

Abstract
Neutrophils, polymorphonuclear (PMN) leukocytes, play a critical role in the innate immune response to Staphylococcus aureus, a pathogen that continues to be associated with significant morbidity and mortality. Neutrophil extracellular trap (NET) formation is involved in ensnaring and killing of S. aureus, but this host-pathogen interaction also leads to host tissue damage. Importantly, NET components including neutrophil proteases are under consideration as therapeutic targets in a variety of disease processes. Although S. aureus lipoproteins are recognized to activate cells via toll-like receptors, specific mechanisms of interaction with neutrophils are poorly delineated. We hypothesized that a lipoprotein-containing cell membrane preparation from methicillin resistant S. aureus (MRSA-CMP) would elicit PMN activation including NET formation. We investigated MRSA-CMP-elicited NET formation, regulated elastase release, and IL-8 production in human neutrophils. We studied PMN from healthy donors with or without a common SNP in TLR1, previously demonstrated to impact TLR2/1 signaling, and utilized CMP from both WT MRSA and a mutant lacking palmitoylated lipoproteins (lgt). MRSA-CMP elicited NET formation, elastase release, and IL-8 production in a lipoprotein-dependent manner. TLR2/1 signaling was involved in NET formation and IL-8 production, but not elastase release suggesting that MRSA-CMP-elicited elastase release is not mediated by tri-acylated lipoproteins. MRSA-CMP also primed neutrophils for enhanced NET formation in response to a subsequent stimulus. MRSA-CMP-elicited NET formation did not require Nox2-derived ROS and was partially dependent on the activity of peptidyl arginine deiminase (PAD). In conclusion, lipoproteins from S. aureus mediate NET formation via TLR2/1 with clear implications for patients with sepsis.
Introduction
Bacterial sepsis continues to be a high prevalence disease responsible for widespread morbidity and significant mortality. Staphylococcus aureus, a common commensal of human skin, remains an important pathogen, causing many subtypes of localized infection as well as disseminated sepsis. Methicillin-resistant S. aureus (MRSA) is of broad significance, both as a hospital-acquired, and a community-associated pathogen. The community-associated strains, USA300 and USA400, emerged at the turn of the century as microbiologically distinct clones from the most common hospital acquired strain, USA100, with USA300 as predominant (1, 2). Despite close to two decades of efforts focused on characterization of the virulence factors for these MRSA strains, and reductions in incidence of hospital-acquired bloodstream infections, the morbidity and mortality remains significant with over 700,000 MRSA infections and 20,000 deaths per year in the US alone (3-5). The primary approach to control of these diseases has been through antibiotic stewardship and targeted antibiotic therapy for MRSA infections. However, the early and exuberant host inflammatory response often leads to organ dysfunction, despite optimal antimicrobials, that can only be managed with supportive care. This underscores the critical importance of elucidating mechanisms and development of immune-based approaches to combat staphylococcal disease (6).
The innate host response to staphylococcal invasion includes rapid deployment and activation of circulating neutrophils to the site of infection. Neutrophil extracellular trap (NET) formation, one component of the neutrophil response to pathogens, is critical for trapping and killing of intact staphylococci. Notwithstanding the critical role in pathogen killing, a growing body of literature demonstrates that NET formation contributes to tissue and organ damage. The role of NETs in host injury is not limited to infectious diseases, with a growing understanding of neutrophil activation and NET formation in the pathogenesis of multiple autoimmune conditions, as well as in cancer (7-13). Antibodies to NET components are present in the circulation of patients with systemic lupus erythematosus (14) and rheumatoid arthritis (15) and are under investigation for use as biomarkers for disease subtype and severity, thus highlighting the broad relevance of understanding the mechanisms driving NET formation.
NET formation was initially described as the extrusion of DNA covered with elastase and other neutrophil proteases in a web like matrix that could ensnare bacteria and facilitate killing (16). Initial studies utilized intact bacteria as phagocytic stimuli (17), with minimal investigation of soluble bacterial products released into the circulation during sepsis that interact with non-phagocytic neutrophil surface receptors (18). In vitro studies of NET formation have primarily employed non-physiologic neutrophil activating agents, including the phorbol ester, PMA, and the calcium ionophore, ionomycin (19). Moreover, NET formation was described as a terminal event leading to the death of the neutrophil, whereas in recent years, non-lytic NET formation has been described (18-20). The breadth of unique agonists and signaling pathways involved in NET formation is beginning to emerge, with significant focus on the role of peptidyl arginine deiminases, or PAD enzymes. These calcium-dependent enzymes (neutrophils express both PAD2 and PAD4) catalyze the conversion of arginine to citrulline in a process that has been implicated in the joint damage associated with rheumatoid arthritis (21, 22). However, the initial receptors and upstream signaling that trigger this process are unclear. A better understanding of disease process specific mechanisms of NET formation is critical to developing effective targeted therapies.
There is a vast body of literature describing the interactions between pathogen-associated molecular patterns and pattern recognition receptors on many cell types, both primary and cell lines (23-25). A minority of these studies utilize freshly isolated human neutrophils given the complexity of employing these cells which are very easily activated during isolation and not amenable to traditional molecular biology techniques. Relevant to the current study, we employ primary human neutrophils from healthy volunteers with or without a common SNP in TLR1 (rs5743618, 1805G>T) to investigate signaling specificity. TLR2 recognizes di- and tri-acylated lipoproteins via heterodimerization with TLR6 and TLR1, respectively (24). Neutrophils from donors with the SNP (1805T) display augmented responses to the synthetic TLR2/1 ligand, Pam3CSK4, when compared with neutrophils from donors with the reference allele (1805G), whereas responses to the TLR2/6 ligand, FSL-1, are no different (26). Therefore, this common SNP in TLR1 provides a useful tool for specifically interrogating the role of TLR2/1 signaling in human neutrophils.
Recognizing the clinical relevance of this SNP and growing evidence for the genetic basis of host variations in immune response (27, 28), we sought to investigate TLR2 signaling in human PMN using a clinically relevant stimulus (29-33). S. aureus expresses dozens of lipoproteins (34) and has been shown to activate cells via numerous receptors including TLR2 (35-38). In the current study, we focused specifically on a preparation of solubilized lipoprotein-containing cell membranes from USA300 MRSA (MRSA-CMP) to simulate the encounter that might occur between circulating neutrophils and staphylococci that had undergone lysis and to investigate neutrophil signaling to a non-phagocytic and clinically relevant stimulus. We found that MRSA-CMP directly elicited NET formation in a concentration-dependent fashion that was dependent upon lipoproteins in that preparation. This MRSA-CMP driven NET formation involved TLR2/1 signaling. Interestingly, MRSA-CMP also elicited regulated elastase release in a process that was distinct from NET formation and did not require TLR2/1 ligation. We also describe the priming of NET formation by MRSA-CMP which has additional clinical implications. The mechanism of NET formation in our studies was ROS-independent and partially PAD-dependent.
Materials and Methods
Materials
HBSS and PBS were purchased from Mediatech (Manassas, VA, USA), dextran from Pharmacosmos (Holbaek, Denmark), Ficoll-Paque from GE Healthcare (Piscataway, NJ, USA), and human serum albumin from CSL Behring (King of Prussia, PA, USA). Antibodies included rabbit polyclonal to histone H3 (citrulline R2+R8+R17) from Cayman Chemical (Ann Arbor, MI, USA), biotinylated anti-human elastase detection antibody from R&D Systems (Minneapolis, MN, USA), mouse monoclonal to IL-8 (clone 8CH) from Invitrogen (Waltham, MA, USA) for flow cytometry, and mouse monoclonal to IL-8 (clone G265-5) from BD Pharmingen (San Diego, CA, USA) for ELISA. Pam3CSK4 and SYTOX™ Green were purchased from InvivoGen (San Diego, CA, USA) and ThermoFisher Scientific (Waltham, MA, USA), respectively. 7-Amino-4-methylcoumarin (AMC) and normal goat serum were from MP Biomedicals (Solon, OH, USA). PMA, fMLF, Cl-amidine, and diphenyleneiodonium chloride (DPI) were from Sigma (St. Louis, MO, USA). NADPH oxidase (Nox) 2 inhibitor (GSK2795039) was from MedChemExpress (Monmouth Junction, NJ, USA). Additional reagents were obtained from Fisher Scientific (Pittsburgh, PA, USA). All buffers and reagents were strictly endotoxin-free.
Human PMN purification
Human PMN were isolated according to standard techniques from acid citrate dextrose anti-coagulated venous blood from healthy consenting adults of known TLR1 genotype following written informed consent and in accordance with a protocol approved by the Institutional Review Board for Human Subjects at the University of Texas Southwestern Medical Center. PMN were isolated using Hypaque-Ficoll density-gradient separation and dextran sedimentation followed by hypotonic lysis of erythrocytes as previously described (39, 40). Neutrophil purity is routinely monitored via cytospin and found to be greater than 98%.
MRSA-Cell Membrane Preparations
USA300 MRSA strain LAC (AH1263) was used as the wild-type bacteria in these studies (41). The lgt mutant was previously constructed in the USA300 genetic background and characterized (42). The lgt mutant lacks the gene encoding the diacylglyceryl transferase enzyme and, therefore, is devoid of palmitoylated lipoproteins and serves as a control for lipoprotein specificity (38). Lipoprotein containing membranes were purified as previously reported (43). Briefly, USA300 MRSA was grown 24hr in rich media, and cells were harvested and washed in a high sucrose buffer. Protoplasts were generated by treatment with lysostaphin, lysed by sonication, and insoluble material was removed. Membranes containing all the lipoproteins were pelleted by ultracentrifugation, washed, and resuspended, and the total protein content was determined by Bradford. The same process was used to purify and quantify membranes from the lgt mutant.
Neutrophil Extracellular Trap Formation
Sytox Green:
1e5 freshly isolated PMN were incubated in a 96-well plate with 5μM Sytox Green and specified stimuli and inhibitors in HBSS containing 0.1% glucose and 1% human serum albumin. The CLARIOstar from BMG Labtech (Cary, NC, USA) was used to measure fluorescence at an excitation of 491 +/− 15nm and emission of 535 +/− 20nm with readings every 5 minutes. In priming experiments, 1μM fMLF or 1nM PMA were added following 30 minutes incubation with the specified stimulus. Kinetic and endpoint readings are expressed as relative fluorescence units (RFU). For each assay, the timing of endpoint readings were selected by evaluation of the kinetic curves and timed prior to the plateau and before the fluorescent substrate might be limited or fully consumed.
Citrullinated H3 (H3CIT)-elastase complexes:
Freshly isolated PMN were incubated with specified stimuli at a PMN concentration of 5e6/ml in RPMI + 10% autologous human serum for 4 hours tumbling in a 37°C/5% CO2 incubator. In addition, a control T0 sample was collected by adding freshly isolated PMN to RPMI + 10% autologous serum and placing immediately on ice. Conditioned media and PMN pellets were collected and stored at −80°C. The 4 hour timepoint was chosen based on our previous experience with cytokine production in neutrophils and the decreased sensitivity of the H3CIT-elastase ELISA. A 96-well NUNC MaxiSorp microplate from ThermoFisher Scientific (Rochester, NY, USA) was coated with anti-histone H3 (citrulline R2+R8+R17) and incubated overnight on a rotator at RT. Following washes (0.05% Tween 20 in PBS) and blocking (1% BSA and 5% sucrose in PBS), samples were added in duplicate wells and allowed to incubate for 2 hours on a rotator at RT. Biotinylated elastase antibody followed by streptavidin-HRP was used to detect the H3CIT-elastase complexes. Wells were washed between steps. The CLARIOstar was used to measure absorbance at 450nm. Results are expressed as the blank corrected OD relative to unstimulated PMN.
Elastase activity
1e5 freshly isolated PMN were incubated in a 96-well plate with 20μM AMC and specified stimuli in PICM-G Buffer (10mM sodium phosphate buffer with 2.7mM KCl, 138mM NaCl, 0.6mM CaCl2, 1.0mM MgCl2 and 0.1% dextrose). The CLARIOstar was used to measure fluorescence at an excitation of 360 +/− 20nm and emission of 450 +/− 30nm with readings every 5 minutes. Kinetic readings are expressed as RFU. Endpoint readings are expressed as the fold change in RFU relative to control, with timing of endpoint readings directed by evaluation of the curves, and prior to full consumption of the substrate.
IL-8 intracellular flow and ELISA
Conditioned media and neutrophil pellets were collected as described above and stored at −80°C. For intracellular flow, pellets were thawed and processed according to the two-step protocol for intracellular proteins developed by Thermo Fisher Scientific (Waltham, MA, USA) and as previously described.(44) Twenty thousand events were acquired on a FACSCalibur from BD Biosciences (San Jose, CA, USA) and analyzed using FlowJo version 9.9.6 from Tree Star (Ashland, OR, USA). We gated on the neutrophil population by forward versus side scatter to exclude debris (Figure S1). Twenty thousand events were collected for analysis. IL-8 ELISA was performed as previously described.(44)
Statistical analysis
Data for each individual experiment are expressed as well as the mean ± standard error of the mean. Statistical analysis was performed using GraphPad Prism 9 for Windows from GraphPad Software (La Jolla, CA, USA). Comparisons between groups were performed using one-way ANOVA with multiple comparison or Student’s t test. Results were considered statistically significant with a p value less than 0.05, and relative p values are noted in the figure legends.
Results
Staphylococcal lipoproteins trigger NET formation
Intact S. aureus-elicited generation of NETs has been well described as a component of host defense (16, 20). However, the NET formation potential of staphylococcal products acting as soluble or non-particulate stimuli has been minimally characterized. Given our interest in TLR2 biology (26, 44) and the importance of MRSA as a sepsis pathogen, we analyzed NET formation by freshly isolated human PMNs in response to MRSA-CMP. MRSA-CMP elicited NET formation in a concentration-dependent fashion, and the kinetics of NET release were different than seen in response to the phorbol ester, PMA, with more rapid onset but prolonged time to peak NET generation. MRSA-CMP lacking mature lipoproteins, an lgt mutant (42), did not elicit NET formation demonstrating that the response was mediated by lipoprotein interactions with PMN (Figure 1A-B). Recognizing the non-specific nature of the Sytox Green assay, we sought to confirm our findings using a more specific assay of NET formation. We adapted an ELISA technique that has been used to measure complexes of cell free DNA and neutrophil proteases in plasma (45, 46) and conditioned media (47, 48). Our H3CIT-elastase complex ELISA is specific for PAD-dependent NET fragments released from the cell. Conditioned media from PMN stimulated with PMA or ionomycin were used as the negative and positive controls, respectively. In contrast to ionomycin-elicited NET formation, PMA-elicited NET formation does not involve citrullination of H3 (19, 49, 50). Consistent with our results using the Sytox Green assay, MRSA-CMP elicited NET formation in a concentration-dependent fashion and required lipoproteins (Figure 1C).
Figure 1. MRSA-CMP elicits NET formation and requires lipoproteins.
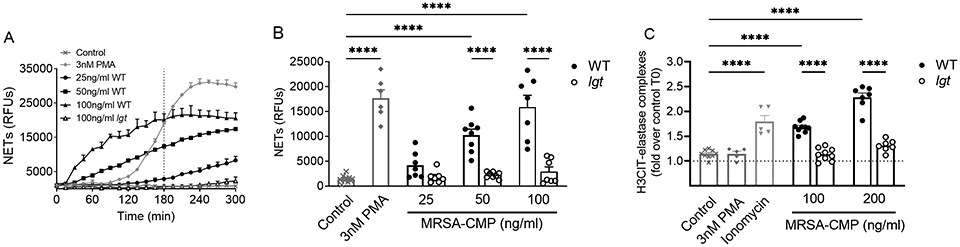
(A) Representative Sytox Green kinetic assay. The dotted vertical line indicates where endpoint readings were taken for comparisons between conditions. (B) 3hr endpoint readings from multiple donors and Sytox Green experiments demonstrating that wild type (WT) MRSA-CMP-elicited NET formation is concentration-dependent. MRSA-CMP lacking all palmitoylated lipoproteins (lgt) does not elicit NET formation. n = 6-10. (C) H3CIT-elastase complexes from PMN conditioned media following 4 hours of stimulation with specified agonists. Data are expressed as the fold change in optical density between conditioned media from PMN stimulated for 4 hours and unstimulated (control) PMN at 0 hours. As expected, 4μM ionomycin elicits release of H3CIT-elastase complexes, whereas 3nM PMA does not. n = 5-9. Each dot represents a unique donor. **** p < 0.0001.
MRSA lipoproteins elicit regulated elastase secretion.
Many assays of NET formation use elastase detection as a surrogate for NET formation. While elastase is one component present in NETs, it is important to note that regulated elastase release also occurs as a distinct neutrophil functional output that may occur independent of NET generation. We measured the activity of extracellular elastase released in response to PMN incubation with MRSA-CMP from WT or the lgt mutant. Elastase activity was stimulated by MRSA-CMP, and was concentration- and lipoprotein-dependent (Figure 2). Importantly, in contrast to NET formation (Figure 1A-B), 3nM PMA does not elicit elastase release (Figure 2A-B).
Figure 2. MRSA-CMP elicits elastase release and requires lipoproteins.
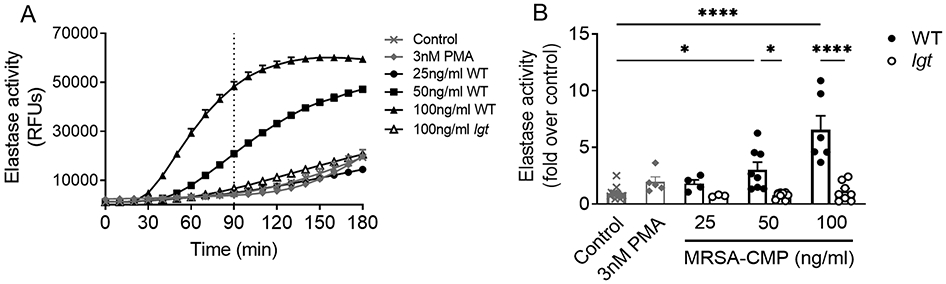
(A) Representative elastase activity assay. The dotted vertical line indicates where endpoint readings were taken for comparisons between conditions. (B) 90min endpoint readings from multiple donors and elastase activity experiments demonstrating that wild type (WT) MRSA-CMP-elicited elastase release is concentration-dependent. MRSA-CMP lacking all palmitoylated lipoproteins (lgt) does not elicit elastase release. In contrast to NET formation, 3nM PMA does not elicit elastase release. Data are presented as the fold change in fluorescence between stimulated and unstimulated (control) PMN at 90min. n = 4-13. Each dot represents a unique donor. * p < 0.05, **** p < 0.0001.
MRSA-CMP elicited NET generation, but not elastase release, involves TLR2/1.
S. aureus expresses both di- and tri-acylated lipoproteins and activates cells via TLR2. Using PMNs from donors with (1805T) or without (1805G) a common SNP in TLR1, we sought to examine TLR2/1 signaling specificity. We found that NET formation in response to MRSA-CMP was significantly reduced in PMNs from 1805G donors (Figure 3A-B), confirming a role for TLR2/1 in MRSA-CMP-elicited PMN activation. As expected, there were no differences in NET generation in response to PMA in 1805G versus T PMN. Pam3CSK4 did not directly elicit NET formation in PMN from either 1805G or 1805T donors (Figure 3A). In contrast, there was no difference in MRSA-CMP-elicited elastase release when comparing the response from 1805G versus 1805T PMN (Figure 3C), providing evidence for distinct mechanisms for NET generation versus elastase release in response to this agonist. Taken together, our data suggest that MRSA-CMP-elicited NET formation is partially TLR2/1-dependent and occurs via a process that is distinct from elastase release.
Figure 3. NET formation elicited by MRSA-CMP is partially TLR2/1 dependent, whereas elastase release is not.
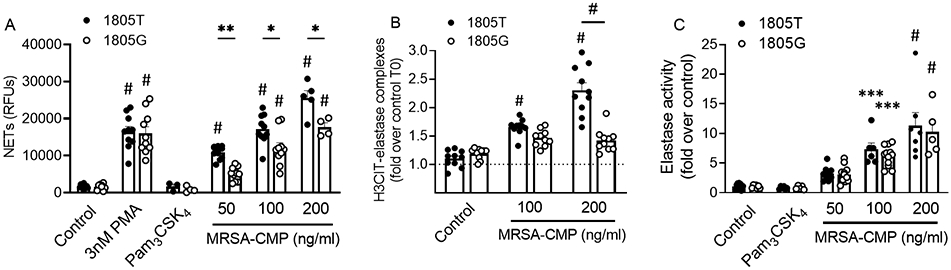
(A) 3hr endpoint readings from multiple donors and Sytox Green experiments demonstrating that PMNs from 1805G donors display reduced MRSA-CMP-elicited NET formation as compared to PMNs from 1805T donors. As expected, there is no difference in NET formation elicited by 3nM PMA between the two genotypes. 1μg/ml Pam3CSK4 fails to elicit NET formation in either genotype. (B) H3CIT-elastase complexes from PMN conditioned media following 4 hours of stimulation with specified agonists. Data are expressed as the fold change in optical density between conditioned media from PMN stimulated for 4 hours and unstimulated (control) PMN at 0 hours. n = 10. (C) 90min endpoint readings from multiple donors and elastase activity experiments demonstrating no difference in MRSA-CMP-elicited elastase release between the two genotypes. 1μg/ml Pam3CSK4 fails to elicit elastase release in either genotype. Data are presented as the fold change in fluorescence between stimulated and unstimulated (control) PMN at 90min. n = 5-12. Each dot represents a unique donor. * p < 0.05, ** p < 0.01, *** p < 0.001, # p < 0.0001 as compared to control unless otherwise indicated.
NET formation can be primed by exposure to MRSA-CMP, but not Pam3CSK4
Neutrophil priming or pre-activation is a critical component of the host immune response in vivo and has been demonstrated in response to numerous TLR ligands in vitro (26, 51-54). Based on our previous studies of neutrophil priming via TLR2, we sought to determine whether NET formation could be primed by MRSA-CMP. Priming of NET formation is highly relevant to the in vivo situation where circulating PMNs might first come in contact with a bacterial product in the circulation, and subsequently reach the site of infection where NETs would be needed for pathogen capture and killing. To detect priming, we employed submaximal concentrations of the agonists PMA and fMLF (1nM and 1μM, respectively) that do not elicit NET formation directly (Figure 4A-B). Importantly, we found that prior exposure to MRSA-CMP, but not to Pam3CSK4, for 30 minutes, primed NET formation in response to both PMA and fMLF (Figure 4C-D). MRSA-CMP-elicited priming of NET formation is lipoprotein dependent as there is minimal output in response to the lgt mutant (Figure 4D).
Figure 4. MRSA-CMP primes PMN for enhanced NET formation in response to a secondary stimulus.
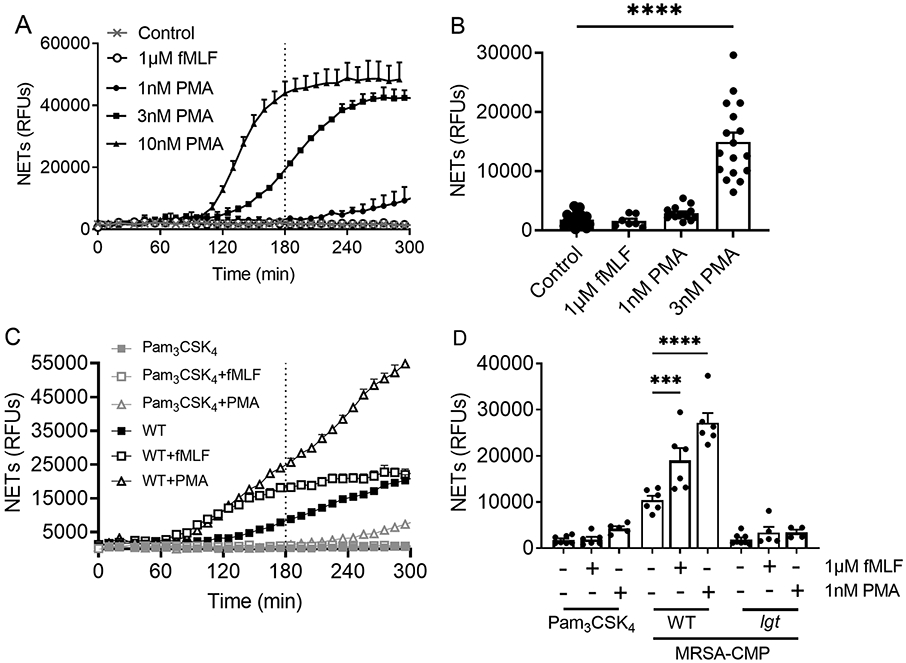
(A) Representative Sytox Green experiment showing concentration-dependent NET formation in response to PMA. The dotted vertical line indicates where endpoint readings were taken for comparisons between conditions. (B) 3hr endpoint readings from multiple donors and Sytox Green experiments demonstrating that 1μM fMLF and 1nM PMA do not elicit NET formation directly. n = 7-21. (C) Representative Sytox Green experiment. (D) PMN were incubated with 1μg/ml Pam3CSK4, 50ng/ml wild type (WT) or 50ng/ml lgt MRSA-CMP for 30 minutes followed by addition of either 1μM fMLF or 1nM PMA, as indicated. 3hr endpoint readings are shown. n = 4-8. Each dot represents a unique donor. *** p < 0.001, **** p < 0.0001.
MRSA-CMP stimulates neutrophil IL-8 production via TLR2/1 and requires lipoproteins
PMN are the most abundant circulating leukocyte, and PMN produce IL-8 in response to numerous stimuli (26, 44, 55). Given our findings that MRSA-CMP elicits NET formation and protease release via distinct mechanisms, we investigated IL-8 generation in response to MRSA-CMP in our healthy donors. Ex vivo stimulation of PMN with MRSA-CMP elicited significant generation of IL-8 as measured both by intracellular flow (Figure 5A-B), demonstrating neutrophil specific IL-8 production, and ELISA (Figure 5C). IL-8 production was lipoprotein-dependent as demonstrated by the lack of production in neutrophils stimulated with the lgt mutant. In addition, as seen with NET formation, IL-8 production in response to MRSA-CMP involves signaling via TLR2/1 as demonstrated by the diminished production of IL-8 in 1805G neutrophils. Importantly, there was no difference in IL-8 production in 1805G and 1805T neutrophils stimulated with TNF-α, as expected (Figure 5B).
Figure 5. MRSA-CMP elicits IL-8 production and release via TLR2/1.
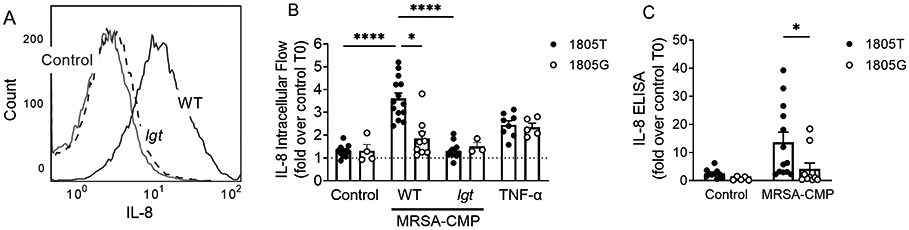
(A) Representative IL-8 intracellular flow histogram showing unstimulated (control) PMN and PMN stimulated with 50ng/ml wild type (WT) or lgt MRSA-CMP for 4 hours. (B) IL-8 intracellular flow following 4 hours of stimulation with no agonist, 50ng/ml WT or lgt MRSA-CMP, or 1ng/ml TNF-α, as specified. 1805T PMN display enhanced IL-8 as compared with 1805G, whereas there are no differences in 1805T vs. G IL-8 abundance in response to TNF-α, as expected. n = 3-14. Data are presented as the fold change in geometric mean intensity between PMN stimulated for 4 hours and unstimulated (control) PMN at 0 hours. (C) IL-8 ELISA of PMN conditioned media following 4 hours of stimulation with no agonist or 50ng/ml WT MRSA-CMP. Data are expressed as the fold change in optical density between conditioned media from PMN stimulated for 4 hours and unstimulated (control) PMN at 0 hours. n = 5-13. Each dot represents a unique donor. * p < 0.05, **** p < 0.0001.
Staphylococcus elicited NETs are Nox2-independent and PAD-dependent
Distinct subtypes and the mechanisms driving NET generation are not fully understood (19). Thus, we sought to elucidate the underlying mechanisms for NET formation in response to MRSA-CMP. In agreement with published data, we found that PMA-elicited NET formation is ROS-dependent, and blocked by both the flavocytochrome inhibitor, DPI (95.63+/−1.1% reduction), and the more specific Nox2 inhibitor, GSK2795039 (90.13+/−1.5% reduction). In contrast, NETs formed in response to MRSA-CMP are not repressed by Nox2 inhibition (Figure 6A-B). Citrullination of histone H3 by PAD4 is a common feature of NET formation in response to numerous stimuli, including ionomycin (19, 49, 56). The PAD inhibitor, Cl-amidine, blocked ionomycin-elicited NET formation (65.57+/−4.1% reduction). Consistent with our H3CIT-elastase complex findings (Figure 1C), inhibition of PAD resulted in reduced NET formation in MRSA-CMP stimulated PMN (Figure 6C-D). In addition, inhibition of PAD significantly diminished NET formation by 1805T PMN (43.4+/−9.0% reduction) as compared with 1805G PMN (9.0+/−6.9% reduction) suggesting that PAD is a required downstream signaling intermediate for the TLR2/1-dependent component of NET formation by MRSA-CMP (Figure 6C-D).
Figure 6. MRSA-CMP elicited NET formation does not require Nox2 and is partially PAD-dependent.
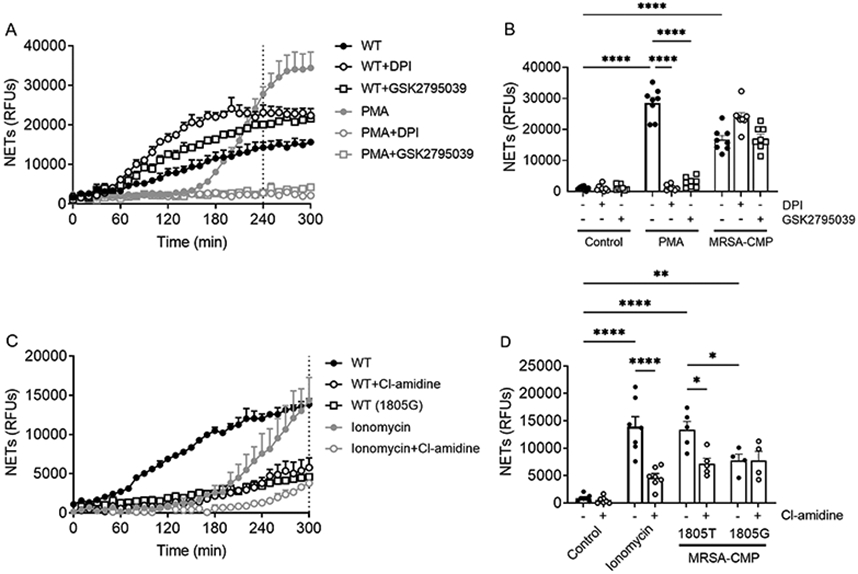
(A) Representative Sytox Green assay. The dotted vertical line indicates where endpoint readings were taken for comparisons between conditions. (B) 4hr endpoint readings from multiple donors and Sytox Green experiments demonstrating that 10μM DPI or 100μM GSK2795039 does not inhibit NET formation elicited by 50ng/ml WT MRSA, whereas PMA-elicited NET formation is inhibited, as expected. n = 5-8. (C) Representative Sytox Green assay. The dotted vertical line indicates where endpoint readings were taken for comparisons between conditions. (D) 5hr endpoint readings from multiple donors and Sytox Green experiments demonstrating that 250μM Cl-amidine inhibits MRSA-CMP-elicited NET formation in 1805T, but not 1805G PMN. Ionomycin (4μM)-elicited NET formation is inhibited by Cl-amidine, as expected. n = 4-7. Each dot represents a unique donor. * p < 0.05, ** p < 0.01, **** p < 0.0001.
Discussion
The ideal host immune response to infection eradicates the pathogen without any damage to the host. Neutrophil-based host defense is essential during infection with Staphylococcus aureus, but the arsenal of anti-microbials produced by the neutrophil are clearly implicated in tissue injury. Given the capacity of this pathogen to develop new mechanisms of resistance to anti-microbials (2, 57, 58), as well as evasion of other targeted immune defenses (57, 59, 60), it is critically important to elucidate the mechanisms of host-pathogen interaction. In this study, we examined the role of MRSA lipoproteins as soluble stimuli for neutrophil activation with a focus on the development of NETs, and the role of TLR2.
Herein we present evidence for several novel findings of relevance to our understanding of MRSA pathogenesis. First, MRSA-CMP elicits generation of NETs in a time and concentration-dependent manner, and this NET formation requires MRSA lipoproteins. This process is partially dependent on an interaction between lipoproteins and the TLR2/1 heterodimer. Second, MRSA-CMP stimulates additional inflammatory outputs from neutrophils including regulated elastase release and generation and secretion of IL-8 in a concentration and lipoprotein-dependent manner. Regulated elastase release is distinct from NET formation and independent of TLR2/1 signaling while IL-8 generation involves TLR2/1 signaling. And finally, the NET formation driven by MRSA-CMP is not dependent on NADPH oxidase-derived ROS, but does involve the PAD enzymes, downstream of TLR2/1.
Although S. aureus elicited neutrophil activation has been extensively studied in the context of phagocytosis, the literature on PMN activation by shedding of soluble components is more limited. There has been some speculation that the TLR2-dependent phase of the neutrophil response follows whole bacteria engulfment and digestion with release of bacterial components to stimulate PRRs (33). Individual lipoproteins have been specifically characterized as TLR2 ligands (36), including a specific role for SitC in the induction of IL-6 and TNF-α by monocytes via TLR2 (61), and the subclass of lipoprotein-like lipoproteins in epithelial cell invasiveness (62). Although it is recognized that lipoproteins control pathogenicity in Gram-positive bacteria, there is very little known about specific lipoprotein interactions with receptors on human PMN.
In S. aureus, lipoprotein biosynthesis and processing requires several enzymes. Diacylglyceryl transferase (Lgt) catalyzes the modification of preprolipoprotein to prolipoprotein, a critical initial step (63). Thus, the lgt mutant lacks lipoproteins and elicited minimal to no response in human neutrophils in our studies. A unique two-component acyl transferase system was recently reported in S. aureus, LnsA/LnsB, that catalyzes the conversion of diacyl to triacyl lipoproteins (64), creating a tool for future dissection of these TLR2-driven pathways. Differences in the lipid moieties on the lipoproteins of unique staphylococcal species control virulence via TLR2 responsiveness (65). Identification of specific MRSA lipoproteins involved in the TLR2/1-driven activation of neutrophils is an area of current investigation in our laboratories.
Additional information about the relative contribution of the unique TLR2 heterodimers can be gleaned from our analyses using PMN from donors with a common SNP in TLR1 (rs5743618, 1805G>T). According to the National Center for Biotechnology Information Allele Frequency Aggregator, the global alternative allele frequency for this SNP is 40.4% with a range of 30%-100% depending on the population (66). 81% of the donors used for this study expressed a T allele. This SNP impacts TLR1 trafficking resulting in greater cell surface expression (67) and enhanced inflammatory potential in neutrophils from individuals heterozygous or homozygous for the T allele (1805T) (26). NET formation in response to MRSA-CMP was significantly abrogated in 1805G PMNs, although these PMNs still displayed a response to MRSA-CMP suggesting involvement of additional receptors, as might be expected using a complex bacterial agonist as a stimulus. The data demonstrating that PAD inhibition significantly reduces MRSA-CMP derived NETs in 1805T PMNs, but has minimal impact on 1805G PMNs highlights the co-existence of unique pathways of NET generation within a given cell type. This heterogeneity in NET formation pathways has been increasingly recognized including the existence of both NET generation as a pathway to PMN cell death and the alternative mechanism termed non-lytic NETosis (7). Our demonstration of TLR2/1 dependent and independent, and PAD-dependent and -independent NET generation in response to MRSA-CMP is novel and likely critical to our understanding of inflammatory amplification as neutrophils encounter components of S. aureus during antimicrobial treatment. Moreover, characterization of co-existing pathways affords the opportunity to seek mechanisms to suppress a subset of NETs in any given infection, without complete loss of neutrophil defense.
The ability of MRSA-CMP to prime NET formation has significant clinical implications and may partially explain the adverse outcomes in sepsis patients with the 1805G>T SNP in TLR1. The TLR1 SNP, rs5743618, has been associated with increased length of stay in pediatric sepsis patients (26) and increased risk of circulatory dysfunction (68) and mortality (69) in adult patients with sepsis. There is limited evidence of priming of NET formation to date (70), and no studies using soluble components of MRSA. Pam3CSK4 did not elicit priming of NET formation suggesting that MRSA-CMP ligation of TLR2/1 results in differential signaling. This is not surprising based on our previous work with lipoarabinomannan from Mycobacterium tuberculosis, and studies with bacterial lipoproteins from Francisella tularensis highlighting the need for further investigation of signaling in human neutrophils using more complex stimuli. Lipoarabinomannan from M. tuberculosis, in contrast to the synthetic Pam3CSK4, does not prime neutrophils for ROS production or elastase release and does not elicit shedding of L-selectin or upregulation of CD11b, but does trigger synthesis and release of cytokines in a TLR2/1-dependent manner (44) that are likely to be involved in amplification of inflammation. Apoptosis is significantly inhibited in PMN from donors with the 1805G>T SNP in TLR1 upon stimulation with bacterial lipoproteins from Francisella tularensis (71). MRSA-CMP is a complex stimulus with the potential to engage numerous surface receptors on PMN. Our data suggest that there is a specific role for tri-acylated lipoproteins in NET formation, and the low level NET generation in 1805G PMN suggests involvement of additional receptors. Additional studies with purified lipoproteins are in progress, and are necessary to further dissect signaling pathways.
Several relevant inhibitors of NETs or NET products are already under investigation in murine models and clinical trials (8, 13). PAD4 inhibition or deficiency improves organ injury and outcomes in rodent models of endotoxin or bleomycin-induced lung injury, ischemic-reperfusion kidney injury and cardiovascular dysfunction after myocardial infarction (72-75). And there is ongoing discovery related to potential reversible inhibitors that might be employed in human trials (76). As mentioned, these enzymes catalyze the post-translational modification of citrullination of histones which is implicated in the early chromatin decondensation, but it is unclear if this event is sufficient to generate NETs (56). It is critical to recognize that these citrullinated histones serve as danger-associate molecular patterns, further amplifying local cell activation (77-79).
The role of elastase and other neutrophil proteases in tissue damage are also unequivocal, with many murine models describing improved outcome, particularly from acute lung injury, in the setting of elastase inhibition or deficiency (80, 81). In human studies, elastase has also been implicated in sepsis associated lung injury (82-84), although these studies cannot distinguish whether this was deposited in the setting of NET formation. Our data demonstrate that MRSA-CMP elicits both regulated elastase release and NETs. There have been a number of human clinical trials using elastase inhibitors to suppress acute lung injury in acute respiratory distress syndrome, sepsis, and after cardiopulmonary bypass with mixed results (85-88). The negative outcome of these trials may reflect the heterogeneity of pathways leading to elastase deposition in the airways, and the need for more targeted repression.
In summary, our understanding of host defense and inflammatory complication management strategies in the setting of sepsis continues to evolve as we probe mechanisms of host-pathogen interactions using physiologically relevant ligands and primary human cells for our investigations. Recognition of the breadth of pathways culminating in NET formation, and elucidation of distinct pathogen and protein-specific mechanisms will be required to facilitate safe and effective therapeutic targeting. The early successes with suppression of NET contents in autoimmunity can be tailored to improve outcomes in sepsis.
Supplementary Material
Key points:
S. aureus lipoproteins drive NET formation and involve TLR2/1 and PAD enzymes.
NET formation can be primed by lipoprotein-containing S. aureus cell membrane.
Acknowledgments
A.R.H. was supported by National Institute of Allergy and Infectious Diseases Grant Number AI141490 and merit award I01 BX002711 from the U.S. Department of Veteran Affairs.
Glossary
- H3CIT
Citrullinated H3
- Lgt
Diacylglyceryl transferase
- MRSA
Methicillin-resistant S. aureus
- MRSA-CMP
MRSA cell membrane preparation
- NET
Neutrophil extracellular trap
- Nox
NADPH oxidase
- PAD
Peptidyl arginine deiminase
- PMN
Polymorphonuclear leukocyte
- RFU
Relative fluorescence units
References
- 1.Carrel M, Perencevich EN, and David MZ. 2015. USA300 Methicillin-Resistant Staphylococcus aureus, United States, 2000-2013. Emerg Infect Dis 21: 1973–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chambers HF, and Deleo FR. 2009. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat Rev Microbiol 7: 629–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kourtis AP, Hatfield K, Baggs J, Mu Y, See I, Epson E, Nadle J, Kainer MA, Dumyati G, Petit S, Ray SM, M. a. g. Emerging Infections Program, Ham D, Capers C, Ewing H, Coffin N, McDonald LC, Jernigan J, and Cardo D. 2019. Vital Signs: Epidemiology and Recent Trends in Methicillin-Resistant and in Methicillin-Susceptible Staphylococcus aureus Bloodstream Infections - United States. MMWR Morb Mortal Wkly Rep 68: 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calfee DP 2017. Trends in Community Versus Health Care-Acquired Methicillin-Resistant Staphylococcus aureus Infections. Curr Infect Dis Rep 19: 48. [DOI] [PubMed] [Google Scholar]
- 5.Hassoun A, Linden PK, and Friedman B. 2017. Incidence, prevalence, and management of MRSA bacteremia across patient populations-a review of recent developments in MRSA management and treatment. Crit Care 21: 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park B, and Liu GY. 2021. Immune-Based Anti-Staphylococcal Therapeutic Approaches. Microorganisms 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Papayannopoulos V. 2018. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol 18: 134–147. [DOI] [PubMed] [Google Scholar]
- 8.Mutua V, and Gershwin LJ. 2020. A Review of Neutrophil Extracellular Traps (NETs) in Disease: Potential Anti-NETs Therapeutics. Clin Rev Allergy Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Castanheira FVS, and Kubes P. 2019. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 133: 2178–2185. [DOI] [PubMed] [Google Scholar]
- 10.Lee KH, Kronbichler A, Park DD, Park Y, Moon H, Kim H, Choi JH, Choi Y, Shim S, Lyu IS, Yun BH, Han Y, Lee D, Lee SY, Yoo BH, Lee KH, Kim TL, Kim H, Shim JS, Nam W, So H, Choi S, Lee S, and Shin JI. 2017. Neutrophil extracellular traps (NETs) in autoimmune diseases: A comprehensive review. Autoimmun Rev 16: 1160–1173. [DOI] [PubMed] [Google Scholar]
- 11.Jorch SK, and Kubes P. 2017. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med 23: 279–287. [DOI] [PubMed] [Google Scholar]
- 12.Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, Friday S, Li S, Patel RM, Subramanian V, Thompson P, Chen P, Fox DA, Pennathur S, and Kaplan MJ. 2013. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med 5: 178ra140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Masucci MT, Minopoli M, Del Vecchio S, and Carriero MV. 2020. The Emerging Role of Neutrophil Extracellular Traps (NETs) in Tumor Progression and Metastasis. Front Immunol 11: 1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, Herrmann M, Voll RE, and Zychlinsky A. 2010. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A 107: 9813–9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Bont CM, Stokman MEM, Faas P, Thurlings RM, Boelens WC, Wright HL, and Pruijn GJM. 2020. Autoantibodies to neutrophil extracellular traps represent a potential serological biomarker in rheumatoid arthritis. J Autoimmun 113: 102484. [DOI] [PubMed] [Google Scholar]
- 16.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, and Zychlinsky A. 2004. Neutrophil extracellular traps kill bacteria. Science 303: 1532–1535. [DOI] [PubMed] [Google Scholar]
- 17.Saffarzadeh M, Cabrera-Fuentes HA, Veit F, Jiang D, Scharffetter-Kochanek K, Gille CG, Rooijakkers SHM, Hartl D, and Preissner KT. 2014. Characterization of rapid neutrophil extracellular trap formation and its cooperation with phagocytosis in human neutrophils. Discoveries (Craiova) 2: e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Twaddell SH, Baines KJ, Grainge C, and Gibson PG. 2019. The Emerging Role of Neutrophil Extracellular Traps in Respiratory Disease. Chest 156: 774–782. [DOI] [PubMed] [Google Scholar]
- 19.Boeltz S, Amini P, Anders HJ, Andrade F, Bilyy R, Chatfield S, Cichon I, Clancy DM, Desai J, Dumych T, Dwivedi N, Gordon RA, Hahn J, Hidalgo A, Hoffmann MH, Kaplan MJ, Knight JS, Kolaczkowska E, Kubes P, Leppkes M, Manfredi AA, Martin SJ, Maueroder C, Maugeri N, Mitroulis I, Munoz LE, Nakazawa D, Neeli I, Nizet V, Pieterse E, Radic MZ, Reinwald C, Ritis K, Rovere-Querini P, Santocki M, Schauer C, Schett G, Shlomchik MJ, Simon HU, Skendros P, Stojkov D, Vandenabeele P, Berghe TV, van der Vlag J, Vitkov L, von Kockritz-Blickwede M, Yousefi S, Zarbock A, and Herrmann M. 2019. To NET or not to NET:current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ 26: 395–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yipp BG, and Kubes P. 2013. NETosis: how vital is it? Blood 122: 2784–2794. [DOI] [PubMed] [Google Scholar]
- 21.Song W, Ye J, Pan N, Tan C, and Herrmann M. 2020. Neutrophil Extracellular Traps Tied to Rheumatoid Arthritis: Points to Ponder. Front Immunol 11: 578129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pratesi F, Dioni I, Tommasi C, Alcaro MC, Paolini I, Barbetti F, Boscaro F, Panza F, Puxeddu I, Rovero P, and Migliorini P. 2014. Antibodies from patients with rheumatoid arthritis target citrullinated histone 4 contained in neutrophils extracellular traps. Ann Rheum Dis 73: 1414–1422. [DOI] [PubMed] [Google Scholar]
- 23.Hayashi F, Means TK, and Luster AD. 2003. Toll-like receptors stimulate human neutrophil function. Blood 102: 2660–2669. [DOI] [PubMed] [Google Scholar]
- 24.Mukherjee S, Huda S, and Sinha Babu SP. 2019. Toll-like receptor polymorphism in host immune response to infectious diseases: A review. Scand J Immunol 90: e12771. [DOI] [PubMed] [Google Scholar]
- 25.Kawai T, and Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11: 373–384. [DOI] [PubMed] [Google Scholar]
- 26.Whitmore LC, Hook JS, Philiph AR, Hilkin BM, Bing X, Ahn C, Wong HR, Ferguson PJ, and Moreland JG. 2016. A Common Genetic Variant in TLR1 Enhances Human Neutrophil Priming and Impacts Length of Intensive Care Stay in Pediatric Sepsis. J Immunol 196: 1376–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwok AJ, Mentzer A, and Knight JC. 2021. Host genetics and infectious disease: new tools, insights and translational opportunities. Nat Rev Genet 22: 137–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Casanova JL 2015. Human genetic basis of interindividual variability in the course of infection. Proc Natl Acad Sci U S A 112: E7118–7127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Basu J, Shin DM, and Jo EK. 2012. Mycobacterial signaling through toll-like receptors. Front Cell Infect Microbiol 2: 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Underhill DM, Ozinsky A, Smith KD, and Aderem A. 1999. Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc Natl Acad Sci U S A 96: 14459–14463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Means TK, Jones BW, Schromm AB, Shurtleff BA, Smith JA, Keane J, Golenbock DT, Vogel SN, and Fenton MJ. 2001. Differential effects of a Toll-like receptor antagonist on Mycobacterium tuberculosis-induced macrophage responses. J Immunol 166: 4074–4082. [DOI] [PubMed] [Google Scholar]
- 32.Krutzik SR, Ochoa MT, Sieling PA, Uematsu S, Ng YW, Legaspi A, Liu PT, Cole ST, Godowski PJ, Maeda Y, Sarno EN, Norgard MV, Brennan PJ, Akira S, Rea TH, and Modlin RL. 2003. Activation and regulation of Toll-like receptors 2 and 1 in human leprosy. Nat Med 9: 525–532. [DOI] [PubMed] [Google Scholar]
- 33.Fournier B 2012. The function of TLR2 during staphylococcal diseases. Front Cell Infect Microbiol 2: 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Graf A, Lewis RJ, Fuchs S, Pagels M, Engelmann S, Riedel K, and Pane-Farre J. 2018. The hidden lipoproteome of Staphylococcus aureus. Int J Med Microbiol 308: 569–581. [DOI] [PubMed] [Google Scholar]
- 35.Nguyen MT, and Gotz F. 2016. Lipoproteins of Gram-Positive Bacteria: Key Players in the Immune Response and Virulence. Microbiol Mol Biol Rev 80: 891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hashimoto M, Tawaratsumida K, Kariya H, Kiyohara A, Suda Y, Krikae F, Kirikae T, and Gotz F. 2006. Not lipoteichoic acid but lipoproteins appear to be the dominant immunobiologically active compounds in Staphylococcus aureus. J Immunol 177: 3162–3169. [DOI] [PubMed] [Google Scholar]
- 37.Schmaler M, Jann NJ, Ferracin F, Landolt LZ, Biswas L, Gotz F, and Landmann R. 2009. Lipoproteins in Staphylococcus aureus mediate inflammation by TLR2 and iron-dependent growth in vivo. J Immunol 182: 7110–7118. [DOI] [PubMed] [Google Scholar]
- 38.Stoll H, Dengjel J, Nerz C, and Gotz F. 2005. Staphylococcus aureus deficient in lipidation of prelipoproteins is attenuated in growth and immune activation. Infect Immun 73: 2411–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nauseef WM 2007. Isolation of human neutrophils from venous blood. Methods in molecular biology 412: 15–20. [DOI] [PubMed] [Google Scholar]
- 40.Kuhns DB, Priel DAL, Chu J, and Zarember KA. 2015. Isolation and Functional Analysis of Human Neutrophils. Curr Protoc Immunol 111: 7 23 21–27 23 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boles BR, Thoendel M, Roth AJ, and Horswill AR. 2010. Identification of genes involved in polysaccharide-independent Staphylococcus aureus biofilm formation. PLoS One 5: e10146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schilcher K, Caesar LK, Cech NB, and Horswill AR. 2020. Processing, Export, and Identification of Novel Linear Peptides from Staphylococcus aureus. mBio 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kavanaugh JS, Thoendel M, and Horswill AR. 2007. A role for type I signal peptidase in Staphylococcus aureus quorum sensing. Mol Microbiol 65: 780–798. [DOI] [PubMed] [Google Scholar]
- 44.Hook JS, Cao M, Weng K, Kinnare N, and Moreland JG. 2020. Mycobacterium tuberculosis Lipoarabinomannan Activates Human Neutrophils via a TLR2/1 Mechanism Distinct from Pam3CSK4. J Immunol 204: 671–681. [DOI] [PubMed] [Google Scholar]
- 45.Zuo Y, Zuo M, Yalavarthi S, Gockman K, Madison JA, Shi H, Woodard W, Lezak SP, Lugogo NL, Knight JS, and Kanthi Y. 2021. Neutrophil extracellular traps and thrombosis in COVID-19. J Thromb Thrombolysis 51: 446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang W, Peng W, and Ning X. 2018. Increased levels of neutrophil extracellular trap remnants in the serum of patients with rheumatoid arthritis. Int J Rheum Dis 21: 415–421. [DOI] [PubMed] [Google Scholar]
- 47.Yoo DG, Floyd M, Winn M, Moskowitz SM, and Rada B. 2014. NET formation induced by Pseudomonas aeruginosa cystic fibrosis isolates measured as release of myeloperoxidase-DNA and neutrophil elastase-DNA complexes. Immunol Lett 160: 186–194. [DOI] [PubMed] [Google Scholar]
- 48.Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, Toy P, Werb Z, and Looney MR. 2012. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest 122: 2661–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kenny EF, Herzig A, Kruger R, Muth A, Mondal S, Thompson PR, Brinkmann V, Bernuth HV, and Zychlinsky A. 2017. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Neeli I, and Radic M. 2013. Opposition between PKC isoforms regulates histone deimination and neutrophil extracellular chromatin release. Front Immunol 4: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moreland JG, Davis AP, Matsuda JJ, Hook JS, Bailey G, Nauseef WM, and Lamb FS. 2007. Endotoxin priming of neutrophils requires NADPH oxidase-generated oxidants and is regulated by the anion transporter ClC-3. J Biol Chem 282: 33958–33967. [DOI] [PubMed] [Google Scholar]
- 52.Lamb FS, Hook JS, Hilkin BM, Huber JN, Volk AP, and Moreland JG. 2012. Endotoxin priming of neutrophils requires endocytosis and NADPH oxidase-dependent endosomal reactive oxygen species. J Biol Chem 287: 12395–12404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Volk AP, Barber BM, Goss KL, Ruff JG, Heise CK, Hook JS, and Moreland JG. 2011. Priming of neutrophils and differentiated PLB-985 cells by pathophysiological concentrations of TNF-alpha is partially oxygen dependent. J Innate Immun 3: 298–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.El-Benna J, Hurtado-Nedelec M, Marzaioli V, Marie JC, Gougerot-Pocidalo MA, and Dang PM. 2016. Priming of the neutrophil respiratory burst: role in host defense and inflammation. Immunol Rev 273: 180–193. [DOI] [PubMed] [Google Scholar]
- 55.Schroder AK, von der Ohe M, Fleischer D, Rink L, and Uciechowski P. 2005. Differential synthesis of two interleukin-1 receptor antagonist variants and interleukin-8 by peripheral blood neutrophils. Cytokine 32: 246–253. [DOI] [PubMed] [Google Scholar]
- 56.de Bont CM, Koopman WJH, Boelens WC, and Pruijn GJM. 2018. Stimulus-dependent chromatin dynamics, citrullination, calcium signalling and ROS production during NET formation. Biochim Biophys Acta Mol Cell Res 1865: 1621–1629. [DOI] [PubMed] [Google Scholar]
- 57.Nasser A, Moradi M, Jazireian P, Safari H, Alizadeh-Sani M, Pourmand MR, and Azimi T. 2019. Staphylococcus aureus versus neutrophil: Scrutiny of ancient combat. Microb Pathog 131: 259–269. [DOI] [PubMed] [Google Scholar]
- 58.Khan A, Wilson B, and Gould IM. 2018. Current and future treatment options for community-associated MRSA infection. Expert Opin Pharmacother 19: 457–470. [DOI] [PubMed] [Google Scholar]
- 59.Jenul C, and Horswill AR. 2019. Regulation of Staphylococcus aureus Virulence. Microbiol Spectr 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thammavongsa V, Kim HK, Missiakas D, and Schneewind O. 2015. Staphylococcal manipulation of host immune responses. Nat Rev Microbiol 13: 529–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Muller P, Muller-Anstett M, Wagener J, Gao Q, Kaesler S, Schaller M, Biedermann T, and Gotz F. 2010. The Staphylococcus aureus lipoprotein SitC colocalizes with Toll-like receptor 2 (TLR2) in murine keratinocytes and elicits intracellular TLR2 accumulation. Infect Immun 78: 4243–4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nguyen MT, Peisl L, Barletta F, Luqman A, and Gotz F. 2018. Toll-Like Receptor 2 and Lipoprotein-Like Lipoproteins Enhance Staphylococcus aureus Invasion in Epithelial Cells. Infect Immun 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nakayama H, Kurokawa K, and Lee BL. 2012. Lipoproteins in bacteria: structures and biosynthetic pathways. FEBS J 279: 4247–4268. [DOI] [PubMed] [Google Scholar]
- 64.Gardiner J. H. t., Komazin G, Matsuo M, Cole K, Gotz F, and Meredith TC. 2020. Lipoprotein N-Acylation in Staphylococcus aureus Is Catalyzed by a Two-Component Acyl Transferase System. mBio 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nguyen MT, Uebele J, Kumari N, Nakayama H, Peter L, Ticha O, Woischnig AK, Schmaler M, Khanna N, Dohmae N, Lee BL, Bekeredjian-Ding I, and Gotz F. 2017. Lipid moieties on lipoproteins of commensal and non-commensal staphylococci induce differential immune responses. Nat Commun 8: 2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Phan L, J. Y, Zhang H, Qiang W, Shekhtman E, Shao D, Revoe D, Villamarin R, Ivanchenko E, Kimura M, Wang ZY, Hao L, Sharopova N, Bihan M, Sturcke A, Lee M, Popova N, Wu W, Bastiani C, Ward M, Holmes JB, Lyoshin V, Kaur K, Moyer E, Feolo M, and Kattman BL. "ALFA: Allele Frequency Aggregator." National Center for Biotechnology Information, U.S. National Library of Medicine, 10 March. 2020, www.ncbi.nlm.nih.gov/snp/docs/gsr/alfa/ [Google Scholar]
- 67.Hart BE, and Tapping RI. 2012. Cell surface trafficking of TLR1 is differentially regulated by the chaperones PRAT4A and PRAT4B. J Biol Chem 287: 16550–16562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pino-Yanes M, Corrales A, Casula M, Blanco J, Muriel A, Espinosa E, Garcia-Bello M, Torres A, Ferrer M, Zavala E, Villar J, Flores C, Grecia, and G.-S. Groups. 2010. Common variants of TLR1 associate with organ dysfunction and sustained pro-inflammatory responses during sepsis. PLoS One 5: e13759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wurfel MM, Gordon AC, Holden TD, Radella F, Strout J, Kajikawa O, Ruzinski JT, Rona G, Black RA, Stratton S, Jarvik GP, Hajjar AM, Nickerson DA, Rieder M, Sevransky J, Maloney JP, Moss M, Martin G, Shanholtz C, Garcia JG, Gao L, Brower R, Barnes KC, Walley KR, Russell JA, and Martin TR. 2008. Toll-like receptor 1 polymorphisms affect innate immune responses and outcomes in sepsis. Am J Respir Crit Care Med 178: 710–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Demers M, Wong SL, Martinod K, Gallant M, Cabral JE, Wang Y, and Wagner DD. 2016. Priming of neutrophils toward NETosis promotes tumor growth. Oncoimmunology 5: e1134073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kinkead LC, Whitmore LC, McCracken JM, Fletcher JR, Ketelsen BB, Kaufman JW, Jones BD, Weiss DS, Barker JH, and Allen LH. 2018. Bacterial lipoproteins and other factors released by Francisella tularensis modulate human neutrophil lifespan: Effects of a TLR1 SNP on apoptosis inhibition. Cell Microbiol 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liang Y, Pan B, Alam HB, Deng Q, Wang Y, Chen E, Liu B, Tian Y, Williams AM, Duan X, Wang Y, Zhang J, and Li Y. 2018. Inhibition of peptidylarginine deiminase alleviates LPS-induced pulmonary dysfunction and improves survival in a mouse model of lethal endotoxemia. Eur J Pharmacol 833: 432–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Du M, Yang W, Schmull S, Gu J, and Xue S. 2020. Inhibition of peptidyl arginine deiminase-4 protects against myocardial infarction induced cardiac dysfunction. Int Immunopharmacol 78: 106055. [DOI] [PubMed] [Google Scholar]
- 74.Suzuki M, Ikari J, Anazawa R, Tanaka N, Katsumata Y, Shimada A, Suzuki E, and Tatsumi K. 2020. PAD4 Deficiency Improves Bleomycin-induced Neutrophil Extracellular Traps and Fibrosis in Mouse Lung. Am J Respir Cell Mol Biol 63: 806–818. [DOI] [PubMed] [Google Scholar]
- 75.Raup-Konsavage WM, Wang Y, Wang WW, Feliers D, Ruan H, and Reeves WB. 2018. Neutrophil peptidyl arginine deiminase-4 has a pivotal role in ischemia/reperfusion-induced acute kidney injury. Kidney Int 93: 365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Aliko A, Kaminska M, Falkowski K, Bielecka E, Benedyk-Machaczka M, Malicki S, Koziel J, Wong A, Bryzek D, Kantyka T, and Mydel P. 2019. Discovery of Novel Potential Reversible Peptidyl Arginine Deiminase Inhibitor. Int J Mol Sci 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Denning NL, Aziz M, Gurien SD, and Wang P. 2019. DAMPs and NETs in Sepsis. Front Immunol 10: 2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cheng Z, Abrams ST, Toh J, Wang SS, Wang Z, Yu Q, Yu W, Toh CH, and Wang G. 2020. The Critical Roles and Mechanisms of Immune Cell Death in Sepsis. Front Immunol 11: 1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tsourouktsoglou TD, Warnatsch A, Ioannou M, Hoving D, Wang Q, and Papayannopoulos V. 2020. Histones, DNA, and Citrullination Promote Neutrophil Extracellular Trap Inflammation by Regulating the Localization and Activation of TLR4. Cell Rep 31: 107602. [DOI] [PubMed] [Google Scholar]
- 80.Sakashita A, Nishimura Y, Nishiuma T, Takenaka K, Kobayashi K, Kotani Y, and Yokoyama M. 2007. Neutrophil elastase inhibitor (sivelestat) attenuates subsequent ventilator-induced lung injury in mice. Eur J Pharmacol 571: 62–71. [DOI] [PubMed] [Google Scholar]
- 81.Li H, Zhou X, Tan H, Hu Y, Zhang L, Liu S, Dai M, Li Y, Li Q, Mao Z, Pan P, Su X, and Hu C. 2018. Neutrophil extracellular traps contribute to the pathogenesis of acid-aspiration-induced ALI/ARDS. Oncotarget 9: 1772–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hayakawa M, Katabami K, Wada T, Sugano M, Hoshino H, Sawamura A, and Gando S. 2010. Sivelestat (selective neutrophil elastase inhibitor) improves the mortality rate of sepsis associated with both acute respiratory distress syndrome and disseminated intravascular coagulation patients. Shock 33: 14–18. [DOI] [PubMed] [Google Scholar]
- 83.Aikawa N, and Kawasaki Y. 2014. Clinical utility of the neutrophil elastase inhibitor sivelestat for the treatment of acute respiratory distress syndrome. Ther Clin Risk Manag 10: 621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zeiher BG, Matsuoka S, Kawabata K, and Repine JE. 2002. Neutrophil elastase and acute lung injury: prospects for sivelestat and other neutrophil elastase inhibitors as therapeutics. Crit Care Med 30: S281–287. [DOI] [PubMed] [Google Scholar]
- 85.Kohira S, Oka N, Inoue N, Itatani K, Hanayama N, Kitamura T, Fujii M, Takeda A, Oshima H, Tojo K, Yoshitake S, and Miyaji K. 2013. Effect of the neutrophil elastase inhibitor sivelestat on perioperative inflammatory response after pediatric heart surgery with cardiopulmonary bypass: a prospective randomized study. Artif Organs 37: 1027–1033. [DOI] [PubMed] [Google Scholar]
- 86.Miyoshi S, Hamada H, Ito R, Katayama H, Irifune K, Suwaki T, Nakanishi N, Kanematsu T, Dote K, Aibiki M, Okura T, and Higaki J. 2013. Usefulness of a selective neutrophil elastase inhibitor, sivelestat, in acute lung injury patients with sepsis. Drug Des Devel Ther 7: 305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Iwata K, Doi A, Ohji G, Oka H, Oba Y, Takimoto K, Igarashi W, Gremillion DH, and Shimada T. 2010. Effect of neutrophil elastase inhibitor (sivelestat sodium) in the treatment of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS): a systematic review and meta-analysis. Intern Med 49: 2423–2432. [DOI] [PubMed] [Google Scholar]
- 88.Zeiher BG, Artigas A, Vincent JL, Dmitrienko A, Jackson K, Thompson BT, Bernard G, and S. S. Group. 2004. Neutrophil elastase inhibition in acute lung injury: results of the STRIVE study. Crit Care Med 32: 1695–1702. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.