

Abstract
Intravesical immunotherapy using Bacillus Calmette-Guerin (BCG) has been the standard of care for patients with high-risk non-muscle invasive bladder cancer (NMIBC) for several decades. Unfortunately, BCG therapy continues to be limited by high rates of disease recurrence and progression, and patients with BCG-unresponsive disease have few effective salvage therapy options besides radical cystectomy, highlighting an urgent need for novel therapies. We find that the immune-stimulatory receptor CD40 is highly expressed on dendritic cells (DC) within the bladder tumor microenvironment (TME) of orthotopic bladder cancer models, recapitulating CD40 expression found in human disease. We demonstrate that local CD40 agonism in the bladder through intravesical delivery of anti-CD40 agonistic antibodies can drive potent anti-tumor immunity and induce significant pharmacodynamic changes in the bladder TME, including the reduction of CD8+ T cells with an exhausted phenotype. We further show that type 1 conventional DCs (cDC1) and CD8+ T cells are required for both bladder cancer immune surveillance and anti-CD40 agonist antibody responses. Using orthotopic murine models humanized for CD40 and Fcγ receptors, we also demonstrate that intravesical treatment with a fully-human, Fc-enhanced anti-CD40 agonist antibody (2141-V11) can induce robust anti-tumor activity in both treatment-naïve and treatment-refractory settings, driving long-term systemic anti-tumor immunity with no evidence of systemic toxicity. Intratumoral 2141-V11 is currently being assessed in a phase I clinical study of metastatic solid tumors, and these findings support targeting CD40-expressing DCs in the bladder TME through a novel intravesical agonistic antibody approach for the treatment of NMIBC.
One Sentence Summary:
Intravesical delivery of Fc-optimized CD40 agonistic antibodies drives potent local and systemic anti-tumor immunity against bladder cancer.
Introduction
Every year approximately 550,000 new cases of bladder cancer are diagnosed worldwide, with nearly 200,000 bladder cancer-associated deaths (1). Although the majority of patients present with earlier-stage, non-muscle invasive bladder cancer (NMIBC), they remain at high risk of disease recurrence and progression despite maximal standard therapy. Current standard of care treatment for intermediate and high-risk NMIBC consists of transuretheral tumor resection followed by treatment with Bacillus Calmette-Guerin (BCG), a live-attenuated mycobacterium delivered intravesically into the bladder. While BCG, one of the oldest forms of cancer immunotherapy, has well-established efficacy in reducing the risk of disease recurrence and progression, up to 75% of patients will ultimately be unresponsive to BCG treatment (2). Despite the recent approval of anti-PD-1 therapy (pembrolizumab) in the BCG-unresponsive setting, the efficacy of this agent remains modest, with no additional effective salvage therapy options aside from complete bladder resection. BCG therapy can be further limited by toxicity from significant non-specific inflammation (3), as well as restricted access due to an ongoing global shortage (4). There is thus an urgent need to develop novel treatments for high-risk NMIBC, either in addition or as an alternative to BCG (5).
Enhancement of anti-tumor immune responses using recombinant human IgG antibodies has emerged as an important paradigm in the treatment of cancer, including bladder cancer. While antibodies blocking inhibitory immune pathways, such as PD-1 and CTLA-4, are capable of driving robust and durable clinical responses in the metastatic setting, they remain effective for only a minority of patients (6). Immune activation by agonism of stimulatory receptors represents another powerful and potentially synergistic approach. In particular, the stimulatory receptor CD40 plays a central role in anti-cancer immunity and the development of tumor-specific T cell responses (7–9). CD40 expression has been shown to be enriched in the human bladder tumor microenvironment (TME), providing a rationale for targeting the CD40 pathway specifically in NMIBC (10).
Several first-generation CD40 agonist antibodies have been developed, but their clinical efficacy has been modest, and their use has been further associated with dose-limiting systemic toxicities (8, 11–14). Antibodies are composed of two ends: one end containing two identical Fab domains conferring target recognition (e.g. CD40), and the other end with an Fc domain engaging a variety of activating and inhibitory Fcγ receptors (FcγRs) on immune cells, which mediate Fc-dependent antibody activity (15). While many anti-tumor therapeutic antibodies have been classically envisioned for their role in stimulating antibody dependent cellular cytotoxicity (ADCC) through the engagement of activating FcγRs, we and others have demonstrated that agonistic antibodies targeting a number of immune-stimulatory receptors (e.g. CD40, GITR) instead require binding to the sole inhibitory FcγR (FcγRIIb) for optimal agonist activity, which facilitates target receptor crosslinking needed for potent agonistic signaling (7, 16, 17). Our group has further demonstrated that selective engineering of the antibody Fc domain to enhance FcγRIIb affinity can significantly potentiate the agonist activity of anti-CD40 antibodies, which results in enhanced CD8+ T cell-dependent anti-tumor immunity (18, 19). However, this was also associated with increased levels of toxicity (thrombocytopenia and transaminitis). Importantly, these toxicities could be circumvented by intratumoral rather than systemic administration, allowing local therapeutic delivery to drive an in situ vaccination effect capable of mediating systemic anti-tumor activity while minimizing systemic toxicity (19).
We now describe a novel approach to localized CD40 agonism relevant to the treatment of bladder cancer through the intravesical delivery of Fc-optimized anti-CD40 agonistic antibodies. Using immunocompetent orthotopic murine models of bladder cancer, including species-matched antibody-FcγR systems humanized for both CD40 and Fcγ receptors, we demonstrate potent in vivo anti-tumor activity in both treatment naïve and BCG-unresponsive settings, with limited associated systemic toxicity. We further demonstrate the ability of local intravesical CD40 agonism to drive the reversal of CD8+ T cell exhaustion phenotypes and induce long-lived protective systemic immunity, and implicate Batf3-dependent dendritic cells (DCs) as a key target mediating this anti-tumor effect. Collectively, these data provide in vivo proof of concept for the therapeutic use of intravesically-delivered Fc-optimized anti-CD40 agonist antibodies for the treatment of bladder cancer, and define important mechanisms underlying CD40-driven bladder cancer immune surveillance.
Results
CD40-expressing dendritic cells are enriched in the bladder tumor microenvironment
We first sought to characterize the immune contexture of the bladder TME using an immunocompetent orthotopic murine model, in which a urethral catheter-based technique is used to intravesically engraft the aggressive syngeneic MB49 bladder cancer cell line into the bladders of C57BL/6J mice (20)’(21). This protocol produces efficient engraftment of tumors within the bladder, which demonstrate rapid growth as seen by bladder weights and histology (Figure 1A, B). Three days following implant, small tumors can be seen in the urothelial layer, progressing at later time points to deeper invasion into the lamina propria and substantial outgrowth into the bladder lumen (Figure 1B). Muscle layer invasion was not observed at the day 6 timepoint.
Figure 1:
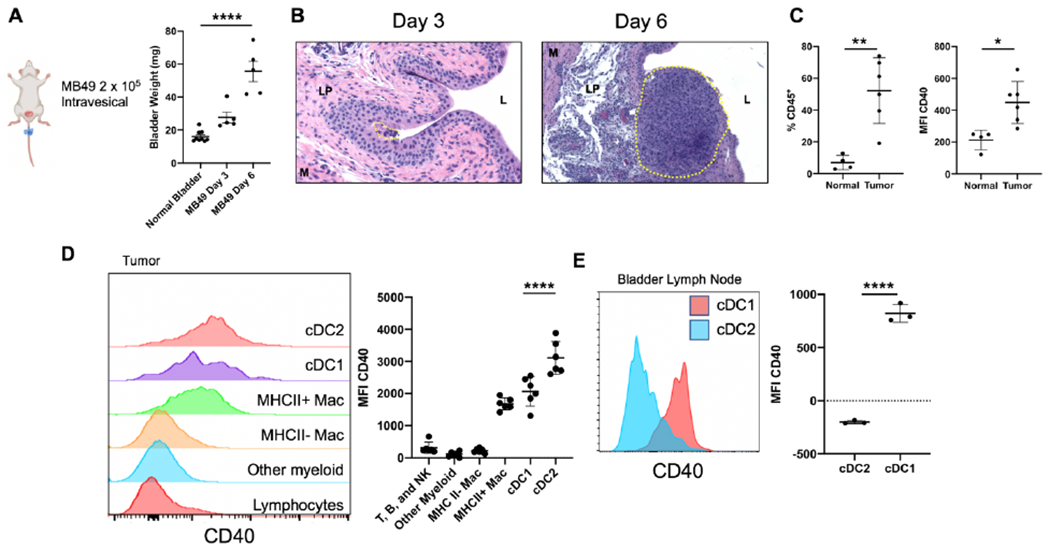
CD40 is expressed on dendritic cells within the bladder tumor microenvironment.
A, Schematic of orthotopic tumor model with bladder weights (mg) at days 3 and 6 post intravesical tumor implantation. B, Histology (H & E) of bladders from days 3 and 6 post tumor implantation. Tumor area is outlined by the yellow dotted line. The bladder lumen (L), lamina propria (LP), and muscle layer (M) are denoted by their respective letters. C-D, Flow cytometry of MB49 bladder tumors. (C) percentage of CD45+ immune cells and mean fluorescent intensity (MFI) of CD40 on CD45+ immune cells in normal (n = 4 independent animals) and tumor-bearing bladders (n = 6 independent animals). (D) histograms and quantification of CD40 expression across immune cell types in the tumor-bearing bladder. E, Flow cytometry histogram and quantification of CD40 expression in bladder-draining lymph nodes across DC subtypes. cDC1 are defined as CD11c+ MHCII+ Xcr1+. cDC2 are defined as CD11c+ MHCII+ Sirpα+. For A and D, One-way ANOVA with Tukey’s test was used to calculate statistical significance, *** p < 0.001, **** p < 0.0001. For C and E, Two-tailed unpaired t-test was used to calculate statistical significance, * p < 0.05, ** p < 0.01, **** p < 0.0001. All data are plotted as mean ± s.d. Data are representative of two independent replicates.
Using this orthotopic bladder implantation model, we set out to characterize the bladder tumor immune microenvironment. Tumor presence in the bladder was accompanied by a substantial increase in bladder CD45+ infiltrating immune cells, with a significant increase in CD40 expression when compared to bladders of non-tumor bearing animals (Figure 1C). Further analysis of immune cell subsets within tumor-bearing bladders identified that dendritic cells (DCs) within the bladder TME expressed the highest levels of CD40 (Figure 1D). Activated tumor-associated macrophages also expressed high levels of CD40, although substantially less than DCs. DCs can be subdivided into two broadly-defined subtypes, conventional type 1 DCs (cDC1), which specialize in cross-presentation of antigens to CD8+ T cells, and conventional type 2 DCs (cDC2), which are better suited to present antigen to CD4+ T cells (22). These DC subtypes can respectively be distinguished by expression of XCR1 (cDC1) and Sirpα (cDC2) (22). Examination of bladder tumor-draining iliac lymph nodes [identified by intravesical administration of Evans Blue dye (Supplemental Figure 1)] demonstrated CD40 expression predominantly on cDC1 compared to cDC2 (Figure 1E), suggesting that cDC1 may be a principal target of CD40 agonist approaches in this disease context.
Intravesical CD40 agonism reverses CD8 T cell exhaustion signatures in the bladder tumor microenvironment
We next evaluated intravesical therapeutic CD40 agonism in this disease model using a murine anti-CD40 IgG1 antibody (1C10), which can agonize CD40 in vivo by virtue of its ability to bind murine FcγRIIb (7), and compared it to standard of care intravesical BCG therapy. Intravesical delivery of 1C10 into day 6 tumor-bearing bladders led to a substantial decrease in bladder tumor weights (Figure 2A, B). BCG treatment also demonstrated a trend for decreased tumor burden (Figure 2B). Previously, our group and others have shown that CD40 agonism leads to a CD8+ T cell-dependent anti-tumor response (19, 23). This led us to hypothesize that phenotypic alterations to CD8+ T cells would occur within the bladder TME following intravesical CD40 agonism. In particular, T cells can express markers associated with exhaustion in response to antigen stimulation (including in the setting of malignancy), defined by a state of functional unresponsiveness and combinatorial expression of T cell co-inhibitory cell surface receptors (24). In response to 1C10-driven CD40 agonism, and to a lesser extent with BCG treatment, tumor-infiltrating CD8+ T cells demonstrated a reduction in exhaustion signatures, with reduced proportions of CD8+ T cells co-expressing the inhibitory cell surface receptors PD-1 and LAG-3 (Figure 2C, D). PD-1lo T cells have been associated with anti-tumor responses and positive clinical outcomes, postulated to represent a population of T effector cells (25, 26) involved in the anti-tumor response. No significant changes in exhaustion marker expression were identified on CD4 T cells or innate lymphocyte populations in response to treatment (Supplemental Figure 2). To confirm a functional role for CD8+ T cells in intravesical anti-CD40 agonist antibody response, anti-CD8 depleting antibody was used to clear CD8+ T cells from the tumor microenvironment during anti-CD40 antibody treatment (Supplemental Figure 3). Bladder weights from CD8-depleted animals treated with the 1C10 anti-CD40 agonist antibody were significantly higher than those treated with 1C10 alone (Figure 2E).
Figure 2:
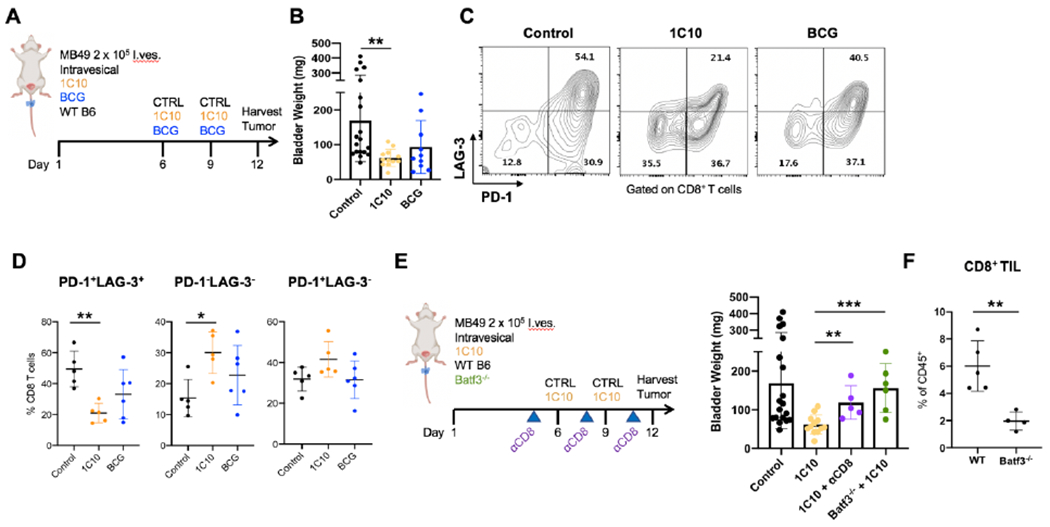
CD40 agonism reverses CD8+ T cell exhaustion signatures through dendritic cell intermediates.
A, Schematic of intravesical treatment of orthotopic bladder tumors with murine anti-CD40 agonist antibody (1C10) and BCG. B, Bladder weights (mg) from day 12 post tumor implantation. C-D, Flow cytometry plots and quantification of LAG-3 and PD-1 expression on tumor-infiltrating CD8+ T cells in mice treated with control (n = 5 independent animals), 1C10 (n = 5 independent animals), and BCG (n = 6 independent animals). E, Schematic of intravesical treatment model with 1C10 in Batf3−/− animals, or in the setting of CD8 depletion using depleting antibodies, with arrowheads indicating dosing times of depleting antibodies. Bladder weights (mg) are shown at day 12 post implantation. F, CD8+ T cells infiltrating bladder tumors were quantified between WT and Batf3−/− animals at day 12 post tumor implantation. For B and D, One-way ANOVA with Tukey’s test was used to calculate statistical significance. For E and F, Two-tailed unpaired t-test was used to calculate statistical significance, * p < 0.05, ** p < 0.01, *** p < 0.001. All data are plotted as mean ± s.d. Data are representative of two independent replicates.
Batf3 is required for CD40 agonist responses and spontaneous bladder tumor growth
Given the observed pharmacodynamic changes to CD8+ T cells in response to intravesical anti-CD40 antibody therapy and the prominent expression of CD40 on cDC1s within the bladder TME, we hypothesized that cDC1s were involved in the therapeutic CD40 agonist response. Batf3−/− mice are known to be deficient in cDC1s (27), and consistent with prior reports (28), we found a defect in the cDC1 compartment in Batf3−/− mice (Supplemental Figure 4). While Batf3 deficiency has also been shown to have pleiotropic effects in certain settings, such as the differentiation of T regulatory cells in the colonic lamina propria (29), we found that T regulatory cell proportions and other immune populations including CD4+ T cells, macrophages, and cDC2 were similar between tumor-bearing Batf3−/− and wild-type control mice (Supplemental Figure 4).
Batf3−/− mice have significantly lower numbers of cDC1s in both the spleen and tumor-draining lymph nodes of tumor-bearing mice, with cDC2 proportions either elevated in the spleen or unchanged in the lymph node (Supplemental Figure 5). We then examined the influence of Batf3 deficiency on tumor growth and progression within the bladder. Following implantation of MB49 tumor cells into the bladders of Batf3−/− mice, we found that mice had significantly decreased survival compared to WT controls (Supplemental Figure 6A). To further interrogate the effect of Batf3 deficiency in an additional spontaneous bladder cancer model, we exposed Batf3−/− or control WT mice to the chemical carcinogen N-butyl-N-(4-hydroxybutyl)-nitrosamine (BBN). BBN administration through drinking water is a well-established carcinogen-induced spontaneous bladder cancer model that results in the development and progression of basal-type bladder tumors over the course of 15 – 20 weeks (30, 31). We found that after 18 weeks of BBN exposure, Batf3−/− mice had significantly increased bladder weights compared to WT mice (Supplemental Figure 6B), with histological analysis revealing more advanced tumor development with multinucleated basal cells and invasion into the lamina propria (Supplemental Figure 6C). A subset of bladders from BBN-treated Batf3−/− mice further showed evidence of tumor invasion into the muscle layer, in contrast to bladders from BBN-treated WT mice demonstrating no evidence of muscle invasion by tumor. This enhanced tumor progression resulted in substantially reduced overall survival in the Batf3−/− background compared to WT controls (Supplemental Figure 6D). Collectively, these data demonstrate that Batf3 deficiency results in a loss of cDC1 in the bladder immune microenvironment and is associated with accelerated tumor growth and progression in the bladder.
We further found that intravesical treatment with the anti-CD40 antibody 1C10 could significantly reduce tumor burden, but that this anti-tumor effect was lost in the Batf3−/− background (Figure 2E), demonstrating a requirement for Batf3 for therapeutic activity of CD40 agonist antibodies in this setting. Accordingly, we found significantly reduced CD8+ T cell infiltration in tumors from Batf3−/− mice (Figure 2F). Similarly, we found that Batf3−/− mice were refractory to 1C10 treatment in subcutaneous models of MB49 and UPPL1541 bladder cancers (Supplemental Figure 7). To assess which cell populations in the bladder are targeted by intravesical anti-CD40 agonist antibody, we fluorescently labeled the 1C10 antibody and administered this intravesically into tumor-bearing bladders. We found that the 1C10 antibody bound primarily to antigen presenting myeloid cells (cDC1, cDC2, and MHCII+ macrophages) in the bladder, while tissues such as the draining lymph node and spleen showed limited 1C10 antibody positivity (Supplemental Figure 8). These data suggest that the cDC1-CD8+ T cell axis, deficient in Batf3−/− mice, plays an important role in the immune response to orthotopic bladder tumors, and given the high level of CD40 expression on cDC1s within the bladder TME, could serve as an attractive target for CD40 agonist therapies.
Intratumoral CD40 agonism using a fully-human Fc-optimized anti-CD40 agonist antibody induces robust anti-tumor immunity in humanized mouse models of bladder cancer
After demonstrating a biological rationale for intravesical CD40 agonism using the murine 1C10 anti-CD40 agonist antibody, we next set out to investigate the anti-tumor therapeutic potential of fully-human anti-CD40 agonist antibodies in a syngeneic, species-matched in vivo setting critical for clinical translation. While not specific to CD40 antibodies, many agonist antibodies translated into clinical practice have been studied in murine models that fail to recapitulate human antibody interaction with the unique human FcγR landscape. Given the substantial differences between mouse and human FcγR systems, species-matched antibodies and Fc receptors are required to accurately assess the in vivo activity and toxicity of candidate therapeutic antibodies. To this end, we crossed humanized FcγR mice, which lack all mouse FcγRs and transgenically express all human FcγRs (32), to a strain expressing human CD40, allowing the development of mice humanized for both CD40 and FcγRs (hCD40/hFcγR). These mice provide a reliable platform for the study of fully human CD40 agonist antibodies in vivo, recapitulating important parameters of biological activity and toxicity observed with CD40 agonist antibodies in the clinical setting (18, 19).
Using this humanized hCD40/hFcγR in vivo platform, we first compared the human CD40 agonist antibody CP-870,893, an investigational clinical product previously used in clinical trials (33), to the Fc-optimized variant 2141-V11. These antibodies have identical Fab regions and differ only in their Fc, with the 2141-V11 variant containing 5 point mutations that enhance FcγRIIB binding (Supplemental Figure 9A) (18, 19). Direct comparison of CP-870,893 to 2141-V11 demonstrated substantially enhanced anti-tumor responses in animals treated with 2141-V11 (Supplemental Figure 9B,C). This was associated with a substantial increase in the DC-associated T cell co-stimulatory factors CD80 and CD86 (Supplemental Figure 9D,E).
We then tested the 2141-V11 antibody for therapeutic responses across several immunocompetent, syngeneic bladder cancer cell line models (MB49, UPPL1541, and BBN963) using a bilateral subcutaneous tumor implantation approach and compared this to BCG therapy. Here, mice are injected intratumorally with 2141-V11 or BCG in a single tumor while the contralateral tumor remains untreated, thus allowing assessment of both a local and systemic response to therapy. Across all models, treatment with 2141-V11 led to both potent primary tumor regression and an additional systemic abscopal effect (Supplemental Figure 10). BCG treatment also demonstrated a therapeutic effect, although none of these BCG responses resulted in durable remission of tumors.
Because there are limited therapeutic options for patients unresponsive to BCG therapy, we next sought to evaluate the therapeutic activity of the 2141-V11 antibody in the setting of BCG-unresponsive disease. In this approach, subcutaneous MB49 tumors were initially treated with BCG, and upon tumor progression, were either transitioned to 2141-V11 or continued on BCG therapy (Figure 3A). Although BCG treatment resulted in slower overall tumor growth compared to controls, BCG was not capable of inducing sustained tumor regression despite continued treatment (Figure 3B). In contrast, treatment with 2141-V11 resulted in significantly improved and sustained local and systemic abscopal anti-tumor activity (Figure 3B–D). These data indicate that prior BCG treatment does not impair subsequent therapeutic efficacy of CD40 agonism, and that 2141-V11 can drive potent, durable anti-tumor responses despite BCG-refractory disease.
Figure 3:
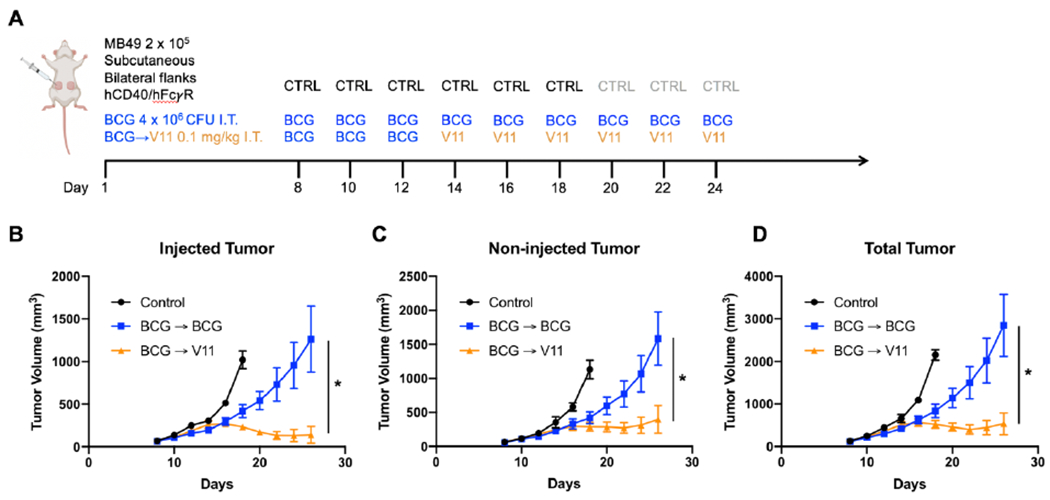
Intratumoral Fc-enhanced anti-CD40 agonist antibody 2141-V11 demonstrates local and systemic anti-tumor activity in BCG-unresponsive bladder cancer.
A, Schematic of bilateral flank subcutaneous tumor implantation model in humanized hCD40/FcγR mice with associated intratumoral treatment schedules of BCG and 2141-V11 (V11). One tumor (injected tumor) was treated, and the contralateral tumor (non-injected tumor) was also measured. BCG was initiated at day 8 for experimental groups and was continued for 3 treatments. Thereafter, progressive tumors were either continued on BCG treatment or switched to V11 on day 14. B-D, Tumor volumes in mice treated with control (n = 5 independent animals), BCG (n = 10 independent animals), and BCG followed by V11 (n = 9 independent animals) for (B) drug injected tumors, (C) non-drug injected tumors, and (D) total tumor burden. For B-D, One-way ANOVA with Tukey’s test was used to calculate statistical significance. * p < 0.05. All data are plotted as mean ± s.d.
Intravesical anti-CD40 agonist antibody therapy is associated with limited systemic toxicity
Clinical experience with CD40 agonist antibodies has demonstrated dose-limiting toxicities associated with systemic administration, including thrombocytopenia and transaminitis (34). These toxicities can be recapitulated in vivo in the humanized hCD40/hFcγR model, as platelets within this model express CD40 (consistent with human platelets), in contrast to platelets from WT mice, which lack CD40 expression (18, 19). Ligation of CD40 on platelets leads to platelet activation, which can result in platelet consumption, intravascular thrombosis, and liver damage that drive the observed clinical toxicities (19, 35). To assess potential toxicities associated with intravesical administration of the 2141-V11 antibody, we first performed a dose titration of intravesical 2141-V11 (from 0.1 to 2 mg/kg) in non-tumor bearing humanized hCD40/hFcγR mice and monitored for parameters of systemic toxicity, including platelet and serum transaminase levels (Supplemental Figure 11A). None of the doses tested resulted in any significant thrombocytopenia or transaminitis, in contrast to that seen with systemic antibody administration (administered intraperitoneal). Of note, intravesical administration of 2141-V11 in the setting of advanced bladder tumors did demonstrate a degree of platelet reduction and serum transaminase elevation at doses above 0.1 mg/kg (Supplemental Figure 11B), potentially driven by systemic absorption associated with advanced, highly-vascular tumors (Figure 1B). Within the orthotopic treatment model, intravesical dose titration of 2141-V11 did not demonstrate a clear relationship between antibody dose and therapeutic efficacy, with the lowest (0.1 mg/kg) treatment dose demonstrating both the highest efficacy (Supplemental Figure 12) and least toxicity (Supplemental Figure 11B). These results highlight the importance of careful dosing of CD40 agonists to achieve an optimal therapeutic index, as well as the need for appropriate humanized models to accurately assess the potential efficacy and toxicity of antibody candidates in the preclinical setting.
Intravesical Fc-enhanced anti-CD40 agonist antibody therapy drives potent and durable bladder cancer immunity in treatment-naïve and treatment-refractory disease
Intravesical treatment of NMIBC in the clinical setting is pursued following maximal transurethral resection of existing disease, where tumor burden at the time of intravesical drug therapy is likely to be low or absent. Thus, while treatment beginning at day 6 post tumor implantation (modeling advanced tumor burden; Figure 1B) demonstrated the ability of intravesical 2141-V11 anti-CD40 agonist antibody therapy (and BCG, to a lesser extent) to induce therapeutic responses in the setting of large established tumors (Supplemental Figure 12), we further investigated the therapeutic activity of intravesical 2141-V11 initiated at day 3 post tumor implantation (Figure 4A), which is likely to be more representative of the lower tumor burden (Figure 1B) present clinically at the time of intravesical drug treatment. In this context, intravesical treatment with 2141-V11 demonstrated anti-tumor activity superior to both BCG and controls. Bioluminescence imaging of luciferase-expressing bladder tumors at early time points post-treatment revealed significantly-reduced tumor burden in mice treated with intravesical 2141-V11, which was not observed in mice treated with intravesical BCG (Figure 4A, B). Survival analysis further demonstrated significantly improved long-term survival of mice treated with intravesical 2141-V11 compared to mice treated with intravesical BCG or controls (Figure 4C). Notably, intravesical 2141-V11 dosing in this setting was not associated with significant toxicities (Supplemental Figure 13). To evaluate the induction of long-term immunity, we subsequently re-challenged mice with a 10-fold higher dose of tumor cells at 60 days post initial tumor challenge in the absence of any additional therapy. Mice previously treated with 2141-V11 rejected tumor re-challenge (Figure 4D), suggesting that even in the context of the unique bladder TME, CD40 agonist therapy with 2141-V11 is capable of inducing an in situ vaccination effect driving protective systemic anti-tumor immunity. While used extensively in the field of bladder cancer, recent data suggest that the orthotopic MB49 model may not be truly representative of human urothelial carcinoma (31). To corroborate our findings in the MB49 model, we utilized an additional immunocompetent orthotopic bladder cancer model using the recently-described syngeneic murine bladder cancer cell line UPPL1541 (Figure 4E). The UPPL1541 cell line is derived from a novel genetically-engineered murine model (Upk3a-CreERT2; Trp53L/L; PtenL/L; Rosa26LSL-Luc) of high-grade bladder cancer, recapitulating the luminal molecular subtype of human high-grade urothelial cancer (31). In this model, intravesical treatment with 2141-V11 demonstrated enhanced anti-tumor activity compared to BCG and controls, as visualized by serial bladder ultrasound and the time to tumor detection (Figure 4F, G). Collectively, these data across orthotopic bladder tumor models suggest that local CD40 agonism via intravesical administration of the Fc-optimized 2141-V11 antibody is capable of driving potent and durable anti-tumor responses, without evidence of systemic toxicity following localized intravesical therapy.
Figure 4:
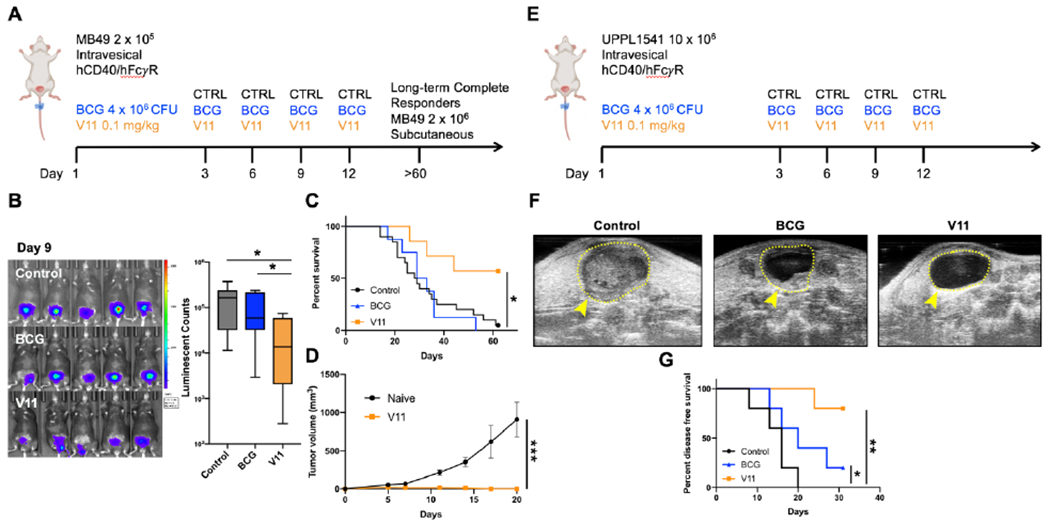
Intravesical Fc-enhanced anti-CD40 agonist antibody 2141-V11 reduces tumor burden, improves survival, and induces long-lasting anti-tumor immunity.
A, Schematic of the intravesical treatment of humanized hCD40/FcγR mice bearing orthotopic MB49 bladder tumors. B, Representative intravital luciferase imaging and quantitation of the bladders of mice treated with control (n = 6 independent animals), BCG (n = 8 independent animals), or 2141-V11 (V11; n = 8 independent animals) day 9 post tumor implantation. C, Survival of mice treated as outlined in (A). D, Long-term treatment responders (mice surviving >60 days after initial tumor challenge) were re-challenged with MB49 tumor cells subcutaneously. Tumor volume was monitored over 3 weeks and compared to naive mice receiving the same MB49 tumor cells. E, Schematic of orthotopic UPPL1541 tumor implantation and treatment with control (n = 5 independent animals), BCG (n = 5 independent animals), and 2141-V11 (V11; n = 5 independent animals). Longitudinal ultrasound imaging twice weekly was used to track bladder tumor growth. F, Representative ultrasound images of bladders at day 20 post tumor implantation. The boundaries of the bladder are outlined by a yellow dashed line. Contrast within the bladder space, marked by yellow arrowhead, indicates abnormal growth. G, Disease-free survival of mice treated as outlined in (E). Detection of enhanced contrast within the bladder space by serial ultrasound imaging was used to determine bladder tumor growth longitudinally. For E, One-way ANOVA with Tukey’s test was used to calculate statistical significance. For C and G, Log-Rank test was used to calculate statistical significance. For D, Two-tailed t-test was used to calculate statistical significance. * p < 0.05, ** p < 0.01, *** p < 0.001. All data are plotted as mean ± s.d. Results shown are representative of 2 independent experiments.
Finally, as up to 75% of patients with NMIBC are ultimately unresponsive to intravesical BCG therapy, we evaluated whether intravesical CD40 agonism with the 2141-V11 antibody would be capable of rescuing orthotopic BCG-unresponsive disease. We established a model where mice bearing progressive bladder tumors despite intravesical BCG therapy were subsequently randomized to treatment with continued BCG or a switch in treatment to intravesical 2141-V11 (Figure 5A). Bioluminescence imaging of luciferase-expressing MB49 tumors at day 9 post tumor implantation confirmed progressive disease despite BCG therapy and comparable tumor burdens in the randomized cohorts (Figure 5B). Subsequent switch to intravesical 2141-V11 therapy induced significant tumor regression by day 18 post tumor implantation (Figure 5B) and significantly improved long-term survival compared to mice continued on BCG therapy (Figure 5C). Likewise, using the orthotopic UPPL1541 model (Figure 5D), a switch in therapy from BCG to 2141-V11 was associated with improved anti-tumor activity, including time to detection of disease on serial bladder ultrasound (Figure 5E, F) and tumor burden by bladder weights (Figure 2G). Overall, these data support the anti-tumor efficacy of intravesical CD40 agonism with 2141-V11 in both treatment naïve and BCG-unresponsive disease settings.
Figure 5:
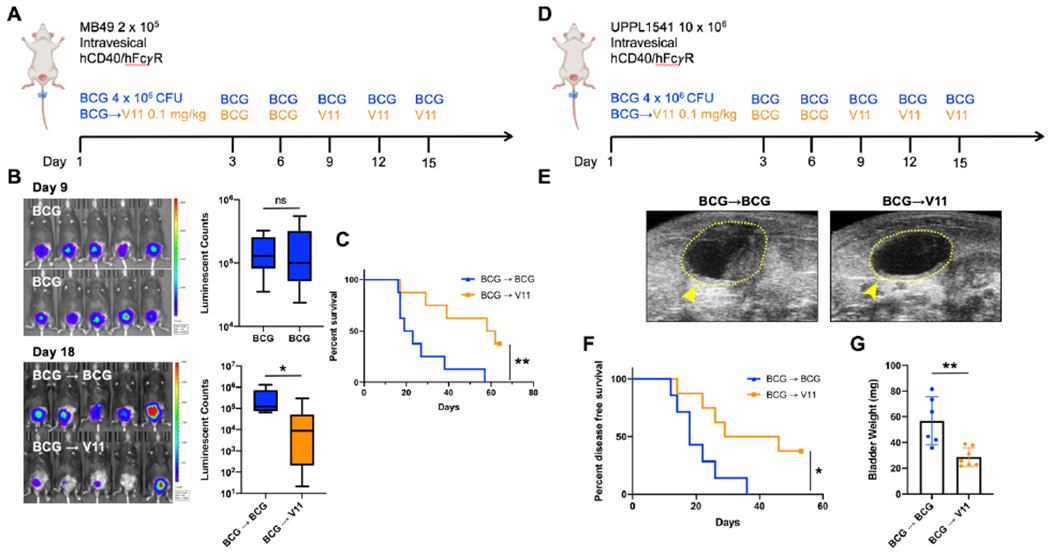
Intravesical Fc-enhanced anti-CD40 agonist antibody 2141-V11 induces tumor regression and improves survival in advanced BCG-unresponsive disease.
A, Schematic of orthotopic MB49 bladder tumor implantation and intravesical treatment with BCG and 2141-V11 (V11). Mice were initially treated with BCG beginning day 3 post tumor implantation, and those with progressive disease as assessed by tumor bioluminescence on day 9 were either continued on BCG (blue line) or switched to V11 treatment (orange line). B, Representative intravital luciferase imaging of the bladders of mice at day 9 post tumor implantation before continued BCG treatment or switch to V11 treatment. C, Quantification of bioluminescence data in B comparing the pre-V11 treatment tumor burden in both BCG continuation (n = 8 independent animals) and V11 (n = 8 independent animals) cohorts. D, Schematic of orthotopic UPPL1541 bladder tumor implantation and intravesical treatment with BCG and 2141-V11 (V11). Mice were initially treated with BCG beginning day 3 post tumor implantation, and at day 9 were either continued on BCG (blue line) or switched to V11 treatment (orange line). E, Representative ultrasound images on day 26 of mice continued on BCG treatment or switched to V11 beginning day 9. F, Disease-free survival of mice treated as outlined in E. Detection of enhanced contrast within the bladder space by serial ultrasound imaging was used to determine bladder tumor growth longitudinally. G, Bladder weights of mice treated as outlined in D were assessed at day 53 post tumor implantation. For B and G, Two-tailed t-tests were used to calculate statistical significance, n.s. = not significant, * p < 0.05, ** p < 0.01. For C and F, Log-Rank test was used to calculate statistical significance, * p < 0.05, ** p < 0.01.
Discussion
Here we provide the rationale and pre-clinical support for a novel approach to the treatment of bladder cancer through intravesical administration of CD40 agonist antibodies. As a proof-of-concept, we utilized multiple immunocompetent orthotopic murine bladder cancer models to demonstrate the therapeutic effect of local CD40 agonism to drive anti-tumor immunity, a process which depends upon CD8+ T cells and the transcription factor Batf3. We then utilized humanized murine models expressing hCD40/hFcγR to show potent in vivo anti-tumor therapeutic activity, long-lived systemic immunity, and minimal toxicity induced by intravesical administration of the fully-human Fc-optimized CD40 agonist antibody 2141-V11 across multiple disease settings and tumor models, including BCG-unresponsive disease. These data provide supportive evidence for the clinical translation of intravesical antibody therapy with CD40 agonist antibodies for bladder cancer therapy, and define important mechanisms underlying CD40-driven bladder cancer immune responses.
Treatment for intermediate and high-risk NMIBC has long relied on intravesical BCG (36). BCG therapy has been shown to depend upon T cell activity for its anti-tumor effects, but can function independently of BCG-specific T cells, suggesting it behaves as an adjuvant to enhance anti-tumor T cell immunity (37). Nevertheless, important limitations exist regarding BCG therapy, including strain-specific differences affecting treatment efficacy (38), toxicity from significant non-specific inflammation (3), recent restricted access due to an ongoing global shortage (4), and perhaps most importantly, high rates of BCG-unresponsive disease (occurring in up to 75% of patients in some series) (2). Pre-clinical models have similarly reflected variable and often marginal rates of BCG efficacy (20, 39), consistent with our current findings. In contrast, our current data demonstrate robust and consistent anti-tumor responses to intravesical anti-CD40 agonist antibody therapy under a range of tumor burdens, as well as in a variety of tumor models and dosage schemes in both the upfront and treatment-refractory disease settings, suggesting a favorable therapeutic profile potentially applicable to multiple clinical bladder cancer disease contexts. Notably, local intravesical CD40 activation with 2141-V11 further induced durable protective systemic anti-tumor immunity, capable of rejecting a 10-fold higher tumor challenge in a distant tissue compartment more than 60 days after initial therapy. Although the intravesical delivery of monoclonal antibodies is not currently used in routine clinical practice, intravesical instillation has been shown to be a viable route of administration for the local delivery of antibody-based therapies in the clinical setting (40)’(41) and is currently being explored in several active trials for the treatment of NMIBC (NCT02808143, NCT03759496).
Prior work from our group and others have shown that anti-tumor immune responses induced by CD40 agonism require cytokines, including IFN-γ (23) and IL-12 (42), as well as the involvement of T cells (19)’(23). Indeed, our analysis of T cell subsets within the bladder TME revealed a significant reduction in the percentage of CD8+ T cells carrying an exhausted phenotype (PD-1+LAG-3+) in response to anti-CD40 agonist antibody therapy, and CD8+ T cells were functionally required for treatment efficacy. This is consistent with findings of other groups in other tumor models, with T cells expressing low exhaustion markers likely representing effector cells involved in the anti-tumor immune response (25, 26, 43).
Our current data now further implicate CD40-expressing DCs within the bladder TME as a critical mediator of intravesical CD40 agonist therapy, as these cells are directly targeted by intravesically-delivered antibodies. CD40 activation rapidly increases the T cell co-stimulatory receptors CD80 and CD86 on DCs, likely driving T cell-dependent effector functions and an in situ vaccination effect inducing both local and systemic immunity. Interestingly, we find that cDC2s express and are activated by CD40 agonism, yet are not essential to treatment efficacy, as they are replete in the tumor microenvironment and draining lymph nodes of Batf3−/− mice that do not respond to CD40 agonism. The specific functions and roles of cDC2s in the tumor microenvironment and their relation to anti-tumor immune responses remains to be elucidated.
Batf3−/− mice lacking cDC1s demonstrate both impaired tumor control in response to CD40 agonist therapy as well as accelerated spontaneous tumor outgrowth in the setting of carcinogen (BBN) exposure. Batf3 deficiency has been shown to have the potential to affect numerous cellular processes in certain settings, such as the differentiation of T regulatory cells in the colonic lamina propria (29). Immunophenotyping in the Batf3−/− tumor models used in the present study demonstrated that T regulatory cell proportions and other immune populations outside of the cDC1-CD8+ T cell axis (including CD4+ T cells, macrophages, and cDC2) were similar between tumor-bearing Batf3−/− and wild-type control mice, suggesting that certain effects associated with Batf3 deficiency may be context dependent. Of note, Batf3 deficiency has recently been described to affect the development of CD8+ T cell memory in infection models, although primary CD8+ T cell responses were found to be unchanged in these settings (44). Importantly, these effects were investigated in a T cell intrinsic manner, in contrast to pervasive Batf3 deficiency, which demonstrate defects in generating primary CD8+ T cell responses (27). The tumor treatment models used in the current study only investigated responses in Batf3−/− mice in the acute setting, where T cell memory responses are unlikely to play a major role. The lack of CD8+ T cell accumulation in tumors from Batf3−/− mice potentially explain the lack of response to CD40 agonist treatment.
Dendritic cells, in particular cDC1s, have been found to be strongly associated with cancer immune surveillance and immunotherapy response (27)’(45)’(46)’(47), and cDC1 exclusion from the TME can act as an immune evasion strategy prohibiting development of anti-tumor T cell responses (48)’(49). Recently published work has also shown that anti-tumor immunity can be blocked by specific deletion of CD40 in the cDC1 lineage using Xcr1-cre; CD40fl/fl conditional knockouts (50). Prior models, however, did not examine DCs within the bladder TME, and did not assess the effects of cDC1s on de novo carcinogenesis. The observation that Batf3 deficiency leads to enhanced chemically-induced carcinogenesis in the bladder argues that Batf3, and possibly the cDC1 subset, is a primary mediator of early bladder cancer growth and progression. However, we cannot formally exclude non-DC based functions of Batf3 on BBN-induced carcinogenesis, which are the subject of ongoing investigations.
In addition to their antigen cross presentation abilities, cDC1s can also recruit T cells into the TME and provide cytokine support signals to enable T cell effector functions (51). cDC1s have also been shown to upregulate CD40 upon activation in response to capturing cell-associated antigens, and are the primary DC population trafficking antigen to lymph nodes (45)’(52). Activated cDC1s are major producers of IL-12, and prior intravital imaging studies of IL-12+ DCs demonstrated that these cells bind CD40 agonist antibodies in vivo (42), consistent with a role for cDC1s in driving the therapeutic effects of CD40 agonist antibodies (23). Given these findings, further characterization of the role of DC subsets in the setting of human NMIBC is warranted.
The studies described herein use species-matched antibody-FcγR systems, including mice humanized for CD40 and FcγRs, to evaluate the in vivo therapeutic activity and toxicity of anti-CD40 antibodies. The use of such systems is essential for accurate assessment of the effects of agonistic antibodies, which critically depend on antibody interaction with target and FcγR landscapes that differ significantly between species. The humanized hCD40/hFcγR murine model has been previously demonstrated to more-accurately model both the efficacy and toxicity of human anti-CD40 antibodies (19), including the fully-human Fc-enhanced CD40 agonist antibody 2141-V11 evaluated in this study. Use of this model has allowed Investigational New Drug-enabling studies supporting the clinical testing of intratumoral 2141-V11 in patients with metastatic solid tumors (NCT04059588).
While the bladder is thought to be relatively impermeable to large macromolecules (e.g. antibodies, 150 kDa), with likely negligible systemic absorption and, therefore, minimal risk of systemic toxicity, use of appropriate humanized models are important to ensure that local intravesical delivery is not accompanied by unexpected toxicities. We previously established that 0.1 mg/kg of 2141-V11 was a safe and effective dose when delivered both systemically and intratumorally in humanized hCD40/hFcγR mice (19). Our current data demonstrate that doses up to 2 mg/kg are not associated with any significant systemic toxicity when administered intravesically into normal bladders of humanized hCD40/hFcγR mice. This is likely to model patients with NMIBC in the clinical setting, where the majority of bladder tissue consists of normal urothelium and intravesical therapy following transuretheral tumor resection is administered only after several weeks to allow for healing of the bladder wall. We did find evidence of systemic pharmacodynamic changes in platelets and transaminases at high doses of 2141-V11 in the setting of advanced in situ orthotopic bladder tumors, although doses up to 0.1 mg/kg remained without significant toxicity even in this setting. Notably, our data also demonstrated maximal anti-tumor efficacy at the 0.1 mg/kg dose, without increased (and potentially even decreased) anti-tumor activity at higher dose levels. This is consistent with an emerging understanding of the limited applicability of traditional cytotoxic chemotherapy-based dose-efficacy and dose-toxicity paradigms to modern clinical immuno-oncology, where maximum tolerated doses may have limited bearing on biologically-effective dosing (53).
Notably, intravesical 2141-V11 treatment across orthotopic bladder tumor models produced a roughly 50-60% complete response rate, whereas intratumoral dosing in the subcutaneous models demonstrated a near 100% complete response rate. Although this could be due to a variety of reasons, one possible explanation could be differential access to the draining lymph node, as we found that intravesical dosing, at least at early time points, had limited lymph node accumulation, while antibodies administered intratumorally can be readily accessible to the draining lymph node (54). Detailed analysis of the spatiotemporal dynamics of antibody tissue distribution after intravesical administration, the importance of the draining lymph node in mediating responses to CD40 agonist therapies, and the potential for further antibody engineering to affect these parameters are important questions to address in future studies.
Within the last several years, anti-PD-1/L1 therapy has become a care standard for locally advanced and metastatic urothelial cancer (55). Most recently, PD-1 blockade with pembrolizumab was approved by the United States Food and Drug Administration (FDA) for patients with high-risk BCG-unresponsive NMIBC ineligible for or electing not to undergo cystectomy. Nevertheless, efficacy with pembrolizumab in this setting is relatively modest, with about 60% of treated patients still having persistent disease at 3 months, and only about half of responding patients (20% of all treated patients) remaining in remission at 1 year (FDA Advisory Committee Briefing Document. 2019. at https://www.fda.gov/media/133542/download). Our current data demonstrate notable PD-1 expression in the bladder TME, which can be significantly reduced (although not completely abrogated) by CD40 agonist therapy. Synergy between CD40 agonist and PD-1 antagonist approaches has been previously observed in multiple preclinical in vivo tumor models, including in settings refractory to PD-1/PD-L1 immune checkpoint monotherapy (56–58). The non-redundant immunologic roles of the CD40 and PD-1/PD-L1 pathways also provide a biologic rationale for combination therapy (34). This suggests a potential additional benefit to rational combination of intravesical CD40 agonist therapy with anti-PD-1 approaches, which will be the subject of future pre-clinical and clinical studies.
Collectively, these data describe a rationally designed immune-based therapeutic approach to the treatment of bladder cancer through intravesically-delivered Fc-optimized anti-CD40 agonist antibodies, which demonstrate robust local and systemic anti-tumor activity and a favorable risk-benefit profile in both treatment-naïve and treatment-refractory disease settings. These data further identify DCs within the bladder TME, particularly the cDC1 subset, as a primary target of CD40 antibody therapy, and describe pharmacodynamic changes in exhausted T cell subsets within the bladder TME driven by treatment with Fc-optimized CD40 agonist antibodies, suggesting additional opportunities for future combination therapy approaches. These data provide a rationale for phase I clinical studies evaluating intravesical administration of the Fc-optimized CD40 agonist antibody 2141-V11 for the treatment of patients with NMIBC.
Materials and Methods
Study Design
The primary objective of this study was to determine the anti-tumor activity and underlying mechanisms associated with intravesically-administered anti-CD40 agonistic antibodies. These studies involved controlled laboratory experiments using humanized orthotopic and subcutaneous implantable mouse models of bladder cancer, as well as spontaneous carcinogen-induced models of bladder cancer, to assess local and systemic effects of CD40 agonism in the bladder TME and the role of DCs in the immune response to bladder cancer. Primary outcomes for most studies included tumor burden (measured by caliper, bladder weight, tumor bioluminescence imaging, ultrasound, or histology) and survival. While we expected CD40 agonism to activate anti-tumor immunity, the observation that low-dose intravesical anti-CD40 agonist antibody therapy can confer durable, systemic anti-tumor immunity in both treatment-naive and treatment-refractory settings was unexpected. Consistent with the requirement for species-matched antibody-FcγR systems to accurately model the activity and toxicity of therapeutic antibodies, these studies used C57BL/6J mice to evaluate the effects of murine anti-CD40 agonist antibodies and humanized hCD40/hFcγR mice to evaluate the effects of the human anti-CD40 agonist antibody 2141-V11. Tumor-bearing mice were either randomized into treatment groups, or baseline tumor burden was measured and groups were assigned with matching tumor burden between experimental conditions. Sample sizes were determined based on initial pilot experiments to estimate likely effect sizes and variation. Data were analyzed using the Grubb’s test for statistical outliers using an α value of 0.01. Data are representative of at least 2 experiments. Tumor measurements and survival monitoring were blinded.
Mouse strains
All mice were bred and housed at Rockefeller University Comparative Bioscience Center under specific pathogen-free conditions. All experiments were performed in compliance with institutional guidelines and were approved by the Rockefeller University Institutional Animal Care and Use Committee (IACUC). C57BL/6J (WT) (Stock# 000664) and Batf3−/− (Stock# 013755) mice were purchased from The Jackson Laboratory. Humanized mice that are fully backcrossed to the C57BL/6J background expressing human Fc receptors (FcgRanull’, FcgRI+, FcgRIIaR131+, FcgRIIb+, FcgRIIaF158+, FcgRIIIb+) and human CD40 were generated and extensively characterized as previously described (18, 32). All mice used were 7-12 weeks old at time of experiment. Bladder implantation experiments used only female mice, due to the specific procedure necessary to engraft tumor cells within the bladder. All other experiments used a mix of male and female mice.
Cell Lines and Cell Culture
The murine tumor cell lines MB49-Luciferase (from M. Glickman, MSKCC), UPP1541 (W. Kim, UNC), and BBN963 (W. Kim, UNC) were cultured inside tissue culture treated flasks with DMEM with 10% FCS (Hyclone) and 1X penicillin-streptomycin (100 units/ml, Gibco) at 37C and 5% CO2. Cells were split twice per week and cell viability was measured using trypan blue staining in the Countess II automated cell counter (Thermo Fisher).
Intravesical Tumor Implantation
Cells were detached from tissue culture plastic using 0.05% Trypsin EDTA (Gibco) for 5 minutes at 37C. Cells were then washed twice using DMEM and cell viability was assessed using the Countess II automated cell counter (Thermo Fisher). Cells were resuspended in DMEM at 4 x 106 cells per ml for MB49-Luciferase or 2 x 108 cells per ml for UPPL1541. Plastic 24.5G catheters were used for intravesical instillation of 7-10 week old female mice. Prior to tumor cell infusion, 50 ul of 10 mg/ml protamine sulfate was instilled into the bladder of anesthetized mice and maintained for 30 minutes. Contents of the bladder were then voided using digital pressure on the bladder. Next, 50 ul of the tumor cell mixture were instilled into the bladder and maintained for 2 hours. Throughout the procedure, mice were kept on inhalational isoflurane (1.5% induction, and 0.5% maintenance) and heating pads to maintain core body temperature. Mice were then monitored regularly after implantation for signs of hematuria and tumor growth. Survival studies were followed up to 60 days after tumor implantation and mice that survived beyond this point were eligible for tumor re-challenge studies to assess immune memory.
Intravesical Treatments
Mice were anesthetized and 24.5G plastic catheters were used to instill treatments into the bladders of mice. A 50 ul volume of murine anti-CD40 (clone 1C10, mouse IgG1) at a dose of 2 mg/kg per mouse was instilled into the bladder and maintained for 1 hour. For labeled 1C10 antibody studies, 1C10 antibody was conjugated to AlexaFluor488 using the Alexa Fluor 488 Antibody Labeling Kit (Thermo Fisher) and then administered intravesically at a dose of 2 mg/kg. Anti-human CD40 (clone 2141-V11, human IgG1 with Fc modifications) was instilled into the bladder at a 0.1 mg/kg dose for most experiments unless indicated otherwise. BCG (TICE strain, Merck) was also instilled into the bladder in a 50 ul volume of PBS, with a dose of 4 x 106 CFU of bacteria. Dosing schemes adopted either a 2 dose (for anti-murine CD40) or 4 dose (for anti-human CD40) schedule for treatment, unless otherwise noted. The 2-dose schedule initiated treatment at day 6 post tumor implantation and a second dose was administered at day 9. The 4-dose schedule initiated treatment at day 2, with follow-up treatments at days 5, 8, and 12. For lymph node tracing experiments, mice were intravesically infused with 50 ul of Evans Blue dye (30 mg/ml in PBS) and sacrificed after 1 hour of dye dwell time.
Subcutaneous Tumor Implantation
MB49-Luciferase, UPPL1541, and BBN963 cells were detached from tissue culture flasks and assessed for viability. Cells were washed twice with PBS, and were resuspended. Mice were injected in their lower flanks with 50 ul of this mixture, corresponding to 2 x 105 (for MB49), 5 x 106 (for UPPL1541), and 10 x 106 (BBN963) cells inoculated per mouse flank. For some experiments, tumors were implanted on both flanks. For tumor re-challenge studies, 2 x 106 cells were implanted into the flanks of mice that were long-term survivors of MB-49-Luciferase tumors in the bladder (>60 days post initial tumor implantation). Tumors were measured 2-3 times per week using an electronic caliper. Volume was calculated using the formula (L12 x L2)/2, where L1 is the longest dimension. Tumors were randomized into treatment groups at day 6 post implantation, or when tumors were approximately 5-6 mm in diameter.
Intratumoral Treatments
Intratumoral injection of either murine anti-CD40 (1C10) or human anti-CD40 (2141-V11) antibodies was performed on subcutaneous tumors. Murine anti-CD40 was dosed at 2 mg/kg, and 2141-V11 was dosed at 0.1 mg/kg in and around the tumor site with care to prevent leakage of the injected dose. For bilateral flank subcutaneous tumor treatment models, one tumor was treated and both the treated tumor and the untreated tumor on the contralateral flank were assessed for growth. Treatments were performed every 3 days, for a total of 4 doses, unless otherwise noted.
Flow Cytometry
Tumor tissues were digested using Mouse Tumor Dissociation kits (Miltenyi) according to the manufacturer’s protocols. Isolated cells were stained for viability using the Aqua Amine fixable live dead dye (Thermo Fisher) using standard protocols. After viability staining, cells were resuspended in FACS buffer (PBS with 0.5% BSA and 2 mM EDTA) and Fc blocked using mouse or human TrueStain FcX (Biolegend). For analysis stains, cells were incubated for 30 minutes at 4C with the following anti-mouse antibodies from Thermo Fisher: CD90.2 (Thy-1.2) SuperBright 645, clone 53-2.1; CD4-AlexaFluor 488, clone GK1.5, CD8a-SuperBright 780, clone 53-6.7; CD3e-PE-Cy5, clone 145-2C11; B220-PE-Cy5, clone RA3-6B2; CD19-PE-Cy5, clone 1D3; CD11c-PE-eFluor610, clone N418; CD45-AlexaFluor 700, clone 30-F11; MHCII-APC-eFluor 780, clone M5/114.15.2; antibodies from Biolegend: NK1.1-PE-Cy5, clone PK136; F4/80-AlexaFluor488; XCR-1-PE, clone ZET; anti-mouse CD80-APC, clone 16-10A1; CD86-Bv785, clone GL-1, PD-1-PE, clone 29F.1A12; LAG-3-PE-Cy7, clone C987W; anti-human CD40-Bv421, clone 5C3; antibodies from BD: anti-mouse CD40-Bv421, clone 3/23; anti-mouse CD172a-Bv605 (Sirpα), clone P84. Samples were analyzed using an Attune NxT flow cytometer (Thermo Fisher) and data was analyzed using FlowJo 10 (TreeStar).
Bioluminescence Imaging
Mice implanted with MB-49-Luciferase tumors were dosed with 3 mg of D-luciferin chemiluminescent substrate diluted in PBS i.p. The lower abdomens of the mice were shaved and subsequently imaged using an IVIS Spectrum in vivo imaging system (Perkin Elmer) with an exposure time of 30 seconds with a wide field setting to image 5 mice simultaneously. Total luminescence counts were quantified by selecting a uniform region of interest (ROI) over the bladder area using the Living Image software package (Perkin Elmer).
Toxicity Testing
Humanized hCD40/hFcγR mice were dosed intravesically with 2141-V11 at the indicated doses and maintained in the bladder for 1 hour. 24 hours after dosing, mice were bled retro-orbitally using heparinized capillary tubes. Complete blood counts were performed using an Element HT5 hematology analyzer (Heska) to assess platelet levels. Blood was also collected in BD SST Microtainer tubes and centrifuged to collect serum. Serum was used to assess AST and ALT levels using serum chemistry panels at the Laboratory of Comparative Pathology at Memorial Sloan Kettering.
Ultrasound Imaging
Mice were anesthetized using inhalation isoflurane and hair was removed from their lower abdomen by shaving and chemical hair removal cream. Ultrasound gel was applied to the abdomen and mouse bladders were imaged using the Vevo 2100 High Resolution In Vivo Micro Imaging System (Fujifilm VisualSonics). Mouse bladders were imaged beginning on day 8 post tumor implantation and were monitored twice per week to track the appearance of bladder tumors to allow calculation of disease-free survival rates.
Statistical Analysis
Data was analyzed using Prism GraphPad software. One-way ANOVA with Tukey’s multiple comparison test was used to compare 3 or more groups. Comparisons between 2 groups used unpaired two-tailed t tests for statistical significance. All data, unless otherwise indicated, are plotted as mean ± standard deviation (s.d.). Kaplan-Meier curves used Log-Rank tests to calculate statistical significance. For all statistical tests, * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, a p value under 0.05 was considered statistically significant, not significant values are denoted as n.s. Lines associated with asterisks indicate groups compared for significance.
Supplementary Material
Supplemental Figure 1: Iliac lymph nodes drain the bladder.
Supplemental Figure 2: CD40 agonism does not reverse exhaustion signatures on CD4+ T cells and innate lymphoid cells.
Supplemental Figure 3: CD8+ T cell depletion in the tumor-bearing bladder.
Supplemental Figure 4: Characterization of immune populations in Batf3-deficient mice.
Supplemental Figure 5: Secondary lymphoid organs of Batf3-deficient mice lack cDC1.
Supplemental Figure 6: Batf3 deficiency enhances tumor growth and progression in the bladder.
Supplemental Figure 7: Batf3 deficiency abrogates anti-CD40 agonist antibody response in multiple subcutaneous models of bladder cancer.
Supplemental Figure 8: Intravesically-administered anti-CD40 agonist antibodies bind to myeloid cells in the tumor-bearing bladder.
Supplemental Figure 9: Fc-enhanced human anti-CD40 agonist antibody 2141-V11 demonstrates improved anti-tumor activity compared to the parental antibody variant and activates dendritic cells in the tumor microenvironment.
Supplemental Figure 10: Fc-enhanced anti-CD40 agonist antibody 2141-V11 demonstrates anti-tumor activity in multiple subcutaneous models of bladder cancer.
Supplemental Figure 11: Toxicity testing of intravesical anti-CD40 agonist antibody 2141-V11.
Supplemental Figure 12: Response to intravesical BCG and anti-CD40 agonist antibody 2141-V11 in orthotopic models of advanced bladder cancer.
Supplemental Figure 13: Intravesical anti-CD40 agonist antibody 2141-V11 demonstrates limited toxicity during treatment of early-stage bladder tumors.
Acknowledgements
We thank members of the J.V.R. laboratory for excellent technical assistance and feedback in construction of this manuscript; G. Redelmen Sidi and M. Glickman for providing bladder cancer cell lines and technical advice; and the Tri-Institutional Laboratory of Comparative Pathology for assistance with toxicology and pathology studies.
Funding
This work was supported in part by the National Institutes of Health grants F32CA250147 to C.S.G., K08CA248966-01 to D.A.K., R01CA244327, R35CA196620 and P01CA190174 to J.V.R, the MSK SPORE in Bladder Cancer (P50CA221745) by a SPORE-Developmental Research Program grant, and the National Center for Advancing Translational Sciences through Rockefeller University grants UL1TR001866 and KL2TR001865 NIH Clinical and Translational Science Award Program to J.L.W and D.A.K. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The authors would also like to acknowledge support from the Bladder Cancer Advocacy Network Innovation Award and the V Foundation for Cancer Research under grant ID #T2017-002. Support was also provided by the Robertson Therapeutic Development Fund and Rockefeller University, the American Society of Hematology from a Research Training Award for Fellows (D.A.K), and the American Society of Clinical Oncology/Conquer Cancer Foundation from a Young Investigator Award (J.L.W.; any opinions, findings, and conclusions expressed in this material are those of the authors and do not necessarily reflect those of the American Society of Clinical Oncology or Conquer Cancer).
Footnotes
Competing Interests
The authors declare no competing interests.
Data and Materials Availability
All data associated with this study are available in the main text or the supplementary materials.
References
- 1.Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Pineros M, Znaor A, Bray F, Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer 144, 1941–1953 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Kamat AM, Colombel M, Sundi D, Lamm D, Boehle A, Brausi M, Buckley R, Persad R, Palou J, Soloway M, Witjes JA, BCG-unresponsive non-muscle-invasive bladder cancer: recommendations from the IBCG. Nat Rev Urol 14, 244–255 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Saluja M, Gilling P, Intravesical bacillus Calmette-Guerin instillation in non-muscle-invasive bladder cancer: A review. Int J Urol 25, 18–24 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Bandari J, Maganty A, MacLeod LC, Davies BJ, Manufacturing and the Market: Rationalizing the Shortage of Bacillus Calmette-Guerin. Eur Urol Focus 4, 481–484 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Jarow JP, Lerner SP, Kluetz PG, Liu K, Sridhara R, Bajorin D, Chang S, Dinney CP, Groshen S, Morton RA, O’Donnell M, Quale DZ, Schoenberg M, Seigne J, Vikram B, Clinical trial design for the development of new therapies for nonmuscle-invasive bladder cancer: report of a Food and Drug Administration and American Urological Association public workshop. Urology 83, 262–264 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Ribas A, Wolchok JD, Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li F, Ravetch JV, Inhibitory Fcgamma receptor engagement drives adjuvant and anti-tumor activities of agonistic CD40 antibodies. Science 333, 1030–1034 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, Torigian DA, O’Dwyer PJ, Vonderheide RH, CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 331, 1612–1616 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beatty GL, Li Y, Long KB, Cancer immunotherapy: activating innate and adaptive immunity through CD40 agonists. Expert Rev Anticancer Ther 17, 175–186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cooke PW, James ND, Ganesan R, Wallace M, Burton A, Young LS, CD40 expression in bladder cancer. J Pathol 188, 38–43 (1999). [DOI] [PubMed] [Google Scholar]
- 11.Chowdhury F, Johnson PW, Glennie MJ, Williams AP, Ex vivo assays of dendritic cell activation and cytokine profiles as predictors of in vivo effects in an anti-human CD40 monoclonal antibody ChiLob 7/4 phase I trial. Cancer Immunol Res 2, 229–240 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Furman RR, Forero-Torres A, Shustov A, Drachman JG, A phase I study of dacetuzumab (SGN-40, a humanized anti-CD40 monoclonal antibody) in patients with chronic lymphocytic leukemia. Leuk Lymphoma 51, 228–235 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Irenaeus SMM, Nielsen D, Ellmark P, Yachnin J, Deronic A, Nilsson A, Norlen P, Veitonmaki N, Wennersten CS, Ullenhag GJ, First-in-human study with intratumoral administration of a CD40 agonistic antibody, ADC-1013, in advanced solid malignancies. Int J Cancer 145, 1189–1199 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Bajor DL, Mick R, Riese MJ, Huang AC, Sullivan B, Richman LP, Torigian DA, George SM, Stelekati E, Chen F, Melenhorst JJ, Lacey SF, Xu X, Wherry EJ, Gangadhar TC, Amaravadi RK, Schuchter LM, Vonderheide RH, Long-term outcomes of a phase I study of agonist CD40 antibody and CTLA-4 blockade in patients with metastatic melanoma. Oncoimmunology 7, e1468956 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pincetic A, Bournazos S, DiLillo DJ, Maamary J, Wang TT, Dahan R, Fiebiger BM, Ravetch JV, Type I and type II Fc receptors regulate innate and adaptive immunity. Nat Immunol 15, 707–716 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.White AL, Chan HT, Roghanian A, French RR, Mockridge CI, Tutt AL, Dixon SV, Ajona D, Verbeek JS, Al-Shamkhani A, Cragg MS, Beers SA, Glennie MJ, Interaction with FcgammaRIIB is critical for the agonistic activity of anti-CD40 monoclonal antibody. J Immunol 187, 1754–1763 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Li F, Ravetch JV, A general requirement for FcgammaRIIB co-engagement of agonistic anti-TNFR antibodies. Cell Cycle 11, 3343–3344 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dahan R, Barnhart BC, Li F, Yamniuk AP, Korman AJ, Ravetch JV, Therapeutic Activity of Agonistic, Human Anti-CD40 Monoclonal Antibodies Requires Selective FcgammaR Engagement. Cancer Cell 29, 820–831 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knorr DA, Dahan R, Ravetch JV, Toxicity of an Fc-engineered anti-CD40 antibody is abrogated by intratumoral injection and results in durable antitumor immunity. Proc Natl Acad Sci U S A 115, 11048–11053 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Biot C, Rentsch CA, Gsponer JR, Birkhauser FD, Jusforgues-Saklani H, Lemaitre F, Auriau C, Bachmann A, Bousso P, Demangel C, Peduto L, Thalmann GN, Albert ML, Preexisting BCG-specific T cells improve intravesical immunotherapy for bladder cancer. Sci Transl Med 4, 137ra172 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Lavelle J, Meyers S, Ramage R, Bastacky S, Doty D, Apodaca G, Zeidel ML, Bladder permeability barrier: recovery from selective injury of surface epithelial cells. Am J Physiol Renal Physiol 283, F242–253 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Guilliams M, Dutertre CA, Scott CL, McGovern N, Sichien D, Chakarov S, Van Gassen S, Chen J, Poidinger M, De Prijck S, Tavernier SJ, Low I, Irac SE, Mattar CN, Sumatoh HR, Low GHL, Chung TJK, Chan DKH, Tan KK, Hon TLK, Fossum E, Bogen B, Choolani M, Chan JKY, Larbi A, Luche H, Henri S, Saeys Y, Newell EW, Lambrecht BN, Malissen B, Ginhoux F, Unsupervised High-Dimensional Analysis Aligns Dendritic Cells across Tissues and Species. Immunity 45, 669–684 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Byrne KT, Vonderheide RH, CD40 Stimulation Obviates Innate Sensors and Drives T Cell Immunity in Cancer. Cell Rep 15, 2719–2732 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McLane LM, Abdel-Hakeem MS, Wherry EJ, CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu Rev Immunol 37, 457–495 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Ngiow SF, Young A, Blake SJ, Hill GR, Yagita H, Teng MW, Korman AJ, Smyth MJ, Agonistic CD40 mAb-Driven IL12 Reverses Resistance to Anti-PD1 in a T-cell-Rich Tumor. Cancer Res 76, 6266–6277 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Kansy BA, Concha-Benavente F, Srivastava RM, Jie HB, Shayan G, Lei Y, Moskovitz J, Moy J, Li J, Brandau S, Lang S, Schmitt NC, Freeman GJ, Gooding WE, Clump DA, Ferris RL, PD-1 Status in CD8(+) T Cells Associates with Survival and Anti-PD-1 Therapeutic Outcomes in Head and Neck Cancer. Cancer Res 77, 6353–6364 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM, Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322, 1097–1100 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, Amigorena S, Van’t Veer LJ, Sperling AI, Wolf DM, Krummel MF, Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell 26, 938 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Lee W, Kim HS, Hwang SS, Lee GR, The transcription factor Batf3 inhibits the differentiation of regulatory T cells in the periphery. Exp Mol Med 49, e393 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vasconcelos-Nobrega C, Colaco A, Lopes C, Oliveira PA, Review: BBN as an urothelial carcinogen. In Vivo 26, 727–739 (2012). [PubMed] [Google Scholar]
- 31.Saito R, Smith CC, Utsumi T, Bixby LM, Kardos J, Wobker SE, Stewart KG, Chai S, Manocha U, Byrd KM, Damrauer JS, Williams SE, Vincent BG, Kim WY, Molecular Subtype-Specific Immunocompetent Models of High-Grade Urothelial Carcinoma Reveal Differential Neoantigen Expression and Response to Immunotherapy. Cancer Res 78, 3954–3968 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith P, DiLillo DJ, Bournazos S, Li F, Ravetch JV, Mouse model recapitulating human Fcgamma receptor structural and functional diversity. Proc Natl Acad Sci U S A 109, 6181–6186 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vonderheide RH, Burg JM, Mick R, Trosko JA, Li D, Shaik MN, Tolcher AW, Hamid O, Phase I study of the CD40 agonist antibody CP-870,893 combined with carboplatin and paclitaxel in patients with advanced solid tumors. Oncoimmunology 2, e23033 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vonderheide RH, CD40 Agonist Antibodies in Cancer Immunotherapy. Annu Rev Med 71, 47–58 (2020). [DOI] [PubMed] [Google Scholar]
- 35.Inwald DP, McDowall A, Peters MJ, Callard RE, Klein NJ, CD40 is constitutively expressed on platelets and provides a novel mechanism for platelet activation. Circ Res 92, 1041–1048 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Pettenati C, Ingersoll MA, Mechanisms of BCG immunotherapy and its outlook for bladder cancer. Nat Rev Urol 15, 615–625 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Antonelli AC, Binyamin A, Hohl TM, Glickman MS, Redelman-Sidi G, Bacterial immunotherapy for cancer induces CD4-dependent tumor-specific immunity through tumor-intrinsic interferon-gamma signaling. Proc Natl Acad Sci U S A 117, 18627–18637 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rentsch CA, Biot C, Gsponer JR, Bachmann A, Albert ML, Breban R, BCG-mediated bladder cancer immunotherapy: identifying determinants of treatment response using a calibrated mathematical model. PLoS One 8, e56327 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suttmann H, Riemensberger J, Bentien G, Schmaltz D, Stockle M, Jocham D, Bohle A, Brandau S, Neutrophil granulocytes are required for effective Bacillus Calmette-Guerin immunotherapy of bladder cancer and orchestrate local immune responses. Cancer Res 66, 8250–8257 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Hughes OD, Bishop MC, Perkins AC, Wastie ML, Denton G, Price MR, Frier M, Denley H, Rutherford R, Schubiger PA, Targeting superficial bladder cancer by the intravesical administration of copper-67-labeled anti-MUC1 mucin monoclonal antibody C595. J Clin Oncol 18, 363–370 (2000). [DOI] [PubMed] [Google Scholar]
- 41.Bamias A, Keane P, Krausz T, Williams G, Epenetos AA, Intravesical administration of radiolabeled antitumor monoclonal antibody in bladder carcinoma. Cancer Res 51, 724–728 (1991). [PubMed] [Google Scholar]
- 42.Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, Engblom C, Pfirschke C, Siwicki M, Gungabeesoon J, Freeman GJ, Warren SE, Ong S, Browning E, Twitty CG, Pierce RH, Le MH, Algazi AP, Daud AI, Pai SI, Zippelius A, Weissleder R, Pittet MJ, Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-gamma and IL-12. Immunity 49, 1148–1161 e1147 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khalil DN, Suek N, Campesato LF, Budhu S, Redmond D, Samstein RM, Krishna C, Panageas KS, Capanu M, Houghton S, Hirschhorn D, Zappasodi R, Giese R, Gasmi B, Schneider M, Gupta A, Harding JJ, Moral JA, Balachandran VP, Wolchok JD, Merghoub T, In situ vaccination with defined factors overcomes T cell exhaustion in distant tumors. J Clin Invest 129, 3435–3447 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ataide MA, Komander K, Knopper K, Peters AE, Wu H, Eickhoff S, Gogishvili T, Weber J, Grafen A, Kallies A, Garbi N, Einsele H, Hudecek M, Gasteiger G, Holzel M, Vaeth M, Kastenmuller W, BATF3 programs CD8(+) T cell memory. Nat Immunol 21, 1397–1407 (2020). [DOI] [PubMed] [Google Scholar]
- 45.Maier B, Leader AM, Chen ST, Tung N, Chang C, LeBerichel J, Chudnovskiy A, Maskey S, Walker L, Finnigan JP, Kirkling ME, Reizis B, Ghosh S, D’Amore NR, Bhardwaj N, Rothlin CV, Wolf A, Flores R, Marron T, Rahman AH, Kenigsberg E, Brown BD, Merad M, A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 580, 257–262 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, Nelson AE, Loo K, Kumar R, Rosenblum MD, Alvarado MD, Wolf DM, Bogunovic D, Bhardwaj N, Daud AI, Ha PK, Ryan WR, Pollack JL, Samad B, Asthana S, Chan V, Krummel MF, A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat Med 24, 1178–1191 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mayoux M, Roller A, Pulko V, Sammicheli S, Chen S, Sum E, Jost C, Fransen MF, Buser RB, Kowanetz M, Rommel K, Matos I, Colombetti S, Belousov A, Karanikas V, Ossendorp F, Hegde PS, Chen DS, Umana P, Perro M, Klein C, Xu W, Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Sci Transl Med 12, (2020). [DOI] [PubMed] [Google Scholar]
- 48.Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, Bigenwald C, Remark R, Sweeney R, Becker CD, Levine JH, Meinhof K, Chow A, Kim-Shulze S, Wolf A, Medaglia C, Li H, Rytlewski JA, Emerson RO, Solovyov A, Greenbaum BD, Sanders C, Vignali M, Beasley MB, Flores R, Gnjatic S, Pe’er D, Rahman A, Amit I, Merad M, Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell 169, 750–765 e717 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spranger S, Bao R, Gajewski TF, Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235 (2015). [DOI] [PubMed] [Google Scholar]
- 50.Ferris ST, Durai V, Wu R, Theisen DJ, Ward JP, Bern MD, Davidson J. T. t., Bagadia P, Liu T, Briseno CG, Li L, Gillanders WE, Wu GF, Yokoyama WM, Murphy TL, Schreiber RD, Murphy KM, cDC1 prime and are licensed by CD4(+) T cells to induce anti-tumour immunity. Nature 584, 624–629 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garris CS, Luke JJ, Dendritic cells, the T cell-inflamed tumor microenvironment and immunotherapy treatment response. Clin Cancer Res, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, Kaisho T, Bogunovic D, Bhardwaj N, Krummel MF, Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 30, 324–336 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baik CS, Rubin EH, Forde PM, Mehnert JM, Collyar D, Butler MO, Dixon EL, Chow LQM, Immuno-oncology Clinical Trial Design: Limitations, Challenges, and Opportunities. Clin Cancer Res 23, 4992–5002 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dammeijer F, van Gulijk M, Mulder EE, Lukkes M, Klaase L, van den Bosch T, van Nimwegen M, Lau SP, Latupeirissa K, Schetters S, van Kooyk Y, Boon L, Moyaart A, Mueller YM, Katsikis PD, Eggermont AM, Vroman H, Stadhouders R, Hendriks RW, Thusen JV, Grunhagen DJ, Verhoef C, van Hall T, Aerts JG, The PD-1/PD-L1-Checkpoint Restrains T cell Immunity in Tumor-Draining Lymph Nodes. Cancer Cell 38, 685–700 e688 (2020). [DOI] [PubMed] [Google Scholar]
- 55.Nadal R, Bellmunt J, Management of metastatic bladder cancer. Cancer Treat Rev 76, 10–21 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Morrison AH, Diamond MS, Hay CA, Byrne KT, Vonderheide RH, Sufficiency of CD40 activation and immune checkpoint blockade for T cell priming and tumor immunity. Proc Natl Acad Sci U S A 117, 8022–8031 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ma HS, Poudel B, Torres ER, Sidhom JW, Robinson TM, Christmas B, Scott B, Cruz K, Woolman S, Wall VZ, Armstrong T, Jaffee EM, A CD40 Agonist and PD-1 Antagonist Antibody Reprogram the Microenvironment of Nonimmunogenic Tumors to Allow T-cell-Mediated Anticancer Activity. Cancer Immunol Res 7, 428–442 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Singh M, Vianden C, Cantwell MJ, Dai Z, Xiao Z, Sharma M, Khong H, Jaiswal AR, Faak F, Hailemichael Y, Janssen LME, Bharadwaj U, Curran MA, Diab A, Bassett RL, Tweardy DJ, Hwu P, Overwijk WW, Intratumoral CD40 activation and checkpoint blockade induces T cell-mediated eradication of melanoma in the brain. Nat Commun 8, 1447 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: Iliac lymph nodes drain the bladder.
Supplemental Figure 2: CD40 agonism does not reverse exhaustion signatures on CD4+ T cells and innate lymphoid cells.
Supplemental Figure 3: CD8+ T cell depletion in the tumor-bearing bladder.
Supplemental Figure 4: Characterization of immune populations in Batf3-deficient mice.
Supplemental Figure 5: Secondary lymphoid organs of Batf3-deficient mice lack cDC1.
Supplemental Figure 6: Batf3 deficiency enhances tumor growth and progression in the bladder.
Supplemental Figure 7: Batf3 deficiency abrogates anti-CD40 agonist antibody response in multiple subcutaneous models of bladder cancer.
Supplemental Figure 8: Intravesically-administered anti-CD40 agonist antibodies bind to myeloid cells in the tumor-bearing bladder.
Supplemental Figure 9: Fc-enhanced human anti-CD40 agonist antibody 2141-V11 demonstrates improved anti-tumor activity compared to the parental antibody variant and activates dendritic cells in the tumor microenvironment.
Supplemental Figure 10: Fc-enhanced anti-CD40 agonist antibody 2141-V11 demonstrates anti-tumor activity in multiple subcutaneous models of bladder cancer.
Supplemental Figure 11: Toxicity testing of intravesical anti-CD40 agonist antibody 2141-V11.
Supplemental Figure 12: Response to intravesical BCG and anti-CD40 agonist antibody 2141-V11 in orthotopic models of advanced bladder cancer.
Supplemental Figure 13: Intravesical anti-CD40 agonist antibody 2141-V11 demonstrates limited toxicity during treatment of early-stage bladder tumors.
Data Availability Statement
All data associated with this study are available in the main text or the supplementary materials.