

Abstract
Background:
Preclinical evidence demonstrates that mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) pathway inhibition increases sensitivity to 5-fluorouracil (5-FU) in colorectal cancer (CRC) cell lines and xenografts. Here, we aimed to investigate how CRC cell sensitivity to this combination is correlated to Kirsten rat sarcoma (KRAS) and proto-oncogene B-rapidly accelerated fibrosarcoma (BRAF) mutation, that are common in CRC and often lead to resistance to chemotherapy.
Materials and Methods:
Wild-type and mutant KRAS/BRAF human CRC cell lines were treated with escalating doses of 5-FU or trifluridine with MEK162 (MEK1/2 inhibitor) for 72 h. Cell viability was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay and synergism expressed by the combination index was calculated using CalcuSyn.
Results:
Evidence of synergistic antitumor activity was observed for the majority of human CRC cell lines treated with MEK162 plus 5-FU (4/6) or trifluridine (7/9). Synergism was greater in KRAS- or BRAF-mutant cell lines compared to wild-type KRAS/BRAF CRC cell lines.
Conclusion:
The combination of MEK inhibition and trifluridine is worthwhile advancing in clinical development, particularly for treatment-refractory KRAS- or BRAF-mutated metastatic CRC.
Keywords: MEK162, 5-fluorouracil, trifluridine, KRAS, colorectal cancer, preclinical
Fluorouracil, or 5-fluorouracil (5-FU), is a fluorinated pyrimidine whose anticancer properties are thought to be due to its ability to inhibit the rate-limiting enzyme in pyrimidine nucleotide synthesis, thymidylate synthase (1). In the current approach to treatment of metastatic CRC (mCRC), 5-FU remains a critical component to combination cytotoxic therapy and forms the backbone of treatment for regimens such as FOLFIRI (5-FU, leucovorin (LV), and irinotecan), FOLFOX (5-FU, LV, and oxaliplatin), and FOLFOXIRI (5-FU, LV, oxaliplatin, and irinotecan) (2–5).
Another nucleoside analog, trifluridine, has been also demonstrated to possess ability for incorporation into DNA and inhibit thymidylate synthase (6). Trifluridine represents the active antitumor component of TAS-102, which is formed by the combination of trifluridine and tipiracil, an inhibitor of thymidine phosphorylase that prevents the degradation of trifluridine (6). Development of this agent has recently led to its approval as a systemic therapy for treatment-refractory mCRC given its overall survival (OS) benefit compared to placebo in the phase III RECOURSE trial (7).
Preclinical evidence demonstrates that mitogen-activated protein kinase (MAPK) kinase (MEK)/extracellular signal-regulated kinase (ERK) signaling is associated with increased resistance to fluoropyrimidines across several tumor types including breast, prostate, and hepatocellular carcinoma (8–11). Furthermore, MEK/ERK pathway inhibition enhanced sensitivity to 5-FU in Kirsten rat sarcoma (KRAS)-mutated or proto-oncogene B-rapidly accelerated fibrosarcoma (BRAF)-mutated CRC cell lines and murine colorectal tumor xenograft models (12–15). In a phase I study, the combination of the MEK1/2 inhibitor trametinib with neoadjuvant 5-FU-based chemoradiation in KRAS/BRAF/neuroblastoma RAS viral oncogene homolog (NRAS)-mutated locally advanced rectal cancer demonstrated a pathological complete response in a patient identified to have the most significant reduction in ERK1/2 activity in post-treatment tumor tissue (16). MEK162 (ARRY-162 or binimetinib) is a novel, potent, and selective ATP-noncompetitive inhibitor of MEK1/2 that has proven anticancer activity against colorectal tumors in vitro and in vivo irrespective of RAS/RAF pathway mutations (17–19). MEK162 has also shown synergistic antitumor activity when combined with cytotoxic chemotherapy paclitaxel and gemcitabine in pancreatic and CRC cell lines with an activated MAPK pathway (17).
Based on the above rationale, we propose that MEK inhibition may enhance the antitumor activity of nucleoside analogs 5-FU and trifluridine in CRC in the preclinical setting. We conducted an in vitro analysis of the antitumor efficacy of MEK162 combined with 5-FU or trifluridine in select CRC cell lines that were KRAS/BRAF wild-type or mutant.
Materials and Methods
Cell lines and culture.
All cell proliferation assays and drug cytotoxicity analyses were performed at the City of Hope National Medical Center (COH) Molecular Pathology Core Laboratory (Duarte, CA, USA). The following KRAS/BRAF-wild-type human CRC cell lines were utilized: CaCO2 obtained from the COH Molecular Pathology Core Laboratory (Duarte, CA, USA) and grown in Eagle’s minimum essential medium (EMEM; Mediatech Inc., Manassas, VA, USA); COLO 320HSR obtained from the American Type Culture Collection (ATCC, cat. no. CCL-220.1) and grown in Roswell Park Memorial Institute (RPMI)-1640 medium (Mediatech Inc.); KM12 courtesy of Dr. YC (Duarte, CA, USA) and grown in Dulbecco’s modified Eagle’s medium (DMEM; Mediatech Inc.); and SNU-C1 obtained from the ATCC (cat.no. CRL-5972) and grown in RPMI-1640 medium. The HT29 human colorectal adenocarcinoma cell line was wild-type for KRAS but BRAFV600E mutated, obtained from the COH Molecular Pathology Core Laboratory (Duarte, CA), and grown in McCoy’s 5A modified medium (Mediatech Inc.).
The following human CRC cell lines were BRAF wild-type but harbored KRAS mutations and were obtained from the COH Molecular Pathology Core Laboratory : LS 174T (G12D), grown in EMEM; DLD-1 (G13D), grown in RPMI-1640 medium; HCT116 (G13D), grown in McCoy’s 5A modified medium; and SW480 (G12V), grown in L-15 medium (Mediatech Inc.). Two human CRC cell lines DLD-1 DKO4 and HCT116 HKE3 had disrupted KRAS mutant alleles as described by Shirasawa et al. (20) and were grown in RPMI-1640 medium and DMEM, respectively. All cell lines were cultured at 37°C and under a humidified atmosphere with 5% CO2 except for SW480, which was cultured at 37°C with standard atmosphere.
Cell treatment.
Prior to 5-FU and MEK162 treatment (day 0), SW480, CaCO2, HCT116, HCT116 HKE3, DLD-1, and DLD-1 DKO4 cells were harvested when 75-80% confluent, seeded into 96-well plates at 1500-2500 cells/well in 100 μL culture medium, and allowed to adhere for 24 hours. MEK162 (ARRY-438162; Array BioPharma Inc., Boulder, CO, USA) and 5-FU (Sigma-Aldrich, St. Louis, MO, USA) were prepared in dimethyl sulfoxide (DMSO) at stock concentrations of 2.27 mM and 38.44 mM, respectively, and stored at −20°C. On day 1, cells were then incubated with 5-FU alone in 50 μL culture medium at final concentrations of 2, 6, 18, and 54 μM, MEK162 alone in 50 μL culture medium at final concentrations of 0.1, 0.3, 0.9, and 2.7 μM, and combination of 5-FU plus MEK162 in 50 μL culture medium at final concentrations of 2 μM 5-FU plus 0.1 μM MEK162, 6 μM 5-FU plus 0.3 μM MEK162, 18 μM 5-FU plus 0.9 μM MEK162, and 54 μM 5-FU plus 2.7 μM MEK162 for 72 hours. On day 4, cell viability was analyzed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.
Prior to trifluridine and MEK162 treatment (day 0), LS 174T, DLD-1, HCT116, SW480, HT29, CaCO2, COLO 320HSR, KM12, and SNU-C1 cells were harvested when 75-80% confluent, seeded into 96-well plates at 1500-8000 cells/well in 100 μL culture medium, and allowed to adhere for 24 hours (suspension for COLO 320 HSR and SNU-C1 cell lines). MEK162 and trifluridine (courtesy of Dr. JW, the COH Animal Tumor Model Program, Duarte, CA, USA) stock solutions were prepared in DMSO at stock concentrations of 2.27 mM and 10 mM, respectively, and stored at −20°C degrees Celsius. On day 1, cells were incubated with trifluridine in 50 μL medium at final concentrations of 2, 6, 18, and 54 μM, MEK162 in 50 μL medium at final concentrations of 0.1, 0.3, 0.9, and 2.7 μM, and combination of trifluridine plus MEK162 in 50 μL medium at final concentrations of 2 μM (trifluridine) plus 0.1 μM (MEK162), 6 μM (trifluridine) plus 0.3 μM (MEK162), 18 μM (trifluridine) plus 0.9 μM (MEK162), and 54 μM (trifluridine) plus 2.7 μM for 72 hours. On day 4, cell proliferation was analyzed using the MTT assay. The cell number plated per well was determined by cell growth doubling time, size, and type of cell proliferation assay used. All experiments were performed using vehicle control in 50 μL culture medium as a negative control.
Cell proliferation.
Cell proliferation assays for 5-FU, trifluridine, and MEK162 treatments were performed using the MTT cell proliferation assay (Promega, Madison, WI, USA) as modified from the original assay described by Mosmann (21). The Promega MTT assay addresses several technical problems from the original method, but in principle remains a rapid colorimetric assay based on the conversion of MTT to its insoluble formazan crystal, which has a purple color specifically in viable cells.
Statistical analyses.
All statistical analyses performed were descriptive and no formal statistical hypotheses were assessed. Drug concentrations were plotted against cell survival rates for all cell lines. The half maximal-inhibitory concentration (IC50) for each cytotoxic treatment was calculated using the FORECAST function in Microsoft Excel. The combination index (CI) for combination treatments was expressed as the mean of the CI at effective dose of 50% (ED50) and calculated using CalcuSyn (Biosoft, Great Shelford, Cambridge, UK). Values of CI ranging from 0.1-0.3 suggest strong synergism, 0.3-0.7 synergism, and 0.7-0.9 slight synergism.
Results
Antitumor activity of 5-FU is greater in KRAS-mutated CRC cell lines than KRAS-wild-type cell lines.
The human CRC cell lines CaCO2, SW480, DLD-1, DLD-1 DKO4, HCT116, and HCT116 HKE3 were all incubated with 2-54 μM 5-FU alone, 0.1-2.7 μM MEK162 alone and their combination for 72 h prior to analysis of cell viability by the MTT assay. The mean IC50 for 5-FU and MEK162 treatments and mean CI at ED50 for 5-FU plus MEK162 across cell lines are summarized in Table I.
Table I.
Half-maximal inhibitory concentrations for 5-flourouracil (5-FU) and mitogen-activated protein kinase kinase 162 (MEK162) and combination index (CI) for their combination for different human colorectal carcinoma cell lines. Data are the mean±SD.
| Cell line | 5-FU (μM) | MEK162 (μM) | CI* |
|---|---|---|---|
| CaCO2 (KRAS/BRAF WT) | 23.59±2.18 | 2.91±0.68 | 0.46 |
| HCT116 HKE3 (KRAS disrupted allele/BRAF WT) | 12.36±2.26 | 2.54±0.46 | 2.53 |
| DLD-1 DKO4 (KRAS disrupted allele/BRAF WT) | 23.47±4.04 | 1.99±0.20 | 1.40 |
| SW480 (KRASG12V MT) | 84.81±11.91 | 2.05±0.16 | 0.13 |
| HCT116 (KRASG13D MT) | 14.08±1.52 | 1.71±0.15 | 0.70 |
| DLD-1 (KRASG13DMT) | 26.35±3.21 | 3.99±0.53 | 0.90 |
WT, Wild-type; MT, mutant.
Mean values at effective dose of 50%, 0.1-0.3: strong synergism, 0.3-0.7: synergism, and 0.7-0.9: slight synergism.
Representative dose–survival curves for all 6 cell lines on treatment with 5-FU, MEK162, and their combination are illustrated in Figure 1. The lowest mean IC50 for 5-FU of 12.36±2.26 μM was seen for HCT116 HKE3 (KRAS disrupted allele/BRAF wild-type) cell line, while the highest mean IC50 of 84.81±11.91 μM was seen for SW480 (KRASG12V mutant/BRAF wild-type) cells. For MEK162, the lowest mean IC50 was 1.71±0.15 μM observed for HCT116 (KRASG13D mutant/BRAF wild-type) cells and the highest mean IC50 was 3.99±0.53 μM seen for DLD-1 (KRASG13D mutant/BRAF wild-type) cells.
Figure 1.
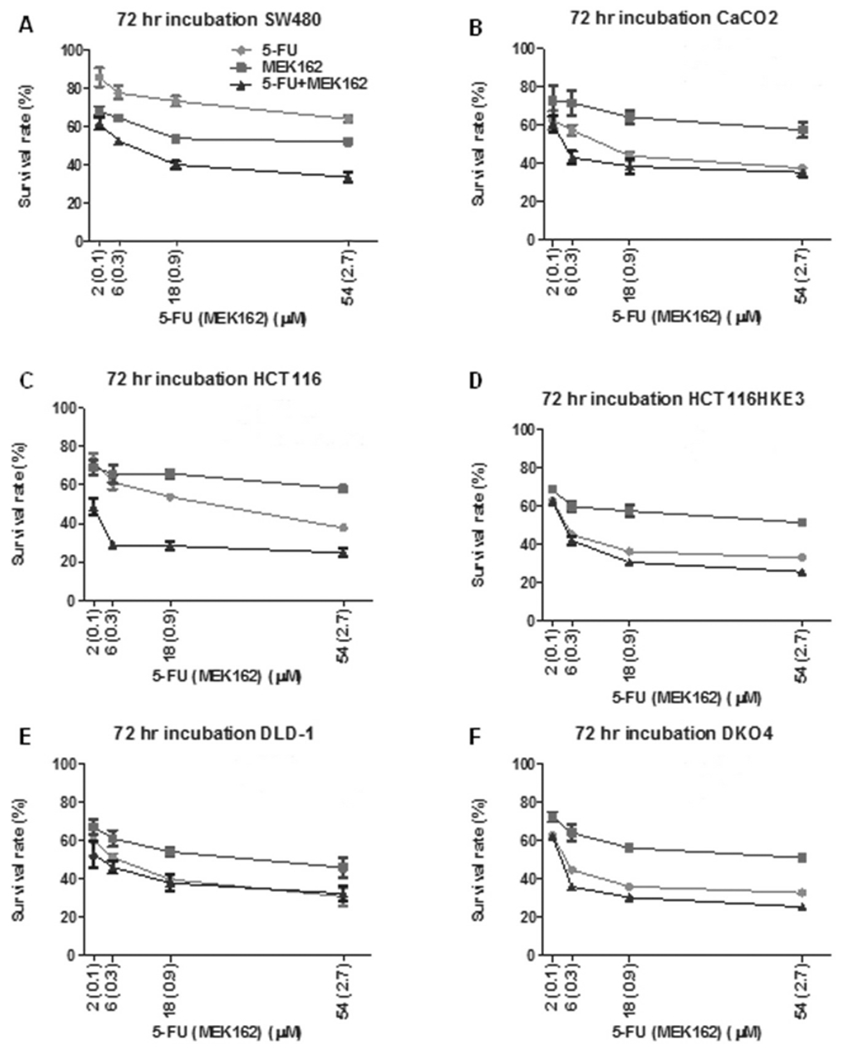
Mitogen-activated protein kinase kinase 162 (MEK162) and 5-fluorouracil (5-FU) demonstrate enhanced antitumor activity in human colorectal cancer cell lines. Dose–survival curves by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay for 5-FU, MEK162, and 5-FU combined with MEK162 for KRAS-mutated SW480 (A), HCT116 (C), and DLD-1 (E) and the wild-type KRAS/BRAF CaCO2 (B) human colorectal carcinoma cell lines. Cell lines with a disrupted KRAS-mutant allele included HCT116 HKE3 (D) and DLD-1 DKO4 (F). X-Axis represents drug concentrations (μM) of 5-FU and MEK162 (in parentheses). Incubation period of 72 hours for all experiments.
The addition of MEK162 to 5-FU showed evidence of enhanced antitumor activity in the majority of cell lines (4/6). Combination 5-FU and MEK162 showed strong synergism in the KRAS-mutated SW480 cell line (CI=0.13) and KRAS-wild-type CaCO2 cell line (CI=0.46). Slight synergism was suggested in KRAS-mutated HCT116 (CI=0.70) and DLD-1 (CI=0.90) CRC cells treated with their combination. Evidence of synergism was seen in all three KRAS-mutated cell lines (SW480, HCT116, and DLD-1) treated with 5-FU plus MEK162. Notably, there was no evidence of synergism with 5-FU plus MEK 162 in the wild-type BRAF HCT116 HKE3 and DLD-1 DKO4 cell lines carrying disrupted KRAS-mutant alleles.
Antitumor activity of trifluridine is greater in CRC cell lines with KRAS or BRAF mutation than in wild-type cell lines.
Nine human CRC cell lines LS 174T, DLD-1, HCT116, SW480, HT29, CaCO2, COLO 320HSR, KM12, and SNU-C1 were treated with trifluridine at concentrations of 2-54 μM, MEK162 at concentrations of 0.1-2.7 μM, and their combination for 72 h followed by evaluation of cell proliferation by MTT assay. Representative dose–survival curves for all nine cell lines treated with trifluridine, MEK162, and their combination are shown in Figure 2. Table II summarizes the mean IC50 for trifluridine and MEK162 treatments and mean CI at ED50 for trifluridine plus MEK162 across all cell lines. The lowest mean IC50 for trifluridine was 16.84±2.67 μM for the wild-type KRAS/BRAF COLO 320HSR cell line, whereas the highest mean IC50 was 88.06±11.04 μM for LS 174T KRASG12D-mutated cancer cells treated with trifluridine. Treatment of the SNU-C1 (KRAS/BRAF wild-type) cell line produced the lowest mean IC50 of 0.043±0.019 μM for MEK162, while the highest mean IC50 for MEK162 was 11.25±1.20 μM seen in COLO 320HSR cells.
Figure 2.
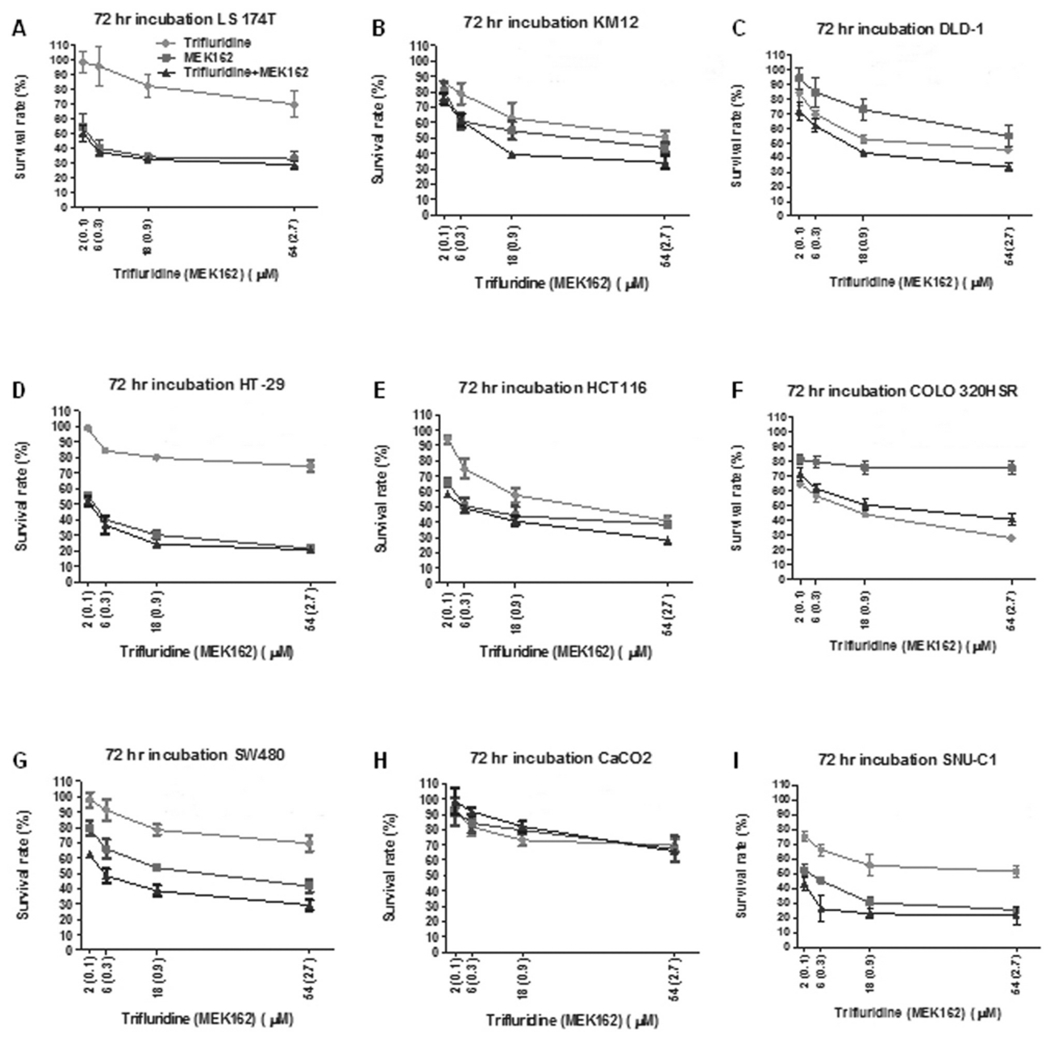
Mitogen-activated protein kinase kinase 162 (MEK162) and trifluridine demonstrate enhanced antitumor activity in human colorectal cancer cell lines. Dose–survival curves by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay for trifluridine, MEK162, and trifluridine combined with MEK162 in KRAS-mutated LS 174T (A), DLD-1 (C), HCT116 (E), SW480 (G), and BRAF-mutated HT-29 (D) human colorectal carcinoma cell lines. Wild-type KRAS/BRAF cell lines included KM12 (B), COLO 320HSR (F), CaCO2 (H), and SNU-C1 (I). X-Axis represents drug concentrations (μM) of trifluridine, MEK162 (in parentheses). Incubation period of 72 hours for all experiments.
Table II.
Half maximal-inhibitory concentrations for trifluridine and mitogen-activated protein kinase kinase 162 (MEK162) and combination index (CI) for their combination for different human colorectal carcinoma cell lines. Data are the mean±SD.
| Cell line | Trifluridine (μM) | MEK162 (μM) | CI* |
|---|---|---|---|
| CaCO2 (KRAS/BRAF WT) | 76.97±2.61 | 4.11±0.52 | 1.35 |
| COLO 320HSR (KRAS/BRAF WT) | 16.84±2.67 | 11.25±1.20 | 1.29 |
| KM12 (KRAS/BRAF WT) | 44.12±13.40 | 1.97±0.63 | 0.83 |
| SNU-C1 (KRAS/BRAF WT) | 44.76±3.12 | 0.043±0.019 | 0.04 |
| HT29 (KRASV600E MT) | 77.45±4.17 | 0.074±0.01 | 0.25 |
| LS 174T (KRASG12D MT) | 88.06±11.04 | 0.21±0.02 | 0.72 |
| SW480 (KRASG12V MT) | 66.28±12.09 | 1.88±0.37 | 0.51 |
| HCT116 (KRASG13D MT) | 34.26±5.65 | 1.21±0.20 | 0.75 |
| DLD-1 (KRASG13DMT) | 29.41±4.95 | 2.48±0.27 | 0.67 |
WT, Wild-type; MT, mutant.
Mean values at effective dose of 50%, 0.1-0.3: strong synergism, 0.3-0.7: synergism, and 0.7-0.9: slight synergism.
Similar to the combination of 5-FU and MEK162, the addition of MEK162 to trifluridine showed evidence of enhanced antitumor activity in the majority of colorectal tumor cell lines (7/9). Evidence of strong synergism was observed with combination trifluridine plus MEK162 in the wild-type KRAS/BRAF SNU-C1 cell line (CI=0.04) and BRAFV600E-mutated HT29 cell line (CI=0.25). The addition of MEK162 to trifluridine also showed synergistic activity in KRAS-mutated SW480 and DLD-1 cells. Slight synergism was demonstrated in 2 KRAS-mutated cell lines LS 174T and HCT 116 and one cell line with wild-type KRAS/BRAF, KM12, treated with the combination. Treatment with trifluridine plus MEK162 did not show evidence of synergism in wild-type KRAS/BRAF COLO 320HSR and CaCO2 cell lines. Evidence of increased activity with trifluridine plus MEK162 was greatest in cell lines carrying KRAS or BRAF mutations. All five KRAS- or BRAF-mutant cell lines (SW480, DLD-1, LS 174T, HT29, and HCT 116) showed evidence of synergism, while only two out of four wild-type KRAS/BRAF cell lines (SNU-C1 and KM12) had enhanced activity when treated with their combination.
Discussion
The objective of this in vitro study was to determine whether addition of the MEK1/2 inhibitor MEK162 to 5-FU or trifluridine enhances antitumor efficacy in CRC cells in a KRAS and/or BRAF mutation status-dependent manner. There is evidence in the literature supporting the role of MEK/ERK signaling in promoting resistance to fluoropyrimidines across several tumor types and the reversal of fluoropyrimidine resistance demonstrated by MEK/ERK pathway inhibition, particularly in CRC cells and animal models (8–15). Consistent with this, in this study, the addition of MEK162 to a nucleoside analog enhanced the antitumor activity in the majority of human CRC cell lines in both 5-FU (4/6) and trifluridine (7/9) experimental arms. The doses of 5-FU used for assessment of tumor cell viability (final concentrations of 2-54 μM) are within the range of those examined in historic analyses of 5-FU (0-50 μM) cytotoxicity in human colorectal tumor cell lines similarly incubated for 72 h (22). Furthermore, our dose range of trifluridine (2-54 μM) is within the range of doses examined in other preclinical cytotoxicity studies involving human CRC cell lines exposed to trifluridine (0-35 μM) for 72 h (23). Treatment with MEK162 at doses of 0.1-2.7 μM are comparable to those explored in human non-small cell lung cancer cell lines similarly treated with MEK 162 (0.04-10 μM) for 72 hours (24). An even wider range of MEK162 dosing (0.0001 μM-10 μM) has been evaluated for cytotoxicity against human pancreatic cancer cell lines, but with an incubation period of 5 days (25). A lower range of MEK162 doses 25-200 nM (72-h treatment) were used in cell proliferation assays of KRAS-mutated CRC cells, although in a combination study with palbociclib, and extrapolation of the dose–response curves of the MEK162 alone arm suggests that doses well beyond 200 nM are needed to reach IC50 for the majority of cell lines (26). Our dose range, as an example, produced an IC50 of 0.074 μM for HT-29 cells that is concordant with the IC50 of 0.1 μM seen in another preclinical study of HT-29 cells treated with escalating doses of MEK162 for 72 h (17).
The enhanced antitumor activity demonstrated with the addition of MEK162 to 5-FU or trifluridine was greater in CRC cell lines carrying KRAS or BRAF mutations than wild-type KRAS/BRAF cell lines. Evidence of synergism was seen in all three KRAS-mutated cell lines (SW480, HCT116, and DLD-1) treated with 5-FU plus MEK162 but only in one wild-type KRAS/BRAF cell line (CaCO2). All five mutant KRAS or BRAF cell lines (SW480, DLD-1, LS 174T, HT29, and HCT 116) showed evidence of synergism, while only two out of four wild-type KRAS/BRAF cell lines (SNU-C1 and KM12) had enhanced activity when treated with trifluridine plus MEK162. Notably, treatment with trifluridine plus MEK162 demonstrated evidence of synergism in KRAS-mutated DLD-1 and HCT116 cell lines. However, in DLD-1 and HCT116 cell lines harboring disrupted KRAS alleles, we observed a loss of synergism when treated with 5-FU plus MEK162. DLD-1 DKO4 and HCT116 HKE3 are two human colon cancer cell lines that have wild-type BRAF with KRASG13D mutant alleles disrupted by homologous recombination; the resultant phenotypes differ from parental KRAS-mutated phenotypes, with loss of anchorage-independent growth, decreased expression of c-MYC, and slower growth in vitro and in vivo (20).
In an initial biomarker analysis of 218 solid cancer cell lines, MEK inhibition demonstrated increased activity across tumor cell lines carrying RAS or RAF mutations compared to cell lines wild-type for these genes (27). Sensitivity to MEK inhibition has also been demonstrated in CRC cell and mouse xenograft models harboring RAS or BRAF mutations (28, 29). Although MEK162 has demonstrated preclinical efficacy against both wild-type and mutant KRAS/BRAF colorectal tumors, synergistic antitumor activity when combined with cytotoxic chemotherapy such as paclitaxel and gemcitabine was demonstrated in pancreatic and CRC cell lines with an activated MAPK pathway (17, 19). In the preclinical studies showing the activity of combined MEK/ERK pathway inhibition and 5-FU therapy, all were carried out on either KRAS-mutated or BRAF-mutated CRC cell lines (12–15). Overall, our findings are consistent with the literature and support the concept that the presence of KRAS or BRAF mutations are critical in eliciting more meaningful responses from the addition of MEK162.
Crosstalk between pathways parallel to, downstream of, or unrelated to the canonical RAS/RAF/MEK/ERK signaling pathway may affect response to the addition of MEK inhibition to combination therapy in cancer cells. Although RAS/RAF mutations are among the primary predictors of sensitivity to MEK inhibition, resistance appears to be mediated by phosphoinositide 3-kinase (PIK3) signaling (27, 28). Furthermore, in KRAS-mutated human colorectal tumor cell lines, activating PIK3CA mutations reduced sensitivity to MEK inhibition, whereas phosphatase and tensin homolog (PTEN) mutations (loss of PTEN function) conferred complete resistance (30). Indeed, in our three KRAS-mutated cell lines (SW480, HCT116, and DLD-1) treated with 5-FU plus MEK162, the strongest synergism was seen in SW480 cells (CI 0.13), which are known to be PIK3CA/PTEN-wild-type, while slight synergism (CI=0.70-0.90) was observed in HCT116 and DLD-1 cells, which are PIK3CA-mutated but PTEN-wild-type (31). In the KRAS or BRAF-mutated cell lines treated with trifluridine plus MEK162, the strongest evidence of synergism was seen in HT29 (CI=0.25) and SW480 cells (CI=0.51). SW480 cells, as described previously, are wild-type for PIK3CA and PTEN. Interestingly, HT29 cells are wild-type for KRAS and PTEN but BRAF/PIK3CA-mutated (31). It has been shown that tumor cells carrying BRAFV600E and PIK3CA mutations have relatively lower expression of resistance genes and more varied sensitivity to MEK inhibition (28). Accordingly, less synergism was seen in our KRAS-mutated cell lines LS 174T, HCT116, and DLD-1 harboring PIK3CA mutations. A separate study described that PI3K/AKT pathway inhibition has a greater effect than MEK/ERK pathway inhibition on reversing resistance to cytotoxics, including 5-FU (9). Intuitively, targeting multiple steps along the MAPK signaling cascade, for example, by dual MEK and PI3K or AKT inhibition, may provide additional avenues of therapeutic benefit (18, 32).
Moreover, the ability of MEK/ERK pathway inhibition to increase sensitivity to 5-FU in human colon cancer cells appears dependent on the presence of wild-type p53 and related mediators p21, p53-up-regulated modulator of apoptosis (PUMA), c-Jun N-terminal kinase (JNK), and p38 MAPK (12). Anti-apoptotic proteins downstream of AKT including B-cell lymphoma XL (BCL-xL) and myeloid cell leukemia 1 (MCL1) have also been implicated in 5-FU resistance (14). Further investigation into molecular phenotypes of potential significance to the efficacy of MEK inhibition and fluoropyrimidine therapy is prudent to understanding resistance and optimizing response to this regimen in CRC.
Lastly, the combination of MEK inhibition and trifluridine can serve an unmet need in the treatment paradigm of mCRC. The development of first-line and second-line regimens involving 5-FU-based combination therapy with the addition of anti-epidermal growth factor receptor and anti-vascular endothelial growth factor A inhibitors in select populations have afforded improved survival in mCRC (33). Despite these advances, it appears that we have reached a plateau in OS in mCRC treatment, and resistance to 5-FU ultimately develops through the treatment course. TAS-102 as a late-line treatment in mCRC offers slight benefit, but progression eventually occurs. The integration of MEK inhibition increases sensitivity to trifluridine in colorectal tumor cell lines and may represent a viable option in treatment-refractory KRAS/BRAF-mutated mCRC that has progressed through all standard therapies.
In conclusion, the addition of MEK162 to 5-FU or trifluridine demonstrated synergistic antitumor activity in the majority of human colorectal tumor cell lines evaluated here. Evidence of synergism appears to be increased in KRAS- or BRAF-mutant cell lines compared to wild-type KRAS/BRAF CRC cell lines. The combination of MEK inhibition and trifluridine represents a potentially viable option in KRAS- or BRAF-mutated mCRC that is worthwhile to advance in clinical development.
References
- 1.Meyerhardt JA and Mayer RJ: Systemic therapy for colorectal cancer. N Engl J Med 352: 476–487, 2005. [DOI] [PubMed] [Google Scholar]
- 2.de Gramont A, Figer A, Seymour M, Homerin M, Hmissi A, Cassidy J, Boni C, Cortes-Funes H, Cervantes A, Freyer G, Papamichael D, Le Bail N, Louvet C, Hendler D, de Braud F, Wilson C, Morvan F and Bonetti A: Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol 18: 2938–2947, 2000. [DOI] [PubMed] [Google Scholar]
- 3.Falcone A, Ricci S, Brunetti I, Pfanner E, Allegrini G, Barbara C, Crinò L, Benedetti G, Evangelista W, Fanchini L, Cortesi E, Picone V, Vitello S, Chiara S, Granetto C, Porcile G, Fioretto L, Orlandini C, Andreuccetti M, Masi G and Gruppo Oncologico Nord Ovest: Phase iii trial of infusional fluorouracil, leucovorin, oxaliplatin, and irinotecan (folfoxiri) compared with infusional fluorouracil, leucovorin, and irinotecan (folfiri) as first-line treatment for metastatic colorectal cancer: The gruppo oncologico nord ovest. J Clin Oncol 25: 1670–1676, 2007. [DOI] [PubMed] [Google Scholar]
- 4.Fuchs CS, Marshall J, Mitchell E, Wierzbicki R, Ganju V, Jeffery M, Schulz J, Richards D, Soufi-Mahjoubi R, Wang B and Barrueco J: Randomized, controlled trial of irinotecan plus infusional, bolus, or oral fluoropyrimidines in first-line treatment of metastatic colorectal cancer: Results from the bicc-c study. J Clin Oncol 25: 4779–4786, 2007. [DOI] [PubMed] [Google Scholar]
- 5.Goldberg RM, Sargent DJ, Morton RF, Fuchs CS, Ramanathan RK, Williamson SK, Findlay BP, Pitot HC and Alberts SR: A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. J Clin Oncol 22: 23–30, 2004. [DOI] [PubMed] [Google Scholar]
- 6.Tanaka N, Sakamoto K, Okabe H, Fujioka A, Yamamura K, Nakagawa F, Nagase H, Yokogawa T, Oguchi K, Ishida K, Osada A, Kazuno H, Yamada Y and Matsuo K: Repeated oral dosing of tas-102 confers high trifluridine incorporation into DNA and sustained antitumor activity in mouse models. Oncol Rep 32: 2319–2326, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mayer RJ, Van Cutsem E, Falcone A, Yoshino T, Garcia-Carbonero R, Mizunuma N, Yamazaki K, Shimada Y, Tabernero J, Komatsu Y, Sobrero A, Boucher E, Peeters M, Tran B, Lenz HJ, Zaniboni A, Hochster H, Cleary JM, Prenen H, Benedetti F, Mizuguchi H, Makris L, Ito M, Ohtsu A and RECOURSE Study Group: Randomized trial of tas-102 for refractory metastatic colorectal cancer. N Engl J Med 372: 1909–1919, 2015. [DOI] [PubMed] [Google Scholar]
- 8.Gomes E, Jennings-Gee J and Gmeiner WH: The mek/erk pathway is required for pc3 cell survival following fdump[10] treatment [abstract]. Cancer Res 70: Abstract nr 1637, 2010. [Google Scholar]
- 9.Jin W, Wu L, Liang K, Liu B, Lu Y and Fan Z: Roles of the pi-3k and mek pathways in ras-mediated chemoresistance in breast cancer cells. Br J Cancer 89: 185–191, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yan Y, Li J, Han J, Hou N, Song Y and Dong L: Chlorogenic acid enhances the effects of 5-fluorouracil in human hepatocellular carcinoma cells through the inhibition of extracellular signal-regulated kinases. Anticancer Drugs 26: 540–546, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Z, Zhou X, Shen H, Wang D and Wang Y: Phosphorylated erk is a potential predictor of sensitivity to sorafenib when treating hepatocellular carcinoma: Evidence from an in vitro study. BMC Med 7: 41,2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pereira DM, Simões AE, Gomes SE, Castro RE, Carvalho T, Rodrigues CM and Borralho PM: Mek5/erk5 signaling inhibition increases colon cancer cell sensitivity to 5-fluorouracil through a p53-dependent mechanism. Oncotarget 7: 34322–34340, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Urick ME, Chung EJ, Shield WP, Gerber N, White A, Sowers A, Thetford A, Camphausen K, Mitchell J and Citrin DE: Enhancement of 5-fluorouracil-induced in vitro and in vivo radiosensitization with mek inhibition. Clin Cancer Res 17: 5038–5047, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watanabe M, Sowa Y, Yogosawa M and Sakai T: Novel mek inhibitor trametinib and other retinoblastoma gene (rb)-reactivating agents enhance efficacy of 5-fluorouracil on human colon cancer cells. Cancer Sci 104: 687–693, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang SY, Miah A, Sales KM, Fuller B, Seifalian AM and Winslet M: Inhibition of the p38 mapk pathway sensitises human colon cancer cells to 5-fluorouracil treatment. Int J Oncol 38: 1695–1702, 2011. [DOI] [PubMed] [Google Scholar]
- 16.Willliams TM, Wuthrick E, Wu C, Robb R, Timmers C and Bekaii-Saab T: A phase i trial combining mek-1/2 inhibition in combination with 5-fluorouracil and radiation for kras, braf, and nras mutant locally advanced rectal adenocarcinoma [abstract]. Cancer Res 76: Abstract nr CT025, 2016. [Google Scholar]
- 17.Woessner R, Winski S, Rana S, Anderson D, Winkler J and Lee P: Arry-162, a potent and selective mek 1/2 inhibitor, shows enhanced efficacy in combination with other targeted kinase inhibitors and with chemotherapy [abstract]. Cancer Res 70: Abstract 2514, 2010. [Google Scholar]
- 18.Raja M, Zverev M, Seipel K, Williams GT, Clarke AR and Shaw PH: Assessment of the in vivo activity of pi3k and mek inhibitors in genetically defined models of colorectal cancer. Mol Cancer Ther 14: 2175–2186, 2015. [DOI] [PubMed] [Google Scholar]
- 19.Winski S, Anderson D, Bouhana K, Rhodes S, Impastato R, Woessner R, Zuzack J, Tunquist B, Garrus J, Pheneger T and Lee P: Mek162 (arry-162), a novel mek 1/2 inhibitor, inhibits tumor growth regardless of kras/raf pathway mutations. Eur J Cancer Suppl 8: 56, 2010. [Google Scholar]
- 20.Shirasawa S, Furuse M, Yokoyama N and Sasazuki T: Altered growth of human colon cancer cell lines disrupted at activated ki-ras. Science 260: 85–88, 1993. [DOI] [PubMed] [Google Scholar]
- 21.Mosmann T: Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immunol Methods 65: 55–63, 1983. [DOI] [PubMed] [Google Scholar]
- 22.Calabro-Jones PM, Byfield JE, Ward JF and Sharp TR: Time-dose relationships for 5-fluorouracil cytotoxicity against human epithelial cancer cells in vitro. Cancer Res 42: 4413–4420, 1982. [PubMed] [Google Scholar]
- 23.Bijnsdorp IV, Peters GJ, Temmink OH, Fukushima M and Kruyt FA: Differential activation of cell death and autophagy results in an increased cytotoxic potential for trifluorothymidine compared to 5-fluorouracil in colon cancer cells. Int J Cancer 126: 2457–2468, 2010. [DOI] [PubMed] [Google Scholar]
- 24.Yao W, Yue P, Zhang G, Owonikoko TK, Khuri FR and Sun SY: Enhancing therapeutic efficacy of the mek inhibitor, mek162, by blocking autophagy or inhibiting pi3k/akt signaling in human lung cancer cells. Cancer Lett 364: 70–78, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamidi H, Lu M, Chau K, Anderson L, Fejzo M, Ginther C, Linnartz R, Zubel A, Slamon DJ and Finn RS: Kras mutational subtype and copy number predict in vitro response of human pancreatic cancer cell lines to mek inhibition. Br J Cancer 111: 1788–1801,2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee MS, Helms TL, Feng N, Gay J, Chang QE, Tian F, Wu JY, Toniatti C, Heffernan TP, Powis G, Kwong LN and Kopetz S: Efficacy of the combination of mek and cdk4/6 inhibitors in vitro and in vivo in kras mutant colorectal cancer models. 2016 7: 39595–39608, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jing J, Greshock J, Holbrook JD, Gilmartin A, Zhang X, McNeil E, Conway T, Moy C, Laquerre S, Bachman K, Wooster R and Degenhardt Y: Comprehensive predictive biomarker analysis for mek inhibitor gsk1120212. Mol Cancer Ther 11: 720–729, 2012. [DOI] [PubMed] [Google Scholar]
- 28.Dry JR, Pavey S, Pratilas CA, Harbron C, Runswick S, Hodgson D, Chresta C, McCormack R, Byrne N, Cockerill M, Graham A, Beran G, Cassidy A, Haggerty C, Brown H, Ellison G, Dering J, Taylor BS, Stark M, Bonazzi V, Ravishankar S, Packer L, Xing F, Solit DB, Finn RS, Rosen N, Hayward NK, French T and Smith PD: Transcriptional pathway signatures predict mek addiction and response to selumetinib (azd6244). Cancer Res 70: 2264–2273, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guinney J, Ferté C, Dry J, McEwen R, Manceau G, Kao KJ, Chang KM, Bendtsen C, Hudson K, Huang E, Dougherty B, Ducreux M, Soria JC, Friend S, Derry J and Laurent-Puig P: Modeling ras phenotype in colorectal cancer uncovers novel molecular traits of ras dependency and improves prediction of response to targeted agents in patients. Clin Cancer Res 20: 265–272, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wee S, Jagani Z, Xiang KX, Loo A, Dorsch M, Yao YM, Sellers WR, Lengauer C and Stegmeier F: Pi3k pathway activation mediates resistance to mek inhibitors in kras mutant cancers. Cancer Res 69: 4286–4293, 2009. [DOI] [PubMed] [Google Scholar]
- 31.Ahmed D, Eide PW, Eilertsen IA, Danielsen SA, Eknæs M, Hektoen M, Lind GE and Lothe RA: Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2: e71, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Migliardi G, Sassi F, Torti D, Galimi F, Zanella ER, Buscarino M, Ribero D, Muratore A, Massucco P, Pisacane A, Risio M, Capussotti L, Marsoni S, Di Nicolantonio F, Bardelli A, Comoglio PM, Trusolino L and Bertotti A: Inhibition of mek and pi3k/mtor suppresses tumor growth but does not cause tumor regression in patient-derived xenografts of ras-mutant colorectal carcinomas. Clin Cancer Res 18: 2515–2525, 2012. [DOI] [PubMed] [Google Scholar]
- 33.Gong J, Cho M and Fakih M: Ras and braf in metastatic colorectal cancer management. J Gastrointest Oncol 7: 687–704, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]