

Abstract
Objective:
Systemic sclerosis (SSc) is characterized by widespread fibrosis and vascular complications. In this study, we utilized an assay for genome-wide chromatin accessibility to examine the chromatin landscape and transcription factor footprints in SSc.
Methods:
Dermal endothelial cells (ECs) and fibroblasts were isolated from healthy controls and patients with diffuse cutaneous (dc) SSc. ATAC-seq was performed to assess genome-wide chromatin accessibility at a read depth of approximately 150 million reads/sample. Transcription factor footprinting and motif binding analysis was performed followed by functional experiments.
Results:
Chromatin accessibility was significantly reduced in dcSSc patients compared to healthy controls. Differentially accessible chromatin loci were enriched in pathways and gene ontologies involved in the nervous system, cell membrane projections and cilia motility, nuclear and steroid receptors, and nitric oxide. In addition, chromatin binding of transcription factors SNAI2, ETV2, and ELF1 was significantly increased in dcSSc ECs, while recruitment of RUNX1 and RUNX2 was enriched in dcSSc fibroblasts. We revealed significant downregulation of the neuronal gene NRXN1 and upregulation of SNAI2 and ETV2 in dcSSc ECs. In dcSSc fibroblasts, downregulation of neuronal gene ENTPD1 and upregulation of RUNX2 were confirmed. Further functional analysis revealed that ETV2 and NRXN1 dysregulation affected angiogenesis in ECs, while ENTPD1 enhanced pro-fibrotic properties in dcSSc fibroblasts.
Conclusion:
Our data uncovered the chromatin blueprint of dcSSc, and suggested that neuronal-related characteristics of SSc ECs and fibroblasts could be a culprit for dysregulated angiogenesis and enhanced fibrosis. Targeting key pathways and transcription factors identified might present novel therapeutic approaches in SSc.
Keywords: scleroderma, ATAC-seq, endothelial cells, fibrosis
Introduction
Systemic sclerosis (scleroderma, SSc) is a chronic debilitating disease characterized by immune activation, vascular injury, and enhanced accumulation of extracellular matrix proteins in many organs. The associated high mortality and morbidity for this disease is due to lack of effective treatment options to modify disease progression, with the exception of autologous hematopoietic stem cell transplant, which is restricted to a small proportion of patients and can be associated with significant adverse events.
Although the etiology of SSc is poorly understood, work from our group and others have generated evidence that strongly implicates epigenetic dysregulation in the pathogenesis of SSc (1). This is supported by the low concordance rate for SSc in twins compared to other autoimmune diseases (2). Indeed, we have shown that in both endothelial cells (ECs) and fibroblasts isolated from SSc skin, epigenetic mechanisms are critically involved in their disease phenotype. Distinct differences in genome-wide DNA methylation pattern were reported in fibroblasts isolated from limited cutaneous and diffuse cutaneous (dc) SSc patients (3). The overexpressed methyl-CpG-binding protein 2 (MeCP2) in SSc fibroblasts appeared to be anti-fibrotic, in part mediated by its effect on target genes including PLAU, NID2, and ADA (4). In addition to DNA methylation, changes in histone-modifying enzymes, such as the histone methyltransferase enhancer of zeste homolog 2 (EZH2), has also been implicated in promoting fibrosis in SSc fibroblasts (5). We also showed that histone deacetylase 5 (HDAC5) and EZH2 were both upregulated in SSc ECs and posed detrimental effects on angiogenesis in these cells (5, 6).
In this study we provide a comprehensive analysis of chromatin accessibility patterns in SSc utilizing an assay for transposase-accessible chromatin with sequencing (ATAC-seq) with high-read density, allowing for a detailed analysis of transcription factor recruitment across the genome in skin-derived SSc ECs and fibroblasts compared to healthy controls.
Methods
Methods are provided in the Online Supplementary Material. Raw and processed sequencing data generated from our ATAC-seq experiments have been deposited in Gene Expression Omnibus (GEO; accession number GSE163199).
Results
Chromatin accessibility is broadly decreased in dcSSc cells.
The ATAC-seq libraries were sequenced to an average of 150 million reads per sample. All ATAC-seq libraries yielded fragment lengths with the expected fragment distribution and clear nucleosome phasing (Supplemental Figure 1A). The accessible regions identified were mostly enriched within 1 kb of TSSs (Supplemental Figure 1B), which is consistent with presence of regulatory elements in these regions. We next compared our ATAC-seq data with ENCODE open chromatin data generated using DNase-seq, as well as ChIP-seq data targeting H3K27ac, which marks active enhancers (Supplemental Figure 1C). There were no publicly accessible data for H3K27ac marks in human dermal microvascular ECs therefore we also included tracks generated from human umbilical vascular ECs (HUVECs). The ATAC-seq peaks from both SSc ECs and fibroblasts overlapped with ENCODE peaks, further supporting the quality of our data. The unsupervised clustering and principal component plots of all samples included in our ATAC-seq experiment are shown in Supplemental Figure 2.
Chromatin accessibility in SSc ECs and fibroblasts was overall lower than that in normal ECs and fibroblasts, potentially reflecting the disease state of SSc cells (Figure 1 and 2). ATAC-seq identified a total of 558 regions that are differentially accessible in SSc ECs compared to normal ECs, among them 97 regions showed increased chromatin accessibility while 461 regions had decreased chromatin accessibility in SSc cells (Figure 1A). This corresponds to 74 and 390 genes in these regions, respectively. Figure 1A depicts the genome tracks of the CDH2 and NLGN1 loci as two examples showing the regions that were differentially less accessible in dcSSc ECs compared to normal ECs, as indicated by the decrease in peak intensity. We found that differentially accessible regions in ECs were enriched in introns (Figure 1B). Less accessible peaks in SSc ECs were also enriched in promotor regions. Among the putative more accessible regions, 11% were located in proximal promoters (within 1 kb of the transcription start site, TSS), 11% in distal promoters (1 to 5 kb of TSS), 24% in exons, 41% in introns, 8% in 5’-UTR, and 5% in 3’-UTR. In the less accessible regions, 21% were located in proximal promoters, 9% in distal promoters, 21% in exons, 28% in introns, 20% in 5’-UTR, and 1% in 3’-UTR.
Figure 1:

ATAC-seq captures chromatin accessibility in dcSSc ECs. (A) Volcano plot of differentially accessible genes between normal and SSc ECs. Values with logFC > 1 or logFC < −1 and adjusted P-value < 0.05 are highlighted in green and blue, respectively (left). Representative ATAC-seq tracks at CDH2 and NLGN1 gene loci in normal and dcSSc ECs (right). Both genes are neuronal-related genes. (B) Genomic distributions of differentially accessible chromatin regions identified by ATAC-seq in ECs. Each color represents a genomic feature, as shown in the right part of the plot. Opening and closing refers to higher and lower chromatin accessibility in dcSSc relative to normal ECs, respectively (C) Pathway enrichment analysis of loci annotated to all differentially accessible regions in dcSSc ECs. (D) Gene regulatory network analysis for transcription factor targets using iRegulon, showing the top 18 most significant enrichments. Normalized Enrichment Score corresponds to a false discovery rate between 3% and 9%. (E) An example of a gene regulatory network showing differentially accessible genes identified by ATAC-seq in dcSSc ECs that are targets of the transcription factor ZBTB33. (F) Network analysis representing genes annotated to differentially accessible chromatin regions in dcSSc ECs compared to controls. The node size corresponds with the significance of the term it represents within the network. Colored portions of the pie charts represent the number of genes identified from the ATAC-seq experiment that are associated with the term.
Figure 2:

Analysis of chromatin accessibility profiles in dcSSc fibroblasts. (A) Volcano plot of differentially accessible genes between normal and SSc fibroblasts. Values with logFC > 1 or logFC < −1 and adjusted P-value < 0.05 are highlighted in green and blue, respectively (left). Representative ATAC-seq tracks at ENTPD1 and ERBB4 loci in normal and dcSSc fibroblasts (right). (B) Genomic distributions of differentially accessible chromatin regions identified by ATAC-seq in fibroblasts. Each color represents a genomic feature, as shown in the right part of the plot. Opening and closing refer to higher and lower chromatin accessibility in dcSSc relative to normal fibroblasts, respectively (C) Gene ontology analysis of loci associated with differentially accessible regions in dcSSc fibroblasts (left). Gene regulatory network analysis for transcription factor targets using iRegulon, showing the top 18 most significant enrichments. Normalized Enrichment Score corresponds to a false discovery rate between 3% and 9% (right). (D) An example of a gene regulatory network showing differentially accessible genes identified by ATAC-seq in dcSSc fibroblasts that are targets of the transcription factor EGR1. (E) Major gene ontology category annotations of genes associated with differentially accessible chromatin in both SSc ECs and fibroblasts. (F) Network analysis of differentially accessible chromatin regions identified in both dcSSc ECs and fibroblasts. The node size corresponds with the significance of the term it represents within the network. Colored portions of the pie charts represent the number of genes identified from the ATAC-seq experiment that are associated with the term. The clusters related to the nervous system are highlighted by arrows.
To gain insight into the functions of the genes that are located in differentially accessible regions in SSc ECs, we performed functional enrichment analyses using Molecular Signatures Database and found that the top 3 pathways that these genes were significantly associated with included nuclear receptor transcription, post-translational protein modification, and nitric oxide (NO)-guanylate cyclase (Figure 1C and Supplemental Table 2). Gene ontology analysis revealed that these genes were significantly associated with cell projection organization, chromosome, transcription regulator activity, and many neuronal-related GO terms (Supplementary Table 3). We also explored potential regulation by transcription factors among differentially accessible genes in SSc ECs, and identified enrichment of several transcription factors including RNF138, ZSCAN9, and BCL11A, to name a few (Figure 1D). An example of transcription factor gene regulatory network is shown in Figure 1E. 31 genes annotated from differential chromatin accessible regions in dcSSc ECs were targets for regulation by the transcription factor ZBTB33, which is a transcription factor also identified in the HINT-ATAC analysis in ECs as shown below.
To further capture the relationships among the pathways and gene ontology terms enriched in differentially accessible regions in SSc ECs, we used ClueGO/CluePedia to generate a network plot comprising a subset of enriched terms with the best p-values (Figure 1F). This analysis highlights gene clusters of enriched pathways and ontology terms related to cell membrane projections assembly and organization, cilia motility, nuclear and steroid receptors, nitric oxide, cilia motility, and the nervous system. The main terms that majority of the genes within the network converge to were “cell projection organization” and “plasma-membrane bounded cell projection assembly”. Several neuronal genes, including NLGN1, NRXN1, and SLIT2 (highlighted by arrows) were among differentially accessible genes shared among three pathways or ontology terms, implicating their possible significance in SSc ECs. Similarly, a group of genes enriched in the three terms related to cilium motility, cell projection assembly, and cell projection organization are highlighted (Figure 1F).
In fibroblasts, we identified 50 genetic loci with differential chromatin accessibility between healthy individuals and patients with dcSSc. 16 and 34 loci demonstrated increased and decreased chromatin accessibility, respectively, in dcSSc compared to normal fibroblasts (Figure 2A). This corresponds to 9 and 24 genes in these regions. These data are consistent with overall reduced chromatin accessibility in dcSSc patients. The genome tracks of two genes, ENTPD1 and ERBB4, were selected to show the reduction in chromatin accessibility in dcSSc fibroblasts when compared to normal fibroblasts (Figure 2A). Similar to the distribution of chromatin regions in SSc ECs, the majority of peaks were detected in the introns in fibroblasts (Figure 2B). Among the more accessible regions, 10% were located in distal promoters, 10% in exons, 80% in introns, while none in proximal promoter, 5’-UTR, and 3’-UTR. In the less accessible regions, 16% were located in proximal promoters, 8% in distal promoters, 13% in exons, 55% in introns, 8% in 5’-UTR, and none in 3’-UTR. The 33 genes that were located in differential chromatin accessibility regions were subjected to gene ontology analysis and they were enriched in “neuron projection”, “post-synapse”, and “postsynaptic membrane” (Figure 2C and Supplemental Table 4). They were also potential targets of transcription factors PBX1, IRF1, NFKB1, and EGR1, among others (Figure 2C). 14 differentially accessible genes in dcSSc fibroblasts are regulated by the transcription factor EGR1 (Figure 2D), which was also independently identified from our HINT-ATAC analysis shown below. CTTNBP2 was the only overlapping gene that was differentially accessible in both cell types and showed reduced accessibility in dcSSc ECs and fibroblasts compared to normal cells.
As shown in our analysis above, the genes located in differentially accessible regions in both ECs and fibroblasts appeared to be enriched in pathways that are involved in the nervous system. Indeed, gene ontology analysis of loci associated with differentially accessible regions from both cell types revealed a significant enrichment in neuron differentiation, neurogenesis, synapse, neuron development, neuron projection, post-synapse, and axon development; 8 out of the top 14 most significant gene ontologies (Figure 2E). Gene network analysis also showed significant clustering of gene ontologies and pathways related to the nervous system, as indicated by the arrows, and highlights cilia-related genes (Figure 2F). To ensure that the enrichment in neuronal pathways and ontologies among differentially accessible genes in dcSSc ECs and fibroblasts were not a result of contaminating neuronal cells, we co-stained dcSSc ECs and fibroblasts with the neuronal marker NEFM and endothelial or fibroblast markers VE-cadherin or α-SMA, respectively. As shown in Supplemental Figure 3, there was no contamination of neuronal cells.
To gain more insights into the relationship between chromatin accessibility and gene transcription, we extracted publicly-available RNA-seq data generated from dermal fibroblasts from healthy controls and dcSSc patients (n=3 each) from Gene Expression Omnibus (GEO; accession number GSE130313). 17 of 33 genes located in differentially accessible regions in dcSSc fibroblasts had detectable expression levels. Among them, only 2 genes showed significant changes in transcription; NAB1 was significantly downregulated, and RELL1 was significantly upregulated. Both genes were located in differentially less accessible regions in dcSSc fibroblasts. NAB1 codes for NGF1A-binding protein, which is a corepressor for early growth response (Egr) transcription factors. RELL1 codes for receptor expressed in lymphoid tissues like protein 1, and it activates the MAPK pathway when overexpressed. Since there is no available RNA-seq data for SSc ECs, we were unable to compare the overlapping genes at a genome-wide transcriptional level with differential chromatin accessibility results.
Transcription factor binding analysis at open chromatin regions in SSc cells.
Within the accessible chromatin regions, putative transcription factors can be detected since the narrow genomic regions occupied by transcription factors are protected from Tn5. We used HINT-ATAC to identify enriched putative transcription factor motifs, which are sequences that bind to specific transcription factors, in the open chromatin regions of both ECs and fibroblasts comparing healthy cells to dcSSc cells. In ECs, TFDP1, ZBTB33, E2F4, NFYA, and HINF, among others, bind more in normal ECs compared to dcSSc ECs, as indicated by their positive activity score (Figure 3A and Supplemental Table 5). In dcSSc ECs, we observed significantly more binding of ETV2, SNAI2, and ELF1 compared to healthy ECs (Figure 3A and Supplemental Table 5). The footprints of these transcription factors are shown in Figure 3B. The putative transcription factors differentially recruited in ECs between dsSSc patients and healthy controls, identified in the HINT-ATAC analysis, were enriched in pathways critical to telomere maintenance, TGFβ-regulated pathways, nerve growth factor (NGF) pathways, and P53 pathways (Figure 3C and Supplemental Table 6).
Figure 3:
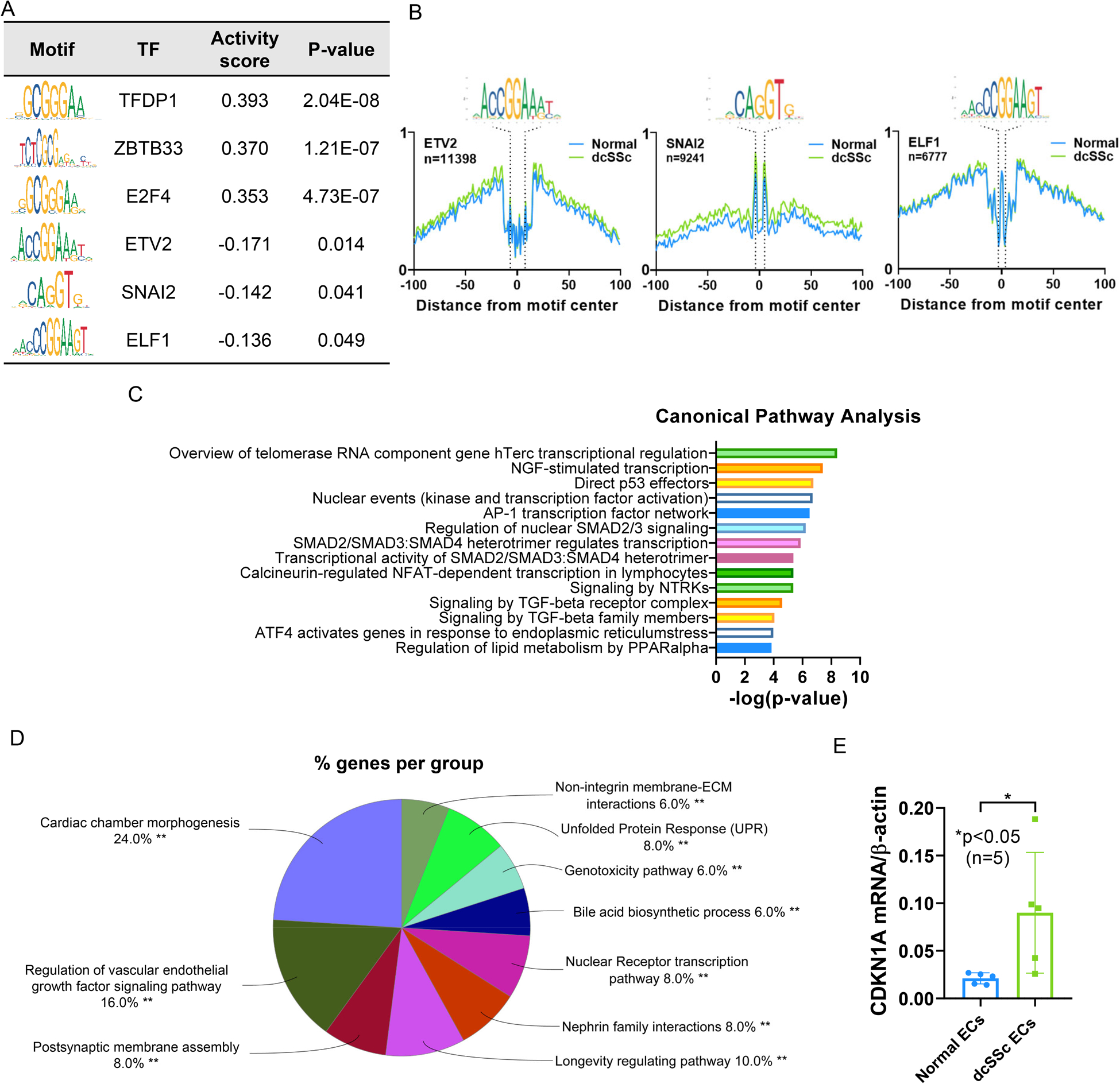
The most enriched transcription factor motifs and their target genes in SSc ECs. (A) HINT-ATAC motif analysis of regions in normal and dcSSc ECs. The top 3 transcription factors with significant differential activity values in ECs are shown. The activity score indicates the difference in activity in normal compared to dcSSc ECs (Normal-dcSSc). A positive score indicates increased transcription factor binding in normal ECs, and a negative score indicates increased binding in dcSSc ECs. The consensus sequences of transcription factor motifs are illustrated, and the height of the letter represents how frequently that nucleotide is observed in that particular position in each motif. (B) The footprint profiles of ETV2, SNAI2, and ELF1 generated from ECs. (C) Pathway analysis of the putative transcription factors identified in ECs. The top 14 are shown. (D) Pathway analysis of differentially accessible genes in dcSSc ECs that overlap with ETV2-target genes reported in Liu et al. (E) The expression of CDKN1A in dcSSc ECs is significantly increased compared to normal ECs. n=number of subjects. Data are shown as mean +/− S.D.
To help us understand the regulatory role of the transcription factors enriched in dcSSc ECs, we extracted gene sets from the TRANSFAC Predicted Transcription Factor Targets and CHEA Transcription Factor Targets databases (7, 8) for SNAI2 and ELF1, respectively. For ETV2, the gene list generated via ChIP-seq in in vitro differentiated embryonic stem cells was used (9). Pathway analysis revealed that SNAI2-target genes are enriched in nuclear receptor transcription and SUMOylation, and that ETV2-target genes are enriched in nervous system development and axon guidance (Supplemental Table 7). We also examined the degree of overlap between differentially accessible genes in ECs between patients and controls and the identified target genes for SNAI2, ETV2, or ELF1. Between the SNAI2-target genes and the differentially accessible ATAC-seq-annotated genes identified, there were 4 genes that overlapped (CACYBP, RXRA, SP4, and THRB). Between the ELF1-target gene set and the differentially accessible genes there were 17 overlapping genes (ARRDC2, AZI2, CCNG1, CD320, EAF2, EDRF1, ESPL1, FAM171A2, GHITM, KCNN1, MYO9B, PDE4A, RAPGEF6, SLC12A4, SPTY2D1, TTC14, and XPC). For ETV2, there were 89 genes overlapping between genes identified by ATAC-seq and the published ChIP-seq data (Supplemental Figure 4). These include genes involved in neural-related pathways such as CDH2, NLGN1, ROBO1, and SLIT2, as well as genes involved in longevity regulating pathway and vascular endothelial growth factor pathway (Figure 3D). Based on these results, the significant enrichment of genes involved in the nervous system identified in the differential chromatin accessibility analysis between dcSSc and normal ECs (Figure 1C and 1F) might be driven by enriched ETV2 binding in dcSSc ECs.
Because our analysis in ECs described above suggests involvement of aging/senescence related pathways, including telomere maintenance, P53, and longevity regulating pathways (Figure 3C and 3D), we examined the expression of CDKN1A (gene encoding for p21) in normal and dcSSc ECs. As shown in Figure 3E, CDKN1A was significantly elevated in dcSSc ECs, suggesting a senescent phenotype.
In normal fibroblasts, HINT-ATAC analysis identified more binding of NRF1, TCFL5, ZBTB33, E2F4, and TFDP1, among other transcription factors, compared to fibroblasts from dcSSc patients (Figure 4A and Supplemental Table 8). In dcSSc fibroblasts, significant enrichment of RUNX1 and RUNX2 was observed compared to normal fibroblasts (Figure 4A and Supplemental Table 8). The transcription factor footprints are shown in Figure 4B. When we performed pathway enrichment analysis, the transcription factors with differential recruitment across the genome between dcSSc and normal fibroblasts were significantly enriched in TGFβ-regulated pathways, NGF pathways, signaling by neurotrophin receptors (NTRKs), and telomere maintenance (Figure 4C and Supplemental Table 9). We extracted the gene sets regulated by RUNX1 and RUNX2 from the CHEA Transcription Factor Targets database (7), and identified 1263 target genes that are regulated by both RUNX1 and RUNX2. Pathway analysis examining RUNX1 and RUNX2-target genes revealed enriched pathways including GPCR signaling and chromatin modification for RUNX1, and infection-related pathways and G alpha signaling for RUNX2 (Supplemental Table 10). We also examined the pathways enriched in the 1263 overlapping genes regulated by both RUNX1 and RUNX2 and found that pathways involved in signal transduction, immune system, and axon guidance were enriched (Figure 4D). Examining the differentially accessible genes in dcSSc fibroblasts compared to controls identified in our ATAC-seq experiments and the gene set from CHEA Transcription Factor Targets databases for RUNX1 revealed 16 genes that overlapped (ABTB2, APBA2, ARHGEF7, ARPP21, ATG7, CNKSR3, CPA5, CTTNBP2, DISC1, FARS2, GABRA4, IPCEF1, NAB1, RELL1, SMARCA2, and TMEM39B). Between the RUNX2 target gene set from the CHEA Transcription Factor Targets databases and genes located in differentially accessible regions from ATAC-seq in fibroblasts, there were 9 that overlapped (ABTB2, CNKSR3, GABRA4, IPCEF1, NAB1, RANBP17, RELL1, SPATA16, and TRIM62). To examine whether the senescent phenotype shown in dcSSc ECs is also present in dcSSc fibroblasts, we measured CDKN1A and show Figure 4E, CDKN1A expression and show significantly higher levels in dcSSc fibroblasts compared to normal fibroblasts (p<0.05).
Figure 4:
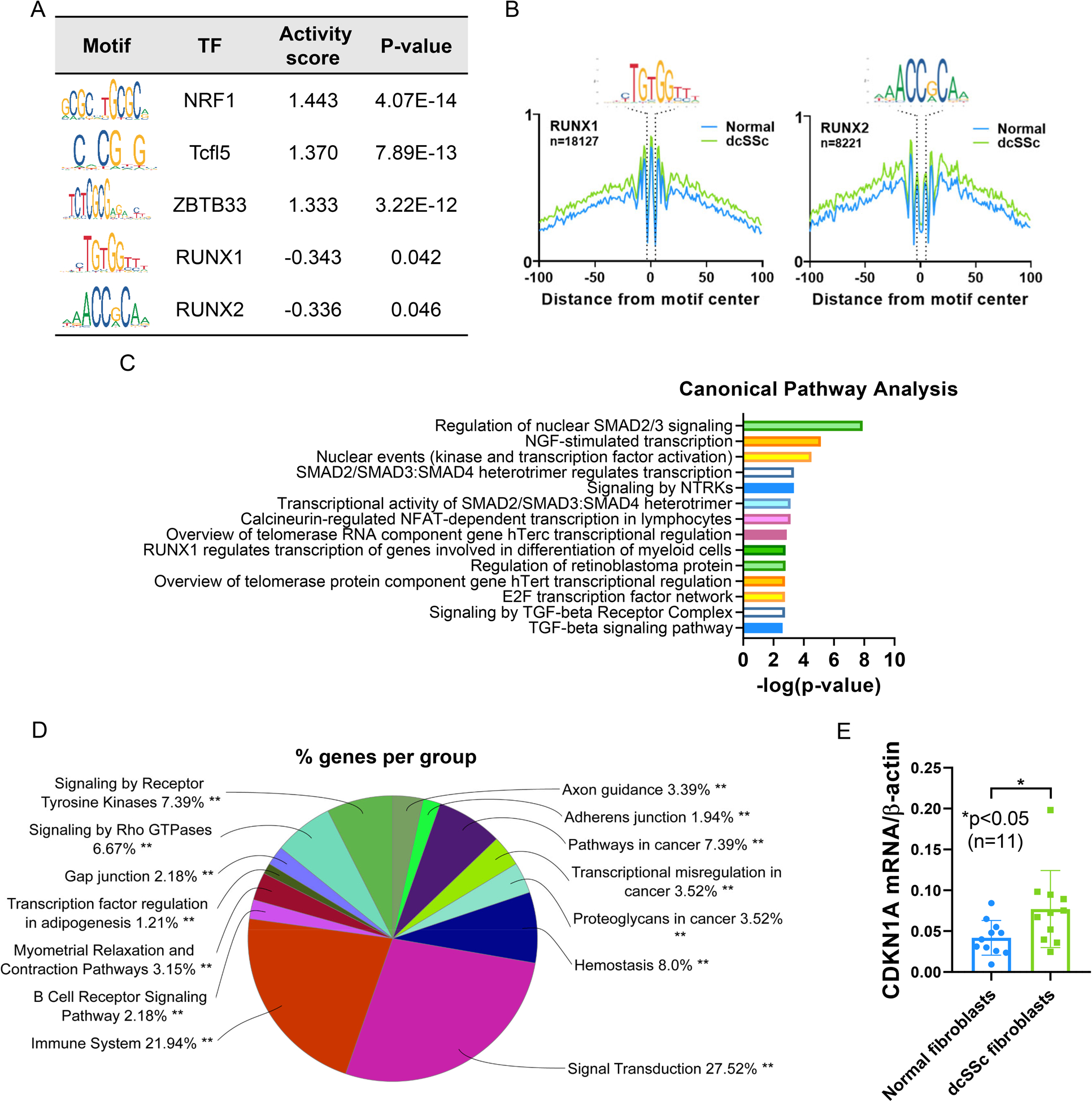
The most enriched transcription factor motifs and their target genes in dcSSc fibroblasts. (A) HINT-ATAC motif analysis of regions in normal and dcSSc fibroblasts. The top 3 transcription factors with significant differential activity values in fibroblasts are shown. The activity score indicates the difference in activity in normal compared to dcSSc fibroblasts (Normal-dcSSc). A positive score indicates increased transcription factor binding in normal fibroblasts, and a negative score indicates increased binding in dcSSc fibroblasts. (B) The footprint profiles of RUNX1 and RUNX2 generated from fibroblasts. (C) Pathway analysis of the putative transcription factors identified in fibroblasts. The top 14 are shown. (D) Pathway analysis of target genes regulated by both RUNX1 and RUNX2 identified using the CHEA Transcription Factor Targets database. (E) The expression of CDKN1A in dcSSc fibroblasts is significantly increased compared to normal fibroblasts. n=number of subjects. Data are shown as mean +/− S.D.
Functional validation studies.
To explore whether the differentially accessible genes or putative transcription factors enriched in dcSSc ECs or fibroblasts were differentially expressed between normal and dcSSc cells, we performed qPCR to examine the mRNA levels of selected genes and transcription factors. We chose neuronal-related genes, including CDH2, NLGN1, and NRXN1, which were differentially less accessible in dcSSc ECs for follow up studies since we observed many enriched pathways in the nervous system from our analyses (Figures 1 and 3). We also examined the expression of ETV2, ELF1, and SNAI2, which were identified as transcriptional factors with more binding in dcSSc ECs compared to healthy ECs in our HINT-ATAC analysis. Among the selected genes and transcription factors in ECs, NRXN1 was significantly downregulated, while ETV2 and SNAI2 were significantly upregulated in dcSSc ECs when compared to normal ECs (Figure 5A). We also examined the expression of SNAI1, which works alongside SNAI2 in many physiological pathways in ECs. The expression of SNAI1 was also significantly increased in dcSSc ECs. To examine whether these differentially expressed genes or transcription factors have any effect on endothelial function, we knocked down either ETV2 or NRXN1 in human dermal microvascular ECs (HMVECs) and performed matrigel tube formation assays. As shown in Figure 5B, NRXN1 knockdown in HMVECs led to significant reduction in tube formation. In contrast, ETV2 knockdown in HMVECs significantly increased tube formation (Figure 5C). These results suggest that upregulation of ETV2 and downregulation of NRXN1 in dcSSc ECs renders these cells to a more anti-angiogenic state.
Figure 5:

Functional relevance of genes and transcription factors identified by ATAC-seq and HINT-ATAC analysis in ECs. (A) The mRNA expression levels of selected genes were analyzed in normal and dcSSc dermal ECs showing significant downregulation of NRXN1 and upregulation of ETV2, SNAI1, and SNAI2 in dcSSc compared to normal controls. (B) Knockdown of NRXN1 significantly decreased tube formation in a matrigel tube formation assay. A representation of 3 independent experiments is shown. (C) Knockdown of ETV2 results in a significant increase in angiogenesis in a matrigel tube formation assay. A representation of 3 independent experiments is shown. Data are shown as mean +/− S.D.
In fibroblasts, we measured the expression of two neuronal-related genes with reduced chromatin accessibility in dcSSc, ERBB4 and ENTPD1. We also measured the expression of the transcription factors RUNX1 and RUNX2, which showed more binding in dcSSc fibroblasts in our HINT-ATAC analysis. ENTPD1 was significantly downregulated in dcSSc fibroblasts, while RUNX2 was significantly upregulated in dcSSc fibroblasts (Figure 6A). To examine whether ENTPD1 plays a role in modulating myofibroblast phenotype, we overexpressed ENTPD1 in dcSSc fibroblasts and measured pro-fibrotic genes ACTA2 and COL1A1. We found that overexpression of ENTPD1 resulted in significant upregulation of ACTA2 and COL1A1 (Figure 6B), suggesting that ENTPD1 is pro-fibrotic in dcSSc. To further validate our observation, we also performed functional studies using dcSSc fibroblasts. Overexpression of ENTPD1 significantly enhanced gel contraction (Figure 6C), and increased cell proliferation and wound closure in dcSSc fibroblasts (Figure 6D and 6E).
Figure 6:

Functional relevance of genes and transcription factors identified by ATAC-seq and HINT-ATAC analysis in fibroblasts. (A) The expression of ENTPD1 is significantly downregulated while RUNX2 is upregulated in dcSSc fibroblasts. (B) Overexpression of ENTPD1 in dcSSc fibroblasts results in upregulation of ACTA2 and COL1A1. (C) Overexpression of ENTPD1 in dcSSc fibroblasts enhanced gel contraction compared to cells transfected with control vectors. (D) Increased cell proliferation in ENTPD1-transfected dcSSc fibroblasts compared to controls. Cell confluence is measured by IncuCyte®. A representation of 4 dcSSc patients is shown. (E) ENTPD1-overexpressing dcSSc fibroblasts migrate faster to close the wound gap compared to control-transfected cells. Wound confluence is measured by IncuCyte®. The analysis of representative pictures at 0 and 44 hours for each group is shown. White color in the pictures denotes cells. Data shown are a representation of 4 dcSSc patients. Data are shown as mean +/− S.D.
Discussion
We report a global reduction in chromatin accessibility in ECs and fibroblasts isolated from dcSSc patients. In addition, we unveiled an unexpected neuronal signature in loci associated with differentially accessible regions from both cell types in dcSSc. By applying HINT-ATAC approaches, we defined the transcription factor footprints in dcSSc ECs and fibroblasts at a genome-wide level. In all, this study uncovered genes and proteins that can serve as potential biomarkers or treatment targets for SSc.
Global changes in chromatin accessibility have been reported in many other diseases. In metastatic cancer cells, genome-wide increase in chromatin accessibility has been reported to drive disease progression (10). In contrast, neurodegenerative diseases such as age-related macular degeneration is associated with reduced chromatin accessibility (11). Interestingly, aging has also been reported to be associated with global decrease in chromatin accessibility (12). Our results raise the possibility that the genome-wide reduction in chromatin accessibility could be a reflection of accelerated aging and senescence in dcSSc. Indeed, senescence is evident in SSc, as telomere shortening, which is a hallmark for senescence and aging has been reported (13). Interestingly, the transcription factors involved in both cell types showed significant enrichment in pathways related to hTERC and hTERT, which are critical for telomerase regulation (Figure 3C and 4C). In addition, the longevity-regulating pathway was enriched in overlapping genes between ones annotated from differentially accessible chromatin regions in dcSSc ECs and reported ETV2-target genes (Figure 3D). We confirmed the presence of senescence and aging phenotype in both dcSSc ECs and fibroblasts, as demonstrated by increased expression of CDKN1A (Figure 3E and 4E).
The global reduction in chromatin accessibility in dcSSc ECs might be due to upregulation of histone deacetylases (HDACs). Histone lysine acetylation alters the charge on histone side chains resulting in an open chromatin configuration that allows transcription factors to bind and enhances gene transcription. We reported previously that in dcSSc ECs, several members of class II HDACs, including HDAC5, were significantly upregulated (6). When we knocked down HDAC5 in dcSSc ECs, an increase in chromatin accessibility, as measured by ATAC-seq, was observed. This mechanism might also be relevant in dcSSc fibroblasts, as various HDACs, including HDAC1 and HDAC6, have been reported to be overexpressed in dcSSc fibroblasts when compared to their normal counterparts (14).
Through pathway enrichment and gene ontology analysis of the genes annotated in differentially accessible chromatin regions in ECs, we observed an enrichment in genes involved in NO-, cilia-, and extracellular matrix (ECM)-related pathways. The NO- and ECM-related pathways observed from our analysis are not surprising, since they have been implicated in the pathogenic phenotype of SSc ECs. It has been shown that NO production is impaired in SSc ECs due to downregulation of endothelial NO synthase (15). These cells also exhibit a mesenchymal-phenotype with enhanced endothelial-to-mesenchymal transition that leads to higher ECM expression (16, 17). Although the role of cilia in fibrosis has been implicated in SSc (18), their involvement in EC function has not been examined. Interestingly, primary cilia, which are microtubule-based organelles, play critical roles in maintaining vascular barrier (19). They also act as mechanosensors in the blood vessels that are critical for vascular functions and development (20). These events are pertinent to SSc vasculopathy, and therefore, further investigation of the involvement of ciliary dysfunction in SSc is warranted.
We observed an enrichment in genes that are involved in the nervous system. This was also one of the top enriched pathways in the transcription factors identified from HINT-ATAC in both ECs and fibroblasts (Figure 3C and 4C). Although this neuronal signature might be surprising at first, the dysregulation of neuro-endothelial mechanisms has been suggested to be critical in initiating microvascular abnormalities such as Raynaud’s phenomenon in SSc (21). Indeed, disruption in the peripheral nervous system, including morphological and functional changes, has been reported in SSc (22). As the nervous and the vascular systems share anatomical patterns and many cellular mechanisms, it is not surprising that ECs express receptors for axon guidance molecules, which are grouped in 4 major families: Slit/Robo, Semaphorin/Plexin/Neuropilin, Netrin/Unc5/DCC, and Ephrin/Eph (23). It has been shown that in dcSSc ECs, dysregulation of these pathways leads to defective angiogenesis (24–26). Specifically, SLIT2, which was located in the less accessible regions in SSc ECs that are reported to be upregulated in these cells, shows potent anti-angiogenic effects (24). The reason for the discrepancy between chromatin accessibility and transcription might be multifactorial, including increased expression of specific activating transcription factors, reduced mRNA decay, or reduced microRNA regulation in SSc. Nonetheless, the studies mentioned above echo our pathway analysis where we detected significant enrichment in pathways involving axon guidance.
In addition to axon guidance pathways, we also observed enrichment in synaptic-related genes, including CDH2, NLGN1, and NRXN1. Besides their role in the nervous system, these genes are also critical in promoting EC angiogenesis. Loss- or gain-of-function experiments showed that CDH2 significantly promoted angiogenesis both in vitro and in vivo (27). In a chicken chorioallantoic membrane system, a monoclonal recombinant antibody against NRXN1 blocked angiogenesis, whereas exogenous NLGN1 promoted angiogenesis (28). We showed that the promoters of these genes are less accessible in dcSSc than normal ECs (Figure 1A), and among them, NRXN1 was significantly downregulated in dcSSc ECs (Figure 5A). We further demonstrated that NRXN1 downregulation inhibits the angiogenic potential of HMVECs (Figure 5B), suggesting that lower levels of NRXN1 might contribute to dysregulated angiogenesis in dcSSc ECs.
Similar to what was observed in ECs, the enrichment of the genes associated with the nervous system in dcSSc fibroblasts might also play relevant a role in fibrosis. ERBB4, which codes for receptor tyrosine-protein kinase ErbB-4, is a key cellular receptor for neuregulin growth factors. In mice, ErbB-4 deletion accelerated renal fibrosis and renal injury (29). In addition, the anti-fibrotic effect of neuregulin growth factor-1/ErbB-4 pathway in the heart, skin, and lungs is linked to its anti-inflammatory activity in macrophages (30). Another example is ENTPD1, which codes for ectonucleoside triphosphate diphosphohydrolase 1 or CD39. In the nervous system, it hydrolyzes ATP to regulate neurotransmission. The deletion of ENTPD1/CD39 in mice promoted biliary injury and fibrosis by acting on gut-imprinted CD8+ T cells and macrophages (31, 32). Since these two genes were annotated in differentially less accessible regions of the chromatin in dcSSc fibroblasts (Figure 2A), it is possible that they are downregulated in SSc and contribute to the exacerbated fibrosis phenotype in this disease. Indeed, when we examined their expression in dcSSc fibroblasts, ENTPD1 was significantly downregulated compared to normal fibroblasts (Figure 6A). Follow-up functional studies showed that ENTPD1/CD39 is pro-fibrotic, as it enhanced myofibroblast functions (Figure 6B–E). The downregulation of this gene in dcSSc fibroblast appears to be a protective mechanism in these cells.
When comparing the transcription factor footprints in dcSSc ECs, we found significant enrichment of SNAI2 and ETV2 in dcSSc ECs when compared to normal ECs. SNAI2 or Slug, is a neurogenic transcription factor that belongs to the Snail family of zinc finger transcription factors. It works with SNAI1 (or Snail) in processes involving epithelial- or endothelial-to-mesenchymal transition (33, 34). In addition, it was found to be critical in angiogenic sprouting in ECs (35). Here we report that both SNAI1 and SNAI2 were upregulated in dcSSc ECs. Since increased in endothelial-to-mesenchymal transition has been observed in dcSSc ECs (16), the enrichment of SNAI2 might play a critical role in this process. ETV2, coding for transcription factor Ets variant 2, appears to be one of the key drivers for the enriched neuronal signal we observed in genes annotated in differentially accessible regions in dcSSc ECs. It is critical in promoting blood vessel formation as well as reprograming non-endothelial cells into cells displaying endothelial phenotypes (36). As the regulatory network of ETV2 includes VEGF signaling, Notch pathways, MAPK signaling, and Ephrin signaling (Supplemental Table 7)(9), its upregulation in dcSSc ECs would be expected to promote healthy EC function. Interestingly, when we knockdown ETV2 in HMVECs, we observed a significant increase in tube formation (Figure 5C), pointing to its role in regulating angiogenesis in SSc.
In the HINT-ATAC analysis, we identified several transcription factors that are less accessible in dcSSc ECs and fibroblasts, including ZBTB33 and EGR. ZBTB33, which codes for Kaiso, is a transcription factor but it also has the ability to bind to methylated DNA. Therefore, ZBTB33 can function as a transcriptional activator or repressor. In ECs, Kaiso protects ECs from apoptosis via regulating BCL2, BAX, and BIK, through its binding partner p120ctn (37). The role of Kaiso in fibroblasts is not clear. However silencing Kaiso in breast cancer cells attenuated TGFβ signaling and TGFβR1 expression (38), suggesting that this transcription factor might play a role in TGFβ-related pathways in fibroblasts. The pro-fibrotic nature of EGR family of transcription factors in SSc has been well documented (39–41). In ECs, EGR3 appears to be an essential downstream mediator of VEGF-mediated endothelial functions (42) while EGR1 supports FGF-dependent angiogenesis (43).
The transcription factor RUNX2, which is enriched and upregulated in dcSSc dermal fibroblasts, belongs to the runt-related transcription factors. These RUNX proteins play critical roles in diverse cellular processes including proliferation, differentiation, epithelial-to-mesenchymal transition, and inflammation. Studies have shown crosstalk between RUNX2 and pro-fibrotic signaling pathways, including TGFβ and Wnt pathways (44, 45). Functionally, the effect of RUNX2 on fibrosis appears to be organ-specific. RUNX2-deficient mice showed exacerbated ureteral obstruction-induced kidney fibrosis, accompanied with enhanced TGFβ-signaling (46). In contrast, RUNX2, which is significantly expressed in human type 2 diabetic aorta, induced aortic fibrosis and stiffening in mice (47). In the lung, the involvement of RUNX2 in fibrogenesis is cell-type dependent (48). Knockdown of RUNX2 in alveolar epithelial type II cells decreased profibrotic signals, whereas fibroblasts deficient in RUNX2 showed increased ECM production. The role of RUNX proteins in SSc has not been fully explored. Our previous studies revealed that RUNX1, RUNX2 and RUNX3 were all hypomethylated in fibroblasts from both dcSSc and limited cutaneous SSc patients compared to healthy individuals (3). However, their functional relevance in these cells were not explored. Recently, lower expression of RUNX3 in dendritic cells was shown to be associated with enhanced fibrosis in mice (49).
While the reduction in chromatin accessibility observed in ECs and fibroblasts in dcSSc might be a hallmark of the disease, it is not clear whether this observation contributes to the pathogenesis of the disease itself, or is a mere representation of adaptive responses in these cells to function in the disease environment. In addition, our study did not account for the cellular heterogeneity and cell subsets of ECs and fibroblasts within the skin. Further functional studies and experiments to identify the molecular mechanisms and specific cell subtypes that mediate the global change in chromatin landscape in SSc are warranted
In summary, we provided novel information identifying open and closed chromatin regions in both ECs and fibroblasts in dcSSc. In addition, we characterized the transcription factor footprints in these cells and revealed the complex networks of transcription factors and their target genes, thereby allowing us to explore the potential mechanisms of dysregulated angiogenesis and enhanced fibrosis in this disease. Indeed, we identified the significant involvement of neuronal genes in SSc, specifically NRXN1 and ENTPD1, that are critical in both EC and fibroblast functions. Through HINT-ATAC analysis followed by functional validation we also showed the impact of ETV2 overexpression in dcSSc ECs. The data presented enhances our understanding of the involvement of epigenetic dysregulation at the chromatin architectural level in SSc.
Supplementary Material
Acknowledgements
This work was supported by the National Institute of Allergy and Infectious Diseases (NIAID) and the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) of the National Institutes of Health (NIH) grants number R01AI097134 and R01AR070148 to Dr. Sawalha. Drs. Tsou and Khanna were supported in part by the Donna and Larry Shelley’s Research Fund. Dr. Tsou was supported by the Edward T. and Ellen K. Dryer Early Career Professorship, and Dr. Khanna was supported by NIH/NIAMS K24AR063120.
Footnotes
Conflict of interest: the authors declare no conflict of interest.
References
- 1.Tsou PS, Sawalha AH. Unfolding the pathogenesis of scleroderma through genomics and epigenomics. J Autoimmun. 2017;83:73–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feghali-Bostwick C, Medsger TA Jr., Wright TM. Analysis of systemic sclerosis in twins reveals low concordance for disease and high concordance for the presence of antinuclear antibodies. Arthritis Rheum. 2003;48(7):1956–63. [DOI] [PubMed] [Google Scholar]
- 3.Altorok N, Tsou PS, Coit P, Khanna D, Sawalha AH. Genome-wide DNA methylation analysis in dermal fibroblasts from patients with diffuse and limited systemic sclerosis reveals common and subset-specific DNA methylation aberrancies. Ann Rheum Dis. 2015;74(8):1612–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He Y, Tsou PS, Khanna D, Sawalha AH. Methyl-CpG-binding protein 2 mediates antifibrotic effects in scleroderma fibroblasts. Ann Rheum Dis. 2018;77(8):1208–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsou PS, Campbell P, Amin MA, Coit P, Miller S, Fox DA, et al. Inhibition of EZH2 prevents fibrosis and restores normal angiogenesis in scleroderma. Proc Natl Acad Sci U S A. 2019;116(9):3695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsou PS, Wren JD, Amin MA, Schiopu E, Fox DA, Khanna D, et al. Histone Deacetylase 5 Is Overexpressed in Scleroderma Endothelial Cells and Impairs Angiogenesis via Repression of Proangiogenic Factors. Arthritis Rheumatol. 2016;68(12):2975–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lachmann A, Xu H, Krishnan J, Berger SI, Mazloom AR, Ma’ayan A. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics. 2010;26(19):2438–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34(Database issue):D108–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu F, Li D, Yu YYL, Kang I, Cha M-J, Kim JY, et al. Induction of hematopoietic and endothelial cell program orchestrated by ETS transcription factor ER71/ETV2. EMBO reports. 2015;16(5):654–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Denny SK, Yang D, Chuang C-H, Brady JJ, Lim JS, Grüner BM, et al. Nfib Promotes Metastasis through a Widespread Increase in Chromatin Accessibility. Cell. 2016;166(2):328–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J, Zibetti C, Shang P, Sripathi SR, Zhang P, Cano M, et al. ATAC-Seq analysis reveals a widespread decrease of chromatin accessibility in age-related macular degeneration. Nat Commun. 2018;9(1):1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ucar D, Marquez EJ, Chung CH, Marches R, Rossi RJ, Uyar A, et al. The chromatin accessibility signature of human immune aging stems from CD8(+) T cells. J Exp Med. 2017;214(10):3123–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lakota K, Hanumanthu VS, Agrawal R, Carns M, Armanios M, Varga J. Short lymphocyte, but not granulocyte, telomere length in a subset of patients with systemic sclerosis. Ann Rheum Dis. 2019;78(8):1142–4. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Fan PS, Kahaleh B. Association between enhanced type I collagen expression and epigenetic repression of the FLI1 gene in scleroderma fibroblasts. Arthritis Rheum. 2006;54(7):2271–9. [DOI] [PubMed] [Google Scholar]
- 15.Romero LI, Zhang DN, Cooke JP, Ho HK, Avalos E, Herrera R, et al. Differential expression of nitric oxide by dermal microvascular endothelial cells from patients with scleroderma. Vascular medicine (London, England). 2000;5(3):147–58. [DOI] [PubMed] [Google Scholar]
- 16.Manetti M, Romano E, Rosa I, Guiducci S, Bellando-Randone S, De Paulis A, et al. Endothelial-to-mesenchymal transition contributes to endothelial dysfunction and dermal fibrosis in systemic sclerosis. Ann Rheum Dis. 2017;76(5):924–34. [DOI] [PubMed] [Google Scholar]
- 17.Tsou PS, Palisoc PJ, Flavahan NA, Khanna D. Dissecting the cellular mechanism of prostacyclin analogue iloprost in reversing vascular dysfunction in scleroderma. Arthritis Rheumatol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teves ME, Strauss JF 3rd, Sapao P, Shi B, Varga J. The Primary Cilium: Emerging Role as a Key Player in Fibrosis. Curr Rheumatol Rep. 2019;21(6):29. [DOI] [PubMed] [Google Scholar]
- 19.Ma N, Zhou J. Functions of Endothelial Cilia in the Regulation of Vascular Barriers. Frontiers in Cell and Developmental Biology. 2020;8(626). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Egorova AD, van der Heiden K, Poelmann RE, Hierck BP. Primary cilia as biomechanical sensors in regulating endothelial function. Differentiation. 2012;83(2):S56–61. [DOI] [PubMed] [Google Scholar]
- 21.Kahaleh B, Matucci-Cerinic M. Raynaud’s phenomenon and scleroderma. Dysregulated neuroendothelial control of vascular tone. Arthritis Rheum. 1995;38(1):1–4. [DOI] [PubMed] [Google Scholar]
- 22.Cerinic MM, Generini S, Pignone A, Casale R. The nervous system in systemic sclerosis (scleroderma). Clinical features and pathogenetic mechanisms. Rheum Dis Clin North Am. 1996;22(4):879–92. [DOI] [PubMed] [Google Scholar]
- 23.Tam SJ, Watts RJ. Connecting Vascular and Nervous System Development: Angiogenesis and the Blood-Brain Barrier. Annual Review of Neuroscience. 2010;33(1):379–408. [DOI] [PubMed] [Google Scholar]
- 24.Romano E, Manetti M, Rosa I, Fioretto BS, Ibba-Manneschi L, Matucci-Cerinic M, et al. Slit2/Robo4 axis may contribute to endothelial cell dysfunction and angiogenesis disturbance in systemic sclerosis. Ann Rheum Dis. 2018;77(11):1665–74. [DOI] [PubMed] [Google Scholar]
- 25.Mazzotta C, Romano E, Bruni C, Manetti M, Lepri G, Bellando-Randone S, et al. Plexin-D1/Semaphorin 3E pathway may contribute to dysregulation of vascular tone control and defective angiogenesis in systemic sclerosis. Arthritis Res Ther. 2015;17(1):221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miao HQ, Soker S, Feiner L, Alonso JL, Raper JA, Klagsbrun M. Neuropilin-1 mediates collapsin-1/semaphorin III inhibition of endothelial cell motility: functional competition of collapsin-1 and vascular endothelial growth factor-165. J Cell Biol. 1999;146(1):233–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhuo H, Zhao Y, Cheng X, Xu M, Wang L, Lin L, et al. Tumor endothelial cell-derived cadherin-2 promotes angiogenesis and has prognostic significance for lung adenocarcinoma. Molecular cancer. 2019;18(1):34-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bottos A, Destro E, Rissone A, Graziano S, Cordara G, Assenzio B, et al. The synaptic proteins neurexins and neuroligins are widely expressed in the vascular system and contribute to its functions. Proc Natl Acad Sci U S A. 2009;106(49):20782–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeng F, Miyazawa T, Kloepfer LA, Harris RC. ErbB4 deletion accelerates renal fibrosis following renal injury. Am J Physiol Renal Physiol. 2018;314(5):F773–f87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vermeulen Z, Hervent A-S, Dugaucquier L, Vandekerckhove L, Rombouts M, Beyens M, et al. Inhibitory actions of the NRG-1/ErbB4 pathway in macrophages during tissue fibrosis in the heart, skin, and lung. American Journal of Physiology-Heart and Circulatory Physiology. 2017;313(5):H934–H45. [DOI] [PubMed] [Google Scholar]
- 31.Peng ZW, Rothweiler S, Wei G, Ikenaga N, Liu SB, Sverdlov DY, et al. The ectonucleotidase ENTPD1/CD39 limits biliary injury and fibrosis in mouse models of sclerosing cholangitis. Hepatol Commun. 2017;1(9):957–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rothweiler S, Feldbrügge L, Jiang ZG, Csizmadia E, Longhi MS, Vaid K, et al. Selective deletion of ENTPD1/CD39 in macrophages exacerbates biliary fibrosis in a mouse model of sclerosing cholangitis. Purinergic Signal. 2019;15(3):375–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Medici D, Hay ED, Olsen BR. Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factor-beta3. Molecular biology of the cell. 2008;19(11):4875–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cooley BC, Nevado J, Mellad J, Yang D, St. Hilaire C, Negro A, et al. TGF-β Signaling Mediates Endothelial-to-Mesenchymal Transition (EndMT) During Vein Graft Remodeling. Science Translational Medicine. 2014;6(227):227ra34–ra34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Welch-Reardon KM, Ehsan SM, Wang K, Wu N, Newman AC, Romero-Lopez M, et al. Angiogenic sprouting is regulated by endothelial cell expression of Slug. Journal of Cell Science. 2014;127(9):2017–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morita R, Suzuki M, Kasahara H, Shimizu N, Shichita T, Sekiya T, et al. ETS transcription factor ETV2 directly converts human fibroblasts into functional endothelial cells. Proc Natl Acad Sci U S A. 2015;112(1):160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xue X, Zhang J, Lan H, Xu Y, Wang H. Kaiso protects human umbilical vein endothelial cells against apoptosis by differentially regulating the expression of B-cell CLL/lymphoma 2 family members. Scientific Reports. 2017;7(1):7116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bassey-Archibong BI, Kwiecien JM, Milosavljevic SB, Hallett RM, Rayner LGA, Erb MJ, et al. Kaiso depletion attenuates transforming growth factor-β signaling and metastatic activity of triple-negative breast cancer cells. Oncogenesis. 2016;5(3):e208–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhattacharyya S, Chen SJ, Wu M, Warner-Blankenship M, Ning H, Lakos G, et al. Smad-independent transforming growth factor-beta regulation of early growth response-1 and sustained expression in fibrosis: implications for scleroderma. Am J Pathol. 2008;173(4):1085–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fang F, Ooka K, Bhattacharyya S, Wei J, Wu M, Du P, et al. The early growth response gene Egr2 (Alias Krox20) is a novel transcriptional target of transforming growth factor-β that is up-regulated in systemic sclerosis and mediates profibrotic responses. Am J Pathol. 2011;178(5):2077–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fang F, Shangguan AJ, Kelly K, Wei J, Gruner K, Ye B, et al. Early growth response 3 (Egr-3) is induced by transforming growth factor-β and regulates fibrogenic responses. Am J Pathol. 2013;183(4):1197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu D, Evans I, Britton G, Zachary I. The zinc-finger transcription factor, early growth response 3, mediates VEGF-induced angiogenesis. Oncogene. 2008;27(21):2989–98. [DOI] [PubMed] [Google Scholar]
- 43.Fahmy RG, Dass CR, Sun L-Q, Chesterman CN, Khachigian LM. Transcription factor Egr-1 supports FGF-dependent angiogenesis during neovascularization and tumor growth. Nat Med. 2003;9(8):1026–32. [DOI] [PubMed] [Google Scholar]
- 44.McCarthy TL, Centrella M. Novel Links among Wnt and TGF-β Signaling and Runx2. Molecular Endocrinology. 2010;24(3):587–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gaur T, Lengner CJ, Hovhannisyan H, Bhat RA, Bodine PVN, Komm BS, et al. Canonical WNT Signaling Promotes Osteogenesis by Directly Stimulating Runx2 Gene Expression. Journal of Biological Chemistry. 2005;280(39):33132–40. [DOI] [PubMed] [Google Scholar]
- 46.Kim JI, Jang HS, Jeong JH, Noh MR, Choi JY, Park KM. Defect in Runx2 gene accelerates ureteral obstruction-induced kidney fibrosis via increased TGF-β signaling pathway. Biochim Biophys Acta. 2013;1832(10):1520–7. [DOI] [PubMed] [Google Scholar]
- 47.Raaz U, Schellinger IN, Chernogubova E, Warnecke C, Kayama Y, Penov K, et al. Transcription Factor Runx2 Promotes Aortic Fibrosis and Stiffness in Type 2 Diabetes Mellitus. Circ Res. 2015;117(6):513–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mümmler C, Burgy O, Hermann S, Mutze K, Günther A, Königshoff M. Cell-specific expression of runt-related transcription factor 2 contributes to pulmonary fibrosis. The FASEB Journal. 2018;32(2):703–16. [DOI] [PubMed] [Google Scholar]
- 49.Affandi AJ, Carvalheiro T, Ottria A, Broen JC, Bossini-Castillo L, Tieland RG, et al. Low RUNX3 expression alters dendritic cell function in patients with systemic sclerosis and contributes to enhanced fibrosis. Ann Rheum Dis. 2019;78(9):1249–59. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.