

Abstract
Mitochondrial stress releases mitochondrial DNA (mtDNA) into the cytosol and triggers the type-I interferon (IFN) response. Mitochondrial outer membrane permeabilization (MOMP), which is required for mtDNA release, has been extensively studied in apoptotic cells, but little is known about MOMP in live cells. Here, we show that oxidatively stressed mitochondria release short mtDNA fragments via pores formed by the voltage-dependent anion channel (VDAC) oligomers in the mitochondrial outer membrane. Furthermore, the positively charged residues in the N-terminal domain of VDAC1 interact with mtDNA and promote VDAC1 oligomerization. The VDAC oligomerization inhibitor VBIT-4 decreases mtDNA release, IFN signaling, neutrophil extracellular traps, and disease severity in a mouse model of systemic lupus erythematosus. Thus, VDAC oligomerization inhibition is a potential therapeutic approach for diseases associated with mtDNA release.
One Sentence Summary:
Inhibiting VDAC oligomerization decreases mtDNA release, type-I interferon response, and ameliorates lupus-like disease.
Mitochondrial stress, such as that triggered by increased mitochondrial reactive oxygen species (mROS), can release mitochondrial DNA (mtDNA) into the cytosol. There, it interacts with and activates a large number of immunostimulatory DNA sensors such as cyclic GMP-AMP synthase (cGAS) that can trigger autoimmunity, including diseases caused by the type-I interferon (IFN) response (1). Mitochondrial outer membrane permeabilization (MOMP) is required for mtDNA release. To date, only BAX/BAK oligomers, which can form extremely large macropores in the mitochondrial outer membrane (MOM), have been shown to mediate mtDNA release (2–4). However, the formation of the BAX/BAK macropore generally occurs under conditions that activate BAX/BAK such as apoptosis or treatment with BAX/BAK activators (2–4). However, the pore(s) that promotes MOMP in live cells or in conditions that do not activate BAX/BAK has not been identified.
Because the voltage-dependent anion channel (VDAC) can oligomerize under oxidative stress conditions, and VDAC oligomers can form large MOM pores (5), we investigated whether VDAC could trigger MOMP in live cells and mediate mtDNA release. VDAC is the most abundant protein in MOM and regulates Ca2+ influx, metabolism, inflammasome activation (6) and cell death (7, 8). Moreover, the expression levels of VDAC1, the most abundant of the three VDAC isoforms, and VDAC3 are increased in the autoimmune disease systemic lupus erythematosus (SLE) (9), which, in some models, is thought to be triggered by released mtDNA (10, 11).
Previous studies on mtDNA release have used cells that are either undergoing apoptosis or that have altered mtDNA content (2, 12, 13), which make studies on the mechanism of mtDNA release difficult to interpret. To avoid these confounding variables, we studied mouse embryo fibroblasts (MEFs) deficient in endonuclease g (Endog), a nuclear-encoded mitochondrial endonuclease (14, 15). Endog−/− MEFs have higher levels of cytosolic mtDNA (cmtDNA) compared to WT MEFs (Fig. 1A), despite having similar levels of total mtDNA (Fig. 1B) and cellular growth rates (fig. S1A). Consistent with this, Endog−/− MEFs (Fig. 1, C and D, and fig. S1, B and C) and plasmacytoid dendritic cells (fig. S1D) have higher mRNA levels of IFN-stimulated genes (ISGs) compared to their WT counterparts. ISG expression and phosphorylation of TANK-binding kinase 1 (TBK1) and IFN regulatory factor 3 (IRF3) were significantly reduced in ρ0 (mtDNA-deficient) cells derived from Endog−/− MEFs, compared with the parental Endog−/− MEFs (Fig. 1, E and F, and fig. S1, E to G). Thus, the cGAS–STING pathway is activated by cmtDNA in Endog−/− MEFs (fig. S2, A to I). Increased mROS appears to cause the mtDNA release in Endog−/− MEFs because mROS was higher in Endog−/− MEFs (fig. S2, J and K), but the mROS scavenger mito-TEMPO decreased ISG expression in Endog−/− MEFs (Fig. 1G).
Fig. 1. Endog-deficiency increases cmtDNA and type-I IFN signaling.
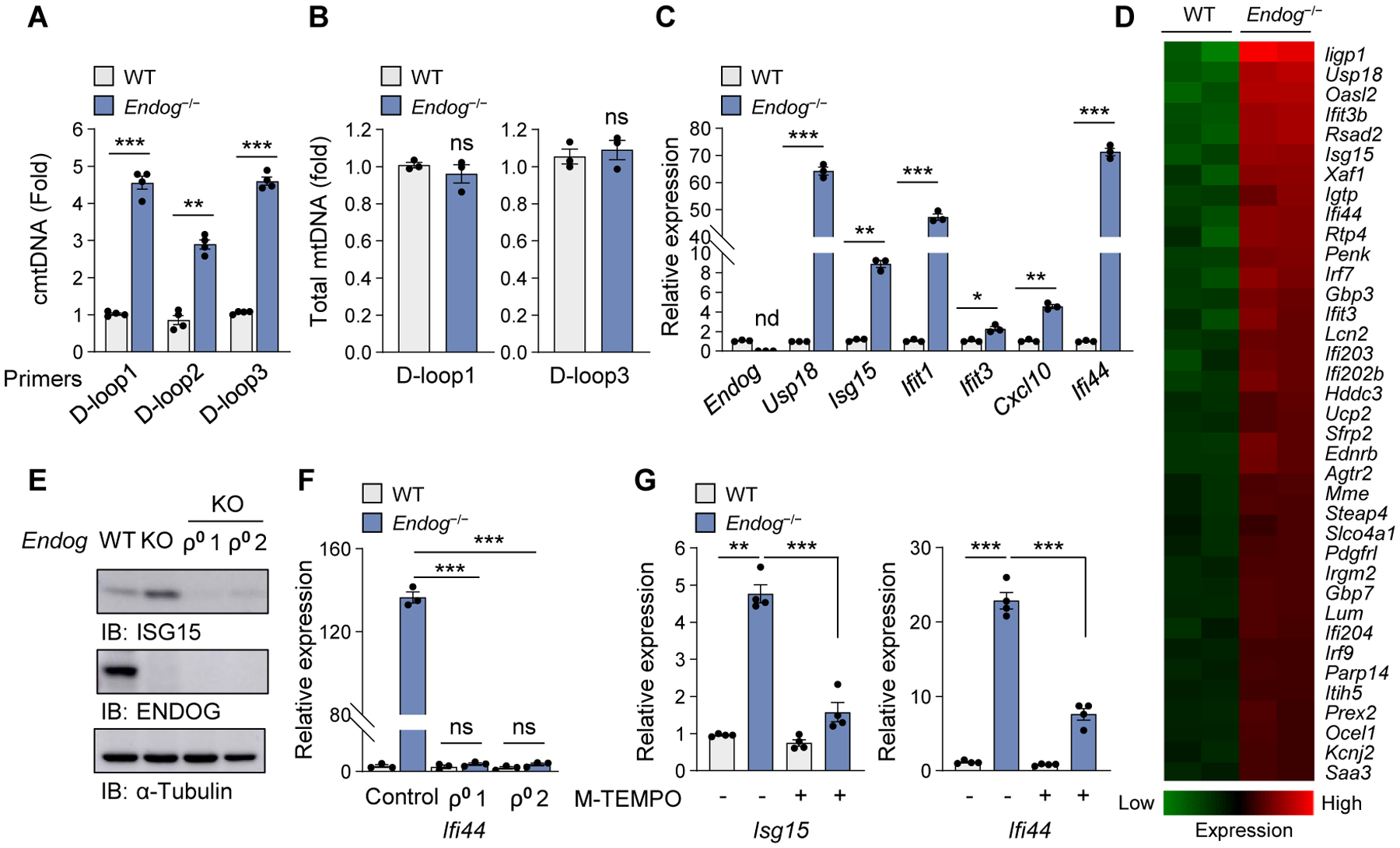
(A and B) Quantification of cmtDNA (A) and total mtDNA (B) in WT and Endog−/− MEFs. (C and D) ISG expression levels (C) and heat map analysis of RNAseq data (D) in WT and Endog−/− MEFs. (E and F) ISG expression levels in WT and Endog−/− MEFs as well as two independently generated ρ0 MEFs (ρ0 1 and ρ0 2) were determined by immunoblotting (E) and RT-qPCR (F). (G) ISG expression was measured in WT and Endog−/− MEFs after treatment with Mito(M)-TEMPO (10 μM) for 48 h. All values are presented as the mean ± SEM of at least three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.005; nd, not detected; ns, not significant.
Under physiological conditions, tissue-culture cells are composed of two populations: live cells which make up the majority of the population and a small minority of cells undergoing spontaneous apoptosis and caspase activation. Prior studies (3, 4, 16) and our data (fig. S3A) show that activation of BAX/BAK with ABT-737 in the presence of caspase inhibition increases ISG expression. However, WT and Bax/Bak−/− MEFs had similar cmtDNA levels (fig. S3B), and knocking down Endog robustly induced ISG expression in Bax/Bak−/− MEFs, albeit slightly less than that in WT MEFs (fig. S3, C and D). Thus, Endog−/− MOMP can occur in the absence BAX/BAK macropores. Moreover, mitochondria in Endog−/− MEFs tended to be slightly longer than those in WT MEFs rather than being fragmented as they would be expected with BAX/BAK activation (fig. S3E) (2, 17).
We then compared the level of apoptosis in WT and Endog−/− MEFs because extremely high levels of mROS can lead to apoptosis, and Endog−/− MEFs have higher mROS compared to WT MEFs (fig. S2, J and K). WT and Endog−/− MEFs had similar levels of apoptotic indicators such as caspase activity (fig. S3F), cell viability (fig. S3G), lactate dehydrogenase (LDH) release (fig. S3H), and ethidium homodimer-1 (EthD-1) staining (fig. S3I) either before or after the induction of apoptosis by BAX/BAK. Thus, Endog−/− MEFs have levels of mROS stress high enough to promote MOMP and mtDNA release in a BAX/BAK-independent manner but insufficient to promote apoptosis at the cellular level (fig. S3I).
An alternative mediator of MOMP may be VDAC, and indeed, Vdac1−/−, Vdac3−/−, and Vdac1/3−/− MEFs had lower levels of ISG mRNA and cmtDNA compared to WT MEFs (Fig. 2, A and B, and fig. S4, A to C) despite having similar total mtDNA levels (fig. S4D). We excluded VDAC2 from our study because VDAC2-deficiency promotes apoptosis (8). Additionally, a knock down of Endog increased ISG expression in WT and Bax/Bak−/− MEFs, but not in Vdac1/3−/− MEFs (Fig. 2C and fig. S3C) and treatment with the VDAC inhibitor DIDS (18) decreased ISG expression in Endog−/− MEFs (fig. S4E). In agreement with their reduced type-I IFN signaling, Vdac1/3−/− MEFs were less resistant to HSV-1 infection compared to WT MEFs (fig. S4, F to H). Thus, both VDAC1 and VDAC3 contribute to both MOMP and mtDNA release.
Fig. 2. VDAC oligomerization is required for mtDNA fragment release.
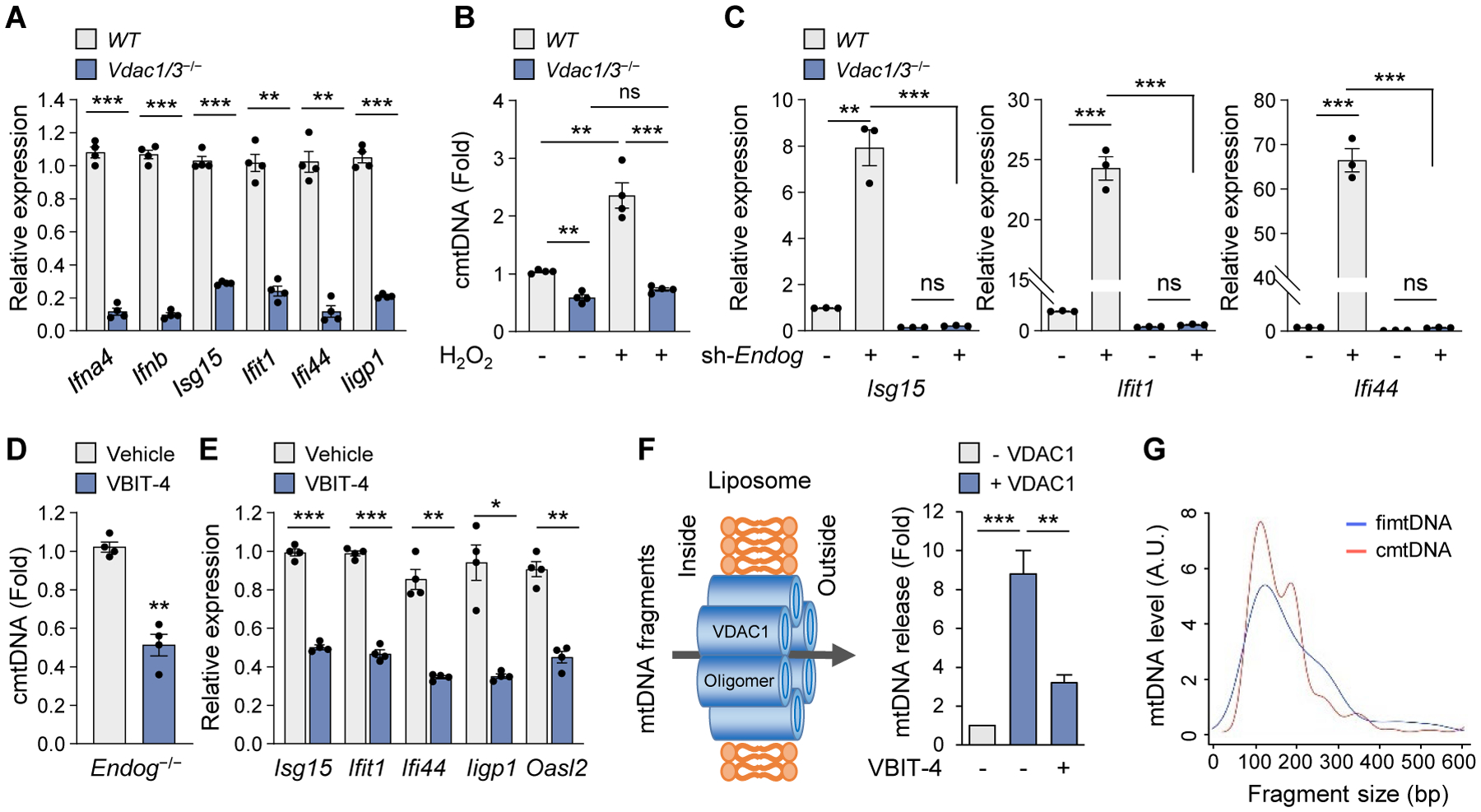
(A) ISG expression levels in WT and Vdac1/3−/− MEFs. (B) cmtDNA levels were determined in WT and Vdac1/3−/− MEFs after treatment with H2O2 (100 μM) for 18 h. (C) ISG expression levels were measured in WT and Vdac1/3−/− MEFs after knocking down Endog. (D and E) cmtDNA (D) and ISG expression (E) levels were measured after treatment with VBIT-4 (10 μM) in Endog−/− MEFs. (F) VDAC1 oligomerization-dependent release of mtDNA from mtDNA-loaded liposomes and inhibition by VBIT-4. (G) Fragment-size distribution of the fimtDNA and cmtDNA. All values are presented as the mean ± SEM of at least three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.005; ns, not significant.
VDAC does not promote mtDNA release by increasing the subpopulation of cells undergoing apoptosis as the total caspase activity was not altered in Vdac1/3−/− MEFs even in the presence of a moderate concentration of H2O2 (fig. S5A), which increases cmtDNA (Fig. 2B). In contrast, BAX/BAK, but not VDAC1/3, was required for ABT-737-induced apoptosis (fig. S5B), indicating that in the presence of BAX/BAK macropores (e.g. apoptosis), VDAC is not required to drive MOMP (fig. S5C).
We initially hypothesized that VDAC regulates mtDNA release by promoting Ca2+ influx (19), which opens the mitochondrial permeability transition pore (mPTP) in the mitochondrial inner membrane (MIM) in a manner that does not trigger apoptosis at the whole cell level (20, 21). This would be possible only if the population of affected mitochondria is very small so that the total caspase activity remains near background levels. Chelation of Ca2+ with BAPTA decreased ISG expression (fig. S6, A to C), and inhibiting mPTP opening with cyclosporin A (CsA) decreased mtDNA release in both Endog−/− MEFs and WT mitoplasts as well as ISG mRNA levels in Endog−/− MEFs (fig. S6, D to F). However, mtDNA release from mitochondria deficient in mitochondrial calcium uptake 1 (MICU1), which had elevated mROS levels and IFN responses (fig. S6, G to I) (22), was still inhibited by DIDS in the absence of Ca2+ (fig. S6J). Thus, VDAC may play a Ca2+ flux-independent role in mtDNA release. In agreement with this, the highly potent VDAC1 oligomerization inhibitor VBIT-4 (23) decreased cmtDNA levels and ISG expression in Endog−/− MEFs (Fig. 2, D and E), without inhibiting either Ca2+ uptake (fig. S6K) or mPTP opening (fig. S6L) in the mitochondria. We then loaded mtDNA fragments into liposomes with or without membrane-reconstituted VDAC1. The presence of VDAC1 in liposomes increased mtDNA passage across the lipid membrane (Fig. 2F), but VBIT-4 decreased VDAC1 oligomerization (fig. S7, A and B) as well as mtDNA efflux (Fig. 2F). Thus, VDAC1 oligomers are sufficient to permeabilize lipid membranes for mtDNA passage.
As the intact mtDNA is large (16–17 kb) and is tethered to the MIM in nucleoid complexes (24), we hypothesized that short and free (untethered) intra-mtDNA fragments (fimtDNA) that can pass through VDAC oligomer pores pre-exist in living cells (fig. S8A). To investigate this possibility, we treated mitochondria purified from WT MEFs with cytoskeleton (CSK) buffer, which gently permeabilizes mitochondrial membranes and releases fimtDNA while leaving mitochondrial nucleoids intact (24). Interestingly, the sequences corresponding to a region within the D-loop in the mitochondrial genome were over represented in the fimtDNA pool (fig. S8, A and B). A size distribution analysis excluding the sequences with 100% homology to both mitochondrial and nuclear genomes indicated that the peak sizes of fimtDNA and cmtDNA are almost identical (~110 bp) (Fig. 2G). Treatment of WT MEFs with mito-TEMPO (fig. S8C) or the mTORC1 inhibitor everolimus (fig. S8D), which promotes mitophagy and elimination of damaged mitochondria, decreased fimtDNA. Thus, fimtDNA accumulates preferentially within a subpopulation of mitochondria with elevated mROS, and cmtDNA is derived largely from fimtDNA in live cells (fig. S5C).
Studies with planar lipid bilayer (PLB) reconstituted with VDAC1 indicate that the N-terminal domain of VDAC1 can interact directly with mtDNA (fig. S9). The N-terminal domain, which is evolutionarily conserved (fig. S10A), is hydrophilic and is thought to translocate out of the channel when VDAC1 is in an oligomerized state (Fig. 3A) (25). This raises the possibility that the negatively charged backbone of mtDNA may interact with multiple VDAC1 molecules simultaneously and act as a scaffold to stabilize the oligomers (Fig. 3A). Indeed, mtDNA increased the formation of VDAC1 trimers and higher order oligomers (Fig. 3, B and C) in vitro. In agreement with higher cmtDNA in Endog−/− MEFs (Fig. 1A), co-immunoprecipitation with anti-VDAC1 antibody pulled down more mtDNA in Endog−/− MEFs compared to WT MEFs (Fig. 3D). VDAC1 oligomerization was also increased in Endog−/− MEFs compared to WT MEFs, but it was reduced by the elimination of mtDNA in Endog−/− MEFs (ρ0) (Fig. 3, E and F). In contrast, BAK oligomerization was not increased in Endog−/− MEFs (Fig. 3G), and BAI, the BAX oligomerization inhibitor (26), did not reduce ISG expression in Endog−/− MEFs (Fig. 3H). Thus, mtDNA may promote further VDAC1 oligomerization, creating a feed-forward cycle.
Fig. 3. mtDNA interacts with VDAC1 and stabilizes its oligomeric state.
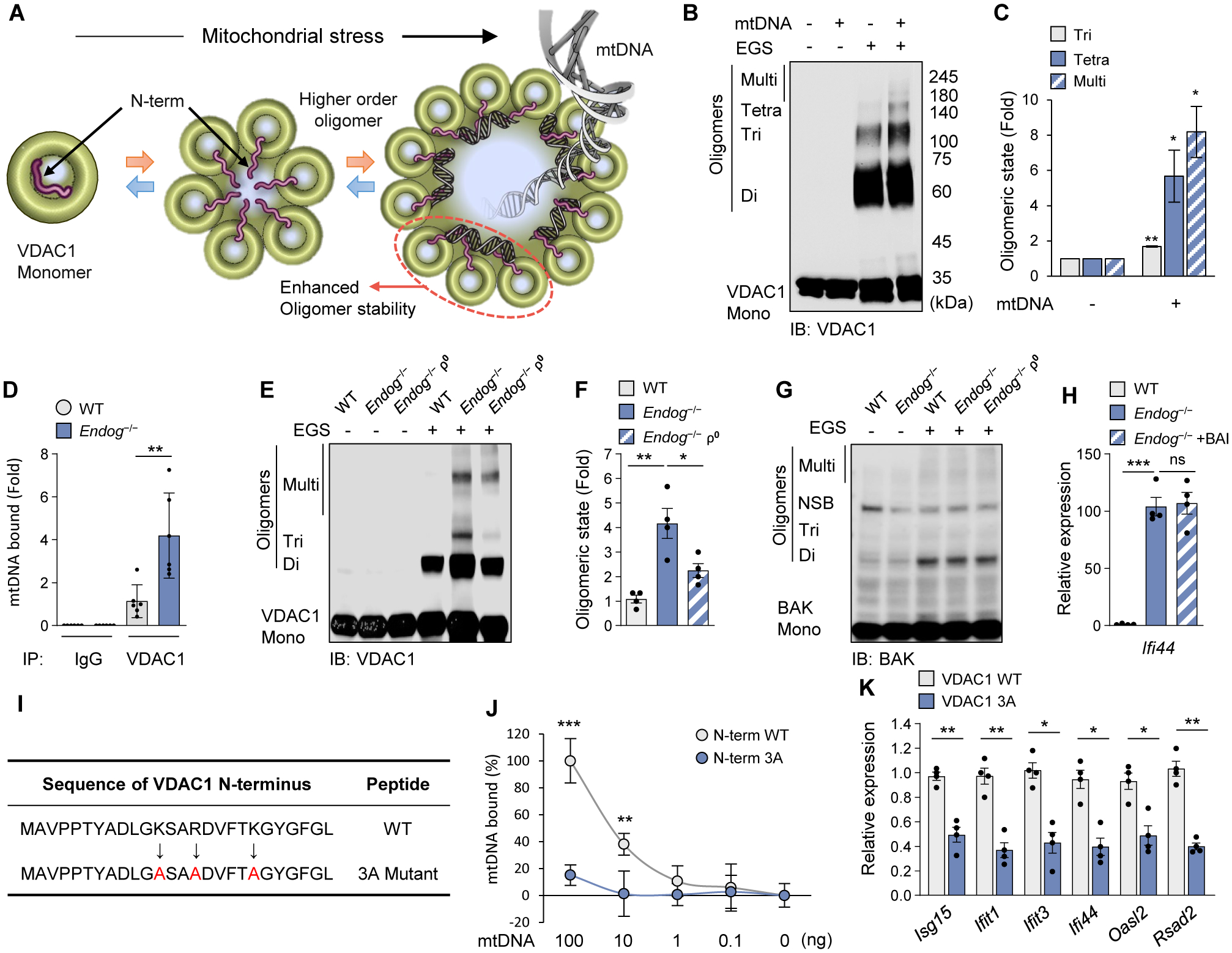
(A) Schematic diagram of VDAC1 oligomerization accompanied by the N-terminal domain (red) translocation into the large oligomer pore. We could not characterize VDAC3 in vitro because it tends to form aggregates. (B and C) mtDNA-induced oligomerization of purified VDAC1 was visualized by immunoblotting after treatment with the cross-linking reagent EGS to stabilize the oligomers during electrophoresis (B). Quantitative analysis of oligomers is shown (C). (D) mtDNA binding to VDAC1 in WT and Endog−/− MEFs. (E to G) VDAC1 (E) and BAK (G) oligomerization in WT, Endog−/− and Endog−/− ρ0 MEFs was visualized by immunoblotting. The positions of VDAC1 monomers (Mono), dimers (Di), trimers (Tri) and multimers (Multi) are indicated. NSB, non-specific band. Quantitative analysis of VDAC1 oligomers is shown (F). (H) ISG expression was measured in WT and Endog−/− MEFs after treatment with the BAX oligomerization inhibitor (BAI) (2 μM) for 24 h. (I) The amino-acid sequence of the VDAC1 N-terminal peptide. The positively charged amino acids were mutated to alanine (A: red color). (J) Direct interaction of mtDNA fragments with WT and 3A N-terminal 26 amino-acid peptides. (K) ISG expression levels were measured by RT-qPCR after H2O2 (100 μM) treatment for 18 h in MEFs expressing either WT or 3A VDAC1. All values are presented as the mean ± SEM of at least three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.005; ns, not significant.
The N-terminal domain of VDAC1 contains three positively charged residues (K12, R15, K20) that could interact with the negatively charged backbone of mtDNA (Fig. 3I). Indeed, mtDNA fragments were pulled down by a 26 a.a. VDAC1 N-terminal peptide but not by a VDAC1 N-terminal mutant peptide (3A) in which K12, R15, K20 residues were replaced by Ala (A) (Fig. 3J). We then examined H2O2-induced expression of ISG in Vdac1/3−/− MEFs with restored expression of either WT VDAC1, the 3A-mutant VDAC1, or ΔN-terminal VDAC1 (fig. S10B). ISG expression was reduced in MEFs expressing either the 3A-mutant VDAC1 (Fig. 3K) or ΔN-terminal VDAC1 (fig. S10C), compared with those expressing WT VDAC1. Thus, direct mtDNA–VDAC interactions appear to promote VDAC oligomerization and increase mtDNA release.
An excessive type-I IFN response is a hallmark of SLE (27). Gene Expression Omnibus (GEO) analysis revealed increased mRNA expression of VDAC1/3 and decreased expression of ENDOG in SLE patients (fig. S11A) (9). However, mRNA levels of BAK, BAX, VDAC2, and HSP60 were not changed in SLE patients (fig. S11A). These findings, combined with the observation that type-I IFN responses in Endog-knock down MEFs were VDAC(1 and 3)-dependent (Fig. 2C), suggest that VDAC oligomerization may be associated with SLE. Indeed, splenocytes from MpJ-Faslpr lupus-prone mice had more VDAC1 oligomers than MpJ control mice (Fig. 4A and fig. S11B) as did peripheral blood mononuclear cells (PBMCs) from SLE patients compared to healthy controls (Fig. 4B and fig. S11C). Splenocytes from MpJ-Faslpr mice also had elevated cmtDNA compared to those from MpJ mice, but this was abrogated with VBIT-4 treatment (Fig. 4C).
Fig. 4. VDAC1 oligomerization inhibitor VBIT-4 ameliorates lupus-like disease.
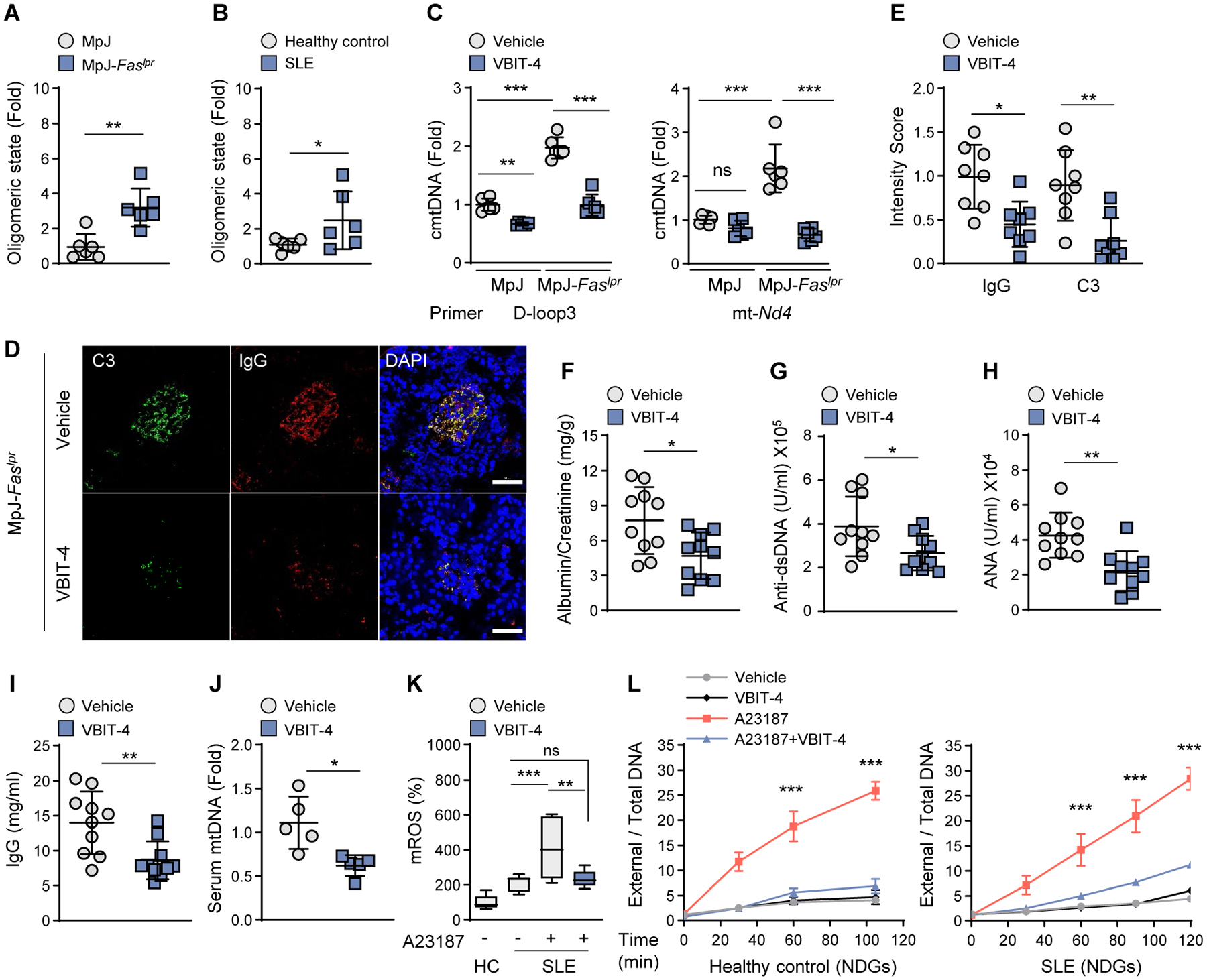
(A) The formation of VDAC1 oligomers in splenocytes of MRL/MpJ-Faslpr lupus-prone mice and MRL/MpJ control mice (see fig. S11B; n=6 in each group). (B) Oligomeric state of VDAC1 in PBMC of healthy control and SLE patients (see fig. S11C; N=6 in each group). (C) cmtDNA levels were measured in splenocytes (A) after treatment with VBIT-4 (10 μM). (D and E) Kidney glomeruli of VBIT-4-treated mice, stained with antibodies against complement C3 (green) and IgG (red). Nuclei were stained with Hoechst (blue). Scale bars, 20 μm (D). Fluorescence intensity of C3 and IgG in (D) (n=8 in each group) (E). (F to I) Urinary albumin:creatinine ratio (F), serum anti-dsDNA levels (G), ANA levels (H), and IgG levels (I) of VBIT-4-treated mice (n=10 in each group). (J) Serum cell-free mtDNA levels of VBIT-4-treated mice (n=5 in each group). (K) mROS levels were measured by mitoSOX in PBMCs of healthy controls (HC) and SLE patients (n = 3 in each group). (L) A23187-induced NET formation by NDGs either from HC or SLE subjects was measured by SYTOX-PicoGreen plate assay (n=3 in each group). All values are presented as the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.005; ns, not significant.
We next investigated whether VBIT-4 could ameliorate lupus-like symptoms in MpJ-Faslpr mice. VBIT-4 blocked the development of skin lesions and the thickening of the epidermis that accompanies leukocyte infiltration, and suppressed facial and dorsal alopecia without affecting mortality or body weight (fig. S12, A to C). VBIT-4 also decreased spleen and lymph node weights (fig. S12D). Furthermore, the levels of ISG mRNA (fig. S12E), renal immune complex deposition (Fig. 4, E and D), proteinuria (Fig. 4F), anti-dsDNA antibody (Fig. 4G), antinuclear antibody (ANA) (Fig. 4H), IgG (Fig. 4I), and cell-free mtDNA (Fig. 4J) were all reduced by VBIT-4. Cell-free mtDNA plays an immunostimulatory role in human and mouse SLE (10, 11). A potential source of cell-free mtDNA in MpJ-Faslpr mice is neutrophil extracellular traps (NETs), in a cell-death process called NETosis (11). mROS is an important trigger for NETosis (11), and VBIT-4 decreased mROS in neutrophils as well as other immune cells from SLE patients (Fig. 4K and fig. S12F). In agreement with this, VBIT-4 suppressed NETosis in low-density granulocytes (LDGs), a distinct class of pro-inflammatory and NETosis-prone neutrophils from SLE patients, and normal-density granulocytes (NDGs) isolated from SLE patients and healthy controls (Fig. 4L and fig. S12G). Thus, VDAC oligomerization increases mROS and NETosis, proposed important triggers of autoimmunity, in both human neutrophils (healthy and SLE) and during lupus-like disease in mice (fig. S12H).
In conclusion, we propose that the MOM pore that mediates mtDNA release and type-I IFN response depends on the level of mitochondrial stress: oligomerized VDAC1 for moderate-level stress and BAX/BAK macropores for extreme-level stress and/or apoptosis (fig. S13). It should be noted that there are other pathways of mtDNA release (10) and that SLE is a very heterogenous disease. Nevertheless, inhibiting VDAC oligomerization may be an alternate therapeutic approach for a wide-range of diseases, like SLE and Parkinson’s disease (28), that are thought to be associated with mtDNA release.
Supplementary Material
Acknowledgements
We thank the NHLBI core facilities, including DNA Sequencing and Genomics core, Bioinformatics and Computational Biology core, Flow Cytometry core, Light Microscopy core, Pathology core, and Biochemistry core. We thank the NIH Fellows Editorial Board for manuscript preparation.
Funding:
This work was supported by the Intramural Research Program, National Heart Lung and Blood Institute, National Institute of Arthritis and Musculoskeletal and Skin Diseases, and National Institute of Allergy and Infectious Diseases, National Institutes of Health and by a grant from the National Institute for Biotechnology in the Negev (NIBN) to V.S.-B. This research was also supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI14C1176) and a grant from the KRIBB Research Initiative Program (Korean Biomedical Scientist Fellowship Program), Korea Research Institute of Bioscience and Biotechnology, Republic of Korea.
Footnotes
Competing interests: None declared.
Supplementary materials.
Materials and Methods
Figs. S1 to S13
Tables S1
References (29–36)
Data and materials availability:
All other data and materials are available from the corresponding author upon request. Gene Expression Omnibus (GEO) database of human SLE patient is supported by national center for biotechnology information (GEO accession number: GSE13887).
References and Notes
- 1.West AP, Shadel GS, Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol 17, 363–375 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McArthur K et al. , BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 359, (2018). [DOI] [PubMed] [Google Scholar]
- 3.Rongvaux A et al. , Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159, 1563–1577 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.White MJ et al. , Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 159, 1549–1562 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keinan N, Tyomkin D, Shoshan-Barmatz V, Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol Cell Biol 30, 5698–5709 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou R, Yazdi AS, Menu P, Tschopp J, A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Shoshan-Barmatz V, Krelin Y, Shteinfer-Kuzmine A, VDAC1 functions in Ca(2+) homeostasis and cell life and death in health and disease. Cell Calcium 69, 81–100 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ, VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 301, 513–517 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Fernandez DR et al. , Activation of mammalian target of rapamycin controls the loss of TCRzeta in lupus T cells through HRES-1/Rab4-regulated lysosomal degradation. J Immunol 182, 2063–2073 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caielli S et al. , Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med 213, 697–713 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lood C et al. , Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med 22, 146–153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakahira K et al. , Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12, 222–230 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.West AP et al. , Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schafer P et al. , Structural and functional characterization of mitochondrial EndoG, a sugar non-specific nuclease which plays an important role during apoptosis. J Mol Biol 338, 217–228 (2004). [DOI] [PubMed] [Google Scholar]
- 15.McDermott-Roe C et al. , Endonuclease G is a novel determinant of cardiac hypertrophy and mitochondrial function. Nature 478, 114–118 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ichim G et al. , Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol Cell 57, 860–872 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Riley JS et al. , Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J 37, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ben-Hail D, Shoshan-Barmatz V, VDAC1-interacting anion transport inhibitors inhibit VDAC1 oligomerization and apoptosis. Biochim Biophys Acta 1863, 1612–1623 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD, Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol 9, 550–555 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia N, Chavez E, Mitochondrial DNA fragments released through the permeability transition pore correspond to specific gene size. Life Sci 81, 1160–1166 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Patrushev M et al. , Mitochondrial permeability transition triggers the release of mtDNA fragments. Cell Mol Life Sci 61, 3100–3103 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Liu JC et al. , MICU1 Serves as a Molecular Gatekeeper to Prevent In Vivo Mitochondrial Calcium Overload. Cell Rep 16, 1561–1573 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ben-Hail D et al. , Novel Compounds Targeting the Mitochondrial Protein VDAC1 Inhibit Apoptosis and Protect against Mitochondrial Dysfunction. J Biol Chem 291, 24986–25003 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iborra FJ, Kimura H, Cook PR, The functional organization of mitochondrial genomes in human cells. BMC Biol 2, 9 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Geula S, Ben-Hail D, Shoshan-Barmatz V, Structure-based analysis of VDAC1: N-terminus location, translocation, channel gating and association with anti-apoptotic proteins. Biochem J 444, 475–485 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Garner TP et al. , Small-molecule allosteric inhibitors of BAX. Nat Chem Biol 15, 322–330 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Obermoser G, Pascual V, The interferon-alpha signature of systemic lupus erythematosus. Lupus 19, 1012–1019 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sliter DA et al. , Parkin and PINK1 mitigate STING-induced inflammation. Nature 561, 258–262 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All other data and materials are available from the corresponding author upon request. Gene Expression Omnibus (GEO) database of human SLE patient is supported by national center for biotechnology information (GEO accession number: GSE13887).