

Abstract
Objective:
The MAST family of microtubule-associated serine-threonine kinases (STK) have distinct expression patterns in the developing and mature human and mouse brain. To date, only MAST1 has been conclusively associated with neurological disease, with de novo variants in individuals with a neurodevelopmental disorder, including a mega corpus callosum.
Methods:
Using exome sequencing we identify MAST3 missense variants in individuals with epilepsy. We also assess the effect of these variants on the ability of MAST3 to phosphorylate the target gene product ARPP-16 in HEK293T cells.
Results:
We identify de novo missense variants in the STK domain in 11 individuals, including two recurrent variants p.G510S (n=5) and p.G515S (n=3). All 11 individuals had developmental and epileptic encephalopathy, with 8 having normal development prior to seizure onset at < 2 years of age. All patients developed multiple seizure types, while 9/11 had seizures triggered by fever and 9/11 had drug-resistant seizures. In vitro analysis of HEK293T cells transfected with MAST3 cDNA carrying a subset of these patient-specific missense variants demonstrated variable but generally lower expression, with concomitant increased phosphorylation of the MAST3 target, ARPP-16, compared to wildtype. These findings suggest the patient-specific variants may confer MAST3 gain-of-function. Moreover, single-nuclei RNA sequencing and immunohistochemistry shows that MAST3 expression is restricted to excitatory neurons in the cortex late in prenatal development and postnatally.
Interpretation:
In summary, we describe MAST3 as a novel epilepsy-associated gene with a potential gain-of-function pathogenic mechanism that may be primarily restricted to excitatory neurons in the cortex.
Summary for social media
Twitter Handle: @CarvillLab
MAST3 de novo variants are a novel cause of developmental and epileptic encephalopathies, and act through a putative gain-of-function mechanism in excitatory neurons.
Introduction
Developmental and Epileptic Encephalopathies (DEEs) encompass a group of disorders featuring frequent epileptic activity and developmental impairment. Development plateaus or regresses when seizures are worse but can also occur independent of seizures and are thus attributable to the underlying cause alone1, 2. DEE causes can be varied, but a genetic etiology is known or suspected in most cases without metabolic or structural causes. The highest yield for genetic testing is in the early onset DEEs where up to 80% of patients have a known underlying genetic etiology3. In patients with DEEs, the goal of genetic investigations is not only to end the diagnostic odyssey for families but ideally, to identify a potentially actionable target that could lead to precision medicine therapies.
Despite the high diagnostic rate of patients with DEEs, many patients still lack a genetic diagnosis, likely at least in part to pathogenic variants in undiscovered genes. Here we describe variants in MAST3, a microtubule-associated serine threonine kinase 3. This gene is a member of a family of serine threonine kinases (MAST1–4 and MAST-like) that is predominantly expressed in the human cortex, hippocampus, and striatum5. The role of MAST3 in the brain is not fully understood, but recent studies in adult rodent striatal medium spiny neurons suggest a role in coordination of cAMP/protein kinase A/protein phosphatase 2A signaling pathways that mediate the effects of dopamine6–9
To date, disease-associations of MAST3 variants are rare; an intronic SNP has been associated with inflammatory bowel disease10 and a single individual has been reported with a de novo MAST3 variant and clinical features of atypical MECP2-associated disorder11. However, pathogenic variants in the MAST3 homolog, MAST1, have been identified in individuals with developmental brain abnormalities that include a particularly striking mega-corpus callosum along with cerebellar hypoplasia and cortical malformations. More recently, three additional patients with additional neurodevelopmental disorders with and without malformations of cortical development and MAST1 de novo variants have been reported12–14. Overall, individuals with de novo MAST1 variants present with a spectrum of developmental impairments and a subset have epilepsy 15. The proposed pathogenic mechanism of MAST1 variants is a dominant negative effect, given the reduced abundance of MAST1 protein to around 20% of wildtype as well as a decrease in MAST homologs (MAST2/3) in a mouse model carrying a heterozygous patient-specific variant15.
As part of an international collaboration, we identified 11 individuals from 4 continents with DEEs and de novo variants in the serine/threonine kinase (STK) domain of MAST3. In vitro modeling of a subset of these variants showed a potential gain of function effect on MAST3 kinase activity. We propose MAST3 as a novel cause of DEEs and implicate a second member of the MAST family in neurodevelopmental disorders though with distinct phenotypes, likely related to gene expression timing and/or localization during neurodevelopment.
Materials and Methods
Study Participants
All patients were identified by either clinical or research investigations to determine the underlying etiology of their DEE. Patients were ascertained through personal communication or GeneMatcher16. Exome sequencing was performed by a commercial clinical laboratory in the USA or Europe (Patients 1, 2, 6, 7, 10, 11)17, in research laboratories/in-house sequencing facilities (Patients 3, 4) or by exome sequencing of the proband by the Epi25 Collaborative (Epi25 Collaborative, in press) (Patients 5, 9). The variant of patient 8 was identified by targeted resequencing in a research laboratory using single-molecule molecular inversion probes designed to cover the exons +5bp into the intron18. Variant allele frequencies were assessed using GnomAD and TopMed (see list of URLs). All variants were classified according to the American College of Medical Genetics and Genomics (ACMG) guidelines for variant interpretation19. This study was approved by the local institutional review board at each site and all patients and parental or legal guardians provided consent for this study and publication of clinical and genetic data. Missense Tolerance Ratios20 were plotted with ggplot2, combined annotation dependent depletion (CADD), and polyphen scores were generated by variant effect predictor. Clinical histories were analyzed and seizure types and epilepsy syndromes were classified according to the International League Against Epilepsy classification criteria21
MAST3 expression in the developing human and mouse brain
To determine the expression of MAST3 in the mature and developing human brain we analyzed RNA-seq and single nucleus-RNA-seq data from the Allen Cell Types Database and the Allen Developing Human Brain Database. In addition to RNA expression analysis, we performed immunohistochemistry in 11-week-old human cerebral organoids generated with the STEMdiff cerebral organoid kit (STEMCELL Technologies), and C57BL/6J mouse brains (Jackson laboratory, Stock No:000664) at E14.5 and E16.5. Cerebral organoids and mouse brains were fixed in 4% paraformaldehyde, then cryoprotected in 30% sucrose until sunk. Samples were frozen in an ethanol/dry-ice bath and 10 to 14-micron thick sections were cut with a cryostat (Leica). Sections were permeabilized and blocked for 1-hour at room temperature with 0.3% Triton X-100 and 10% normal donkey serum, respectively, and then overnight at 4ºC with the following primary antibodies: MAST3 (Novus Biologicals, Cat # NBP1–82993), CUX1 (Santa Cruz Biotechnology, sc-514008), SATB2 (Abcam, ab51502), TBR1 (Proteintech Group, 66564–1-Ig) and CTIP2 (Abcam, ab18465). After a 2-hour incubation at room temperature with secondary antibodies (Donkey anti-rabbit AlexaFluor 594, anti-rat AlexaFluor 488, anti-mouse AlexaFluor 647), slides were counterstained with Hoechst 33342 (ThermoFisher Scientific) and mounted with ProLong Gold Antifade (ThermoFisher Scientific). Images were acquired with a Nikon A1R laser scanning confocal microscope.
Analysis of effect of MAST3 variants on phosphorylation of ARPP-16 in HEK293 cells
To assess their kinase activity, MAST3 wild-type and a subset of variants were individually expressed in human embryonic kidney (HEK) 293T cells (ATCC CRL-3216) with the established MAST3 substrate protein ARPP-166, 7. All MAST3 constructs were cloned into the pCMV plasmid backbone and fused to a 6× histidine tag on the C-terminus. MAST3 mutations identified in this study, as well as a kinase-dead control (K396H)22,were generated by single-site mutagenesis (Agilent 210518) according to the manufacturer’s instructions. ARPP-16 was expressed under the control of a CMV promoter and fused to a C-terminus hemagglutinin (HA) tag. HEK 293T cells (300,000/well) were plated in 12-well plates and grown in DMEM (Thermo 11995–065) supplemented with 5% FBS and 1% penicillin/streptomycin in a humidified incubator at 37°C and 5% CO2 one day prior to transfection. Cells were co-transfected with 250–750 ng each of pCMV MAST3 and pCMV ARPP-16 DNA using Lipofectamine 2000 (Invitrogen 52887) according to the manufacturer’s instructions and incubated overnight. A pCMV GFP construct was used in place of MAST3 for the control well expressing only ARPP-16. The next day, cells were collected in lysis buffer (TBS + 1% Triton X-100) containing protease inhibitors (Complete Mini EDTA-Free, Roche 04693159001), briefly sonicated, and centrifuged at 14,000 × g for 5 min at 4°C. Supernatants were transferred to a new tube and protein concentration measured using the bicinchoninic acid (BCA) assay (Pierce 23225). Protein concentrations were adjusted to 1 mg/L, and 15 g was used for analysis by immunoblot. Samples were mixed with Laemmli sample buffer, denatured at 98°C for 5 min, then separated by SDS-PAGE using Criterion 4–20% Tris-HCl gels (BioRad 3450033). Proteins were transferred to nitrocellulose membranes and probed with HA-tag antibody (1:5000, mouse monoclonal, CST 2367S), MAST3 antibody (1:2000, rabbit polyclonal, Bioworld BS5790), or pS46 ARPP-16 antibody (RU1102, rabbit polyclonal, 1:3000)6, 7. Antibodies were diluted in 2% non-fat dry milk in TBS and incubated overnight at 4°C. The next day, blots were incubated for 1 hour at room temperature with IRDye 680RD goat-anti-rabbit (1:20,000, LiCor Biosciences 926–68071) and IRDye 800CW goat-anti-mouse (1:20,000, LiCor Biosciences 926–32210). Blots were imaged using a LiCor Odyssey system. Band signal intensities were analyzed using ImageJ software and the pS46 ARPP-16 band intensities were normalized to the MAST3 band intensities.
Results
Genetic characterization of individuals with MAST3 variants
Twelve individuals with variants in MAST3 were identified; 3 females and 9 males with a median age of 10.5 years (range 7 to 44 years). 11 individuals carried variants in the Serine Threonine kinase (STK) domain (Table 1, Table S1 and S2, Fig 1A) and one outside this domain. In this study we focus on the variants in the STK domain, all of whom exhibit DEE as described below. The twelfth individual is a 3 year-old boy with a diagnosis of Autism Spectrum Disorder (ASD), but with no history of seizures. He has a similarly affected brother. We identified a de novo c.1963T>C, p.F655L variant (CADD = 24.3 and polyphen = 0.604) in the proband only. Language development was normal until a regression at 15–18 months leading to both receptive and expressive language delays. Similarly, there was a regression in social development leading to poor eye contact, smile, and joint attention. Given that this variant was associated with an atypical presentation as compared to other individuals in this cohort, and the variant was outside of the STK domain, we interpret this case as a variant of uncertain significance (VUS).
Table 1.
Genetic details and seizure-specific clinical features for individuals with MAST3 STK domain de novo variants
| Patient | Variant (CADD, Polyphen) |
Degree of ID | Epilepsy Diagnosis | Age at sz onset in months | Sz type at onset | Subsequent sz types (age of onset if known) | Current sz control (age if applicable) |
|---|---|---|---|---|---|---|---|
| 1 | c.1217G>C, p.R406P (34.0, 1.00) |
Severe | DEE | 18 | Convulsive status epilepticus with fever | Atonic, tonic, GTCS; often in clusters with fever | Moderate-control |
| 2 | c.1528 G>A, p.G510S (32.0, 0.999) |
Severe | DEE | 11 | Febrile tonic-clonic |
Tonic-clonic Focal (motionless staring, oroalimentary automatisms, diffuse tonic) Absences Often in clusters with fever |
Seizure control since age 2 y 9 mo |
| 3 | c.1528 G>A, p.G510S (32.0, 0.999) |
Severe | DEE | 6–7 | Myoclonic | Absence (14 mo) Atonic (Drop attacks) (14 mo) Tonic-clonic (14 mo) |
Well-controlled (2.5 yrs) |
| 4* | c.1528 G>A, p.G510S (32.0, 0.999) |
Severe | DEE | 2 | Generalized tonic clonic |
Atonic Tonic TCS Often in clusters with fever |
Drug-resistant |
| 5 | c.1528 G>A, p.G510S (32.0, 0.999) |
Severe | DEE | 24 | Generalized tonic-clonic (<24 hours of vaccination) |
TCS Absence, FIAS |
Drug-resistant at age of last follow-up (27 yrs) |
| 6 | c.1528 G>A, p.G510S (32.0, 0.999) |
Moderate | DEE | 10 | Generalized tonic-clonic in context of viral illness |
GTCS Drop attacks Absence, FIAS Focal status |
Drug-resistant |
| 7 | c.1543 G>A, p.G515S (27.3, 0.989) |
Moderate | DEE | 11–12 | Myoclonic-atonic |
Myoclonic absence (eye flutter) Myoclonic atonic Myoclonic Atonic (head drops) GTCS (17 mo) Tonic (vibratory) |
Drug-resistant |
| 8 | c.1543 G>A, p.G515S (27.3, 0.989) |
Moderate | DEE | 20 | Tonic-clonic | Tonic (20 mo) FIAS with manual automatisms evolving to tonic seizure (30 years) FBTCS |
Moderate-control |
| 9 | c.1543 G>A, p.G515S (27.3, 0.989) |
Mild-moderate | DEE | 7 | Febrile convulsion | Absence (1 year) Myoclonic Tonic-clonic Convulsive status epilepticus (single event at 7 years) |
Moderate control |
| 10 | c.1547 T>C, p.L516P (26.4, 0.999) |
Severe-Profound | DEE | 24 | ND | FIAS (mouth automatism, head and eye deviation to one side and generalized shaking – nocturnal) GTC |
Drug-resistant |
| 11 | c.1651G>T, p.V551L (25.3, 0.915) |
Severe | DEE | 11 months | Febrile convulsion | FIAS (11 years), myoclonic, absence, atonic, GTCS, tonic | Moderate control |
Abbreviations: Sz – seizure, FBTCS, focal to bilateral tonic clonic seizures, FIAS – focal impaired awareness, GTCS – generalized tonic clonic seizures, TCS -tonic clonic seizures, ND – not determined
patient previously described in Iwama et al, 2019
Figure 1. MAST3 patient-specific variants occur at conserved sites that are intolerant to genetic variation.

(A) MAST3 patient-specific variants (number of individuals in parenthesis) in relation to ST kinase (amino acids 367–640) and PDZ domains (amino acids 958–1038). (B) Corresponding graph with missense tolerance ratio (MTR, y-axis) and protein position (x-axis). The tolerance ratio measures cDNA intolerance to missense variants with 5% representing the cutoff for more intolerant segments20. Regions of the ST kinase domain which are intolerant to variations portions of the ST kinase domain overlap with the distribution of pathogenic variants. (C) Multispecies alignment of the STK domain harboring the MAST3 patient variants (highlighted in green) show all pathogenic variants are highly conserved across 11 species (identical/similar amino acids in red). (D) Alignment of the MAST1 and MAST3 protein sequences. The serine threonine kinase (STK) domain has high homology and the MAST1 p. G517S and MAST3 p.G510S (highlighted in green) occur at the same position in the STK domain. Overall, these protein sequences have 61% identity.
Six unique missense variants were identified in 11 individuals occurring in the STK domain and two variants were recurrent (Fig 1A): c.1528G>A, p.G510S (patients 2–6) and c.1543G>A, p.G515S (patients 7–9). Three additional individuals had unique missense variants in the STK domain (patient 1: c.1217G>C, p.R406P; patient 10: c.1547T>C, p.L516P; patient 11: c.1651G>T, p.V551L).
Of the variants in the STK domain, 10/11 arose de novo, while segregation analysis is pending in proband 8 with the recurrent p.G515S variant. All variants were highly conserved (Fig 1C and D) and were predicted to be deleterious or damaging by in silico tools (CADD and polyphen respectively) (Table 1) and were absent in controls (GnomAD or TOPMed). MAST3 is intolerant to variation with a loss-of-function observed/expected upper bound fraction (LOEUF) of 0.6 for missense variation (range 0–1 denoting intolerant to tolerant to missense variation). Moreover, the STK domain is highly conserved and intolerant to variation, with missense tolerance ratio (MTR) scores all below the 25th percentile (Fig 1B)20.
Epilepsy phenotype of patients with MAST3 STK domain variants
All 11 patients had DEE with seizure onset at or before 2 years of age (median: 11 months, range: 2 to 24 months), with 7/11 patients having seizure onset before 12 months of age. The first seizure was a febrile convulsion, in 4 patients, an afebrile, tonic-clonic seizure in 4, myoclonic seizure in 1 and a myoclonic-atonic seizure in 1 patient (Table 1, Table S1). Three of 11 patients (1, 2 and 4) were reported to have a Dravet-like phenotype, primarily due to the febrile-induced seizures and status epilepticus associated with fever. However, in contrast with those with Dravet Syndrome, the hemiclonic/myoclonic seizures that are hallmarks of this DEE were not present.
All patients developed multiple seizure types, including atonic (3), myoclonic (2) myoclonic absence with eye flutter, myoclonic-atonic (1), tonic (5), generalized tonic-clonic with or without fever (11) and focal with impaired awareness (6). Seizures evolved to include other types in most patients, and these included atonic, myoclonic, myoclonic absence with eye flutter, myoclonic, atonic, tonic, tonic-clonic, and focal with impaired awareness. All patients experienced generalized tonic-clonic seizures with and without fevers (n = 11). Ten of 11 patients reported provoking factors for seizures (such as illness or fever and sleep deprivation). Eight patients experienced episodes of convulsive status epilepticus of whom 3 also experienced episodes of non-convulsive status epilepticus.
Two patients were seizure free at their most recent follow up: patient 3, now 8 years old, has been seizure free since age 2.5 years and is on no medications while patient 2, now seven years old, has been seizure-free since 2 years nine months on valproate alone. The remaining 9 patients had drug-resistant seizures and were taking 1–3 (mean: 2) antiepileptic medication with a variety of pharmacological targets represented (Table S1 and S2); 3 patients are currently taking sodium channel blockers. Antiepileptic medications that elicited some improvement in seizure control included valproic acid (5), topiramate (1), zonisamide (2), phenobarbital (1), felbamate, oxcarbazepine (1), lamotrigine (1), lacosamide (1), levetiracetam (2), briavaracetam, clobazam and cannabidiol. The ketogenic diet was trialed in 3 patients with improvement in seizure control for patient 7 but not for patient 6 or 10.
Initial EEGs performed within 12 months of seizure onset were available for 8/11 patients; 6 were reported as normal whilst background slowing was reported in patient 1 and generalized and focal abnormalities were reported in patient 3. Follow up EEGs were available for 9/11 patients; 3 were reported as normal whilst 4 patients had background disorganized or slowing, while patient 9 had bisynchronous frontal spikes and polyspikes and patient 7 had generalized seizures with eyelid flutters captured on EEG. Where this information was available, neither burst suppression nor hypsarrhythmia were features of the EEG (n=8).
Neurodevelopmental phenotype of patients with MAST3 STK pathogenic variants
Early development was normal in all 8 individuals with data available (Table S2). All individuals except patient 5, experienced developmental regression or plateau before 2 years of age (mean: 16 months, range: 11–24 months) coinciding with seizure in 6 patients and status epilepticus in 5. Data was unavailable for patient 5. In 4 individuals, regression was persistent, while in 3 individuals (patient 3,4 and 6) there was a period of recovery after initial regression associated with seizure control. Regression was associated with seizure onset in three individuals, and status epilepticus in seven individuals (Table S2). Developmental regression most commonly affected language (7) followed by gross motor (4), social (1), fine motor (1) and toilet training (1). The majority of patients (10/11) are described as non-verbal or with limited speech with expressive speech affected more than receptive speech. Eight patients exhibited delayed gross motor milestones with walking attained at a mean age of 17.5 months (range 14–31 months). Intellectual disability was severe in 6 individuals, moderate in 3, mild-moderate in patient 9 and severe-profound in patient 10 (data not available for patient 5).
Nine patients were normocephalic whilst patient 3 had macrocephaly and patient 9 had microcephaly. Three of 11 patients had dysmorphic features; patients 10 and 11 had dysmorphic facies, patient 10 also had short stature and patient 7 had 2–3rd toe syndactyly. Six patients had hypotonia with patient 1 developing progressive axial hypotonia and appendicular hypertonia. Eight patients had an abnormal gait; 5 were unsteady, had poor balance or fell frequently, 2 had knee flexion, patient 1 uses a wheelchair and patient 9 had wide-based walking. Sleep difficulties were reported in 7 patients, movement disorder in 2, a high pain threshold in 2, drooling in 2, stereotypies in 2 and hypothyroidism in 2. Three patients reported gastrointestinal difficulties including celiac disease in patient 3, cyclical vomiting in patient 7 and gastroesophageal reflux (resolved) – in patient 11.
Neuroimaging for patients with MAST3 pathogenic variants
Brain MRI was reported as normal in all patients with reports available (n=5) except for patient 4 who had a smaller left anterior hippocampus. MR images for 5 patients were reviewed independently by a single pediatric neuroradiologist (S.M.); all 5 were abnormal demonstrating a small pituitary (2), thin corpus callosum (1) or both (1) (Fig 2) Patients showed no evidence of clinical features associated with a hypo-active pituitary.
Figure 2. MRI changes in 5 individuals with MAST3 variants.
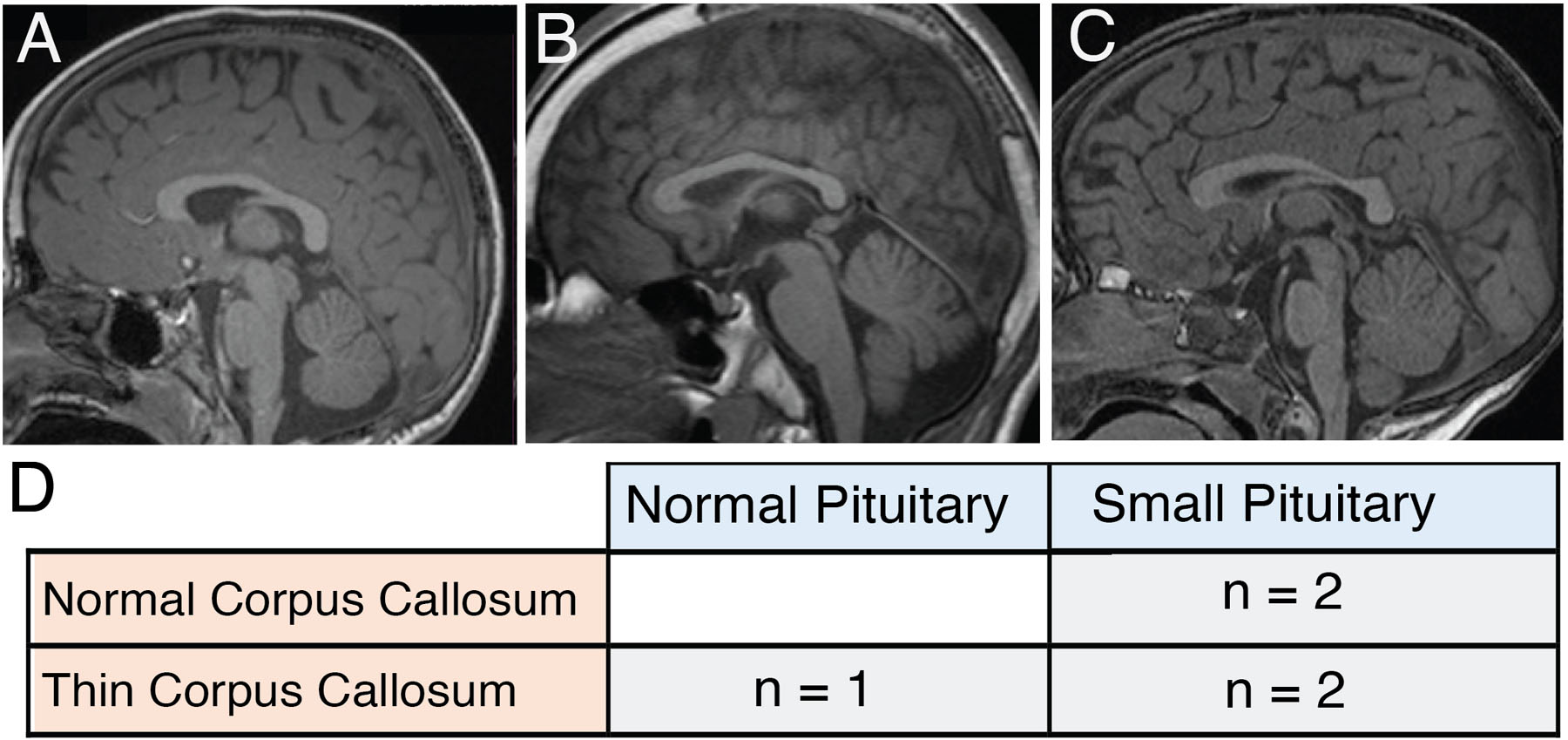
(A-C) - Sagittal FLAIR images () Patient 7 (p.G515S) 4 years old, normal corpus callosum and small pituitary. (B) Patient 11, 15 years old, thing corpus callosum and normal pituitary. (C) Patient 3, 2 years 9 months old, thin corpus callosum and small pituitary. D. Table summarizing changes found in corpus callosum and pituitary gland in the 5 individuals in whom we had access to MRIs. All patients had either a thin corpus callosum and/or a small pituitary gland, but not the extensive abnormalities observed in in individuals with MAST1 pathogenic variants.
Expression of MAST3
We generated a heat map of MAST1/MAST3 expression, as well as other epilepsy associated genes (SCN1A/SCN3A), neuronal marker genes using bulk RNA-seq data from the Brainspan atlas of the developing human brain (see list of URLs) (Fig 3A). MAST3 expression is low over the period of human fetal neurodevelopment, with expression increasing at 26 weeks post-conception and steadily increasing postnatally (Fig 3A). Single-nuclei RNA-seq of post-mortem motor cortex from the Allen Brain Map demonstrates that MAST3 expression is restricted to excitatory neurons in the human cortex (Fig 3B). At the protein level, MAST3 is present in a fraction of early-born (CTIP2+) neurons, and in the majority of late-born (CUX1+) excitatory neurons in 11-week-old human cerebral organoids (Fig 3C). In mouse cortex, MAST3 is primarily cytosolic (Fig 3D) and present in postmitotic excitatory neurons, mostly colocalizing with CTIP2 and TBR1 at E14.5, and mainly with the upper-layer neuron marker SATB2 at E16.5 (Fig 3E).
Figure 3: Expression of MAST3 and MAST1 in whole brain throughout prenatal development and postnatally.

(A) Heatmap of bulk RNA-seq data from the Brainspan atlas of the developing human brain (see list of URLs). MAST3 expression begins at about 26 weeks in prenatal development. In contrast, expression of MAST1 is highest early in pretnatal development (9 weeks) and then is reduced throughout the lifespan. This pattern is similar in other DEE-associated genes, SCN1A and SCN3A, where only the latter is associated with malformation of cortical development28. Key neuronal and developmental markers NESTIN, EOMES, SOX2 and TUBB3) are also shown. FPKM = Fragments Per Kilobase of transcript per Million mapped reads. (B) Heatmap of single-nucleus RNA-seq data from post-mortem adult human motor cortex for MAST3 and MAST1 (see list of URLs). MAST3 is expressed exclusively in excitatory neurons, while MAST1 is expressed in both inhibitory and excitatory neurons in the cortex. (C) Immunofluorescence in 11-week-old cerebral organoids shows the presence of MAST3 in a fraction of excitatory CTIP2-positive neurons (upper panels, arrows) and in the majority of CUX1-positive neurons (lower panels, arrowheads). (D) Immunofluorescence performed in E14.5 (left panels) and E16.5 (right panels) mouse coronal brain sections show MAST3 is mainly cytosolic, surrounding Tbr1+ cell nuclei (arrowheads). And some cells do present some MAST3 nuclear speckles (arrows). Scale bar=20 microns.(E) Immunofluorescence performed in E14.5 (left panels) and E16.5 (right panels) mouse coronal brain sections confirm MAST3 expression in postmitotic upper layer neurons, co-markers evolving as cerebral development progresses. TBR1 and CTIP2 are neuronal markers specific to deep cortical layers. SATB2 is a neuronal marker for upper cortical layers. Scale bars = 50 µm.
Assessment of kinase activity of MAST3 variants
Wild-type and four of the MAST3 variants described in this study were expressed in HEK293T cells together with its substrate, ARPP-16 (Fig 4). A kinase-dead control (p.K396H) was also co-expressed with ARPP-16. Compared to wild-type, the expression levels of the MAST3 variants was consistently lower, with the exception of the p.V551L variant which exhibited intermediate expression (Fig 4A). As expected, wild-type MAST3 was active as demonstrated by robust phosphorylation at Ser46 of ARPP-16, while there was minimal phosphorylation by kinase-dead MAST3. Despite lower expression, the levels of Ser46 phosphorylation for each of the variants (p.G510S, p.G515S, p.L516P, p.V551L) was similar to that of wild-type MAST3. When normalized to expression levels of MAST3 protein, the activities of each variant was therefore higher than wild-type, reaching significance for the p.G510S-containing mutant (*p = 0.0159, one-way ANOVA with Dunnett’s posthoc test). The reason for the lower expression of the MAST3 variants is not clear but could involve decreased protein synthesis or increased protein turnover. Nevertheless, these initial studies suggest the possibility of some sort of gain-of-function phenotype for these MAST3 variants through more effective phosphorylation of ARPP-16 and possibly other substrates.
Figure 4. Phosphorylation of ARPP-16 at Ser46 by MAST3 kinase.

(A) HA-tagged ARPP-16 alone, or together with MAST3, were expressed in HEK293T cells. Cell lysates were separated by SDS-PAGE and analyzed by immunoblotting for MAST3 protein, phospho-Ser46-ARPP-16, and HA-ARPP-16. Representative blots show that WT and all MAST3 mutant phosphorylate ARPP-16. There was minimal, background, phosphorylation of ARPP-16, as shown by the expression of ARPP-16 alone (lane 2) or when co-expressed with the dead-kinase mutant K396H (lane 4). Lanes: 1) GFP Only, 2) ARPP-16 Only, 3) Wild-Type MAST3 + ARPP-16, 4) K396H dead kinase control + ARPP-16, 5) G510S MAST3 + ARPP-16, 6) G515S MAST3 + ARPP-16, 7) L516P MAST3 + ARPP-16, 8) V551L MAST3 + ARPP-16. (B) Quantification of immunoblots revealed an increase in ARPP-16 phosphorylation when co-expressed with the G510S mutant compared to WT MAST3 (*p = 0.0159, one-way ANOVA with Dunnett’s posthoc test). Note that the GFP control, ARPP-16 alone control, and the dead kinase control quantifications were omitted from the graph due to the presence of zero values
Discussion
In this study, we describe de novo missense variants located in the STK domain of MAST3 as a novel cause for DEE in 11 patients. There were two recurrent variants (p.G510S and p.G515S present in 8 individuals collectively), and an additional three variants (p.R406P, p.L516P, p.V551L) located nearby11. A twelfth patient harboured a genetic change outside of the STK domain; he did not have epilepsy but rather a diagnosis of ASD and additional patients and functional studies are required to interpret the pathogenicity of variants outside the STK domain. Moreover, the first variant is located within the third position of the “Asp-Phe-Gly+1” (DFG+1) domain, which determines the specificity of the target residue that will be phosphorylated and is highly conserved among protein kinases11, 23. Modulation of MAST3 by Protein Kinase A (PKA) involves phosphorylation of position 512, again close to the variants’ location in this series6. Alteration of a subset of these specific variants in the STK domain resulted in lower expression in an in vitro cell culture model, but more effective phosphorylation of the MAST3 target, ARPP-16, consistent with a potential gain-of-function phenotype. Notably, a gain-of-function Gly to Ser mutation (p.G2019S) is found in an analogous position in the LRRK2 kinase and is the most common genetic cause of Parkinson’s disease24–26.
Patients with pathogenic variants in the STK domain of MAST3 typically presented with DEE, including 3 patients with Dravet syndrome-like phenotype. However, while these three individuals all presented with febrile-induced seizures and status epilepticus, age of onset was more variable, and individuals did not present with hemiclonic/myoclonic seizures typical of Dravet-syndrome. All patients had seizure onset before 2 years of age, with 7/11 having onset before 12 months of age. Most patients had normal development prior to the onset of seizures but then experienced developmental regression or plateau which coincided with seizure onset or episodes of status epilepticus. All patients developed multiple seizure types: all 11 patients had generalized tonic clonic seizures with tonic (n=5) and focal impaired awareness seizures (n=6) also common. Triggers for seizures such as fever, illness or lack of sleep were seen in 10/11 patients. Six patients were drug-resistant whilst 3 had achieved moderate seizure control on 2–3 antiepileptic drugs and 2 had been seizure free since age 2.5–2.75 years. All patients had intellectual disability, with most (5/11) having severe impairment. Most patients developed comorbidities including 6 patients with ASD or autistic features, 8 with gait disturbances, 7 experienced sleep issues, 6 had hypotonia, 3 had dysmorphic features and 3 had gastrointestinal problems.
MAST3 is the third of the five-member MAST family of serine/threonine kinases, which share a STK and a postsynaptic density protein-95/discs large/zona occludens-1 domain (PDZ)5. These genes have an overlapping but unique expression in the brain, with MAST3 expressed predominantly in the striatum, hippocampus, and cortex5. It has been studied most extensively in the rodent striatum, were the majority of MAST3 expression is localized to medium spiny inhibitory GABAergic projection neurons5. Conversely, our transcriptomic analysis illustrates that MAST3 expression is primarily restricted to late neurodevelopment and may be restricted to cortical excitatory neurons in humans. This pattern contrasts to MAST1, which is expressed early and at high levels in the developing brain and all cell types, including excitatory, inhibitory, and non-neuronal cells. De novo MAST1 variants were previously described as a cause of a mega-corpus callosum syndrome with cerebellar hypoplasia15. Intriguingly, this includes a p.Gly517Ser variant identified in three individuals, the same amino acid position as the MAST3 p.Gly510Ser variant described in five individuals here (Fig 1D). The authors suggest MAST1 variants result in a dominant-negative mechanism due to reduced expression of MAST2 and MAST3 expression in a mouse model. However, here we show MAST3 variants may act in a gain-of-function manner as demonstrated by more efficient phosphorylation of its substrate, ARPP-16. Therefore, another possible explanation for the reduced expression of MAST2 and MAST3 in MAST1 knockout models is compensation for a hyperactive MAST1. Finally, the MRI abnormalities in individuals with MAST1 pathogenic variants are extensive, while those with MAST3 pathogenic variants we analyzed revealed very mild abnormalities (Fig 2). These differences/ likely lie in the expression patterns of the two genes, where MAST1 is expressed throughout development with disruption resulting in extensive malformations of cortical development while MAST3, expressed later and exclusively in excitatory neurons, is associated with DEE. This expression pattern is similar in sodium channels, where SCN3A, which is expressed early in development, is associated with malformations of cortical development. In contrast, SCN1A is expressed much later and is associated with DEE generally devoid of MRI findings (Fig 3A)27, 28. Moreover, the restriction in MAST3 expression to excitatory neurons in the cortex (but not striatum) is somewhat reminiscent of SCN8A-associated DEE, where SCN8A is expressed in excitatory neurons, with gain-of-function variants leading to excessive excitatory neuron activity29. We speculate that MAST3 gain-of-function variants may lead to the same excessive excitatory neuronal activity, though this requires further investigation in model systems. For instance, the study of patient-specific induced pluripotent stem cells (iPSCs) and isogenic controls to glutamatergic and GABAergic neurons could be used as a model system to address this question, as well as assess the functional impacts of the variants on mutant MAST3 stability, MAST homolog abundance and phosphorylation of putative targets6–9.
In both in vitro and in vivo models of striatum, MAST3 has been shown to phosphorylate ARPP-16 at Ser466, 7, and this shifts the balance of its downstream target, protein phosphatase 2A (PP2A), from active to inactive8. The in vitro work from this study suggests that four MAST3 variants result in a potential gain-of-function phenotype and therefore increased phosphorylation of ARPP-16. This would subsequently lead to a greater decrease in PP2A activity and a concomitant overall increase in phosphorylation of substrates for PP2A. Changes in PP2A have been reported in individuals with intellectual disability and developmental delays, and more specifically, the catalytic subunit of PP2A, PPP2CA (Catalytic Cα Subunit), has been implicated in patients with language delays, hypotonia, epilepsy, and brain abnormalities reminiscent of our patients30.
In summary, we propose MAST3 as a novel gene for DEE, thereby expanding the genetic landscape of DEEs. Notably, CDKL5, which is one of the most common causes of DEE, also functions like MAST3 as a kinase in the brain. Patients with pathogenic MAST3 variants present with DEE with normal development prior to seizure onset at < 2 years. Status epilepticus and fever-sensitive seizures are common, severe intellectual disability was seen in most as were significant comorbidities. We also identified a single individual with a neurodevelopmental disorder, including ASD but without epilepsy, with a de novo variant outside the kinase domain. With the addition of MAST3 to clinical diagnostic sequencing panels, the prevalence of de novo kinase variants in DEE will be determined, along with the spectrum of neurodevelopmental phenotypes associated with this gene. MAST3 is the second member of MAST family of kinases to be implicated in CNS dysfunction. We propose a potential gain-of-function pathogenic mechanism that is restricted to MAST3 dysfunction in cortical excitatory neurons, reminiscent of SCN8A-associated epilepsy.
Supplementary Material
Acknowledgements
We would like to thank the patients and families for their participation in this research study. This work was sponsored by NIH NINDS R00NS089858 (GLC), National Health and Medical Research Council of Australia, CURE, Australian Epilepsy Research Fund, March of Dimes and NIH/NINDS (IES), the Japan Agency for Medical Research and Development (AMED) under grant numbers JP20ek0109280, JP20dm0107090, JP20ek0109301, JP20ek0109348, JP20kk0205012 (NM), JSPS KAKENHI under grant numbers JP17H01539 (NM), JP20K16862 (KI); a pilot award from the Swebilius Foundation at Yale University (ACN), NIH Common Fund, through the Office of Strategic Coordination/Office of the NIH Director under Award Numbers U01HG007690 and U01HG007530 (LHR, The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health), KMC is an employee of GeneDx, Inc. Research reported in this manuscript was supported by the NIH Common Fund, through the Office of Strategic Coordination/Office of the NIH Director under Award Numbers U01HG007690 and U01HG007530. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Undiagnosed Diseases Network (UDN) members: https://hmsharvard.box.com/udnmemberlist
Footnotes
URLs:
Gene Matcher: https://genematcher.org/
gnomAD: https://gnomad.broadinstitute.org/ for both allele frequencies and LOEUF scores
TOPMed/Bravo: https://bravo.sph.umich.edu/freeze3a/hg19/
MTR: http://mtr-viewer.mdhs.unimelb.edu.au/
Brainspan atlas of the developing human brain: https://www.brainspan.org/
Allen Brain Map single-nucleus transcriptome: https://portal.brain-map.org/atlases-and-data/rnaseq/human-m1–10x
Potential conflicts of interest
The authors report no competing interests.
References
- 1.McTague A, Howell KB, Cross JH, Kurian MA, Scheffer IE. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2015. March;15(3):304–16. [DOI] [PubMed] [Google Scholar]
- 2.Scheffer IE. A new classification and class 1 evidence transform clinical practice in epilepsy. Lancet Neurol. 2017. January;17(1):7–8. [DOI] [PubMed] [Google Scholar]
- 3.Shellhaas RA, Wusthoff CJ, Tsuchida TN, et al. Profile of neonatal epilepsies. Neurology. 2017;89(9):893–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.EpiPM_Consortium. A roadmap for precision medicine in the epilepsies. Lancet Neurol. 2015. December;14(12):1219–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garland P, Quraishe S, French P, O’Connor V. Expression of the MAST family of serine/threonine kinases. Brain Research. 2008;1195:12–9. [DOI] [PubMed] [Google Scholar]
- 6.Musante V, Li L, Kanyo J, et al. Reciprocal regulation of ARPP-16 by PKA and MAST3 kinases provides a cAMP-regulated switch in protein phosphatase 2A inhibition. eLife. 2017. June 14;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andrade EC, Musante V, Horiuchi A, et al. ARPP-16 Is a Striatal-Enriched Inhibitor of Protein Phosphatase 2A Regulated by Microtubule-Associated Serine/Threonine Kinase 3 (Mast 3 Kinase). The Journal of neuroscience : the official journal of the Society for Neuroscience. 2017. March 8;37(10):2709–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leslie SN, Nairn AC. cAMP regulation of protein phosphatases PP1 and PP2A in brain. Biochim Biophys Acta Mol Cell Res. 2019. January;1866(1):64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christensen KR, Nairn AC. cAMP-regulated phosphoproteins DARPP-32, ARPP16/19, and RCS modulate striatal signal transduction through protein kinases and phosphatases. Adv Pharmacol. 2021;90:39–65. [DOI] [PubMed] [Google Scholar]
- 10.Labbé C, Boucher G, Foisy S, et al. Genome-wide expression profiling implicates a MAST3-regulated gene set in colonic mucosal inflammation of ulcerative colitis patients. 2012;18(6):1072–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iwama K, Mizuguchi T, Takeshita E, et al. Genetic landscape of Rett syndrome-like phenotypes revealed by whole exome sequencing. J Med Genet. 2019. June;56(6):396–407. [DOI] [PubMed] [Google Scholar]
- 12.Rodríguez-García ME, Cotrina-Vinagre FJ, Gómez-Cano M, Martínez de Aragón A, Martín-Hernández E, Martínez-Azorín F. MAST1 variant causes mega-corpus-callosum syndrome with cortical malformations but without cerebellar hypoplasia. American journal of medical genetics Part A. 2020. June;182(6):1483–90. [DOI] [PubMed] [Google Scholar]
- 13.Hecher L, Johannsen J, Bierhals T, Buhk JH, Hempel M, Denecke J. The Clinical Picture of a Bilateral Perisylvian Syndrome as the Initial Symptom of Mega-Corpus-Callosum Syndrome due to a MAST1-Gene Mutation. Neuropediatrics. 2020. December;51(6):435–9. [DOI] [PubMed] [Google Scholar]
- 14.Ben-Mahmoud A, Al-Shamsi AM, Ali BR, Al-Gazali L. Evaluating the Role of MAST1 as an Intellectual Disability Disease Gene: Identification of a Novel De Novo Variant in a Patient with Developmental Disabilities. J Mol Neurosci. 2020. March;70(3):320–7. [DOI] [PubMed] [Google Scholar]
- 15.Tripathy R, Leca I, Van Dijk T, et al. Mutations in MAST1 Cause Mega-Corpus-Callosum Syndrome with Cerebellar Hypoplasia and Cortical Malformations. Neuron. 2018;100(6):1354–68.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015. October;36(10):928–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Retterer K, Juusola J, Cho MT, et al. Clinical application of whole-exome sequencing across clinical indications. Genetics in Medicine. 2016;18(7):696–704. [DOI] [PubMed] [Google Scholar]
- 18.Myers CT, Hollingsworth G, Muir AM, et al. Parental Mosaicism in “De Novo” Epileptic Encephalopathies. N Engl J Med. 2018. April 26;378(17):1646–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015. May;17(5):405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Traynelis J, Silk M, Wang Q, et al. Optimizing genomic medicine in epilepsy through a gene-customized approach to missense variant interpretation. Genome research. 2017. October;27(10):1715–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017. April;58(4):512–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iyer GH, Moore MJ, Taylor SS. Consequences of lysine 72 mutation on the phosphorylation and activation state of cAMP-dependent kinase. The Journal of biological chemistry. 2005. March 11;280(10):8800–7. [DOI] [PubMed] [Google Scholar]
- 23.Chen C, Ha BH, Thévenin AF, et al. Identification of a major determinant for serine-threonine kinase phosphoacceptor specificity. Mol Cell. 2014. January 9;53(1):140–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Di Fonzo A, Rohé CF, Ferreira J, et al. A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson’s disease. Lancet. 2005. Jan 29-Feb 4;365(9457):412–5. [DOI] [PubMed] [Google Scholar]
- 25.Gilks WP, Abou-Sleiman PM, Gandhi S, et al. A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet. 2005. Jan 29-Feb 4;365(9457):415–6. [DOI] [PubMed] [Google Scholar]
- 26.Nichols WC, Pankratz N, Hernandez D, et al. Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet. 2005. Jan 29-Feb 4;365(9457):410–2. [DOI] [PubMed] [Google Scholar]
- 27.Harkin LA, McMahon JM, Iona X, et al. The spectrum of SCN1A-related infantile epileptic encephalopathies. Brain. 2007;130(Pt 3):843–52. [DOI] [PubMed] [Google Scholar]
- 28.Zaman T, Helbig KL, Clatot J, et al. SCN3A -Related Neurodevelopmental Disorder: A Spectrum of Epilepsy and Brain Malformation. Annals of neurology. 2020;88(2):348–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bunton-Stasyshyn RKA, Wagnon JL, Wengert ER, et al. Prominent role of forebrain excitatory neurons inSCN8Aencephalopathy. Brain. 2019;142(2):362–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reynhout S, Jansen S, Haesen D, et al. De Novo Mutations Affecting the Catalytic Cα Subunit of PP2A, PPP2CA, Cause Syndromic Intellectual Disability Resembling Other PP2A-Related Neurodevelopmental Disorders. American journal of human genetics. 2019. February 7;104(2):357. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.