

Abstract
Bioactive peptides derived from food proteins are becoming increasingly popular due to the growing awareness of their health-promoting properties. The structure and mechanism of anti-cancer action of pentapeptide Glu-Gln-Arg-Pro-Arg (EQRPR) derived from a rice bran protein are not known. Theoretical and experimental methods were employed to fill this gap. The conformation analysis of the EQRPR pentapeptide was performed first and the obtained lowest energy conformer was optimized. The experimental structural data obtained by FTIR and CD spectroscopies agree well with the theoretical results. d-isomer introduced one-by-one to each position and all D-isomers of the peptide were also examined for its possible anti-proteolytic and activity enhancement properties. The molecular docking revealed avid binding of the pentapeptide to the integrins α5β1 and αIIbβ3, with Kd values of 90 nM and 180 nM, respectively. Moreover, the EQRPR and its D-isomers showed strong binding affinities to apo- and holo-forms of Mpro, spike glycoprotein, ACE2, and dACE2. The predicted results indicate that the pentapeptide may significantly inhibit SARS-CoV-2 infection. Thus, the peptide has the potential to be the leading molecule in the drug discovery process as having multifunctional with diverse biological activities.
Keywords: Anti-cancer peptides, Molecular modeling, Molecular docking, FTIR, Circular dichroism, Cationic peptides
Graphical abstract
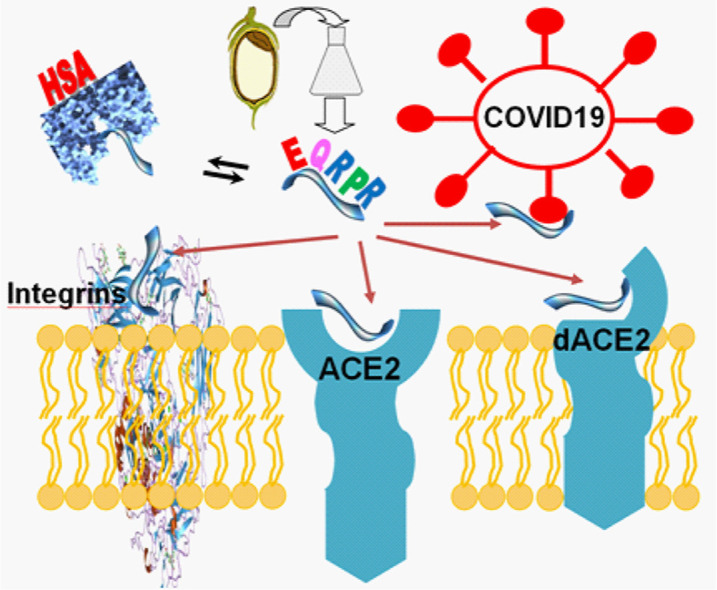
1. Introduction
Rice bran is rich in proteins, lipids, dietary fibers, vitamins and minerals. Components of rice bran show significant biological activities that make it a promising raw material for various medical applications [[1], [2], [3]]. Particularly, proteins and small molecular weight peptides derived from rice bran exhibit a wide variety of biological activities, including anti-cancer, anti-inflammatory, anti-diabetic and angiotensin-converting enzyme (ACE) inhibitory properties and may be valuable against certain chronic diseases [[4], [5], [6], [7], [8]]. Because of having high activity and low toxicity properties, some of these peptides are in clinical trials or are considered to have the great potential to be drugs [9,10].
Significant advances in cancer therapy by short peptides lead to a new class of peptides, namely anti-cancer peptides (ACPs) [11]. ACPs are classified into three groups (inhibitor activity, necrosis activity and apoptosis activity) with respect to their biological actions [11,12]. The peptides that adhere to the cell surface are linked to the inhibitory activity. For anti-cancer activity, some of ACPs interact with integrins to inhibit migration and metastatic processes of the cancer cells [11,13]. Direct interaction of ACPs with cancer cell membrane that destabilizes and/or disrupts the cell membrane stimulates necrotic processes [[14], [15], [16]]. For apoptosis activity, it is considered that some class of ACPs is internalized inside the cancer cell and then permeates and swells the mitochondria membrane. The result of this action may result in the release Cyt c promoting apoptosis in cancer cells [17,18]. ACPs inhibit tumor angiogenesis and participate in immune regulation to fight cancer cells by inducing the production of cytokines [19].
The cationic pentapeptide Glu-Gln-Arg-Pro-Arg (EQRPR) derived from rice bran by enzyme hydrolysis has been characterized to be an anti-cancer agent. The peptide shows inhibitory effects on cell proliferation and apoptosis activity in many cancer types [20,21].
Structural information about the cationic pentapeptide EQRPR as well as the mechanism of action in anti-cancer activity is absent. The most ACPs and other bioactive peptides are cationic with alpha-helical structures, anti-cancer activity of which higher compared to that of beta-pleated, beta-sheet and random coil APCs [12]. From sequence consideration, the EQRPR is not likely to have an alpha-helical conformation. Proline (Pro) residue located in the N-terminus can initiate alpha-helix formation. However, in positions distant from the N-terminus, as in EQRPR, Pro residue destabilizes alpha-helical conformation [22,23].
We have employed theoretical and experimental methods to elucidate the conformational features of the cationic pentapeptide EQRPR. Advanced molecular modeling indicates that the peptide mainly forms beta-structure and beta-turn conformations in the lowest energy state. Assuming that the peptide may function in the excited states, conformations having energies higher than the lowest energy were also analyzed. Fourier transform infrared (FTIR) and circular dichroism (CD) spectroscopies agree well with the theoretical analysis. Molecular docking was performed to test binding of the cationic pentapeptide EQRPR to integrins α5β1 and αIIbβ3 that may implicate the peptide in anti-proliferation activity for a particular anti-cancer function. High binding affinities of the peptide to the integrins suggest an anti-cancer activity. Docking studies of the peptide with integrin αIIbβ3 and ACE 2 receptor demonstrate that the peptide has a potential against SARS-CoV-2 infection. Altogether, the cationic pentapeptide EQRPR shows multifunctional properties that demand further investigation of this peptide both experimentally and theoretically.
d-amino acid substitution in the peptide may increase proteolytic stability and/or biological activity compared to that of the native peptide [[24], [25], [26], [27], [28]]. Therefore, d-isomer modified peptides, d-amino acid substituted one-by-one at each position and all D-isomers were also examined.
2. Materials and methods
The peptide EQRPR and its sequential d-isomer substituted and all d-isomer peptides were custom synthesized by Synpeptide Co., Ltd, Shanghai, China. Common chemical reagents for the preparation of buffer were purchased from Sigma-Aldrich.
2.1. Experimental studies
2.1.1. FTIR spectroscopy
The FTIR spectra of peptide solutions were recorded using a BioATR cell of the spectrometer (VERTEX 70v, Bruker, Inc., Germany) with a liquid nitrogen-cooled mercury-cadmium telluride detector. All FTIR spectra were measured with 2 cm−1 resolution in the spectral range from 400 cm−1 to 4000 cm−1. For each sample, 256 scans were collected. Spectral contributions of water were removed from the sample spectra by spectral subtraction procedure to obtain a flat recording around 2130 cm−1 associated only with water. After baseline correction spectral regions that cover Amide I and Amide II bands were used to analyze the secondary structure of the peptide solutions. Fitting the spectra to multiple Gaussian components was performed using OriginLab software (OriginLab Corp., Northampton, MA). Because of variations of intensity ratios of Amide I and Amide II bands, there will be some uncertainties in choosing a baseline for Amide I. Therefore, all peptide FTIR spectra (Amide I + Amide II) were subjected to Global analysis where only width and peak positions of Amide II band were global parameters. A certain restriction was applied in multiple Gaussian component fitting of Amide I bands. The band position and width values associated with various secondary structures were allowed to vary in a fixed range [29,30]. Fixing the number of the peaks, their spectral positions and limiting the peak width range, significantly decreases the number of floating parameters and uncertainties. The application of this methodology in FTIR spectral fitting shows consistent results [29,30].
The amide III bands of the peptides were further analyzed for the confirmation of the obtained results on the secondary structure. The amide III bands of the FTIR spectra of proteins and peptides cover a spectral region of 1200–1350 cm−1 [31]. The six Gaussian components were sufficient to fit the Amide III spectral regions of the peptides. As in the case of Amide I, band positions and width values were allowed to vary in a fixed range. One component outside of secondary structure elements was added to cover the long wavenumber tail of the spectra.
2.1.2. Circular dichroism (CD) spectroscopy
The CD spectra of the peptide solutions were recorded using a Chirascan V100 (Applied Photophysics, UK) circular dichroism spectrometer. Far-UV CD measurements were recorded from 190 to 260 nm with a step of 0.5 nm and bandwidth of 1 nm. A path length of a quartz cell was 0.2 mm.
Secondary structure analyses of the peptide solutions from far-UV CD spectra were performed using the DICHROWEB server (http://dichroweb.cryst.bbk.ac.uk/html/home.shtml) [32,33]. The CDSSTR method with SMP 180 library dataset was used for the calculation of secondary structure content [34,35] NRMSD values in the structure analysis of the peptide solutions were less than 0.02.
2.2. Computational studies
2.2.1. Conformational analysis and structure optimization
The method of molecular mechanics was applied to study the spatial structure and conformational properties of the cationic pentapeptide EQRPR and its sequentially substituted single d-isomer and all d-isomer analogs. Table 1 shows the amino acid sequences of all L-pentapeptide and its analogs. The application of the method of molecular mechanics permits the determination of a set of possible stable spatial forms of the peptide and its analogs.
Table 1.
Amino acid sequence of the anticancer pentapeptide and its single and all d-isomer substituted analogs.
| № | Designated name of the peptides | Amino acid sequence of the peptide and its analogs |
|---|---|---|
| 1. | EQRPR | Glu1 – Gln2 – Arg3 – Pro4 – Arg5 |
| 2. | [DGlu1]-EQRPR | DGlu – Gln-Arg-Pro – Arg |
| 3 | [DGln2]-EQRPR | Glu – DGln – Arg-Pro-Arg |
| 4. | [DArg3]-EQRPR | Glu – Gln – DArg – Pro – Arg |
| 5. | [DPro4]-EQRPR | Glu-Gln-Arg – DPro – Arg |
| 6. | [DArg5]-EQRPR | Glu-Gln-Arg – Pro – DArg |
| 7. | [DX1-5]-EQRPR | DGlu - DGln - DArg -DPro – Darg |
The molecular geometry of the L-pentapeptide and its various D-isomers were investigated by conformational analysis by using the program written by Ref. [36], benefiting from the Ramachandran maps [37,38].
The lowest energy conformations of the pentapeptide and its D-isomers obtained after the conformational analyses were optimized using the Amber force field molecular mechanics method [39] available in the Gaussian16 program package [40]. Moreover, for docking studies, the 310 helix, alpha helix, antiparallel beta, parallel beta, beta-sheet, left-hand alpha, and pi helix secondary structures of the pentapeptide EQRPR were formed by altering the dihedral angles. The energies of the obtained structures were obtained by PM3MM calculations.
2.2.2. Molecular docking
For docking studies the crystal structure of DNA, ACE-2, COVID-19 main protease apo- and holo-forms, spike glycoprotein, α5β1 and αııbβ3 integrins and HSA were obtained from the protein data bank (PDB IDs: 1bna; 6m0j; 6m03; 6lu7; 6vxx; 4wk0; 3zdx; 5z0b, respectively) [[41], [42], [43], [44], [45], [46], [47]]. Molecular docking simulations were performed by the Autodock Vina program [48] and binding affinities were calculated. The binding free energies of the most stable ligand-DNA and ligand-protein systems, determined by molecular docking analysis, were calculated by the programs developed by Ref. [49] and the ACFIS 2.0 [[50], [51], [52], [53]].
The active sites of receptors were screened by using the CAVER program [54].
2.2.3. The toxicological and physicochemical properties prediction of EQRPR pentapeptide
The toxicological risk and physicochemical properties of the EQRPR pentapeptide were obtained using the OSIRIS [55]. The ADMET (absorption, distribution, metabolism, excretion and toxicity) properties of the pentapeptide, from its 2D structure, were predicted using admetSAR [56].
3. Results and discussion
3.1. Theoretical study on the tertiary structure of the pentapeptide EQRPR
The structure of the most stable conformer of EQRPR obtained by optimization by AMBER force field molecular mechanics method embedded in GAUSSIAN 16 program package is shown in Fig. 1 . The dihedral angles of the optimized structures of the EQRPR pentapeptide and various D-isomers are tabulated in Table 1S (in Supplementary Data). The comparison of the molecular structure of the most stable conformer (I) to those of the molecular models of EQRPR peptide in 310helix (II), alpha-helix (III), antiparallel-beta (IV), beta-sheet (V), lefthand-alpha (VI), parallel-beta, and pi-helix (VII) structures are given in Fig. 1S (in Supplementary Data).
Fig. 1.

The molecular structure of the most stable conformer of EQRPR obtained by optimization with the AMBER force field molecular mechanics method (a) and its wireframe representation (b).
The energies, obtained by PM3MM calculations of the optimized structures of all L- and various D-analogs of EQRPR, together with its non-optimized various conformations were given in Table 2 S, comparatively. As seen in Table 2S, among all L analogs of EQRPR, the antiparallel-beta plate sheet conformation (−132.288 kcal/mol) is the second-lowest energy conformation after the most stable conformer (−183.268 kcal/mol) obtained by optimization with the AMBER force field. Moreover, the d-isomer substitution of the first amino acid gives the lowest energy (−202.311 kcal/mol), and the d-isomer substitution of the 4th amino acid gives the second lowest energy (−183.971 kcal/mol), in comparison to all optimized isomers of EQRPR pentapeptide (see Table 2S). The results revealed that the substitution of the first or fourth amino acid residue with its d-isomer increases the stabilization of the pentapeptide.
Table 2.
The binding affinities (ΔG) of optimized all L- and various D-analogs of EQRPR pentapeptide, docked into the target DNA or proteins in kcal/mol.
| Target | EQRPR (all L-) | D-EQRPR (all D-) | [DGlu1]-EQRPR | [DGln2]-EQRPR | [DArg3]-EQRPR | [DPro4]-EQRPR | [DArg5]-EQRPR |
|---|---|---|---|---|---|---|---|
| 1BNA-DNA | −6.2 | −6.8 | −6.4 | −6.4 | −6.4 | −6.1 | −6.6 |
| 3zdx-integrin | −9.2 | −8.5 | −8.5 | −9.1 | −8.0 | −8.6 | −8.9 |
| 4wk0-integrin | −9.6 | −10.3 | −9.8 | −8.7 | −9.7 | −9.6 | −7.4 |
3.2. FTIR spectroscopy of the peptide EQRPR
FTIR spectroscopy gives quantitative information regarding the secondary structures of proteins and peptides. IR absorption bands do not depend on the L/D isomeric state of the constituent amino acids of peptides. Therefore, FTIR spectroscopy is best suited to estimate the secondary content of the peptide EQRPR and its d-isomer analogs.
Amide I band components cover the 1700 cm−1- 1600 cm−1 region and arise primarily from the C O stretching vibration with a minor contribution of the NH in-plane bend, the CCN deformation and the out-of-phase CN stretching vibration [57,58]. Amide I vibration exhibits a strong signal by protein and peptides and is sensitive to the secondary structure. In FTIR spectroscopy Amide I band is widely used to estimate secondary structure elements. The frequency of this vibration band depends on the geometry of hydrogen bonding involving the backbone amide group [57,58]. In the fitting procedure, difficulties arise from the fact that Amide I components responsible for alpha-helix, beta-sheet, beta-turn and random coil conformations are overlapping. The optimal peak parameters established in the work [30] were used in the fitting procedure of the Amide I bands of the peptides. This method was successfully tested for proteins having various beta-sheet content.
The FTIR spectra of the peptides and their best fitting curves using the methodology presented in Ref. [30] are shown in Fig. 2 A. Usually, the amide I band of the peptides and proteins are used to calculate their secondary structures. To eliminate possible errors that may be resulted from the uncertainties in baseline levels of amide I, amide I and amide II bands were analyzed together. All FTIR spectra show adequate fit to the multiple Gaussian components with restricted parameters (see the method for details). The secondary structure fractions derived from the fitting procedure are demonstrated in Fig. 2B. The common feature for all peptides is almost the lack of alpha-helix structure. This result is consistent with the position of Pro in the peptides, as mentioned above. The peptides show from 36% to 47% secondary structure related to beta-structures. The beta-sheet aggregates (1611 cm−1 and 1696 cm−1) indicate the possibility of forming the sheet structure from several peptide molecules, particularly in high concentrations. Indeed, to get high-quality FTIR spectra, a high concentration of the peptide (about 0.64 mM) were used. Calculated values for beta-turn structure vary within 28%–39% interval. It is undeniable that the single pentapeptide molecule may not contain the secondary structures derived from the fitting procedure. Instead, data indicate that each peptide molecule samples structures with different secondary structures.
Fig. 2.

A) Normalized amide I and amide II bands of FTIR spectra of the peptide EQRPR and its d-isomer analogs. Thick black and thin white lines represent experimental and best fitting spectra, respectively. The best fitting spectra are results of eight Gaussian component fitting (See Methods for details). Two Gaussian components around 1730 cm−1 and 1545 cm−1 that represent the contribution from side-chains and amid II, respectively, were not considered in the calculation of the secondary structures of the peptides. B) and C) Secondary structure content of the peptide EQRPR and its d-isomer analogs. A and B are data derived from FTIR and CD spectroscopies, respectively.
Amide III bands that cover the 1200 cm−1 - 1350 cm−1 region were also examined to estimate the secondary structure elements of the peptide. Amide III band arises from N–H in-plane bending coupled with C–N stretching. The C–H and N–H deformation vibrations also contribute to the Amide III band [31]. The Amide III vibration exhibits a weak signal and, therefore, is not widely used for the analysis of secondary structure elements of peptides and proteins. Because of being a weak signal, baseline uncertainties in the region of 1200 cm−1 – 1350 cm−1 influence estimation of the secondary structure [31]. However, in the Amide III region, the overlap of the bands responsible for secondary structure elements is much less compared to that of Amide I. Therefore, the inspections of the Amide III bands together with Amide I bands are valuable to validate the estimation of secondary structure content of proteins and polypeptides.
The FTIR spectra of the peptide in the Amide III region and their best fitting curves to Gaussian components are shown in Fig. 2S. The assignment of the peak frequencies and the secondary structure content of the peptides calculated from the fitting procedure are shown in Table 3 S. These data corroborate the result of the Amide I analysis to show that the peptides do not assume alpha-helix conformation and mostly adopt beta-sheet conformation. However, as indicated above the quantitative values for secondary structure content estimated by Amide III bands are less accurate than from Amide I bands.
Table 3.
The binding affinities and the molecular interactions between EQRPR and target DNA, and proteins, obtained by molecular docking simulationsa.
| Ligand/Target | DNA (1BNA) | α5β1 (4WK0) | αIIBβ3 (3ZDX) | apo-form (6M03) | holo-form |
spike glycoprotein (6VXX) | ACE2 |
Truncated ACE2 | dACE2 | Albumin |
|---|---|---|---|---|---|---|---|---|---|---|
| (6LU7) | (6M0J) | (5Z0B) | ||||||||
| EQRPR (all L-) | DG2 | PHE21 | LEU23 | THR24 | THR26 | THR(B)912 | PHE40 | ASN599 | TYR454 | ARG114 |
| DG4 | SER22 | ASP24 | LEU141 | HIS41 | ARG(C)1091 | TRP69 | LYS600 | ARG482 | LEU115 | |
| DA5 | VAL23 | PHE25 | ASN142 | TYR54 | GLU(A)1092 | ALA348 | ASN601 | GLU483 | TYR138 | |
| DA6 | ARG106 | ALA95 | SER144 | PHE140 | GLU(B)1092 | ASP350 | SER602 | ILE484 | GLU141 | |
| DT7 | PHE168 | SER96 | CYS145 | ASN142 | GLY(B)1093 | ASP382 | PHE603 | VAL485 | HIS146 | |
| DT20 | GLU171 | SER99 | HIS163 | SER144 | ARG(B)1107 | LEU391 | LYS676 | ASN599 | LYS190 | |
| DC21 | SER234 | PHE171 | GLN189 | CYS145 | GLN(B)1113 | ASN394 | ARG678 | ASN601 | LYS519 | |
| DG22 | VAL235 | SER173 | HIS163 | ASN(A)1119 | HIS401 | ASN682 | GLY605 | |||
| DC23 | LYS269 | TYR237 | GLU166 | ASN(B)1119 | PHE684 | SER680 | ||||
| TYR287 | SER238 | GLN189 | TYR781 | THR803 | ||||||
| LEU355 | ARG261 | SER804 | ||||||||
| SER417 | HIS291 | |||||||||
| ARG420 | VAL293 | |||||||||
| ILE360 | ||||||||||
| SER420 | ||||||||||
| LEU421 | ||||||||||
| ARG422 | ||||||||||
| Binding affinity (kcal/mol) | −6.2 | −9.6 | −9.2 | −6.6 | −7.5 | −8.0 | −7.6 | −6.7 | −8.0 | −8.4 |
Truncated ACE2 target is the optimized truncated protein that lacks 356 amino acids from the N-terminal of ACE2 and has 449 amino acids.
The secondary structure of the peptides was also investigated by CD spectroscopy.
3.3. CD spectroscopy of the peptide EQRPR
Far-UV CD spectroscopy is a powerful tool to determine the secondary structure of peptides and proteins [59,60]. Far-UV CD spectra of basic secondary structure components (beta-sheet, alpha-helix and random coil) are very distinct. However, the CD spectra of peptides and proteins are different for L – and D-isomers. In fact, CD spectra of D-isomers have ellipticities equivalent to that of matching l-isomer but of opposite sign. Therefore, the CD spectrum of each peptide with single d-isomer amino acid will contain a CD band that is a mirror-image of the corresponding l-isomer. The CD spectra of all peptides considered in this study are shown in Fig. S2. Although FTIR spectroscopy reveals a similar secondary structure for peptides, CD spectra of the peptides, predominantly determined from the electronic transition in the amide groups, exhibit very different patterns (Fig. S2). D-isomers, all- and sequentially substituted single d-isomer, are the origin of the differences in CD spectra. The contribution of d-isomer amino acid to the far-UV CD spectra depends on its position in the sequence. The contribution of the “mirror-image type” spectral component coming from the single d-isomer in peptides is very difficult to estimate. However, the CD spectrum of the peptide with all d-isomer can be used for calculation by taking the mirror-image of the CD spectrum. The mirror-image of mirror-imaged spectra of d-isomer will produce an l-isomer type CD spectrum. The CD spectra of the peptide EQRPR and its all d-isomer analog are demonstrated in Fig. 3 A. It is evident that the mirror-image of the CD spectrum of all d-isomer analog produces a spectrum very similar to that of all l-isomer peptide. CDSSTR derived curves of the peptides are consistent with the corresponding experimental spectrum. The results of the calculation are shown in Fig. 2C. The lack of the alpha-helix fraction obtained from CD spectra is very consistent with the results of FTIR analysis. Thus, secondary structure content obtained by analysis of CD spectra shows that the peptide mainly samples beta-sheet, beta-turn and random coil conformations. Despite the qualitative agreement between the results of the two methods, there are some quantitative discrepancies in the estimation of secondary structure fractions. Estimation of secondary structure content from CD spectra is based on the use of various protein libraries. The accuracy of the estimate is high if the protein library contains data of proteins that are structurally closely related to examined one. Currently, the short peptides are not well represented in the protein library data. Therefore, it is reasonable to speculate that the secondary structure estimate from CD data is affected by lack of the data for short peptides in the protein library.
Fig. 3.

A) Far-UV CD spectra of the peptide EQRPR and derived curves calculated by CDSSTR method. Positive and negative sign dashed black curves represent the peptides with all d-isomer and its mirror-image, respectively. Solid black line is CD spectrum of all l-isomer peptide. CDSSTR derived curves of the peptides conform well to the respective experimental spectra. B) Secondary structure transition in the peptide EQRPR induced by TFE. Far-UV CD spectra of the peptide in buffer pH 7.3 (open cirles), in 50% (v/v) TFE (solid circles) and their difference (open triangles). The minimum observed around 218 nm in the difference spectrum indicates beta-sheet formation.
The most cationic anti-cancer peptides possess alpha-helix structure. Therefore, the secondary structure of the peptides was investigated in 2,2,2- trifluoroethanol (TFE) to reveal a propensity for alpha-helix formation. It is well established that TFE induces alpha-helix formation in peptides and proteins [61,62]. TFE titration of the peptide has been used to determine the hidden propensity for alpha-helix formation of small peptides [61]. In all cases, alpha-helix formations in peptides are saturated at 50% (v/v) TFE. Far-UV CD spectra of the peptide in the buffer with and without 50% TFE are shown in Fig. 3B. Contrary to the most expected transition induced by TFE, the difference CD spectrum reveals beta-sheet formation for a small portion of the peptide. Thus, theoretical and experimental data indicate a lack of alpha-helix structure in the peptide EQRPR. Moreover, this peptide does not have any hidden propensity for alpha-helix formation.
3.4. In silico molecular docking analysis of EQRPR as anticancer agent
To evaluate the anticancer activity of EQRPR pentapeptide, molecular docking studies were performed and the interactions of EQRPR ligand with DNA and target proteins α5β1 and αIIbβ3 integrins were revealed.
For the docking simulations of EQRPR pentapeptide with DNA, the crystal structure of DNA was obtained from the protein data bank (PDB ID: 1BNA) [41], and, was prepared for the docking by removing water molecules in DNA and adding polar hydrogens to it. The optimized structure of EQRPR was adapted for the docking. The partial charges of the EQRPR molecule were calculated using the Geistenger method. The active site of DNA was defined within the grid size of 40Å × 40Å × 40 Å.
The EQRPR pentapeptide is found to interact with DG2, DG4, DA5, DA6, DC21, DG22, DC23 and DT20 residues of DNA via the intermolecular hydrogen bonds. These hydrogen bonds are between DG2 and Glu (2.59 Å); DG4 and Glu (two hydrogen bonds; 2.43 ve 2.82 Å); DG4 and Gln (1.99 Å); DA5 and Gln (3.48 Å); DA6 and Arg5 (3.40 Å); DC21 and Arg5 (2.54 Å); DG22 and Glu (3.08 and 2.78 Å); DG22 and Gln (2.53 Å); DG22 and Pro (3.40 Å); DC23 and Gln (2.01 Å); DT20 and Arg5 (2.09 ve 2.74 Å). Moreover, there is an attractive charge interaction between DA5 and Glu residue of the pentapeptide, at 4.74 Å length, and an unfavorable negative-negative interaction between DT7 and Arg5 residue at 3.98 Å length. The result shows that the binding affinity (ΔG) is −6.2 kcal/mol (See Fig. 4 ). The docking simulations of d-isomer analogs of the EQRPR pentapeptide into DNA were also investigated, and it was revealed that the pentapeptide, all residues of which were d-amino acids, showed the highest binding affinity to DNA (−6.8 kcal/mol) (see Table 2).
Fig. 4.

Molecular docking results of EQRPR bound to DNA (a), the doted lines present the interactions of EQRPR with the nucleic acids of DNA (b).
The anti-proliferation effect of the EQRPR pentapeptide for the anticancer function was revealed by molecular docking studies of the pentapeptide into target proteins α5β1 and αIIbβ3 integrins. After the α5β1 integrin (PDB ID: 4WK0) was prepared for molecular docking, the binding probability of EQRPR pentapeptide in the two different active sites was examined. The molecular model of α5β1 integrin was shown in Fig. 3S. The circles 1 and 2 shown in Fig. 3S have roughly indicated the two active sites of α5β1. The docking simulations of EQRPR to the α5β1 integrin were performed for both active sites. The most effective binding was found in the active site 2 of α5β1 integrin with −9.6 kcal/mol binding energy. Fig. 5 a-c shows the 3D docked view of EQRPR in the active site 2 of α5β1 integrin. The interaction diagrams of EQRPR- α5β1 integrin complex are also indicated in the figure.
Fig. 5.

The 3D docked views of the most stable conformer of EQRPR in active site 2 of α5β1 integrin (a–c) and in αIIBβ3 integrin (d–f). The ligand interaction diagrams of receptor ligand complexes are show.
The molecular docking of αIIBβ3 integrin (PDB ID: 3ZDX) with EQRPR resulted in a binding affinity of −9.2 kcal/mol. The 3D docked view of EQRPR in αIIBβ3 integrin and the estimated interactions are shown in Fig. 5 d-f.
The effect of the d-isomer substitutions on the binding free energy of pentapeptide-DNA and pentapeptide-integrin interaction complexes was evaluated by docking simulations using the most stable conformers of all-L, all-D and d-amino acid substituted one-by-one at each position conformers. The results are tabulated in Table 2. The docking results were also evaluated by sorting the docked conformations of EQRPR according to their predicated binding free energy. The best predicted binding affinity for DNA was found for the pentapeptide (−6.8 kcal/mol), all of which are D isomers. The best predicted binding affinities for DNA and for α5β1 integrin, with −6.8 kcal/mol and −10.3 kcal/mol, respectively, were found for the pentapeptide, which has all D isomers. However, the EQRPR pentapeptide that has all L-isomers is found to have the best binding affinity for αIIBβ3 integrin (−9.2 kcal/mol).
The high binding affinity between the investigated molecule and the integrins prompted us to investigate the relationship between binding energy and conformation of the pentapeptide. Table 4S shows the binding affinities and binding free energies, calculated by Poisson Boltzmann ΔG (PB), and generalized Born ΔG (GB) approximations of EQPRP pentapeptide in various conformations. As can be seen from Table 4S, the pentapeptide in the 310 helix conformation has the highest binding affinity (−10.7 kcal/mol) to the α5β1 integrin (in the active site 2), followed by parallel beta (−9.6 kcal/mol) and optimized structure (−9.6 kcal/mol). Despite the highest affinity to the integrin αIIBβ3, the peptide does not assume the 310 helix structure that has high unfavorable energy (see Table 2S). On the other hand, the binding affinity of the pentapeptide in the optimized structure has the highest binding affinity to αIIBβ3 integrin (−9.2 kcal/mol) followed by the antiparallel beta structure of the EQRPR (- 9.1 kcal/mol).
3.5. In silico molecular docking analysis of EQRPR as AntiCOVID-19 agent
In this study, we performed docking-based virtual screening on three SARS-CoV-2 targets (the proteases Mpro and the spike glycoprotein) using investigated pentapeptide to suggest as a potential compound that may act as antivirals. SARS-CoV-2 that causes COVID-19 infection uses angiotensin-converting enzyme 2 (ACE2) for entry into target cells. For this reason, the interaction between the investigated pentapeptide and ACE2 enzyme was also investigated.
The crystal structures of spike glycoprotein (PDB ID: 6VXX), apo-form of COVID-19 Mpro (PDB ID: 6M03) and holo-form of COVID-19 Mpro (PDB ID: 6LU7), ACE2 (PDB ID: 6M0J) were obtained from the protein database [[42], [43], [44], [45]]. The docking for molecules was adapted by removing the water molecule from the receptors and adding polar hydrogens. The active sites of the target proteins were defined in the grid size of 40 Å × 40 Å × 40 Å.
It was determined that the investigated pentapeptide bound to the spike glycoprotein, most strongly; the binding affinities of all L- and all D-isomers analogs of EQRPR for spike glycoprotein are −8.0 and −7.9 kcal/mol, respectively. This is followed by ACE2: The binding affinities of all L- and all d-isomer analogs of EQRPR for ACE2 are −7.6 and −7.5 kcal/mol, respectively. The binding affinities of L- and all d-isomer analogs of EQRPR for the holo- and apo-forms of COVID-19 Mpro are found to be −7.5 and −7.5 kcal/mol, and −6.6 and −6.9 kcal/mol, respectively. Fig. 6, Fig. 7 shows the molecular docking results of EQRPR to target proteins (Apo and holo forms of Mpro, Spike glycoprotein and ACE2). Table 5S gives the binding affinities (kcal/mol) of the optimized structures of all L- and all D-isomers of EQRPR with the target proteins and DNA.
Fig. 6.

The 3D docked views of the most stable conformer of EQRPR in Apo (a) and Holo (b) forms of Mpro. The ligand interaction diagrams of receptor-ligand complexes are shown.
Fig. 7.

The 3D docked views of the most stable conformer of EQRPR in the Spike glycoprotein (a), in ACE2 (b). The ligand interaction diagrams of receptor-ligand complexes are shown.
In the study conducted by Onabajo et al., in 2020, it was proposed that a truncated isoform of ACE2, which was designated as deltaACE2 (dACE2) acts as an interferon-stimulated gene. Onabajo et al. proposed that the dACE2, which lacks 356 amino acids from the N-terminal of ACE2, was non-functional in binding the SARS-CoV-2 spike protein and as a carboxypeptidase. In the study of Onabajo et al. [63], not ACE2, but dACE2 has been reported to be an interferon-stimulated gene. Since the interferon-induced variability in ACE2 expression levels is thought important for susceptibility to COVID-19, dACE2 has importance. For this reason, in this study, the interaction between EQRPR and dACE2 is also investigated by docking simulations. The dACE2 is a novel inducible isoform of ACE2 [63], so its crystal structure is not available in the database. But its structure for docking purposes is prepared as described [63]. The full-length ACE2 protein, with 805 amino acids was taken from the study conducted by Ref. [64] and by using CABS-flex 2.0 online server, the first 356 amino acids from the N-terminal were taken out. Since, by Onabajo et al. it was proposed that the remaining part, which lacks 356 amino acids from the N-terminal of ACE2, was non-functional in binding the SARS-CoV-2 spike protein and as a carboxypeptidase, we also investigated this truncated ACE2. The remaining truncated protein (which lacks 356 amino acids from the N-terminal of ACE2), with 449 amino acids, was then optimized and prepared for docking with EQRPR. The docking results are shown in Fig. 8 a. Binding affinity is found to be reduced to −6.7 kcal/mol. It must be noted that the binding affinity of EQRPR to ACE2 was −7.6 kcal/mol (See Fig. 7b). Then dACE2 was prepared, by adding ten amino acids (MREAGWDKGG) to the N terminal of the remained truncated ACE2, which lacks 356 amino acids from its N terminal. In other words, to prepare dACE2, the first 356 amino acids of ACE2 were replaced by 10 amino acids in the MREAGWDKGG sequence and then optimized by AMBER. The docking of the optimized dACE2 with EQRPR pentapeptide resulted in −8.0 kcal/mol binding affinity (Kd = 1.4 μM). The molecular docking of EQRPR with dACE2 and the interaction diagrams are shown in Fig. 8b. This preliminary result reveals the antiCOVID-19 activity of the EQRPR molecule.
Fig. 8.

The 3D docked views of the most stable conformer of EQRPR in optimized truncated ACE2, which lacks 356 amino acids from the N-terminal of ACE2 (a) and in dACE2 (b). The ligand interaction diagrams of receptor-ligand complexes are shown.
As a summary, the binding affinities and interactions of EQRPR ligand-target complexes were tabulated in Table 3.
One of the difficulties in the biomedical application of the natural or derived short peptides is a rapid renal clearance in part owing to the low molecular weight. Since the peptides do not show a renal tubular re-absorption property, they have a high clearance rate and short half-life [65]. Several approaches have been used to increase the half-life of the peptide. However, binding to the plasma proteins is a well-known mechanism to retard renal clearance. To decide what approach is the most practical for biomedical application, in silico evaluation was performed for binding of the EQRPR to the human serum albumin.
3.6. Molecular docking analysis of EQRPR with plasma-derived human serum albumin (HSA)
The crystal structure of plasma-derived HSA was taken from the Protein Data Bank (PDB ID: 5Z0B) and, was confirmed to the docking by removing water molecules and adding polar hydrogens in it. The active sites of HSA were searched by the CAVER program [54]. As a result of the docking simulations of EQRPR pentapeptide with HSA, binding affinity is found as −8.4 kcal/mol (Kd = 0.7 μM). The Albumin-EQRPR interaction is given in Fig. 9 . The molecular interaction between EQRPR and HSA (5Z0B) is given in Table 3. High-affinity binding to HSA suggests that as the first approach EQRPR can be used in vitro without any modification and expect to get similar results obtained in vivo. A webserver “SuperCYPsPred” predicts that the modifications of the peptide by cytochrome P450 enzymes are unlikely [66].
Fig. 9.

The 3D docked view of the most stable conformer of EQRPR in albumin. The ligand interaction diagrams of receptor ligand complex are show.
3.7. Back to COVID-19 and cancer
Possible biomedical application of the peptide EQRPR without any modification makes it more valuable for COVID-19 treatment. Evidence-based data suggest neuronal dissemination of SARS-CoV-2 invasion into the central nervous system (CNS) [67]. SARS-CoV-2 RNA was detected in the cerebrospinal fluid (CSF) in the patient diagnosed with viral meningitis [68]. It was suggested that COVID-19 may enter the CNS via retrograde neuronal diffusion and then, infection swiftly spread to other brain regions by trans-neuronal path [69]. Consistent with that SARS-CoV particles were observed in CNS neurons and brain samples from patients diagnosed with SARS [70]. ACE2 was identified in both neurons and glial cells [71]. As in the lung, ACE2 in the brain also provides viral entry for SARS-CoV-2. ADMET profile predicts that the peptide EQRPR having a molecular weight less than 1000 Da is permeable to the blood-brain barrier (BBB). Data imply that the peptide EQRPR may also effectively work in the brain against the viral infection.
Integrins play a significant role in tumor invasion and progression in glioblastomas [72]. Therefore bioavailability of the peptide EQRPR in the brain environment could be beneficial for brain cancer treatment.
Despite the significant success in chemo- and radio-therapy, rare subpopulations of cancer cells that show chemo-, radio- and immuno-resistance capacity are major obstacles to effectively treat cancer patients. This population of cancer cells identified as cancer stem cells is believed to be the source of relapses after intensive therapies [73,74]. Varies approaches have been developed to increase the susceptibility of cancer stem cells to various therapies. Interferons with strong antiviral properties have also been effectively used in cancer treatment. One of the many effects of interferon actions is to force cancer stem cells to differentiate. Loss of stem cell like properties upon differentiation increases the effectiveness of radio- and chemotherapy. The peptide EQRPR has immense potential to be effective with combinatorial use with interferon. As indicated above interferon also stimulate the production of dACE2 receptors on epithelial cells including cancer cells. Increased expression of dACE2 was identified in squamous tumors of the respiratory, gastrointestinal and urogenital tracts [63]. Effective interaction of the peptide EQRPR with dACE2 could be very beneficial to use it in combination with interferon in cancer treatments.
Altogether, comprehensive in silico analysis reveals that the peptide has the potential to be the leading molecule in the drug discovery process for multiple therapeutic applications involving multiple organs.
3.8. Drug-likeness prediction studies of EQRPR pentapeptide
Considering the potential use of the studied pentapeptide as a drug, it is important to estimate the Absorption, Distribution, Metabolism, Excretion and Toxicity (ADMET) properties of EQRPR in humans. The drug-likeness properties of EQRPR pentapeptide were calculated using OSIRIS [55] and the results were summarized in Table 6S. The toxicity risk predictor shows that the compound does not present risks of mutagenicity, tumorigenicity, irritant or reproductive effects. A significant determinant of the pharmacokinetic behavior of a drug is lipophilicity. LogP {logP value = log (Coctanol/Cwater)}) is a measure of the lipophilicity of a compound [75]. The EQRPR pentapeptide shows low lipophilicity, which reflects a good aqueous solubility.
The silico module admetSAR [56] has been used to characterize the various ADMET parameters for EQRPR pentapeptide and the results are given in Table 7S. The expected probabilities are given between 0 and 1 as a numerical value.
The blood brain barrier (BBB) is the brain's microvascular endothelial cell layer which plays a very significant role in the separation of blood from the brain [75]. While most central nervous system (CNS) targeting drugs require high penetration, BBB penetration should be reduced for non-CNS drugs to prevent unintended effects. The BBB permeability of EQRPR pentapeptide is found to be positive with 0.9448 probability value, which has importance for acting as an antiCOVID-19 agent.
The cytochrome P450 (CYP) enzyme is the most effective enzyme in drug metabolism. The probability value of the CYP3A4 substrate for EQRPR is estimated as 0.6724, which means that it can be metabolized in the liver, as Cytochrome P450s is an essential enzyme system for liver drug metabolism. In addition, when we look at the estimation of outflow by P-glycoprotein (P-gp), EQRPR is a P -glycoprotein's substrate with a probability of 0.7122 and its inhibitor with a probability of 0.6470.
Thyroid hormone receptors regulate gene expression by binding to DNA through (HREs) [76]. When activated by estrogen, the Estrogen Receptor (ER) can settle in the nucleus and bind to DNA (i.e., it is a DNA binding transcription factor) to regulate the activity of different genes [77]. The primary function of the androgen receptor is to control gene expression and activate the DNA-binding transcription factor by binding to some of the androgenic hormones in the cytoplasm, including testosterone and dihydrotestosterone [78]. The probability values of Estrogen receptor binding, Androgen receptor binding and Thyroid hormone receptor for EQRPR were found to be 0.6846, 0.6182 and 0.5400 respectively.
4. Conclusions
The FTIR and CD experimental results and theoretical conformational analysis presented here establish that the cationic pentapeptide EQRPR derived from rice bran and its d-isomer analogs mostly adopt beta-sheet conformations. Molecular docking studies provided the multiple ways by which the peptide may act as anti-cancer as well as an anti-viral agent. The results reveal strong interaction between the pentapeptides and integrins (α5β1 and αIIbβ3) important for anti-cancer activity.
The present study demonstrates the mechanism of binding of the pentapeptides to apo- and holo-forms of Mpro, spike glycoprotein, ACE2 and dACE2. Data indicate that the peptide can use multiple ways to prevent SARSCoV-2 (COVID-19) infection. The docking studies show the effectiveness of the use of the pentapeptides in combination with interferons. Furthermore, the present study highlights the importance of d-isomer substitutions in the peptides to control selected functions.
The important conclusion from this study is that the examined pentapeptide is not predicted. Our recent experimental data support some of our findings. The peptides have been found incorporated into the phospholipid monolayer (DPPC) and the membranes of human lung cancer cells. The results of the studies will be published elsewhere.
Funding
This study is supported by TUBITAK-ANAS international project (Project numbers TUBITAK 118F445 and ANAS PH05-01).
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data related to this article can be found online at https://doi.org/10.1016/j.jmgm.2021.107999
Appendix A. Supplementary data
The following is the supplementary data to this article:
References
- 1.Nagendra Prasad Mn N.P., Kr S., Khatokar M S. Health benefits of rice bran - a review. J. Nutr. Food Sci. 2011:1000108. doi: 10.4172/2155-9600.1000108. 01. [DOI] [Google Scholar]
- 2.Sharif M.K., Butt M.S., Anjum F.M., Khan S.H. Rice bran: a novel functional ingredient. Crit. Rev. Food Sci. Nutr. 2014;54:807–816. doi: 10.1080/10408398.2011.608586. [DOI] [PubMed] [Google Scholar]
- 3.Abdul-Hamid A., Luan Y.S. Functional properties of dietary fibre prepared from defatted rice bran. Food Chem. 2000;68:15–19. doi: 10.1016/S0308-8146(99)00145-4. [DOI] [Google Scholar]
- 4.Wang M., Hettiarachchy N.S., Qi M., Burks W., Siebenmorgen T. Preparation and functional properties of rice bran protein isolate. J. Agric. Food Chem. 1999;47:411–416. doi: 10.1021/jf9806964. [DOI] [PubMed] [Google Scholar]
- 5.Cicero A.F.G., Derosa G. Rice bran and its main components: potential role in the management of coronary risk factors. Curr. Top. Nutraceutical Res. 2005;3:29–46. [Google Scholar]
- 6.Zaky A.A., Abd El-Aty A.M., Ma A., Jia Y. An overview on antioxidant peptides from rice bran proteins: extraction, identification, and applications. Crit. Rev. Food Sci. Nutr. 2020:1–13. doi: 10.1080/10408398.2020.1842324. [DOI] [PubMed] [Google Scholar]
- 7.Phongthai S., Rawdkuen S. Fractionation and characterization of antioxidant peptides from rice bran protein hydrolysates stimulated by in vitro gastrointestinal digestion. Cereal Chem. 2020;97:316–325. doi: 10.1002/cche.10247. [DOI] [PubMed] [Google Scholar]
- 8.Ngamsuk S., Hsu J.L., Huang T.C., Suwannaporn P. Ultrasonication of milky stage rice milk with bioactive peptides from rice bran: its Bioactivities and absorption. Food Bioprocess Technol. 2020;13:462–474. doi: 10.1007/s11947-019-02371-2. [DOI] [Google Scholar]
- 9.Shobako N., Ohinata K. Anti-hypertensive effects of peptides derived from rice bran protein. Nutrients. 2020;12:3060. doi: 10.3390/nu12103060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu Y., Zhang J., Wang J., Sun B. The anti-cancer activity and potential clinical application of rice bran extracts and fermentation products. RSC Adv. 2019;9:18060–18069. doi: 10.1039/c9ra02439e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu C., Chen X., Zhao W. Design and modification of anticancer peptides. Drug Des. Open Access. 2016:1000138. doi: 10.4172/2169-0138.1000138. 05. [DOI] [Google Scholar]
- 12.Xie M., Liu D., Yang Y. Anti-cancer peptides: classification, mechanism of action, reconstruction and modification: anticancer peptides. Open Biol. 2020;10 doi: 10.1098/rsob.200004rsob200004. 200004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruoslahti E. RGD and other recognition sequences for integrins. Annu. Rev. Cell Dev. Biol. 1996;12:697–715. doi: 10.1146/annurev.cellbio.12.1.697. [DOI] [PubMed] [Google Scholar]
- 14.Hoskin D.W., Ramamoorthy A. Studies on anticancer activities of antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2008;1778:357–375. doi: 10.1016/j.bbamem.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang Y., Feng Q., Yan Q., Hao X., Chen Y. Alpha-helical cationic anticancer peptides: a promising candidate for novel anticancer drugs. Mini Rev. Med. Chem. 2015;15:73–81. doi: 10.2174/1389557514666141107120954. [DOI] [PubMed] [Google Scholar]
- 16.Huang Y.B., Wang X.F., Wang H.Y., Liu Y., Chen Y. Studies on mechanism of action of anticancer peptides by modulation of hydrophobicity within a defined structural framework. Mol. Canc. Therapeut. 2011;10:416–426. doi: 10.1158/1535-7163.MCT-10-0811. [DOI] [PubMed] [Google Scholar]
- 17.Mai J.C., Mi Z., Kim S.H., Ng B., Robbins P.D. A proapoptotic peptide for the treatment of solid tumors. Canc. Res. 2001;61:7709–7712. [PubMed] [Google Scholar]
- 18.Li H., Kolluri S.K., Gu J., Dawson M.I., Cao X., Hobbs P.D. Cytochrome c release and apoptosis induced by mitochondrial targeting of nuclear orphan receptor TR3. Science. 2000;289:1159–1164. doi: 10.1126/science.289.5482.1159. [DOI] [PubMed] [Google Scholar]
- 19.Chiangjong W., Chutipongtanate S., Hongeng S. Anticancer peptide: physicochemical property, functional aspect and trend in clinical application. Int. J. Oncol. 2020;57:678–696. doi: 10.3892/ijo.2020.5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kannan A., Hettiarachchy N.S., Lay J.O., Liyanage R. Human cancer cell proliferation inhibition by a pentapeptide isolated and characterized from rice bran. Peptides. 2010;31:1629–1634. doi: 10.1016/j.peptides.2010.05.018. [DOI] [PubMed] [Google Scholar]
- 21.Li R., Hettiarachchy N., Mahadevan M. Rice bran derived pentapeptide-induced apoptosis in human breast cancer cell models (MCF-7 and MDA-MB-231) Int. J. Biomed. Res. 2014;5:599–605. doi: 10.7439/ijbr. [DOI] [Google Scholar]
- 22.Kim M.K., Kang Y.K. Positional preference of proline in α-helices. Protein Sci. 1999;8:1492–1499. doi: 10.1110/ps.8.7.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yun R.H., Anderson A., Hermans J. Proline in α‐helix: stability and conformation studied by dynamics simulation. Proteins Struct. Funct. Bioinforma. 1991;10:219–228. doi: 10.1002/prot.340100306. [DOI] [PubMed] [Google Scholar]
- 24.Carmona G., Rodriguez A., Juarez D., Corzo G., Villegas E. Improved protease stability of the antimicrobial peptide pin2 substituted with d-Amino acids. Protein J. 2013;32:456–466. doi: 10.1007/s10930-013-9505-2. [DOI] [PubMed] [Google Scholar]
- 25.Melchionna M., Styan K., Marchesan S. The unexpected advantages of using D-amino acids for peptide self-assembly into nanostructured hydrogels for medicine. Curr. Top. Med. Chem. 2016;16:2009–2018. doi: 10.2174/1568026616666160215155136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H., Anuwongcharoen N., Malik A.A., Prachayasittikul V., Wikberg J.E.S., Nantasenamat C. Roles of D-amino acids on the bioactivity of host defense peptides. Int. J. Mol. Sci. 2016;17:1–27. doi: 10.3390/ijms17071023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feng Z., Xu B. Inspiration from the mirror: D-amino acid containing peptides in biomedical approaches. Biomol. Concepts. 2016;7:179–187. doi: 10.1515/bmc-2015-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grishin D.V., Zhdanov D.D., Pokrovskaya M.V., Sokolov N.N. D-amino acids in nature, agriculture and biomedicine. Front. Life Sci. 2020;13:11–22. doi: 10.1080/21553769.2019.1622596. [DOI] [Google Scholar]
- 29.Gasymov O.K., Botta C., Ragona L., Guliyeva A.J., Molinari H. Silk fibroin-based films enhance rhodamine 6G emission in the solid state: a chemical-physical analysis of their interactions for the design of highly emissive biomaterials. Macromol. Chem. Phys. 2019;220:1800460. doi: 10.1002/macp.201800460. [DOI] [Google Scholar]
- 30.Belton D.J., Plowright R., Kaplan D.L., Perry C.C. A robust spectroscopic method for the determination of protein conformational composition – application to the annealing of silk. Acta Biomater. 2018;73:355–364. doi: 10.1016/j.actbio.2018.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh B.R., DeOliveira D.B., Fu F.-N., Fuller M.P. In: Biomol. Spectrosc. III. Nafie L.A., Mantsch H.H., editors. 1993. Fourier transform infrared analysis of amide III bands of proteins for the secondary structure estimation; pp. 47–55. [DOI] [Google Scholar]
- 32.Whitmore L., Wallace B.A. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004;32:W668–W673. doi: 10.1093/nar/gkh371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whitmore L., Wallace B.A. Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers. 2008;89:392–400. doi: 10.1002/bip.20853. [DOI] [PubMed] [Google Scholar]
- 34.Compton L.A., Johnson W.C. Analysis of protein circular dichroism spectra for secondary structure using a simple matrix multiplication. Anal. Biochem. 1986;155:155–167. doi: 10.1016/0003-2697(86)90241-1. [DOI] [PubMed] [Google Scholar]
- 35.Manavalan P., Johnson W.C. Variable selection method improves the prediction of protein secondary structure from circular dichroism spectra. Anal. Biochem. 1987;167:76–85. doi: 10.1016/0003-2697(87)90135-7. [DOI] [PubMed] [Google Scholar]
- 36.Maksumov I.S., Ismailova L.I., Godjaev N.M. A program for the semiemprical calculation of the conformations of molecular-complexes on a computer. J. Chem. Struct. 1983;24:647–648. [Google Scholar]
- 37.Ramachandran G.N., Sasisekharan V. Conformation of polypeptides and proteins. Adv. Protein Chem. 1968;23:283–437. doi: 10.1016/S0065-3233(08)60402-7. [DOI] [PubMed] [Google Scholar]
- 38.Ramachandran G.N. Noriplunar Peptide Units in Polypeptide Chains. 1968;6:1494–1496. doi: 10.1002/bip.1968.360061013. [DOI] [PubMed] [Google Scholar]
- 39.Cornell W.D., Cieplak P., Bayly C.I., Gould I.R., Merz K.M., Ferguson D.M. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995;117:5179–5197. doi: 10.1021/ja00124a002. [DOI] [Google Scholar]
- 40.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R. Inc.; Wallingford CT: 2016. Gaussian 16; p. 2016. [Google Scholar]
- 41.Drew H.R., Wing R.M., Takano T., Broka C., Tanaka S., Itakura K. Structure of a B-DNA dodecamer: conformation and dynamics. Proc. Natl. Acad. Sci. U.S.A. 1981;78:2179–2183. doi: 10.1073/pnas.78.4.2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 43.Zhang B., Zhao Y., Jin Z., Liu X., Yang H., Rao Z. Publ. Online.; 2020. The Crystal Structure of COVID-19 Main Protease in Apo Form. [DOI] [Google Scholar]
- 44.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 45.Walls A.C., Park Y.J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181:281–292. doi: 10.1016/j.cell.2020.02.058. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xia W., Springer T.A. Metal ion and ligand binding of Integrin α5β1. Proc. Natl. Acad. Sci. U.S.A. 2014;111:17863–17868. doi: 10.1073/pnas.1420645111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu J., Zhu J., Springer T.A. Complete integrin headpiece opening in eight steps. J. Cell Biol. 2013;201:1053–1068. doi: 10.1083/jcb.201212037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trott O., Olson A.J., Vina AutoDock. Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Z., Wang X., Li Y., Lei T., Wang E., Li D. FarPPI: a webserver for accurate prediction of protein-ligand binding structures for small-molecule PPI inhibitors by MM/PB(GB)SA methods. Bioinformatics. 2019;35:1777–1779. doi: 10.1093/bioinformatics/bty879. [DOI] [PubMed] [Google Scholar]
- 50.Hao G.F., Jiang W., Ye Y.N., Wu F.X., Zhu X.L., Guo F.B. ACFIS: a web server for fragment-based drug discovery. Nucleic Acids Res. 2016;44:W550–W556. doi: 10.1093/nar/gkw393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hao G.F., Wang F., Li H., Zhu X.L., Yang W.C., Huang L.S. Computational discovery of picomolar Q o site inhibitors of cytochrome bc 1 complex. J. Am. Chem. Soc. 2012;134:11168–11176. doi: 10.1021/ja3001908. [DOI] [PubMed] [Google Scholar]
- 52.Chéron N., Jasty N., Shakhnovich E.I. OpenGrowth: an automated and rational algorithm for finding new protein ligands. J. Med. Chem. 2016;59:4171–4188. doi: 10.1021/acs.jmedchem.5b00886. [DOI] [PubMed] [Google Scholar]
- 53.Yang J.F., Wang F., Jiang W., Zhou G.Y., Li C.Z., Zhu X.L. PADFrag: a database built for the exploration of bioactive fragment space for drug discovery. J. Chem. Inf. Model. 2018;58:1725–1730. doi: 10.1021/acs.jcim.8b00285. [DOI] [PubMed] [Google Scholar]
- 54.Jurcik A., Bednar D., Byska J., Marques S.M., Furmanova K., Daniel L. CAVER Analyst 2.0: analysis and visualization of channels and tunnels in protein structures and molecular dynamics trajectories. Bioinformatics. 2018;34:3586–3588. doi: 10.1093/bioinformatics/bty386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Osiris . Actelion Pharmaceuticals Ltd; 2010. OSIRIS Property Explorer.http://www.organic-chemistry.org/prog/peo/ [Google Scholar]
- 56.Cheng F., Li W., Zhou Y., Shen J., Wu Z., Liu G. AdmetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012;52:3099–3105. doi: 10.1021/ci300367a. [DOI] [PubMed] [Google Scholar]
- 57.Kong J., Yu S. Fourier transform infrared spectroscopic analysis of protein secondary structures. Acta Biochim. Biophys. Sin. 2007;39:549–559. doi: 10.1111/j.1745-7270.2007.00320.x. [DOI] [PubMed] [Google Scholar]
- 58.Barth A. Infrared spectroscopy of proteins. Biochim. Biophys. Acta Bioenerg. 2007;1767:1073–1101. doi: 10.1016/j.bbabio.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 59.Fasman G.D., editor. Circular Dichroism and the Conformational Analysis of Biomolecules. Plenum Press; New York: 1996. [Google Scholar]
- 60.Sreerama N., Woody R.W. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 2000;287:252–260. doi: 10.1006/abio.2000.4880S0003-2697(00)94880-2. ([pii]) [DOI] [PubMed] [Google Scholar]
- 61.Luo P., Baldwin R.L. Mechanism of helix induction by trifluoroethanol: a framework for extrapolating the helix-forming properties of peptides from trifluoroethanol/water mixtures back to water. Biochemistry. 1997;36:8413–8421. doi: 10.1021/bi9707133. [DOI] [PubMed] [Google Scholar]
- 62.Jasanoff A., Fersht A.R. Quantitative determination of helical propensities from trifluoroethanol titration curves. Biochemistry. 1994;33:2129–2135. doi: 10.1021/bi00174a020. [DOI] [PubMed] [Google Scholar]
- 63.Onabajo O.O., Banday A.R., Stanifer M.L., Yan W., Obajemu A., Santer D.M. Interferons and viruses induce a novel truncated ACE2 isoform and not the full-length SARS-CoV-2 receptor. Nat. Genet. 2020;52:1283–1293. doi: 10.1038/s41588-020-00731-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ahmad W., Shabbiri K., Islam N. 2020. O-β-GlcNAcylation, Chloroquine and 2-Hydroxybenzohydrazine may hamper SARS-CoV-2 entry to human via inhibition of ACE2 phosphorylation at Ser787 but also induce disruption of virus-ACE2 binding. [DOI] [Google Scholar]
- 65.Di L. Strategic approaches to optimizing peptide ADME properties. AAPS J. 2015;17:134–143. doi: 10.1208/s12248-014-9687-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Banerjee P., Dunkel M., Kemmler E., Preissner R. SuperCYPsPred-a web server for the prediction of cytochrome activity. Nucleic Acids Res. 2020;48:W580–W585. doi: 10.1093/nar/gkaa166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jarrahi A., Ahluwalia M., Khodadadi H., Da Silva Lopes Salles E., Kolhe R., Hess D.C. Neurological consequences of COVID-19: what have we learned and where do we go from here? J. Neuroinflammation. 2020;17:1–12. doi: 10.1186/s12974-020-01957-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moriguchi T., Harii N., Goto J., Harada D., Sugawara H., Takamino J. A first case of meningitis/encephalitis associated with SARS-Coronavirus-2. Int. J. Infect. Dis. 2020;94:55–58. doi: 10.1016/j.ijid.2020.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Butowt R., Bilinska K. SARS-CoV-2: olfaction, brain infection, and the urgent need for clinical samples allowing earlier virus detection. ACS Chem. Neurosci. 2020;11:1200–1203. doi: 10.1021/acschemneuro.0c00172. [DOI] [PubMed] [Google Scholar]
- 70.Xu J., Zhong S., Liu J., Li L., Li Y., Wu X. Detection of severe acute respiratory syndrome coronavirus in the brain: potential role of the chemokine mig in pathogenesis. Clin. Infect. Dis. 2005;41:1089–1096. doi: 10.1086/444461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xiao L., Haack K.K.V., Zucker I.H. Angiotensin II regulates ACE and ACE2 in neurons through p38 mitogen-activated protein kinase and extracellular signal-regulated kinase 1/2 signaling. Am. J. Physiol. Cell Physiol. 2013;304:1073–1079. doi: 10.1152/ajpcell.00364.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Malric L., Monferran S., Gilhodes J., Boyrie S., Dahan P., Skuli N. Interest of integrins targeting in glioblastoma according to tumor heterogeneity and cancer stem cell paradigm: an update. Oncotarget. 2017;8:86947–86968. doi: 10.18632/oncotarget.20372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Moserle L., Ghisi M., Amadori A., Indraccolo S. Side population and cancer stem cells: therapeutic implications. Canc. Lett. 2010;288:1–9. doi: 10.1016/j.canlet.2009.05.020. [DOI] [PubMed] [Google Scholar]
- 74.Doherty M.R., Jackson M.W. The critical, clinical role of interferon-beta in regulating cancer stem cell properties in triple-negative breast cancer. DNA Cell Biol. 2018;37:513–516. doi: 10.1089/dna.2018.4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chandrasekaran B., Abed S.N., Al-Attraqchi O., Kuche K., Tekade R.K. Elsevier Inc.; 2018. Computer-Aided Prediction of Pharmacokinetic (ADMET) Properties. [DOI] [Google Scholar]
- 76.Ortiga-Carvalho T.M., Sidhaye A.R., Wondisford F.E. Thyroid hormone receptors and resistance to thyroid hormone disorders. Nat. Rev. Endocrinol. 2014;10:582–591. doi: 10.1038/nrendo.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Levin D.E. Cell wall integrity signaling in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2005;69:262–291. doi: 10.1128/mmbr.69.2.262-291.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Roy A.K., Lavrovsky Y., Song C.S., Chen S., Jung M.H., Velu N.K. Vitam. Horm. 1998. Regulation of androgen action; pp. 309–352. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.