

Abstract
Autism spectrum disorder (ASD) is characterized by a triad of behavioural impairments including social behaviour. Neuroligin, a trans-synaptic adhesion molecule, has emerged as a penetrant genetic determinant of behavioural traits that signature the neuroatypical behaviours of autism. However, the function of neuroligin in social circuitry and the impact of genetic variation to this gene is not fully understood. Indeed, in animal studies designed to model autism, there remains controversy regarding the role of neuroligin dysfunction in the expression of disrupted social behaviours. The model organism, Caenorhabditis elegans, offers an informative experimental platform to investigate the impact of genetic variants on social behaviour. In a number of paradigms, it has been shown that inter-organismal communication by chemical cues regulates C. elegans social behaviour. We utilize this social behaviour to investigate the effect of autism-associated genetic variants within the social domain of the research domain criteria. We have identified neuroligin as an important regulator of social behaviour and segregate the importance of this gene to the recognition and/or processing of social cues. We also use CRISPR/Cas9 to edit an R-C mutation that mimics a highly penetrant human mutation associated with autism. C. elegans carrying this mutation phenocopy the behavioural dysfunction of a C. elegans neuroligin null mutant, thus confirming its significance in the regulation of animal social biology. This highlights that quantitative behaviour and precision genetic intervention can be used to manipulate discrete social circuits of the worm to provide further insight into complex social behaviour.
Introduction
Autism spectrum disorder (ASD) is a pervasive developmental disorder (1). It is clinically characterized by a triad of neuroatypical behaviours that include impaired verbal communication, repetitive behaviours and impaired social interactions. Sensory processing deficits span across the autism spectrum (2). For example, recognition of social cues and multi-sensory integration of those cues are often impaired (2,3). There is a strong genetic association in ASD with hundreds of genes implicated in the disorder (4). Many of these genes encode synaptic proteins (5–7), suggesting that synaptic dysfunction underpins the expression of ASD-associated neuroatypical behaviours (5). However, it is still unclear how genetic variants lead to changes in neural circuits that result in the spectrum of characteristics that are represented in individuals with a recognizable ASD diagnosis. The role of individual genes and the wider complex interaction between polygenic loci make the genetic architecture of ASD complex. This complexity requires investigation that will ultimately delineate the weight of contribution in genetic backgrounds that range from a single penetrant gene to ones with multiple common variants at multiple loci (8).
One of the synaptic proteins that have been implicated in ASD is neuroligin (NLGN). NLGN is a post-synaptic cell adhesion protein, which aids the stabilization of synaptic function (9). One highly penetrant variation, which has been shown to affect the NLGN3 gene of autistic individuals is the R451C mutation (10). This missense variant causes a substitution of arginine for cysteine in the extracellular domain of NLGN3, which leads to its misfolding and failure to traffic to the cell surface. It has been suggested that the R451C mutation may result in a loss of function and disruption of synaptic function and plasticity within a number of central neural circuits (11). Despite broad investigation, it remains unclear as to how penetrant this mutation and further neuroligin mutants are in disrupting social behaviour in animal models. There have been differing reports of disrupted social behaviour in mice where the mutation has been introduced into strains with different genetic backgrounds (12,13).
The model organism, Caenorhabditis elegans, is a good experimental platform to investigate the effect of ASD-associated genetic variants. C. elegans facilitate systems level analysis due to their genetic tractability, simple nervous system and gene homology to humans (14,15). Included within this is the conservation of genes involved in synapse maturation and function (16–18). C. elegans encode a single orthologue of mammalian NLGN3 called nlg-1, which has been shown to share key structural and functional domains with the human protein (19,20). This conservation is reinforced by observations showing human NLGN is able to provide functional rescue of an nlg-1 deficiency in C. elegans (19).
In order to investigate the potential function of nlg-1 within the social domain, we have utilized a paradigm of inter-organismal communication between C. elegans and progeny. The effect of chemosensory stimuli on worm behaviour can be investigated using an assay in which the propensity of an adult worm to leave its food source, a bacterial lawn, is monitored over time. This food-leaving assay scores food-leaving events in which each event is defined as an occasion when the whole of the worm’s body comes off food. This is a behavioural output, which can be modulated in response to different cues (21). For example, when exposed to ad libitum source of food, C. elegans will remain on the food lawn and perform infrequent food-leaving events (21). However, an increase in the number of progeny populating an otherwise replete food lawn causes a population-dependent increase in food-leaving events of adult worms. This progeny-induced leaving is not observed in a daf-22 loss of function mutant (22) showing that offspring-produced ascaroside pheromones modulate adult behaviour. This suggests that inter-organismal signalling from progeny to parents on food-replete lawns provides a quantifiable behaviour to investigate the genetic determinants of social circuits (22).
We use this assay to show that the ASD-associated gene, NLGN, is an important determinant in regulating C. elegans social circuitry. We identify that a genetic change that models the human R451C phenocopies the functional null. In doing this, we facilitate further insight into the genetic underpinnings of social behaviour, a key diagnostic Research Domain Criteria in autism and other psychiatric disorders (https://www.nimh.nih.gov/research/research-funded-by-nimh/rdoc/index.shtml).
Results
The importance of nlg-1 in modulating social communication in adult worms
Two of the major characteristics of ASD are impaired social behaviour and deficits to multi-sensory integration (2). It has been previously shown that C. elegans elicit a social response to progeny populating a food lawn (22). More specifically, in response to increasing numbers of progeny on food, wild-type (N2) worms show a population-dependent increase in food leaving in the presence of an otherwise replete food lawn. This progeny-enhanced food-leaving behaviour was shown to be the result of chemosensory social signalling between progeny and adult worms (22). We have used this social behaviour to investigate the autism-associated gene, NLGN3. To investigate the behaviour of nlg-1(ok259) null mutants in response to progeny, worms were picked onto the centre of a bacterial lawn and food-leaving events were counted at 2 and 24 h. At 2 h, no progeny are present on the food lawn. In comparison, at 24 h progeny will have accumulated on the food lawn. Therefore, quantifying food-leaving events at these time points allows for a direct comparison of food-leaving behaviour in the absence and presence of progeny.
Observing food leaving in N2 adults showed an increase in leaving events over time. After 2 h, when no progeny were present, worms remained on the food lawn (Fig. 1A). At 24 h, when the food lawn is populated with progeny, N2 display progeny-enhanced food-leaving behaviour, in which they show a 12-fold increase in the number of food-leaving events (Fig. 1A). nlg-1(ok259) mutants remained on food, similar to N2, after 2 h on the food lawn (Fig. 1A). However, at 24 h, when the food lawn is dense with progeny, nlg-1(ok259) leaves the food patch less than N2 (Fig. 1A). nlg-1(ok259) did not show the same degree of progeny-enhanced food leaving as N2, suggesting that their social interaction with progeny may be impaired.
Figure 1 .
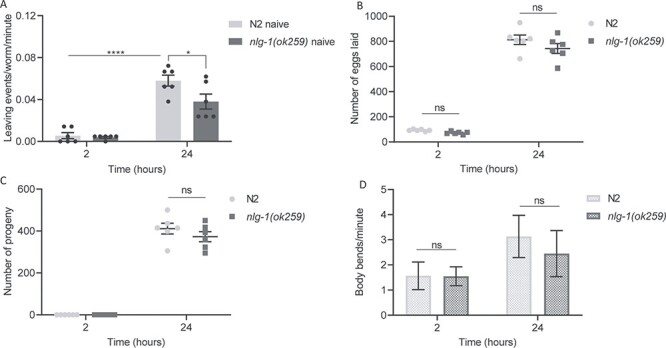
nlg-1(ok259) adults show deficient food leaving in response to progeny. (A) A food-leaving assay was performed with N2 and nlg-1(ok259) null mutant. N2 and nlg-1(ok259) adults were picked onto the centre of a bacterial lawn before food-leaving events were counted at 2 and 24 h. N2 and nlg-1(ok259) n = 6. (B and C) the number of eggs (B) and progeny (C) for N2 and nlg-1(ok259). N2 and nlg-1(ok259) n = 6. (D) The number of body bends per minute performed by N2 and nlg-1(ok259) worms on food at 2 and 24 h. N2 n = 6. nlg-1(ok259) n = 4. All data shown as mean ± SEM. Statistical analysis performed using Two-way ANOVA with Sidak’s multiple comparison test; ns P ≥ 0.05, *P < 0.05, ****P ≤ 0.0001.
We next wanted to understand if the reduced food-leaving behaviour of nlg-1(ok259) was due to impaired social interaction with progeny. We reasoned that reduced food leaving could also result if nlg-1(ok259) had an egg-laying deficiency. Previous studies had shown that the amount of food leaving was directly related to the number of progeny, with a reduced progeny accumulation giving a reduced food leaving (22). To test this, the number of eggs laid and the number of progeny present on the food lawn was counted after each food-leaving assay. For both N2 and nlg-1(ok259), the total number of eggs laid (Fig. 1B) during the assay, and number of progeny present at 24 h were counted (Fig. 1C), and there was no difference between the strains. These data show that the reduced food leaving of nlg-1(ok259) cannot be explained by reduced progeny available to drive adult food leaving. Furthering this, no difference was seen in the number of body bends for nlg-1(ok259) (Fig. 1D) indicating that locomotion per se does not underlie the reduced food leaving of nlg-1(ok259). Thus, the deficit in progeny-enhanced food-leaving behaviour of nlg-1(ok259) appears due to impaired social interaction of the mutant with progeny.
Next, we wanted to investigate whether the reduced food leaving described above for nlg-1(ok259) was due to an impaired social interaction with progeny. To this end, we used a pre-conditioned food lawn, as previously described (22). Pre-loading progeny onto a food lawn before assaying the adult food-leaving events preconditions the lawn with chemosensory cues released by progeny. This recapitulates that the progeny-dense conditions worms are exposed to after 24 h on the food lawn, but the assayed adults undergo more acute exposure to the progeny (Fig. 2A).
Figure 2 .
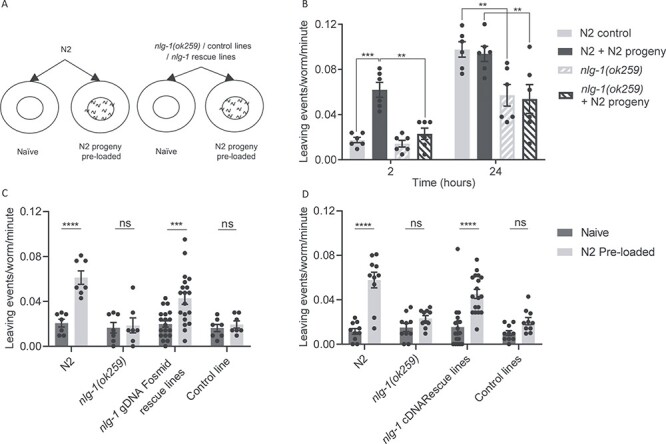
nlg-1(ok259) show a deficit in social interaction when exposed to N2 progeny. (A) Cartoon showing the principles of the pre-conditioned food-leaving assay. In order to test the effect of progeny on food-leaving behaviour, C. elegans adults were picked onto either a naïve or pre-conditioned food lawn. A naïve lawn contains no progeny whereas a pre-conditioned food lawn contains ~140 N2 progeny. (B) N2 and nlg-1(ok259) were picked onto naïve and pre-conditioned food lawns and their food-leaving behaviour observed at 2 and 24 h. N2 and nlg-1(ok259) n = 6. Two-way ANOVA with Tukey’s multiple comparison test; **P ≤ 0.01, ***P ≤ 0.001. (C) The number of food-leaving events of N2, nlg-1(ok259), nlg-1 gDNA fosmid rescue nlg-1(ok259) X, Ex [WRM0610dD09; Pmyo-3::gfp] and nlg-1(ok259) X, Ex [pPD95.77; Pmyo-3::gfp] control adults on naïve and pre-conditioned food lawns. N2, nlg-1(ok259) and nlg-1(ok259) X, Ex [pPD95.77; Pmyo-3::gfp] control line n = 7, nlg-1(ok259) X, Ex [WRM0610dD09; Pmyo-3::gfp] n = 19. Two-way ANOVA with Sidak’s multiple comparison test; ns P ≥ 0.05, ***P ≤ 0.001, ****P ≤ 0.0001. (D) The number of food-leaving events of N2, nlg-1(ok259), nlg-1 cDNA rescue nlg-1(ok259) X, Ex [pPD95.77 (Pnlg1::nlg-1 Δ#14); Pmyo-3::gfp] and nlg-1(ok259) X, Ex [pPD95.77; Pmyo-3::gfp] control line on naïve and pre-conditioned food lawns. N2, nlg-1(ok259) and nlg-1(ok259) X, Ex [pPD95.77; Pmyo-3::gfp] control line n = 10. nlg-1(ok259) X, Ex [pPD95.77 (Pnlg-1::nlg-1 Δ#14); Pmyo-3::gfp] n = 18. Conditioning assays were performed as paired experiments and data obtained from independent replicas is presented. Two-way ANOVA with Tukey’s multiple comparison test; ns P ≥ 0.05, ****P ≤ 0.0001. All data shown as mean ± SEM
The pre-conditioning with N2 progeny results in enhanced food leaving in N2 adults compared to the naive control at 2 h (Fig. 2B). This is consistent with previous findings, which showed that social interaction of adult worms and progeny on pre-conditioned food-lawn results in enhanced food leaving in N2 C. elegans (22). In comparison, nlg-1(ok259) adults when exposed to plates preconditioned with N2 progeny did not show enhanced food leaving. They left infrequently on both naïve and pre-conditioned food lawns at 2 h (Fig. 2B). This confirms that nlg-1(ok259) has reduced food leaving in response to progeny compared to N2. Therefore, this suggests that NLG-1 may be an important regulator of chemosensory-driven social interaction in C. elegans. To confirm the importance of NLG-1 in the social circuit of the worm, we generated two transgenic rescue lines expressing either nlg-1 gDNA or nlg-1 cDNA in the nlg-1(ok259) background. In response to pre-loaded N2 progeny, both the rescue lines expressing nlg-1 gDNA and cDNA showed progeny-enhanced food leaving, similar to that of N2 (Fig. 2C and D). This shows that expression of either nlg-1 in its genomic or cDNA form can rescue the reduced food leaving of nlg-1(ok259) mutants in response to progeny. Together, these data suggest that nlg-1(ok259) mutants are impaired in their ability to modulate food-leaving behaviour in the presence of progeny and that NLG-1 may play an important role in regulating this social behaviour.
We next wanted to segregate whether NLG-1 is important for the production and/or release of social cues from progeny or the recognition and/or integration of the social cue in the adult worm. To do this, we pre-conditioned food lawns with either N2 or nlg-1(ok259) progeny. Food-leaving behaviour was then observed for N2 and nlg-1(ok259) adults in response to either N2 or nlg-1 progeny (Fig. 3A). In this way, we were able to investigate whether nlg-1(ok259) mutant progeny are capable of driving enhanced food-leaving behaviour in adult worms, hence informing on their ability to produce/release chemical social cues.
Figure 3 .
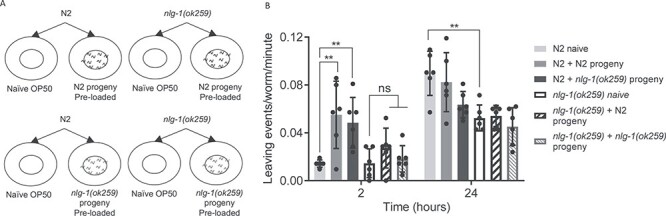
Parental nlg-1 is required for recognition and/or processing of progeny-derived social cues. (A) Experimental setup. N2 and nlg-1(ok259) were placed onto a naïve OP50 bacterial lawn or a lawn preconditioned with N2 or nlg-1(ok259) progeny. (B) The number of food-leaving events of N2 and nlg1(ok259) adults on naïve and pre-conditioned food lawns. Data are mean ± SEM. N2 and nlg-1(ok259) n = 6. Conditioning assays were performed as paired experiments and data obtained from independent replicas is presented. Two-way ANOVA with Tukey’s multiple comparison test; ns P ≥ 0.05, **P ≤ 0.01.
N2 adults showed enhanced food-leaving behaviour in the presence of both N2 and nlg-1(ok259) progeny (Fig. 3B). This suggests that both N2 and nlg-1(ok259) progeny are capable of driving-enhanced food-leaving behaviour in N2 adults. In turn, this suggests that nlg-1(ok259) mutant progeny are capable of producing and releasing chemical social cues in order to stimulate enhanced food leaving in adults. In comparison, nlg-1(ok259) adults do not show enhanced food leaving in response to either progeny relative to the naïve control at 2 h (Fig. 3B). Taken together, these results suggest that N2 and nlg-1(ok259) progeny are not impaired in the production and release of social cues. Furthermore, these results are consistent with the hypothesis that nlg-1(ok259) adults have impaired social communication with progeny, which may involve deficits in recognition and/or integration of progeny derived social cues.
C. elegans carrying R433C mutation phenocopy the social impairment of nlg-1 null
So far, we have shown that the nlg-1(ok259) null mutant does not show progeny-driven-enhanced food-leaving behaviour and we hypothesize that this behavioural deficit is specific to the recognition and/or integration of social cues in the adult worm. In this way, nlg-1(ok259) mutants are modelling neuroatypical behaviour in the social domain. Considering this, we next wanted to know whether C. elegans could model the same social impairment but in response to a specific human genetic variation implicated in autism. We investigated a penetrant missense variant, which has been identified in the NLGN3 gene of individuals on the autistic spectrum. The R451C mutation results in an R-C amino acid substitution within the extracellular cholinesterase-like domain of NLGN3 (10). C. elegans encode a conserved arginine within the cholinesterase-like domain of NLG-1, which is present in human NLGN1–4 (Fig. 4A). Using CRISPR/Cas9 the R451C mutation was generated in C. elegans by editing an R-C substitution at position 433 within the cholinesterase-like domain of NLG-1 (Fig. 4A). The bona fide nature of this CRISPR event was confirmed by genomic sequencing, which identified that the CGA codon of the wild-type was converted to TGC encoding cysteine in the mutant line (Supplementary Figure 1). The conservation in sequence suggests that the same disruption in NLGN structure that arises in the human protein with this mutation at position 451 will be replicated in C. elegans with the orthologs mutation at 433 (based on NLG-1 isoform C40C9.5e). The social behaviour of the CRISPR generated line, nlg-1(qa3780), was investigated using lawns that were pre-conditioned with N2 progeny in order to compare the response of CRISPR generated nlg-1(qa3780) to nlg-1(ok259) null mutant in response to N2-derived social cues.
Figure 4 .
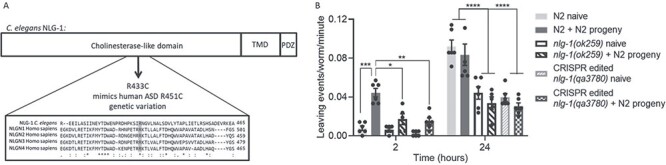
R433C mutation in nlg-1 phenocopies social impairment of nlg-1 null. (A) Domain structure of NLG-1 indicating the arginine to cysteine (R-C) amino acid substitution at position 433 generated within the cholinesterase like domain following CRISPR/Cas9. Protein sequence alignment of C. elegans NLG-1 and human NLGN1–4 indicates the arginine residue involved in ASD and its conservation in C. elegans (shaded in grey). ‘*‘Indicates conservation of a single amino acid residue, ‘:’ indicates conservation between amino acid groups with similar properties and ‘.’ indicates conservation between amino acid groups with weakly similar properties. (B) The number of food-leaving events were counted for N2, nlg-1(ok259) and CRISPR generated nlg-1(qa3780) on naïve and N2 progeny conditioned food lawns. Data are mean ± SEM. N2, nlg1(ok259) and nlg-1(qa3780) n = 6. Two-way ANOVA with Tukey’s multiple comparison test; *P < 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Consistent with our previous findings, N2 adults show enhanced food leaving in response to progeny on pre-conditioned food lawns (Fig. 4B). Furthermore, nlg-1(ok259) does not show enhanced food leaving in the presence of progeny (Fig. 4B). The response of nlg-1(qa3780) mutants, carrying the R433C mutation, is very similar to that of nlg-1(ok259) null mutants. Nlg-1(qa3780) showed no enhanced food leaving when exposed to progeny (Fig. 4B). This suggests that the single R433C missense variant to nlg-1 results in social impairment, which phenocopies that of the nlg-1 null mutant. Furthering this, these results suggest that C. elegans can model social impairment in response to autism-associated human genetic variants.
Discussion
ASD causes neuroatypical social behaviour and deficits in sensory processing (2). ASD has a well-characterized genetic dependence, with the underlying determinants becoming increasingly well resolved with the use of quantitative genetics. There is a range of determinants that imply polygenic interaction between common variations of the genome. In addition, there are a number of more penetrant single gene mutations that are identified as contributing to the expression of behavioural traits that signature neuroatypical behaviour of ASD. Many of these penetrant genes encode synaptic proteins (4) highlighting synaptic dysfunction as orchestrating the phenotypes associated with ASD. In the case of penetrant genes, animal experiments utilizing functional nulls or engineered mutants designed to mimic human mutations have facilitated investigation of the cellular circuit and system-level mechanisms that disrupt behavioural domains that model ASD-associated behaviour (23). In this study, we have shown that C. elegans can be used to model social impairment in response to genetic models of the human variants identified in individuals with an ASD diagnosis. This is highlighted by our investigation and comparison of the single neuroligin gene in C. elegans that has high conservation with the five human neuroligin genes, particularly the NLGN3 gene that has been strongly implicated in ASD (19,24).
NLGNs are a family of synaptic adhesion proteins required for synaptic maturation through interaction with synaptic adhesion partners. Of these, the most-studied interaction is between neuroligin and neurexin, which can co-ordinate the localization of receptors at inhibitory synapses (25). It has been shown in C. elegans that this interaction and its function in the organization of receptors is conserved at the neuromuscular junction of the worm (26). The neuroligin functional null used in our study has been previously reported as having defects in synaptic transmission at the neuromuscular junction, in the absence of an obvious locomotory phenotype (26). We reached a similar finding that the neuroligin null mutation did not impair locomotion. It has previously been shown that the nlg-1 knockout has a hyporeversal phenotype (20). Hence, we do not think that the decreased food-leaving behaviour observed in the nlg-1 mutants in our study can be explained by a hyper-reversal phenotype or excessive dwelling. We propose that the difference in food-leaving behaviour of the null cannot be explained by impaired motor control but are mindful that there may be differences in the relative profile of more subtle locomotory sub-behaviours that underpin the food-leaving behaviour in response to social cues.
The clinical data for the NLGN3 R451C mutation suggest it causes severe autism and Aspergers’ syndrome in a sibling carrying the mutation (10). This motivated animal experiments in which the functional null and R451C mutation were compared. These mutants exhibit cellular changes selective for inhibitory synapses that disrupt plasticity and cause changes in learned behaviours associated with social interaction and motor control (12,13,27). In the former case, there is a controversy as to how penetrant the mutations that mimic the human mutation are in causing a mutation-dependent change in social interaction. The differential conclusions derived from mice strains raised in distinct genetic backgrounds suggest that background modifiers can affect the phenotypic output of the genetic mutation being investigated (12,13). In this study, we used a neuroligin null mutant and a CRISPR/Cas9 line in the same wild-type N2 genetic background to compare the effects of the mutation on social behaviour.
Distinct cues and neural circuits regulate social behaviour in humans, both in terms of their nature and complexity (28). The behavioural observations reported in this study are underpinned by a previously characterized progeny-dependent inter-organismal communication that drives changes in adult behaviour and provides a basis for understanding the molecular determinants of a social behaviour in C. elegans (22). Our results show that nlg-1(ok259) adults have impaired food leaving, caused by impaired social communication with progeny. We are aware that food leaving is a complex behaviour and consider that the reduced food-leaving response observed in our experimental system is not defined by deficient responses to other environmental sensory cues described elsewhere (21,29). This deficit in nlg-1(ok259) shows NLGN’s synaptic organizing function may play a key role in the neural circuits that underlie social interaction. We provide evidence that neuroligin-dependent signalling is required in the adult for their ability to respond to the chemical cues generated by the progeny. This assay thus provides a quantitative measure for neuroligin function in the adult. We used this to probe the integrity of the circuits driving this output utilizing a CRISPR/Cas9 mutant that models the penetrant human ASD mutation R451C. Our data show that this mutation phenocopies the deficit in food leaving seen in the null mutation, which is consistent with the widely held view that this mutation is a loss of function (30). Taken together our data imply that nlg-1 plays an important role in the circuits that organize social interaction in C. elegans. This would reinforce a conserved role for this class of adhesion molecule in organizing circuits that underpin social behaviour. Previous work has identified nlg-1 function in the animals response to a number of environmental cues including food, chemicals and temperature. Interestingly, disruption at the input level of sensory cues is an important emerging theme in human autism and future work will be required to address if the sensitivity to pheromones per se is the major determinant of the observed disruption. Equally, work in C. elegans has implicated changes in the balance of excitation and inhibition downstream of sensory inputs generating behavioural disruption in C. elegans so the results here may arise through an essential role for neuroligin at several levels of the social circuit responsible for the adult response to progeny induced food leaving (31,32).
Previous studies have used C. elegans morphology and locomotion as readouts of disrupted behaviour in response to mutation to ASD-associated genes (33,34). We have extended this analysis to inform on the effect of a neuroligin variant on a phenotype that relates to one of the triad of impairments that make up the diagnostic criteria of ASD. The social interaction assay used in this study allows for quantification of ASD-associated variants and their effect on social behaviour. The gene homology of C. elegans with mammalian systems, conservation of synaptic architecture and requirement for sensory-motor integration in the face of environmental cues provides a tractable paradigm for these investigations. Overall, the assay of social interaction in C. elegans has the potential to provide crucial insight into the neural circuits that underpin social behaviour and how genetic variants impact on those circuitries. This suggests it provides a robust platform to screen other genes implicated in autism and complex psychiatric disorders that exhibit underpinning disruption of the social domain.
Materials and Methods
C. elegans culturing and strains used
All C. elegans strains were maintained using standard conditions (35). C. elegans were age synchronized by picking L4-day-old hermaphrodites onto a new plate 18 h prior to the behavioural assay. Strains used are as follows: Bristol N2; VC228 nlg-1(ok259) X (x6 outcrossed); nlg-1(ok259) X, Ex [WRM0610dD09; Pmyo-3::gfp]; nlg-1(ok259) X, Ex [pPD95.77 (Pnlg-1::nlg-1 Δ#14); Pmyo-3::gfp]; nlg-1(ok259) X, Ex [pPD95.77; Pmyo-3::gfp]; CRISPR/Cas9 edited XA3780 nlg-1(qa3780) (x2 outcrossed). For transgenic animals, stable lines were selected for behavioural analysis.
Rescue construct and transgenic methods
The nlg-1 fosmid, WRM0610dD09, was provided by SourceBioScience. The nlg-1 cDNA rescue construct was designed as previously described (36). Briefly, 2.5 kb Pnlg-1 was cloned into the pPD95.77 vector. Subsequently, nlg-1 Δ#14 cDNA sequence was fused to Pnlg-1 (37). nlg-1(ok259) L4 + 1 day old worms were microinjected with nlg-1 fosmid WRM0610dD09 (0.3 ng/μl) or nlg-1 cDNA rescue plasmid (50 ng/μl) and the marker plasmid Pmyo-3::gfp (30 ng/μl). Control lines were microinjected with marker plasmid Pmyo-3::gfp (30 ng/μl).
Behavioural assays
Food leaving
In total, 5 cm NGM plates were prepared using a standard protocol (35). Plates were seeded with OP50 E.coli as described previously (22). About, 50 μL of OP50 E. coli at OD600 of 0.8 was gently spotted on the middle of an unseeded plate the day prior to the assay. For a naive food-leaving assay, seven age synchronized L4 + 1 day old worms were gently picked onto the centre of the bacterial lawn on the assay plate. To pre-condition the food lawn, progeny were loaded onto the bacterial lawn as previously described (22). About, 10 gravid adults were picked onto the bacterial lawn and left to lay 140–200 eggs before being picked off. Approximately, 18 h following this, seven L4 + 1 day old worms were picked onto the centre of the bacterial lawn. In all food-leaving assays, the number of food-leaving events were counted during 30 min observations at 2 h only or 2 and 24 hs. A food-leaving event is defined as when the whole of the worm’s body comes off the bacterial lawn. For all assays, N2 animals were systematically observed in parallel with the strain under investigation. For all behavioural analysis, investigators were blind to the genotypes being observed.
Body bends
Following a food-leaving assay 5–7 worms were selected for body bend quantification. Each worm was observed for 1 min on food. A body bend was defined as a muscle contraction that resulted in a dorsal or ventral bend of the worm’s body.
Genome editing
CRISPR/Cas9 editing was generated using a previously described method (38). N2 L4 + 1 day old hermaphroidtes were microinjected with expression vectors for Cas9 and sgRNA’s targeting nlg-1, unc-58 and dpy-10. The sequence of the nlg-1 sgRNA, synthesized by Integrated DNA Technologies (IDT) was: 5’-GATTTCGAATTGATTTCGGGTGG-3′. The sequence for unc-58 and dpy-10 sgRNA were as previously described (39). The repair templates used for co-CRISPR genes unc-58 and dpy-10 were as previously described (38). The repair template targeted to nlg-1 was: 5’-ACGCCTCAAAAATTAAAGGTAGAACATTTATTTCATCATTATAGGACCACCCGAAATCAATTTGCAATGGAGTTCTGAATGCTCTTAGCGACGTACTTTACACCGCACCTCTCATTGAAACATTGCGAAG-3′. The mutated codon is underlined. Repair templates were purchased from IDT. All injection reagents were diluted in water and injected at a final concentration of 50 ng/μl. Worms were screened using the co-CRISPR phenotype then recombination of the repair template was screened using restriction digest and finally recombinant worms were sequenced over the targeted region to identify the mutation. The CRISPR-edited line nlg-1(qa3780) was outcrossed against the wild-type background used in paired behavioural assays twice.
Protein sequence alignment
Multiple protein sequence alignment of C. elegans NLG-1, and human NLGN1–4 was performed using the Clustal W method. C. elegans NLG-1 sequence was downloaded from WormBase version WS274. The longest NLG-1 isoform (C40C9.5e) was used for analysis. Human NLGN1–4 sequences were downloaded from NCBI. Accession numbers for the sequences used are as follows: NLGN1, NP_001352856; NLGN2, XP_005256801; NLGN3, NP_061850; NLGN4, AAQ88925.
Supplementary Material
{kind=link}
Acknowledgements
Cas9 plasmid, sgRNA’s targeting unc-58 and dpy-10 and unc-58 and dpy-10 repair templates were kindly provided by Thomas Boulin.
Conflict of Interest statement. None declared.
Contributor Information
Helena Rawsthorne, School of Biological Sciences, Highfield Campus, University of Southampton, Southampton SO17 1BJ, UK.
Fernando Calahorro, School of Biological Sciences, Highfield Campus, University of Southampton, Southampton SO17 1BJ, UK.
Emily Feist, School of Biological Sciences, Highfield Campus, University of Southampton, Southampton SO17 1BJ, UK.
Lindy Holden-Dye, School of Biological Sciences, Highfield Campus, University of Southampton, Southampton SO17 1BJ, UK.
Vincent O’Connor, School of Biological Sciences, Highfield Campus, University of Southampton, Southampton SO17 1BJ, UK.
James Dillon, School of Biological Sciences, Highfield Campus, University of Southampton, Southampton SO17 1BJ, UK.
Funding
nlg-1(ok259) was provided by CGC funded by NIH Office of Research Infrastructure Programs (P40 OD010440); the Gerald Kerkut Charitable Trust.
References
- 1. Baio, J., Wiggins, L., Christensen, D.L., Maenner, M.J., Daniels, J., Warren, Z., Kurzius-Spencer, M., Zahorodny, W., Rosenberg, C.R., White, T. et al. (2018) Prevalence of autism Spectrum disorder among children aged 8 years-autism developmental disabilities monitoring network, 11 sites, United States, 2014. MMWR-Morb. Mortal. Wkly. Rep., 67, 1280–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sharma, S.R., Gonda, X. and Tarazi, F.I. (2018) Autism Spectrum disorder: classification, diagnosis and therapy. Pharmacol. Ther., 190, 91–104. [DOI] [PubMed] [Google Scholar]
- 3. Endevelt-Shapira, Y., Perl, O., Ravia, A., Amir, D., Eisen, A., Bezalel, V., Rozenkrantz, L., Mishor, E., Pinchover, L., Soroka, T. et al. (2018) Altered responses to social chemosignals in autism spectrum disorder. Nat. Neurosci., 21, 111–119. [DOI] [PubMed] [Google Scholar]
- 4. De Rubeis, S., He, X., Goldberg, A.P., Poultney, C.S., Samocha, K., Cicek, A.E., Kou, Y., Liu, L., Fromer, M., Walker, S. et al. (2014) Synaptic, transcriptional and chromatin genes disrupted in autism. Nature, 515, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bourgeron, T. (2015) From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat. Rev. Neurosci., 16, 551–563. [DOI] [PubMed] [Google Scholar]
- 6. Giovedi, S., Corradi, A., Fassio, A. and Benfenati, F. (2014) Involvement of synaptic genes in the pathogenesis of autism spectrum disorders: the case of synapsins. Front. Pediatr., 2, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pinto, D., Delaby, E., Merico, D., Barbosa, M., Merikangas, A., Klei, L., Thiruvahindrapuram, B., Xu, X., Ziman, R., Wang, Z.Z. et al. (2014) Convergence of genes and cellular pathways dysregulated in autism Spectrum disorders. Am. J. Hum. Genet., 94, 677–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huguet, G., Ey, E. and Bourgeron, T. (2013) The genetic landscapes of autism Spectrum disorders. Annu. Rev. Genomics Hum. Genet., 14, 191–213. [DOI] [PubMed] [Google Scholar]
- 9. Sudhof, T.C. (2008) Neuroligins and neurexins link synaptic function to cognitive disease. Nature, 455, 903–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jamain, S., Quach, H., Betancur, C., Rastam, M., Colineaux, C., Gillberg, I.C., Soderstrom, H., Giros, B., Leboyer, M., Gillberg, C. et al. (2003) Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet., 34, 27–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Etherton, M., Foldy, C., Sharma, M., Tabuchi, K., Liu, X.R., Shamloo, M., Malenka, R.C. and Sudhof, T.C. (2011) Autism-linked neuroligin-3 R451C mutation differentially alters hippocampal and cortical synaptic function. Proc. Natl. Acad. Sci. U. S. A., 108, 13764–13769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chadman, K.K., Gong, S.C., Scattoni, M.L., Boltuck, S.E., Gandhy, S.U., Heintz, N. and Crawley, J.N. (2008) Minimal aberrant Behavioral phenotypes of Neuroligin-3 R451C Knockin mice. Autism Res., 1, 147–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tabuchi, K., Blundell, J., Etherton, M.R., Hammer, R.E., Liu, X.R., Powell, C.M. and Sudhof, T.C. (2007) Neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science, 318, 71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kaletta, T. and Hengartner, M.O. (2006) Finding function in novel targets: C. elegans as a model organism. Nat. Rev. Drug Discov., 5, 387–398. [DOI] [PubMed] [Google Scholar]
- 15. Sonnhammer, E.L.L. and Durbin, R. (1997) Analysis of protein domain families in Caenorhabditis elegans. Genomics, 46, 200–216. [DOI] [PubMed] [Google Scholar]
- 16. Hobert, O. (2013) The neuronal genome of Caenorhabditis elegans. Worm Book, ed. The C. elegans Research Community, Worm Book. doi: 10.1895/wormbook.1.161.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bargmann, C.I. (1998) Neurobiology of the Caenorhabditis elegans genome. Science, 282, 2028–2033. [DOI] [PubMed] [Google Scholar]
- 18. Cherra, S.J. and Jin, Y.S. (2015) Advances in synapse formation: forging connections in the worm. Wiley Interdiscip. Rev. Dev. Biol., 4, 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Calahorro, F. and Ruiz-Rubio, M. (2012) Functional phenotypic rescue of Caenorhabditis elegans Neuroligin-deficient mutants by the human and rat NLGN1 genes. PLoS One, 7, e39277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hunter, J.W., Mullen, G.P., McManus, J.R., Heatherly, J.M., Duke, A. and Rand, J.B. (2010) Neuroligin-deficient mutants of C. elegans have sensory processing deficits and are hypersensitive to oxidative stress and mercury toxicity. Dis. Model. Mech., 3, 366–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shtonda, B.B. and Avery, L. (2006) Dietary choice behavior in Caenorhabditis elegans. J. Exp. Biol., 209, 89–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Scott, E., Hudson, A., Feist, E., Calahorro, F., Dillon, J., de Freitas, R., Wand, M., Schoofs, L., O’Connor, V. and Holden-Dye, L. (2017) An oxytocin-dependent social interaction between larvae and adult C. elegans. Sci. Rep., 7, 10122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Crawley, J.N. (2012) Translational animal models of autism and neurodevelopmental disorders. Dialogues Clin. Neurosci., 14, 293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Calahorro, F., Alejandre, E. and Ruiz-Rubio, M. (2009) Osmotic avoidance in Caenorhabditis elegans: synaptic function of two genes, orthologues of human NRXN1 and NLGN1, as candidates for autism. J. Vis. Exp., 34, 1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Graf, E.R., Zhang, X.Z., Jin, S.X., Linhoff, M.W. and Craig, A.M. (2004) Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell, 119, 1013–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maro, G.S., Gao, S.B., Olechwier, A.M., Hung, W.L., Liu, M., Ozkan, E., Zhen, M. and Shen, K. (2015) MADD-4/Punctin and Neurexin Organize C. elegans GABAergic Postsynapses through Neuroligin. Neuron, 86, 1420–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rothwell, P.E., Fuccillo, M.V., Maxeiner, S., Hayton, S.J., Gokce, O., Lim, B.K., Fowler, S.C., Malenka, R.C. and Sudhof, T.C. (2014) Autism-associated Neuroligin-3 mutations commonly impair striatal circuits to boost repetitive Behaviors. Cell, 158, 198–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bergan, J.F. (2015) Neural computation and neuromodulation underlying social behavior. Integr. Comp. Biol., 55, 268–280. [DOI] [PubMed] [Google Scholar]
- 29. Milward, K., Busch, K.E., Murphy, R.J., de Bono, M. and Olofsson, B. (2011) Neuronal and molecular substrates for optimal foraging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A., 108, 20672–20677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Singh, S.K. and Eroglu, C. (2013) Neuroligins provide molecular links between syndromic and nonsyndromic autism. Sci. Signal., 6, re4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou, K.M., Cherra, S.J., Goncharov, A. and Jin, Y.S. (2017) Asynchronous cholinergic drive correlates with excitation-inhibition imbalance via a neuronal Ca2+ sensor protein. Cell Rep., 19, 1117–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chalasani, S.H., Chronis, N., Tsunozaki, M., Gray, J.M., Ramot, D., Goodman, M.B. and Bargmann, C.I. (2016) Dissecting a circuit for olfactory behaviour in Caenorhabditis elegans. Nature, 533, 130–130. [DOI] [PubMed] [Google Scholar]
- 33. McDiarmid, T.A., Belmadani, M., Liang, J., Meili, F., Mathews, E.A., Mullen, G.P., Hendi, A., Wong, W.-R., Rand, J.B., Mizumoto, K. et al. (2019) Systematic phenomics analysis of autism-associated genes reveals parallel networks underlying reversible impairments in habituation. Proc. Natl. Acad. Sci. U. S. A., 117, 656–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wong, W.R., Brugman, K.I., Maher, S., Oh, J.Y., Howe, K., Kato, M. and Sternberg, P.W. (2019) Autism-associated missense genetic variants impact locomotion and neurodevelopment in Caenorhabditis elegans. Hum. Mol. Genet., 28, 2271–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brenner, S. (1974) The genetics of Caenorhabditis elegans. Genetics, 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Calahorro, F., Keefe, F., Dillon, J., Holden-Dye, L. and O'Connor, V. (2019) Neuroligin tuning of pharyngeal pumping reveals extrapharyngeal modulation of feeding in Caenorhabditis elegans. J. Exp. Biol., 222, 1–11. [DOI] [PubMed] [Google Scholar]
- 37. Calahorro, F., Holden-Dye, L. and O'Connor, V. (2015) Analysis of splice variants for the C. elegans orthologue of human neuroligin reveals a developmentally regulated transcript. Gene Expr. Patterns, 17, 69–78. [DOI] [PubMed] [Google Scholar]
- 38. El Mouridi, S., Lecroisey, C., Tardy, P., Mercier, M., Leclercq-Blondel, A., Zariohi, N. and Boulin, T. (2017) Reliable CRISPR/Cas9 genome engineering in Caenorhabditis elegans using a single efficient sgRNA and an easily recognizable phenotype. G3-Genes Genomes Genet., 7, 1429–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Arribere, J.A., Bell, R.T., Fu, B.X.H., Artiles, K.L., Hartman, P.S. and Fire, A.Z. (2014) Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics, 198, 837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.