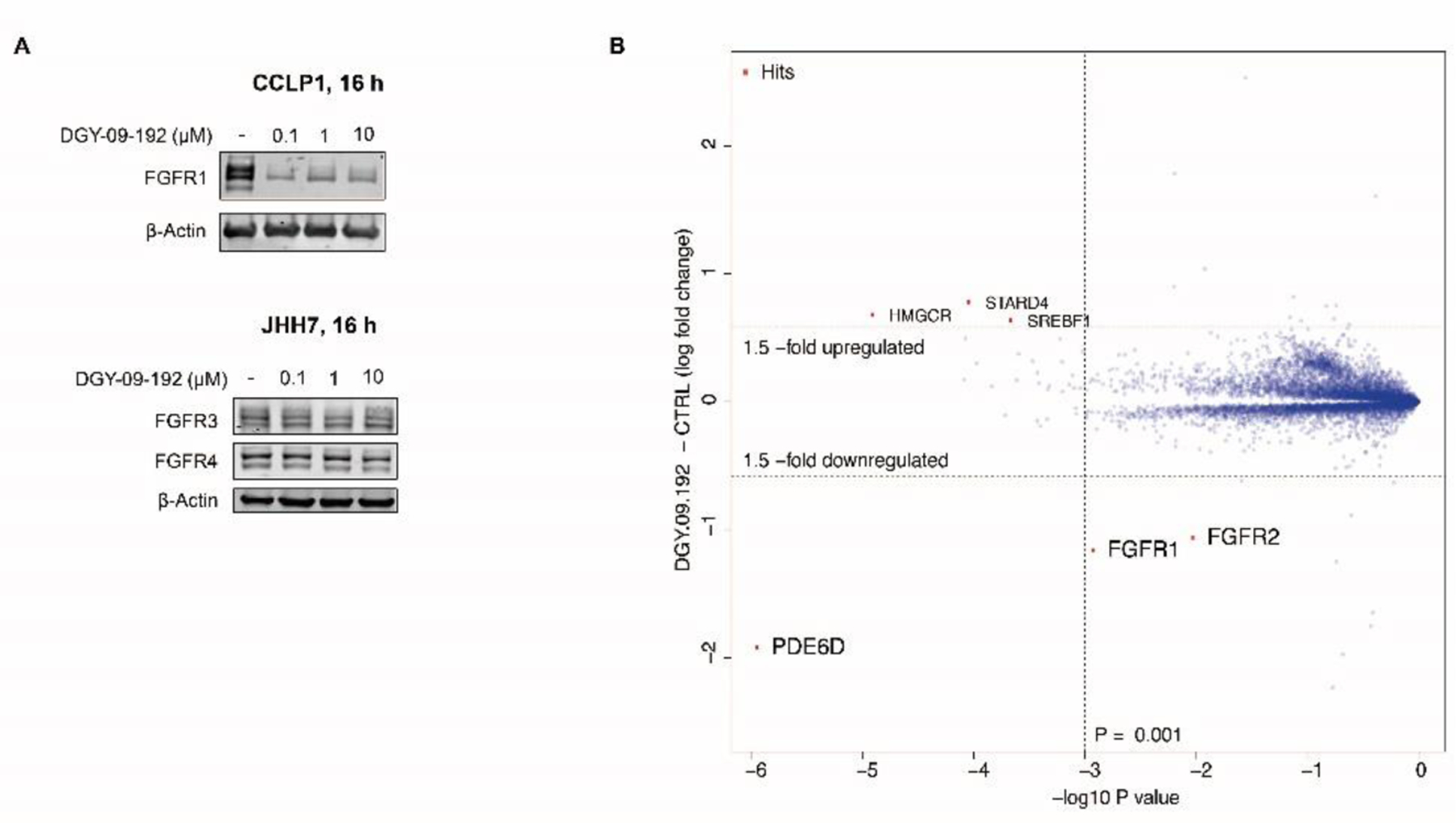
Selectivity of DGY-09-192. A. Immunoblot analysis of FGFR1 in CCLP1 cells or FGFR3/4 in JHH7 cells treated with DGY-09-192 for 16h. B. Quantitative proteomics showing relative abundance of proteins in Kelly cells treated for 5h with 1 µM.
As the proliferation of KATO III cells is highly dependent on FGFR2 kinase activity, we next investigated the anti-proliferative effects of DGY-09-192 in KATO III cells. CellTiter-Glo analysis revealed that DGY-09-192 had an anti-proliferative IC50 of 1 nM after 72-hour treatment (Figure 5A). In contrast, the negative control compound DGY-09-192-Neg exhibited a 70-fold loss in anti-proliferative activity (IC50 = 77 nM), indicating that FGFR2 degradation contributes significantly to the cell growth inhibition induced by DGY-09-192. In addition, DGY-09-192 also exhibited anti-proliferative activities against the FGFR1 overexpressing CCLP1 cells, FGFR2 fusion-positive ICC13–7 cells and engineered FGFR2 fusion CCLP-FP cells, with IC50s of 17 nM, 40 nM and 8 nM, respectively, versus 232 nM, 689 nM and 70 nM upon DGY-09-192-Neg treatment. Importantly, control biliary cancer cell lines lacking FGFR activation (RBE and SNU1079) were completely insensitive to DGY-09-192 (IC50>10 uM for both) (Figure 5A). In addition, downstream FGFR2 signaling, evaluated by phosphorylation of the scaffolding protein FRS2 or ERK, was durably suppressed by DGY-09-192, whereas DGY-09-192-Neg had incomplete and transient effects on FGFR signaling (Figure 5B). AKT phosphorylation was not as strongly affected, consistent with studies of FGFR kinase inhibitors in this cell line [17].
Figure 5.
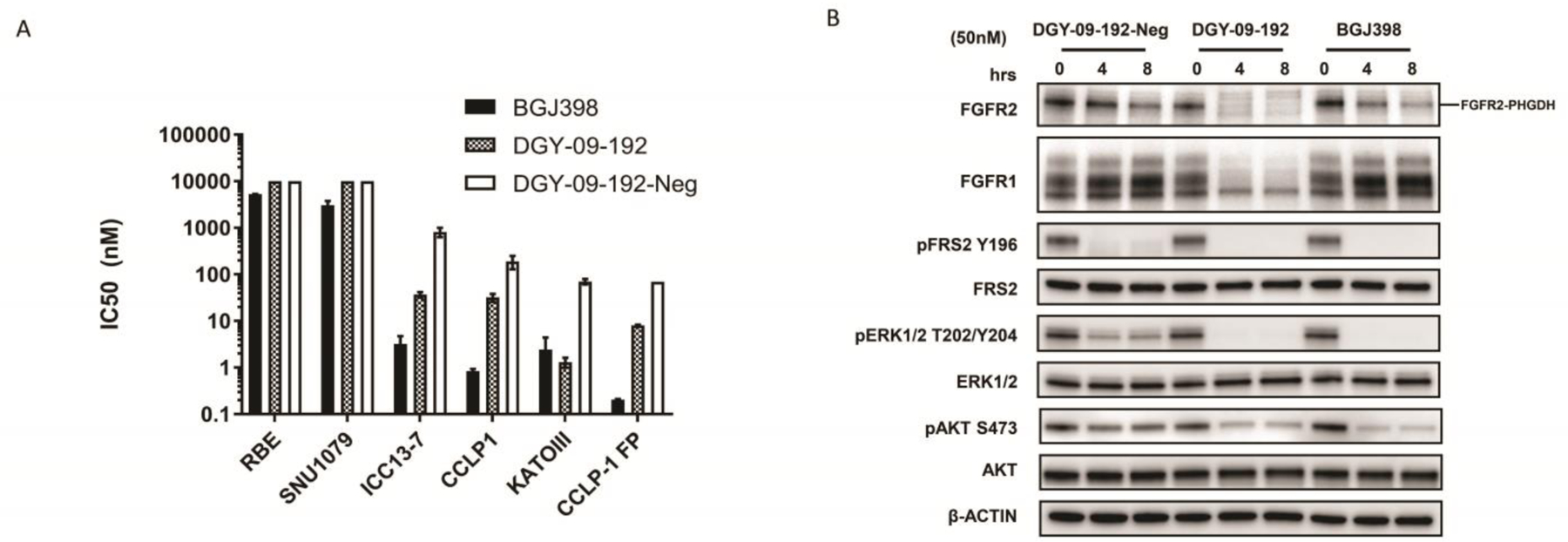
Evaluation of DGY-09-192 in FGFR-dependent and independent cell lines. A. RBE and SNU1079 cells (no FGFR abnormalities), Kato III cells (FGFR2 amplication), CCLP1 cells (FGFR1 overexpression) or CCLP-FP (FGFR2-PHGDH fusion) were treated with BGJ398[17], DGY-09-192 and DGY-09-192-Neg for 3d (ICC13–7 cells for 10d). Cell viability was assessed with CellTiter-Glo. Data are presented as the mean ± SEM (n = 3).(Data see Table S2, Figure S3) B. Immunoblot analysis of FGFR2-PHGDH fusion and signaling inhibition in CCLP1-FP cells treated with DGY-09-192, DGY-09-192-Neg and BGJ398 with 50 nM for 4,or 8 hours.
Next, we sought to evaluate target engagement and inhibition of signaling following in vivo administration of DGY-09-192. We conducted a pharmacokinetics (PK) study in mice following a single dose of DGY-09-192 via intravenous (IV, 1 mg/kg), intraperitoneal (IP, 3 mg/kg) injection or oral administration (10 mg/kg). DGY-09-192 exhibited a fairly long half-life (T1/2 of 5h) with low clearance via IV and IP administration, but negligible oral bioavailability (Table 4). We then chose to use a CCLP1-FGFR2-PHGDH xenograft model to assess in vivo degradation. Once tumors reached ~200 mm3, mice were administered DGY-09-192 (20 or 40 mg/kg, IP QD) for 6d. Immunoblot analysis of tumor samples isolated 4h after the last dose revealed reduction in both FGFR2-PHGDH protein levels and phosphorylation of downstream markers FRS2 and ERK1/2 in a dose-dependent manner (Figure 6).
Table 4.
Pharmacokinetics property of DGY-09-192.
| Parameter | Unit | iv (1 mg/kg) | ip (3 mg/kg) | po (10 mg/kg) |
|---|---|---|---|---|
| T1/2 | h | 5.45 | 4.25 | 5.10 |
| Tmax | h | 0.08 | 1.33 | 1.67 |
| Cmax | µM | 1.60 | 1.49 | 0.01 |
| AUClast | µM·h | 2.94 | 7.33 | 0.07 |
| CL | mL/min/kg | 5.58 | 6.84 | 1550.77 |
| Vss | L/kg | 1.41 | ||
| Fpo | 0.2 |
Figure 6.
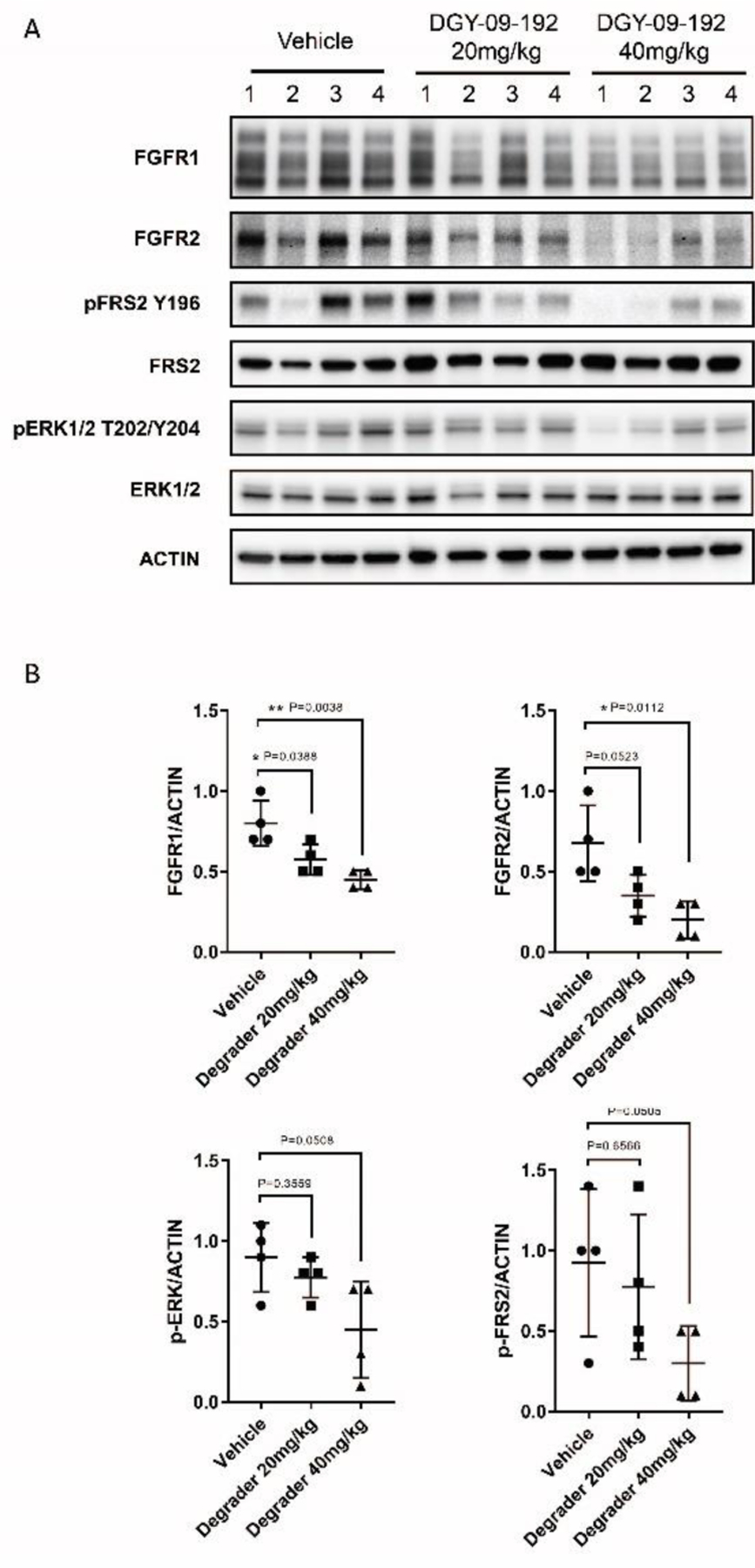
In vivo pharmacodynamics study of DGY-09-192. A. Immunoblot analysis of lysates from four independent tumors from each treatment group. B. Quantification of FGFR1, FGFR2, p-FRS2 and p-ERK. (Significantly different: P < 0.05; tumor size data shown in Figure S8.)
Conclusion
Small molecule-induced protein degradation is an emerging strategy in drug discovery. In contrast to conventional inhibitors, in which pharmacology is predicated on receptor occupancy, bivalent degraders demonstrate event driven pharmacology, and highly potent degraders that act sub-stoichiometrically relative to their protein targets have been successfully developed for numerous targets, including BRD4[30] and BTK.[31]
Mutant FGFRs are validated targets in several cancer types but current drugs not only lack selectivity amongst the closely related FGFR1, 2, and 3 but also are unable to distinguish between wild-type and mutant FGFRs. Thus, in this study we sought to explore: 1) whether FGFR and its fusion variants are degradable targets; and 2) whether degraders can achieve selectivity for specific FGFR isoforms. Our initial imide-based degraders achieved degradation of TEL-FGFR2 expressed in murine Ba/F3 cells but failed to degrade full length FGFR2 protein in KATO III cells. This limitation was overcome by switching to recruitment of a VHL E3 ligase, which resulted in the discovery of DGY-09-192, a low nanomolar degrader for both wild type and fusion mutant FGFR2 proteins. In addition, despite retaining equipotent biochemical inhibition of all four FGFR isoforms, DGY-09-192 displayed highly selective degradation of FGFR1 and 2, and degradation of either full-length FGFR2 or FGFR2 fusion proteins led to profound degradation-dependent antiproliferative activity in multiple FGFR2-dependent cells. Additionally, our lead degrader DGY-09-192 features a short, 2-carbon linker, which may present an advantage with respect to cellular permeability and bioavailability. We demonstrated that DGY-09-192 has an acceptable PK profile and induced degradation of FGFR2 fusion protein in vivo. However, as a prototype molecule, DGY-09-192 has some limits that will need to be overcome. For example, DGY-09-192 lacks the superiority to the FGFR parental inhibitor BGJ398 with respect to antiproliferation activity and is unlikely to overcome BGJ398-induced point mutation on FGFR proteins that confer resistance (Table S3). In addition, DGY-09-192 still potently inhibits all FGFR isoforms, so its antiproliferative activity cannot be attributed solely to degradation alone. Further optimization will be necessary to reduce FGFR binding, enhance oral bioavailability, improve selectivity for a particular FGFR, decrease off-target binding to PDE6D, and increase potency against drug resistant mutants. Additionally, we envision that degraders that exhibit selectivity for oncogenic mutants or fusions relative to wild-type FGFR to achieve enhanced therapeutic index, could also be developed with further optimization.
Supplementary Material
Acknowledgements
We thank Milka Kostic, Eric Wang and Inchul You for editing the manuscript. We thank Zhen-Yu Jim Sun for his assistance on 1H NMR and 13C NMR data collection. We acknowledge funding from NIH 5 R01 CA218278-02 (NSG and ESF), Pediatric Low-Grade Astrocytoma Fund at the Pediatric Brain Tumor Foundation (NSG and ESF), SPORE NIH P50 CA127003 (NSG and NB), V Foundation for Cancer Research (NSG, NB and TZ) and CCF award, supported by a Research Grant from the Cholangiocarcinoma Foundation (TZ).
Footnotes
Supporting information for this article is given via a link at the end of the document
Conflict of interest
G.D., N.J.H., T.Z., J.J., and N.S.G. are inventors on FGFR2 degrader patents. E.S.F. is a founder, science advisory board member, and equity holder in Civetta, Jengu (board member), and Neomorph, an equity holder in C4, and a consultant to Astellas, Novartis, Deerfield, and EcoR1. The Fischer lab receives or has received research funding from Novartis, Astellas, and Deerfield. N.S.G. is a Scientific Founder, member of the Scientific Advisory Board (SAB) and equity holder in C4 Therapeutics, Syros, Soltego (board member), B2S, Allorion, Gatekeeper, Aduro, Jengu and Inception. The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Janssen, Kinogen, Voroni, Arbella, Deerfield, and Sanofi. N.B. has received research funding from Taiho. J.C. is a consultant to Soltego, Jengu, Allorion, and equity holder for Soltego, Allorion, M3 bioinformatics & technology Inc. The other authors declare no competing interests.
Institute and/or researcher Twitter usernames: @Dana-Farber, @GuangyanD
References
- [1].Beenken A, Mohammadi M, Nat. Rev. Drug Discov 2009, 8, 235–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Chae YK, Ranganath K, Hammerman PS, Vaklavas C, Mohindra N, Kalyan A, Matsangou M, Costa R, Carneiro B, Villaflor VM, Cristofanilli M, Giles FJ, Oncotarget 2017, 8, 16052; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Farrell B, Breeze AL, Biochem. Soc. Trans. 2018, 46, 1753–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Katoh M, Nat. Rev. Clin. Oncol 2019, 16, 105–122. [DOI] [PubMed] [Google Scholar]
- [4].a) Cheng W, Wang M, Tian X, Zhang X, Eur. J. Med. Chem 2017, 126, 476–490; [DOI] [PubMed] [Google Scholar]; b) Porta R, Borea R, Coelho A, Khan S, Araújo A, Reclusa P, Franchina T, Van Der Steen N, Van Dam P, Ferri J, Sirera R, Naing A, Hong D, Rolfo C, Crit. Rev. Oncol. Hematol 2017, 113, 256–267; [DOI] [PubMed] [Google Scholar]; c) Marseglia G, Lodola A, Mor M, Castelli R, Expert Opin. Ther. Pat 2019, 29, 965–977. [DOI] [PubMed] [Google Scholar]
- [5].a) Renhowe PA, Pecchi S, Shafer CM, Machajewski TD, Jazan EM, Taylor C, Antonios-McCrea W, McBride CM, Frazier K, Wiesmann M, Lapointe GR, Feucht PH, Warne RL, Heise CC, Menezes D, Aardalen K, Ye H, He M, Le V, Vora J, Jansen JM, Wernette-Hammond ME, Harris AL, J. Med. Chem 2009, 52, 278–292; [DOI] [PubMed] [Google Scholar]; b) André F, Bachelot T, Campone M, Dalenc F, Perez-Garcia JM, Hurvitz SA, Turner N, Rugo H, Smith JW, Deudon S, Shi M, Zhang Y, Kay A, Porta DG, Yovine A, Baselga J, Clin. Cancer Res 2013, 19, 3693–3702. [DOI] [PubMed] [Google Scholar]
- [6].a) Roth GJ, Heckel A, Colbatzky F, Handschuh S, Kley J.r., Lehmann-Lintz T, Lotz R, Tontsch-Grunt U, Walter R, Hilberg F, J. Med. Chem 2009, 52, 4466–4480; [DOI] [PubMed] [Google Scholar]; b) Roth GJ, Binder R, Colbatzky F, Dallinger C, Schlenker-Herceg R, Hilberg F, Wollin S-L, Kaiser R, J. Med. Chem 2015, 58, 3, 1053–1063. [DOI] [PubMed] [Google Scholar]
- [7].a) Huang W-S, Metcalf CA, Sundaramoorthi R, Wang Y, Zou D, Thomas RM, Zhu X, Cai L, Wen D, Liu S, Romero J, Qi J, Chen I, Banda G, Lentini SP, Das S, Xu Q, Keats J, Wang F, Wardwell S, Ning Y, Snodgrass JT, Broudy MI, Russian K, Zhou T, Commodore L, Narasimhan NI, Mohemmad QK, Iuliucci J, Rivera VM, Dalgarno DC, Sawyer TK, Clackson T, Shakespeare WC, J. Med. Chem 2010, 53, 4701–4719; [DOI] [PubMed] [Google Scholar]; b) Gozgit JM, Wong MJ, Moran L, Wardwell S, Mohemmad QK, Narasimhan NI, Shakespeare WC, Wang F, Clackson T, Rivera VM, Mol. Cancer Ther 2012, 11, 690–699. [DOI] [PubMed] [Google Scholar]
- [8].Hilberg F, Roth GJ, Krssak M, Kautschitsch S, Sommergruber W, Tontsch-Grunt U, Garin-Chesa P, Bader G, Zoephel A, Quant J, Heckel A, Rettig WJ, Cancer Res 2008, 68, 4774–4782. [DOI] [PubMed] [Google Scholar]
- [9].a) Zhou W, Hur W, McDermott U, Dutt A, Xian W, Ficarro SB, Zhang J, Sharma SV, Brugge J, Meyerson M, Settleman J, Gray NS, Chem. Bio 2010, 17, 285–295; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tan L, Wang J, Tanizaki J, Huang Z, Aref AR, Rusan M, Zhu S-J, Zhang Y, Ercan D, Liao RG, Capelletti M, Zhou W, Hur W, Kim N, Sim T, Gaudet S, Barbie DA, Yeh JJ, Yun CH, Hammerman PS, Mohammadi M, A Jänne P, Gray NS, Proc. Natl. Acad. Sci 2014, 111, E4869–E4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Brameld KA, Owens TD, Verner E, Venetsanakos E, Bradshaw JM, Phan VT, Tam D, Leung K, Shu J, LaStant J, Loughhead DG, Ton T, Karr DE, Gerritsen ME, Goldstein DM, Funk JO, J. Med. Chem 2017, 60, 15, 6516–6527. [DOI] [PubMed] [Google Scholar]
- [11].Lin X, Yosaatmadja Y, Kalyukina M, Middleditch MJ, Zhang Z, Lu X, Ding K, Patterson AV, Smaill JB, Squire CJ, ACS Med. Chem. Lett 2019, 10, 1180–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kalyukina M, Yosaatmadja Y, Middleditch MJ, Patterson AV, Smaill JB, Squire CJ, ChemMedChem 2019, 14, 494–500. [DOI] [PubMed] [Google Scholar]
- [13].Gavine PR, Mooney L, Kilgour E, Thomas AP, Al-Kadhimi K, Beck S, Rooney C, Coleman T, Baker D, Mellor MJ, Brooks AN, Klinowska T, Cancer Res 2012, 72, 2045–2056. [DOI] [PubMed] [Google Scholar]
- [14].a) Guagnano V, Furet P, Spanka C, Bordas V, Le Douget M, Stamm C, Brueggen J, Jensen MR, Schnell C, Schmid H, Wartmann M, Berghausen J, Drueckes P, Zimmerlin A, Bussiere D, Murray J, Porta DG, J. Med. Chem 2011, 54, 7066–7083; [DOI] [PubMed] [Google Scholar]; b) Pal SK, Rosenberg JE, Hoffman-Censits JH, Berger R, Quinn DI, Galsky MD, Wolf J, Dittrich C, Keam B, Delord J-P, Schellens JHM, Gravis G, Medioni J, Maroto P, Sriuranpong V, Charoentum C, Burris HA, Grünwald V, Petrylak D, Vaishampayan U, Gez E, De Giorgi U, Lee J-L, Voortman J, Gupta S, Sharma S, Mortazavi A, Vaughn DJ, Isaacs R, Parker K, Chen X, Yu K, Porter D, Porta DG, Bajorin DF, Cancer Discov 2018, 8, 812–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Loriot Y, Necchi A, Park SH, Garcia-Donas J, Huddart R, Burgess E, Fleming M, Rezazadeh A, Mellado B, Varlamov S, Joshi M, Duran I, Tagawa ST, Zakharia Y, Zhong B, Stuyckens K, Santiago-Walker A, De Porre P, O’Hagan A, Avadhani A, Siefker-Radtke AO, N. Engl. J. Med 2019, 381, 338–348; [DOI] [PubMed] [Google Scholar]; b) Tabernero J, Bahleda R, Dienstmann R, Infante JR, Mita A, Italiano A, Calvo E, Moreno V, Adamo B, Gazzah A, Zhong B, Platero SJ, Smit JW, Stuyckens K, Chatterjee-Kishore M, Rodon J, Peddareddigari V, Luo FR, Soria JC, J. Clin. Oncol 2015, 33, 3401–3408. [DOI] [PubMed] [Google Scholar]
- [16].Liu PC, Koblish H, Wu L, Bowman K, Diamond S, DiMatteo D, Zhang Y, Hansbury M, Rupar M, Wen X, Collier P, Feldman P, Klabe R, Burke KA, Soloviev M, Gardiner C, He X, Volgina A, Covington M, Ruggeri B, Wynn R, Burn TC, Scherle P, Yeleswaram S, Yao W, Huber R, Hollis G, PLoS One 2020, 15, e0231877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Goyal L, Shi L, Liu LY, de la Cruz FF, Lennerz JK, Raghavan S, Leschiner I, Elagina L, Siravegna G, Ng RW, Vu P, Patra KC, Saha SK, Uppot RN, Arellano R, Reyes S, Sagara T, Otsuki S, Nadres B, Shahzade HA, Dey-Guha I, Fetter IJ, Baiev I, Van Seventer EE, Murphy JE, Ferrone CR, Tanabe KK, Deshpande V, Harding JJ, Yaeger R, Kelley RK, Bardelli A, Iafrate AJ, Hahn WC, Benes CH, Ting DT, Hirai H, Getz G, Juric D, Zhu AX, Corcoran RB, Bardeesy N, Cancer Discov. 2019, 9, 1064–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Woyach JA, Furman RR, Liu T-M, Ozer HG, Zapatka M, Ruppert AS, Xue L, Li DH-H, Steggerda SM, Versele M, Dave SS, Zhang J, Yilmaz AS, Jaglowski SM, Blum KA, Lozanski A, Lozanski G, James DF, Barrientos JC, Lichter P, Stilgenbauer S, Buggy JJ, Chang BY, Johnson AJ, Byrd JC, N. Engl. J. Med 2014, 370, 2286–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, Ercan D, Matthews SE, Cantarini M, Barrett JC, Janne PA, Oxnard GR, Nat. Med 2015, 21, 560–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gattineni J, Alphonse P, Zhang Q, Mathews N, Bates CM, Baum M, Am. J. Physiol. Renal Physiol 2014, 306, F351–F358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cromm PM, Crews CM, Cell Chem. Bio 2017, 24, 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ, Proc. Natl. Acad. Sci 2001, 98, 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Huang H-T, Dobrovolsky D, Paulk J, Yang G, Weisberg EL, Doctor ZM, Buckley DL, Cho J-H, Ko E, Jang J, Shi K, Choi HG, Griffin JD, Li Y, Treon SP, Fischer ES, Bradner JE, Tan L, Gray NS, Cell Chem. Bio 2018, 25, 88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jiang B, Wang ES, Donovan KA, Liang Y, Fischer ES, Zhang T, Gray NS, Angew. Chem. Int. Ed 2019, 58, 6321–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Olson CM, Jiang B, Erb MA, Liang Y, Doctor ZM, Zhang Z, Zhang T, Kwiatkowski N, Boukhali M, Green JL, Haas W, Nomanbuhoy T, Fischer ES, Young RA, Bradner JE, Winter GE, Gray NS, Nature Chem. Bio 2018, 14, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Roy RD, Rosenmund C, Stefan MI, BMC Syst. Biol 2017, 11, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].a) Nakamura H, Arai Y, Totoki Y, Shirota T, Elzawahry A, Kato M, Hama N, Hosoda F, Urushidate T, Ohashi S, Hiraoka N, Ojima H, Shimada K, Okusaka T, Kosuge T, Miyagawa S, Shibata T, Nat. Genet 2015, 47, 1003–1010; [DOI] [PubMed] [Google Scholar]; b) Arai Y, Totoki Y, Hosoda F, Shirota T, Hama N, Nakamura H, Ojima H, Furuta K, Shimada K, Okusaka T, Kosuge T, Shibata T, Hepatology 2014, 59, 1427–1434; [DOI] [PubMed] [Google Scholar]; c) Wu Y-M, Su F, Kalyana-Sundaram S, Khazanov N, Ateeq B, Cao X, Lonigro RJ, Vats P, Wang R, Lin S-F, Cheng AJ, Kunju LP, Siddiqui J, Tomlins SA, Wyngaard P, Sadis S, Roychowdhury S, Hussain MH, Feng FY, Zalupski MM, Talpaz M, Pienta KJ, Rhodes DR, Robinson DR, Chinnaiyan AM, Cancer Discov 2013, 3, 636–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zengerle M, Chan K-H, Ciulli A, ACS Chem. Bio 2015, 10, 1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].a) Winzker M, Friese A, Koch U, Janning P, Ziegler S, Waldmann H, Angew. Chem. Int. Ed 2020, 59, 5595–5601; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li Y, Meng Q, Wang P, Liu X, Fu Q, Xie Y, Zhou Y, Qi X, Huang N, bioRxiv 2020.; c) Popow J, Arnhof H, Bader G, Berger H, Ciulli A, Covini D, Dank C, Gmaschitz T, Greb P, Karolyi-Özguer J, Koegl M, McConnell DB, Pearson M, Rieger M, Rinnenthal J, Roessler V, Schrenk A, Spina M, Steurer S, Trainor N, Traxler E, Wieshofer C, Zoephel A, and Ettmayer JP. Med. Chem 2019, 62, 5, 2508–2520. [DOI] [PubMed] [Google Scholar]
- [30].Zhao Y, Zhou B, Bai L, Liu L, Yang C-Y, Meagher JL, Stuckey JA, McEachern D, Przybranowski S, Wang M, Ran X, Aguila A, Hu Y, Kampf JW, Li X, Zhao T, Li S, Wen B, Sun D, Wang S, J. Med. Chem 2018, 61, 6110–6120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dobrovolsky D, Wang ES, Morrow S, Leahy C, Faust T, Nowak RP, Donovan KA, Yang G, Li Z, Fischer ES, Treon SP, Weinstock DM, Gray NS, Blood 2019, 133, 952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.