

Abstract
To evaluate its potential as a ligand discovery tool, we compare a newly developed 1D protein-observed fluorine NMR (PrOF NMR) screening method with the well-characterized ligand-observed 1H CPMG NMR screen. We selected the first bromodomain of Brd4 as a model system to benchmark PrOF NMR because of the high ligandability of Brd4 and the need for small molecule inhibitors of related epigenetic regulatory proteins. We compare the two methods’ hit sensitivity, triaging ability, experiment speed, material consumption, and the potential for false positives and negatives. To this end, we screened 930 fragment molecules against Brd4 in mixtures of five and followed up these studies with mixture deconvolution and affinity characterization of the top hits. In selected examples, we also compare the environmental responsiveness of the 19F chemical shift to 1H in 1D-protein observed 1H NMR experiments. To address concerns of perturbations from fluorine incorporation, ligand binding trends and affinities were verified via thermal shift assays and isothermal titration calorimetry. We conclude that for the protein understudy here, PrOF NMR and 1H CPMG have similar sensitivity, with both being effective tools for ligand discovery. In cases where an unlabeled protein can be used, 1D protein-observed 1H NMR may also be effective; however, the 19F chemical shift remains significantly more responsive.
Graphical Abstract
Fragment-based ligand discovery (FBLD) and fragment-based drug design (FBDD) have been rapidly gaining traction in the chemical probe development and drug discovery process for difficult targets such as RNA,1 transcription factors,2 and epigenetic regulatory proteins.3,4 Using molecules with lower molecular weight and lower functional group density than molecules found in traditional high throughput screening libraries, fragment screens seek to sample chemical space more efficiently. With vemurafenib5 and venetoclax6 as approved drugs that originated from FBDD and several other drug candidates in phase III clinical trials that also started as fragments, FBDD is becoming a validated technique for drug discovery. These success stories highlight the impact that new methods can have in the drug discovery process.
Fragment screening facilitates drug/chemical probe development at the earliest stages of the discovery process. However, because of their small size (molecular weight typically less than 300), fragments are likely to bind with low affinity, necessitating sensitive methods to detect protein–ligand interactions during a screen. Among these techniques are thermal shift analysis,7 surface plasmon resonance,8 X-ray crystallography,9 and NMR techniques such as saturation transfer difference (STD),10 Carr–Purcel–Meiboom–Gill (CPMG),11 WaterLOGSY,12 and HSQC NMR.13 Remarkably, some studies show little overlap in hits detected by different screening methods.14–16 With the potential for low overlap, it is prudent and common practice to use an orthogonal biophysical method to verify an active.17 Using protein-observed and ligand-observed NMR experiments allows sensitive follow-up with different assays that utilize similar conditions. The goal of this study is to compare the effectiveness of a new protein-observed 19F NMR (PrOF NMR) screening method with an established ligand-observed 1H CPMG screen to provide insight on the appropriate way to implement PrOF NMR in a ligand discovery setting as a complementary assay.
Ligand-observed NMR techniques take advantage of protein-specific properties (e.g, relaxation rates, diffusion coefficients, and nuclear Overhauser effects) being transferred to bound small molecules. The ligand-observed 1H CPMG method used here detects ligand-binding interactions as a function of a drop in signal intensity due to a transfer of the protein’s short transverse relaxation time (T2) properties to the ligand. 19F CPMG or 1H CPMG are most common due to the high gyromagnetic ratios of 19F and 1H nuclei and thus lead to readily acquired spectra. 19F CPMG is widely used and effective due to the large chemical shift range exhibited by fluorine, the reduced spectral overlap due to its much simplified spectra, and the absence of background signals. However, because we wanted to assess a wide array of compounds without being limited to ligands containing fluorine, we used 1H CPMG as the ligand-observed experiment for our benchmark study.
Protein-observed NMR experiments monitor perturbations to protein resonances. These protein resonances provide an in situ quality control since protein aggregation or precipitation events are readily detected by reduced intensity of the NMR resonances. Because different regions of the protein give rise to unique NMR resonances, protein-observed experiments also provide information on changes in the chemical environment on the protein surface which can be used to characterize ligand binding sites and binding-induced conformational changes. Typically, these experiments monitor the amide backbone in the case of 1H–15N HSQC NMR or the sidechain through selective side-chain labeling for 1H–13C HSQC NMR, but this approach can be material-intensive, time-consuming, and expensive due to the need for isotopic labeling.13,18 The fluorine nucleus, as an alternative, is highly responsive to changes in the chemical environment. 19F is 83% as sensitive as 1H and 100% isotopically abundant (thus inexpensive,19 e.g., at $52 per gram, 5-fluoroindole is over 10–20-fold cheaper for either 13C- or 15N-labeled indole), facilitating detection of 19F at low concentrations (40–50 μM) for small and medium-sized proteins.20
Although alternative protein-observed NMR methods are available, PrOF NMR benefits from both a simplified NMR spectrum as well as being a sensitive reporter of weak binding interactions. As such, 19F NMR has been used in ligand discovery for both ligand-observed and protein-observed experiments.21–30 In some cases, protein-observed 19F NMR has proven advantageous when used in conjunction with other protein-observed NMR methods. In studying Equinatoxin II, Anderluh et al. found that 1H–13C HSQC NMR resulted in longer experiment times and a lower response than 19F NMR.31 Harner et al. reported a 7 min experiment using SOFAST 1H–15N HSQC on a comparable bromodomain at 70 μM protein, compared with the 2 min PrOF NMR experiments reported here at 50 μM protein.32 Additionally, Richards et al. reported in studies of Δ-somatostatin that 19F NMR offered improved Kd precision due to higher spectrum resolution and greater chemical environment sensitivity.33
As an alternative to 2D-NMR methods, chemical shift perturbation experiments using 1D protein-observed NMR can be valuable using an unlabeled protein.34 For the protein under study here, we found the chemical shift response from the 1H of the N–H of tryptophan34 to be 6–20-times less responsive than the equivalent PrOF NMR shift with acetaminophen and two compounds uncovered in this screen which will be described below (Figures S1 and S2). We have previously applied PrOF NMR for fragment-based screening of over 500 small molecules and characterized the bromodomains Brd4, BrdT, and BPTF using fluorine labeled aromatic amino acids 3-fluorotyrosine and 5-fluorotryptophan (5FW).24 Using this method, we reported the first molecule selective for BPTF over Brd4 using a simultaneous dual protein screening approach.23 However, due to a reduced surface coverage from labeling only a few side chains, the potential for false negatives was a concern which we sought to evaluate in this study.
The first bromodomain of Brd4 represents an important model protein because of its high ligandability and the need for small molecule inhibitors of other bromodomains due to a growing understanding of their importance in regulation of disease. Bromodomains are epigenetic regulatory proteins, recognizing acetylated lysine residues present on histone tails. Of the 61 human bromodomains, Brd4 in particular has been shown to be involved in cancer, inflammation, and heart disease.35 However, many other bromodomains lack specific chemical probes for evaluating the pharmacological inhibition effects on their biology. In this study, we sought to compare PrOF NMR with the well-established 1H CPMG NMR method to evaluate the effectiveness of this technique as a screening method, as well as the complementarity between ligand-observed and protein-observed NMR techniques. 1H CPMG was chosen because it is frequently used as a screening technique,36 and we found it to be robust for this protein system. We sought to compare assay speed, hit overlap, and the potential for false negatives and false positives with PrOF NMR. To this end, we screened 930 fragments against Brd4 using each method and discuss our findings regarding the potential advantages and limitations of the two methods.
RESULTS
PrOF NMR Results.
In the PrOF NMR screen, 930 fragments were tested in mixtures of five. Induced chemical shift perturbations and resonance broadening were used to rank-order hits. The PrOF NMR screen was performed in the presence of 2% DMSO, and because DMSO is a known bromodomain ligand that affects fluorine chemical shifts,24 all PrOF NMR screening experiments are compared with a 2% DMSO control sample. For assessing the speed and reproducibility of the experiment the screen was conducted twice with different numbers of scans leading to a 13 and 2 min NMR experiment time. The comparison of resonance perturbations between the long and short PrOF NMR experiments indicates that 2 min is a sufficient experiment time for acquiring screening data under these conditions (Figures S3 and S4). Chemical shift perturbations from fragment binding between the 13 and 2 min experiments were very similar, with modest concordance for the line width broadening between the two screening times (Figure S3). In shorter experiments resulting in lower signal-to-noise ratios (S/N = 7:1), the ability to accurately measure changes in line width and integration values of resonances is diminished, while the induced chemical shift changes are still readily interpreted. When chemical shift is used as the primary indicator of a binding interaction, as is the case for this study, S/N ≥ 7:1 is sufficient.
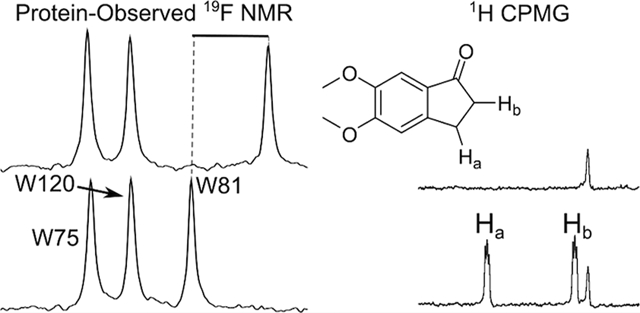
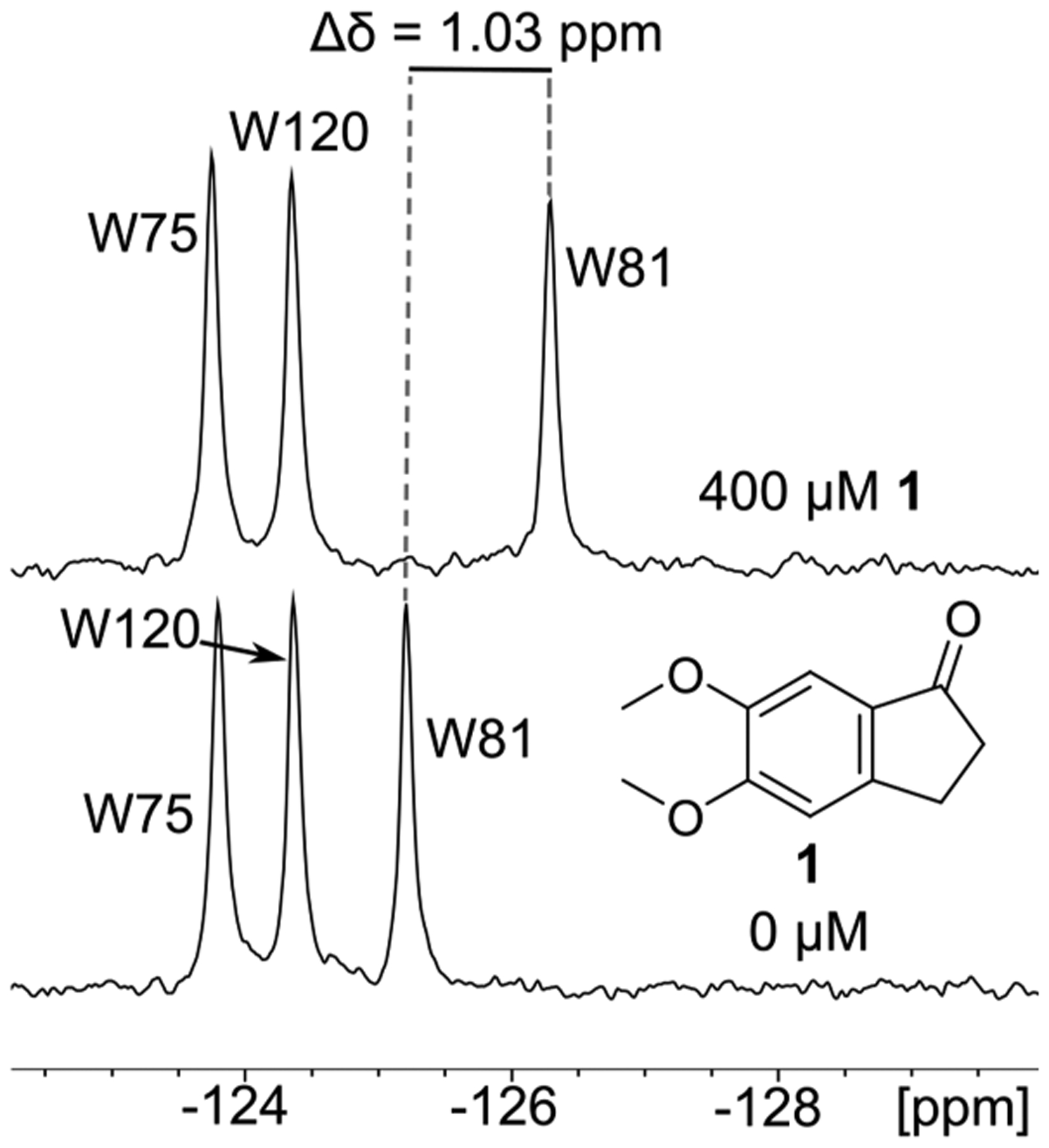
Perturbation of chemical shift is used as the initial indicator of a binding interaction because we expect most of the active fragments to be relatively weak ligands (Figure 1). Weak ligand interactions (Kd > 100 μM) typically fall into the fast exchange regime on the NMR time scale, with highly reproducible chemical shifts. Broadening of a resonance is also an indication of a binding event, potentially in the intermediate exchange regime, and was used as a secondary measure. The first bromodomain of Brd4 has three tryptophan residues: W81 (11 Å from N140, a conserved residue in bromodomains) is at the binding site, W75 is 18 Å from N140, and W120 is on the opposite end of the protein, 36 Å from N140 (Figure 2).37–39 Because of the distance of W120 from the binding site, it is assumed that W120 is not perturbed by ligands during the screen. Thus, when the 19F resonances corresponding to W81 or W75 are perturbed more than the greatest observed perturbation of W120, we conclude that a binding interaction is taking place. To capture as many potential binding interactions as possible for the analysis, we take a perturbation of 0.03 ppm for either W75 or W81 to be the cutoff value indicating the lowest limit for a binding interaction because this was the highest perturbation observed for W120 (Figure 3). In this screen, 59.1% of mixtures yielded a perturbation of W81 greater than 0.03 ppm. As an example of seeking a more stringent hit-rate, the percentage of detected hits drops to 21% with a cutoff perturbation of 0.14 ppm, as illustrated in Figure 3.
Figure 1.
PrOF NMR spectrum of 5FW-Brd4 and 1. In the presence of 1, the resonance for W81 is significantly shifted upfield consistent with binding near the acetylated lysine interaction site of Brd4.
Figure 2.

First bromodomain of Brd4 with all three tryptophan residues displayed in blue and labeled by residue number. Red spheres indicate the acetylated lysine binding site (generated by SiteMap). PDB ID: 3UVW.
Figure 3.
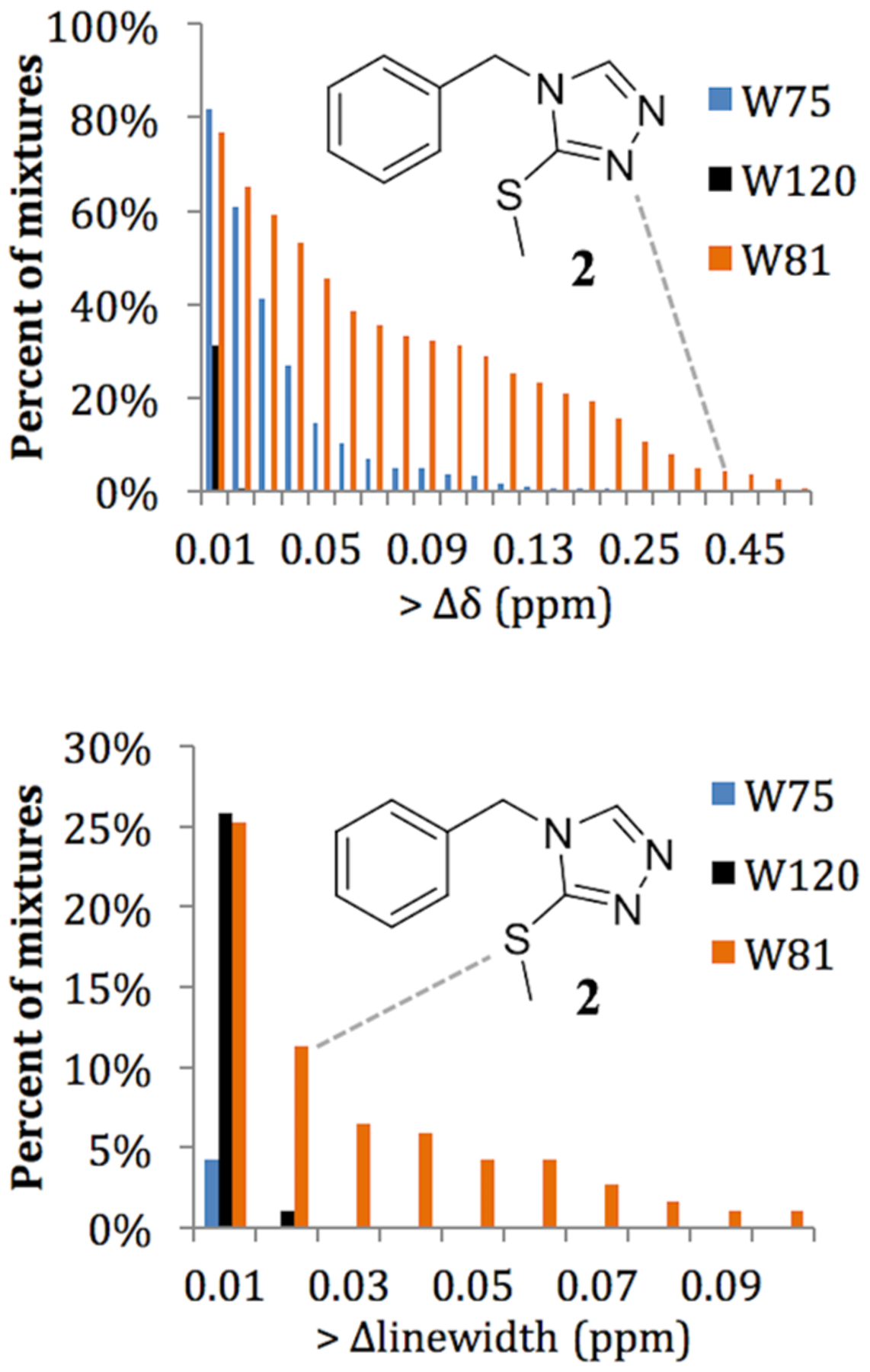
Analysis of the chemical shifts of the three resonances of 5FW-Brd4, comparing the percentage of mixtures perturbing the chemical shift of a particular resonance. Compound 2 is shown in each graph with a dashed line indicating its chemical shift and broadening in the screen. (Top) With a chemical shift perturbation cutoff of 0.01 ppm or higher, W120 is perturbed in over 30% of mixtures, but with a 0.03 ppm cutoff, W120 is not perturbed. Thus, 0.03 ppm perturbations of either W75 or W81, which are closer to the binding site, are used as the hit cutoff for PrOF NMR. (Bottom) A 0.03 ppm line width change for the resonances for W75 and W81 was also considered a hit cutoff.
Because PrOF NMR is a protein-observed technique, it cannot be determined which compound is binding to the protein in the mixture of five without further deconvolution in which each compound is tested individually. Because of the high hit-rate with Brd4, 20% of the mixtures were deconvoluted in order to reduce protein consumption while assessing a large representative sample of the hits in the screen. A total of 10% of the mixtures in the screen were selected to be deconvoluted due to broadening of the W81 resonance in their corresponding PrOF NMR spectra, and an additional 10% were chosen to span a range of PrOF NMR chemical shift perturbations (0.03–0.31 ppm) so as to capture both low- and high-affinity interactions, as well as assess five mixtures causing global loss of protein signals, potentially indicating the presence of aggregators.
A valuable strength of protein-observed NMR methods is their ability to quantify ligand affinities by titration. While chemical shift perturbation is a standard method for detecting ligand interactions, the magnitude of the perturbation also varies due to local chemical environment, so a larger shift does not necessarily correspond to a stronger interaction. To quickly rank-order hits from the PrOF NMR screen, we took 22 ligands (11 of which are publically available) and titrated each ligand into a single NMR tube containing 5-fluorotryptophan-labeled Brd4 (5FW-Brd4; Figure 4, Figure S5). Using a single tube reduces protein consumption but results in error propagation from pipetting and does not control for the effect of DMSO (a known bromodomain ligand) or protein dilution. Additionally, the concentration of the ligands was not verified by quantitative NMR. We term these values Δδ50, whereas we ascribe dissociation constants to titrations that use distinct NMR samples for each titration point to keep the DMSO concentration consistent, with each sample made from fresh ligand stocks with a concentration verified by quantitative NMR. Using the more rigorous titration method, we obtained dissociation constants for five actives found in the screen.
Figure 4.

Selected molecules that were hits in the PrOF NMR screen. The Δδ50 was measured by PrOF NMR titration. Percent reduction in signal in the 1H CPMG experiment is indicated by percent drop, and the change in chemical shift upon the addition of ligand at 400 μM is indicated with Δδ.
1H CPMG Results.
In the 1H CPMG screen, the same 930 compounds were screened in the same mixtures of five. Some of the more potent compounds showed a 100% signal intensity reduction (Figure 5). Due to the signal-to-noise from using 100 μM ligand, for a reliable measurement we assume the limit of detection for change in resonance signal intensity is 10%. A hit was thus defined by a 20% reduction in ligand signal intensity upon the addition of the protein to allow for a competition experiment. For the 1H CPMG screen, a competition step with a known potent ligand can significantly reduce false positive rates and at the same time provide valuable information on the inferred binding site for the hit. For this reason, we performed the competition step using (+)-JQ1 as the competitor, and a competitive hit required a 50% recovery of the signal intensity upon the addition of (+)-JQ1 (i.e., a 10% reduction of the original signal intensity in the limiting case).40 Of the 930 compounds screened, 230 of the compounds were identified as hits (24.7%), with 190 (20.4%) compounds being competitive hits against (+)-JQ1.
Figure 5.
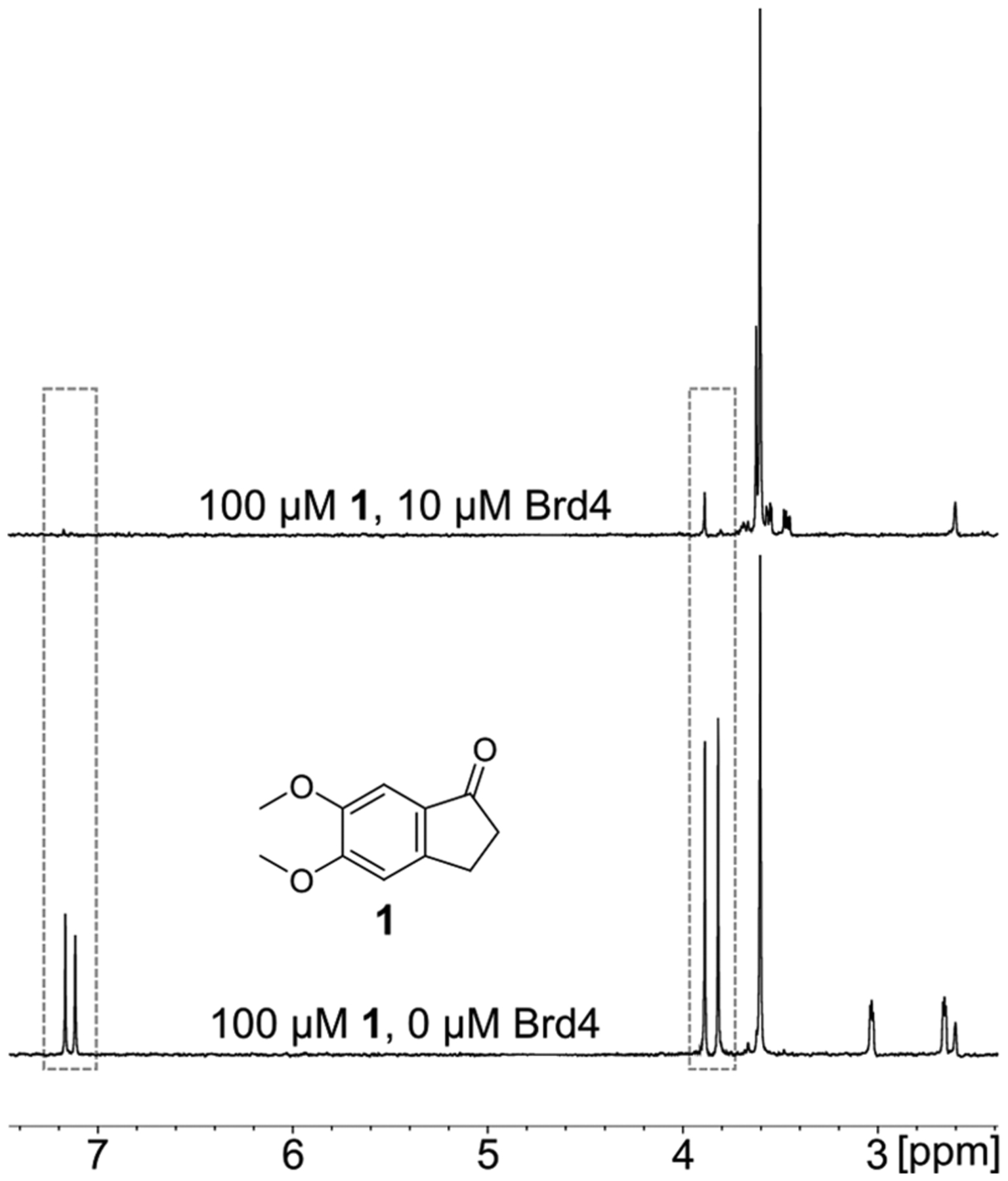
A 1H CPMG experiment of 1 and Brd4. Upon the addition of Brd4, the 1H resonances of fragment 1 signal intensity are reduced up to 100% in the top spectrum. Examples of reduced resonances are indicated by the dashed lines.
Comparison of Results.
Due to the high hit rate in the PrOF NMR screen that is in part ascribed to the conservative chemical shift cutoff, not every mixture which contained a hit was deconvoluted. Because the same mixtures were used for both the PrOF NMR and 1H CPMG screens, we offer a comparison of the data as mixtures as well as the 190 compounds individually tested by PrOF NMR. When comparing the fragment mixture results between 1H CPMG and PrOF NMR, the two assays are 77% similar when using a competition 1H CPMG experiment (74% when results from the competition step are omitted). From the data for the 190 individual compounds deconvoluted by PrOF NMR (38 mixtures), there was 85% similarity between the two assays when using a competition 1H CPMG experiment (80% when omitting the competition experiments; Figure 6). After deconvolution, nearly every mixture that was a hit by PrOF NMR contained at least one individual compound that bound, with 17 of the 33 mixtures containing more than one hit. The only mixture that was a hit by PrOF NMR that did not identify a small molecule binder after deconvolution of the mixture had an initial W81 chemical shift perturbation of 0.033 ppm, barely above the cutoff of what is considered a hit in this screen.
Figure 6.
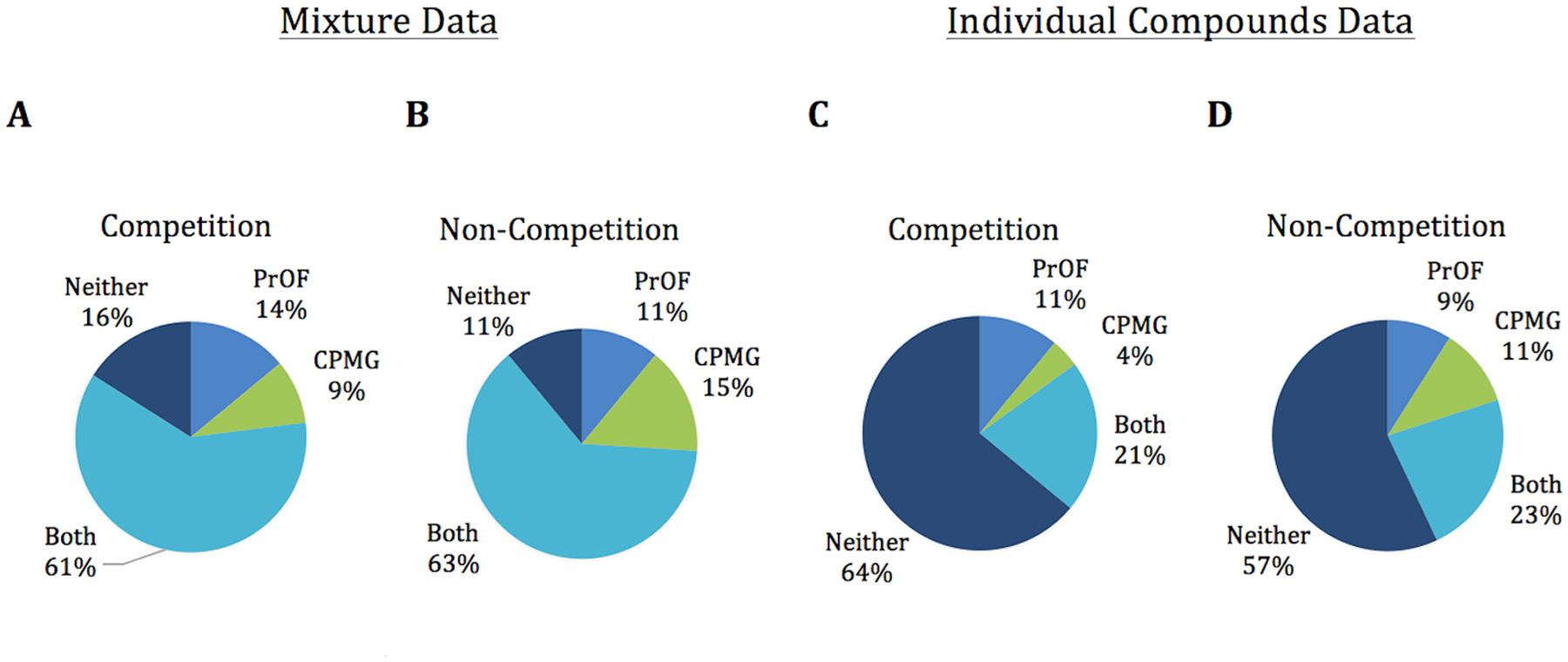
Comparison of hit data between PrOF NMR and 1H CPMG from the fragment mixture data (A,B) as well as the individual deconvoluted compounds (C,D).
While both assays have a large overlap in detected hits, we analyzed disagreements between the two screens to identify sources of potential false positives and negatives. For weak binding ligands, there were 10 discrepancies exhibiting only minor perturbation in one assay (i.e., chemical shift changes of 0.03–0.05 ppm in PrOF NMR or signal intensity drop of 20–30% in 1H CPMG) and no perturbation in the other. In this case, the discrepancies can be explained by the weakest binders being missed by one of the two assays as the sensitivity limits of these assays are approached. In the case of stronger binding compounds, there were 13 cases where there was a moderate to strong perturbation in one assay that was a nonbinder by the other assay (Table S1, 0.05–0.2 ppm, PrOF NMR; 30–100% signal drop, 1H CPMG). In the case of 1H CPMG, seven ligands were detected as competitive hits but not detected by PrOF NMR, though only two of those exhibit a signal drop of greater than 50%. These two compounds may bind deeply in the binding site, or at an allosterically coupled location, such that PrOF NMR may not have a fluorine probe close enough to detect the binding event and could thus be false negatives in the context of PrOF NMR. Additionally, there were two PrOF NMR hits that were noncompetitive hits by 1H CPMG, indicating that the ligands bound but were not competitive at the acetylated lysine binding site. These molecules may be consistent with either nonspecific binders or binding to a second site on the protein. By PrOF NMR, both of these hits perturbed W81 as well as W75, but because there are more than 10 fragments that perturbed both W81 and W75 in the PrOF NMR screen that were competitive hits by 1H CPMG, it is unlikely that W75 could be used to ascertain differential binding. From this analysis, we conclude that although a small fraction of false positives and negatives were identified between the two methods, the majority of compounds that were missed by one of the two screening methods correspond to low- to moderate-affinity fragments. Importantly, only two fragments (1% of competitive 1H CPMG hits, 0.2% of total molecules in the screen) that scored as strong and competitive hits by 1H CPMG are potential false negatives by PrOF NMR (Table S1).
Small molecule-induced protein aggregation is a common artifact in many screens41 and can be a significant cause of false positives. Therefore, we also evaluated this effect in our study. In our screen, there were five mixtures where the resonances in the PrOF NMR spectrum were no longer visible, and these five mixtures were deconvoluted to reveal one compound from each mixture responsible for the signal suppression, potentially acting as protein aggregators (Figure S6). These molecules were not detected as 1H CPMG hits because of insufficient signal-to-noise ratios to analyze the data. However, if a competitive ligand-observed experiment was conducted detecting the displacement of a “spy molecule,”11 these aggregators could result in false positives. Adding detergents to the screening buffer can be one way to reduce such effects. Due to the absence of fluorine in many commercial detergents, they are compatible with 19F-based ligand-observed and protein-observed experiments, although they were not used in our current study.
Prioritizing compounds based on affinity is important for carrying compounds forward in the lead discovery process. This is particularly true in cases of high hit rates in a fragment screen, such as described here. From our screening data and follow-up affinity determination, we found that both methods are fairly effective at triaging hits from the screening data. We used PrOF NMR titrations to determine the Δδ50 for 22 compounds. We then compared several parameters with Δδ50 and found that chemical shift perturbation in PrOF NMR and signal intensity reduction in 1H CPMG correlate modestly with Δδ50 measured by PrOF NMR (Figure 7). Despite broadening being related to chemical exchange kinetics in PrOF NMR, there is a poor correlation between broadening and Δδ50 with Brd4. Interpretation of broadening in terms of relative affinity is complicated by the differences in chemical shifts between bound and unbound states which vary between ligands, as well as additional protein dynamics such as restricted mobility of side-chains due to binding interactions. Additionally, with higher affinity fragments, there are cases where the W81 resonance is sharpening out of the baseline, approaching a fully bound state. This can result in situations where a broader resonance does not necessarily indicate a tighter binder. Thus, resonance broadening was not an effective method for ranking compounds in this PrOF NMR screen. However, when screening proteins with a lower ligandability, broadening of resonances in PrOF NMR could potentially be a useful parameter for rank-ordering compounds as well.
Figure 7.

A comparison of 20 different hits ranked by Δδ50 by PrOF NMR and different assay data (signal reduction in 1H CPMG in panel A or PrOF NMR chemical shift perturbation in panel B). A correlation is noticeable between Δδ50 and 1H CPMG signal drop, as well as Δδ50 and W81 chemical shift perturbation. While one could expect broadening of W81 to also correlate well, with Brd4 this is not the case. Using a protein with a lower ligandability could result in better rank-ordering with broadening, but with Brd4 many of the tighter binders are already coalescing at 400 μM ligand.
As further support for the successful identification of true bromodomain binders, several of the compounds identified in this screen have similar pharmacophores to known bromodomain inhibitors: oxazoles,42 triazoles,40,43 amidines,23 and ureas23,44 have all been previously reported.3 Additionally, we identified several ligands containing ketones, potentially acting as acetylated lysine mimetics. We thus conclude, that the conditions for screening fragments against Brd4 by both PrOF NMR and 1H CPMG were effective for uncovering new ligands.
Dissociation Constant Determination.
Protein-observed NMR techniques, including PrOF NMR, have been utilized to quantify dissociation constants via ligand titration at a fixed protein concentration. These derived dissociation constants rely on chemical shift perturbations from a titration experiment. While titrations can be performed with 1H CPMG ligand-observed experiments, these experiments require rigorous controls to prevent offsetting effects from convoluting the measured dissociation constants and can also be time-consuming.27,45 These titrations are easily accomplished with PrOF NMR and can be used to prioritize compounds based on affinity and/or ligand efficiency. The dissociation constants obtained by this method were used to establish ligand affinities described above for comparing with resonance perturbations. Original affinity estimates were obtained by titrating a small molecule from the library stock directly into the protein solution, which can introduce errors as described above. To verify the rank ordering and more accurately measure the dissociation constants, the affinities of the top five compounds (molecules 1, 2, 3, 4, and 12) were determined by PrOF NMR (Figures S7–S11). The affinities shown in Table 1 ranged from 6.2 to 256 μM and were consistent with the rank ordering by Δδ50. Using the same samples, a 1D protein-observed experiment was conducted monitoring the protein indole N–H resonances, however due to the lack of significant chemical shift perturbation upon ligand binding only two of five Kd values could be obtained but were in agreement with PrOF NMR values (32 vs 39 μM for 1 and 148 vs 142 μM for 2).
Table 1.
| compound | ITC - Kd (μM; ligand efficiency, LE) | 5FW-Brd4 ITC (μM) | PrOF NMR Kd (μM) | 1H NMR Kd (μM) | Δδ50 PrOF NMR (μM) | thermal shift (Δ °C) |
|---|---|---|---|---|---|---|
| 12 | 3.4 (LE: 0.53) | 2.8 | 6.2 | b | <1 | 4.53 |
| 1 | 18.6 (LE: 0.46) | 14.7 | 39 | 32 | <1 | 2.23 |
| 2 | 37.4 (LE: 0.42) | a | 142 | 148 | 3.6 | 0.77 |
| 3 | 42.1 (LE: 0.40) | a | 132 | b | 3.6 | 1.1 |
| 4 | 54.6 (LE: 0.41) | a | 256 | b | 6.4 | 0.72 |
| 5 | a | a | a | a | 9.9 | a |
| 6 | b | a | a | a | 7.6 | 1.4 |
| 7 | a | a | a | a | 14 | a |
| 8 | b | a | a | a | 9.1 | 1.3 |
| 9 | b | a | a | a | 9.6 | 0.57 |
| 10 | a | a | a | a | 21.3 | a |
| 11 | a | a | a | a | 24.3 | a |
Values not determined.
Results inconclusive.
To verify the rank-ordering of NMR hits by additional methods, we measured the top eight fragments by Δδ50 with thermal shift analysis (TSA) and isothermal titration calorimetry (ITC) with unlabeled Brd4. All eight showed an increased melting temperature of Brd4 by 0.5 °C or greater by TSA (Figure S12), and five led to quantifiable dissociation constants by ITC (Figures S13–S15). Two of the molecules for which we were unable to quantify the affinities showed an incomplete binding isotherm trend, consistent with weak binding. One of the fragments that showed incomplete binding was 9. Seeking to verify binding measurements by another method, we attempted to measure the Ki of 9 and 1 by fluorescence anisotropy. While 1 resulted in a Ki of 10.8 μM (comparable to ITC measurements), the fluorescence anisotropy experiments with 9 still led to an incomplete binding curve supporting a weak binding affinity (Figure S16). In summary of these results, the rank-ordering of the hits by ITC and TSA matches the rank-ordering of hits by PrOF NMR Δδ50 determined by the initial titrations.
In comparison of Kd values from PrOF NMR titration and ITC, variations of 1.8- to 4.7-fold were observed. To determine the effect of fluorine incorporation on binding to Brd4, the dissociation constants of the two strongest binders, 1 and 12, were measured by ITC with 5FW-Brd4 (Figures S13 and S14). In this case, the Kd values by ITC were similar (12, 3.4 vs 2.8 μM; 1, 18.6 vs 14.7 μM). Besides error in fitting the data for the weak binding fragments, additional origins of these effects may be the 4-fold higher concentration of DMSO used in the PrOF NMR experiment (2% versus 0.5%) which can attenuate ligand binding. Comparison of the affinities obtained for the fluorinated proteins by PrOF NMR and ITC suggest this could exert a 2.2- to 2.6-fold effect. The 4.7-fold difference supports an added perturbation in binding beyond differences in experimental conditions. Therefore, we conclude in the small sample of molecules studied that binding interactions seem to be minimally to modestly perturbed by fluorine incorporation (Table 1). These results are consistent with our prior studies.24
DISCUSSION
Because 1H CPMG (see Table 2) is a ligand-observed technique, the method can report on ligand solubility as well as ligand binding interactions. However, ligand-observed methods can suffer substantially when there are no known ligands for the desired target for optimizing the experiment. The known ligand allows the tuning of screening parameters, such as ligand and protein concentration, as well as the CPMG filter length. Acetaminophen was used as a test compound for this screen, which we previously determined to bind to Brd4 with a Kd between 230 and 290 μM. With unoptimized parameters, the signal change upon binding was negligible, but optimized ligand concentration (100 μM) and CPMG filter length (800 ms) resulted in a nearly 30% drop in signal intensity (Figure S17). Without a reference compound, one could screen an entire library while unknowingly using poorly optimized conditions. With protein-observed techniques, once the labeled protein has been obtained, the NMR parameters can all be optimized using only the protein. The effects of different ligand concentrations are more straightforward with protein-observed techniques (greater ligand concentration results in greater response) and can be initially estimated based on protein ligandability prediction. Importantly, a high-affinity reference ligand allows for competition CPMG experiments, which could help to avoid false positives resulting from nonspecific binding or protein denaturants. This highlights the utility of broad spectrum inhibitors for a class of proteins, such as bromosporine for bromodomains.47 As an alternative to a competition experiment to eliminate nonspecific binding with ligand-observed NMR experiments, an STD NMR method exists that tests for specific binding by epitope mapping.48
Table 2.
A General Comparison of PrOF NMR with 1H CPMG as Screening Methods
| 1H CPMG | PrOF NMR |
|---|---|
| Difficult to optimize without known ligand | Does not require reference compound for optimization of experimental conditions |
| More prone to false positives without competition experiments | Less prone to false positives |
| Low concentration of unlabeled protein (2–10 μM) needed | Requires moderate concentration of fluorinated protein (≤40 μM) |
| Low concentration of ligand needed (low to mid μM), solubility of ligands can be observed by resonance height | High concentration of ligand needed (high μM to mM), no information on solubility of ligands is provided |
| No experimental deconvolution needed, but data analysis is time-consuming | Data analysis can be readily automated, but experimental deconvolution required |
| Faster with larger proteins, no theoretical upper bound size limitation | Faster with smaller to medium proteins, approaches a size limitation (<65 kDa for aromatic amino acid labeling)46 |
| NMR time for this 1H CPMG screen (15 kDa protein, three 10 min NMR experiments): 93 h | NMR time for this PrOF NMR screen (15 kDa protein, 2 min NMR experiment): 29 h (23 h of which would be used to deconvolute 140 mixtures; note: only 38 mixtures were deconvoluted to conserve protein) Facile Kd determination with titration experiment |
A potential difficulty with using PrOF NMR is the ability to obtain fluorinated protein in sufficient yields, as well as the potential for the fluorinated protein to behave differently than the unlabeled protein, although neither of these have been problematic for bromodomains. Fluorine substitutions on aromatic rings tend to increase π–π stacking interactions by 0.5 kcal/mol per fluorine atom.49 Work by Dougherty et al. found that incorporation of 5-fluorotryptophan into proteins can reduce cation–π interactions by 0.6–0.8 kcal/mol.50,51 These energetic differences result in a 4-fold change in binding affinity or less and would only occur if the fluorinated amino acid is directly involved in the binding interaction. ITC studies with 1 and 12 indicate no significant change in binding affinities between Brd4 and 5FW-Brd4, consistent with prior studies using different ligands.24 However, the 4.7-fold difference in binding between the unlabeled protein and fluorine labeled protein for molecule 4 may reflect such a case.
Because only certain amino acids are fluorinated for PrOF NMR, some structural information is useful to determine which amino acids are close to the binding site of interest. Acetaminophen binds weakly with Brd4 and has been crystallized with the highly homologous bromodomain of Brd2. Assuming a similar binding mode between Brd4 and Brd2, the fluorine of W81 is 6.1 Å from the nearest heavy atom of acetaminophen (Figure S18). However, with Brd4 bound to (+)-JQ1, W75 is also slightly affected (0.13 ppm) with the fluorine atom 11.3 Å away from the nearest heavy atom of (+)-JQ1. This latter effect may occur from a subtle conformational change not observed in the X-ray structure. Several other ligands in the PrOF NMR screen perturb both W81 and W75, so the distance dependence of the fluorine atom can vary. Aromatic amino acids tend to be enriched at protein–protein interfaces,52 and with bromodomains we found high conservation of aromatic amino acids near the binding site.24 Therefore, for these types of interactions it is highly likely that at least one aromatic amino acid will be close by to monitor.
Protein size will play a large role in the speed with which spectra can be acquired. A smaller protein results in rapid PrOF NMR experiments with lengthy 1H CPMG experiments, while for larger proteins the ligand-observed becomes the faster of the two methods. For a protein the size of Brd4, a PrOF NMR screen is substantially faster than a 1H CPMG screen (2 min vs 20–30 min for a single experiment, though experimental deconvolution is necessary by PrOF NMR), but as protein size increases, PrOF NMR will require longer experiment times due to a longer rotational correlation time leading to resonance broadening. Rule et al. has shown that a 65 kDa protein labeled with 5-fluorotryptophan can lead to broad but resolved resonances.46 In this case, experiment times would be significantly longer unless a more dynamic side chain is labeled.53,54 Because CPMG exploits the differences in rotational correlation between a small ligand and large protein, it exhibits the opposite trend; i.e., the responsiveness of CPMG increases with larger proteins. With larger proteins, higher concentrations of ligand can be used, dramatically reducing experiment time. One difficulty with 1H CPMG that could be resolved with software is the ability to work up screening data. Because of the potential for overlapping resonances, and the need to correlate resonances from the screen with baseline spectra of the individual ligands, automated data processing can greatly expedite the process.55
Finally, protein-observed techniques are inherently less prone to false positives as long as more than one resonance is present. Because there are NMR resonances in a specific pattern due to the local chemical environment of each observed nucleus, the spectrum will reveal whether or not a protein is in solution and well-folded. If the protein is denatured or degraded, the resonances coalesce, disappear, or sharpen. This is essentially an in situ quality control for every protein-observed experiment, reducing potential false positives. Additionally, because there are multiple resonances corresponding to amino acids at different positions on the protein, the perturbed resonances frequently correlate with the rough identification of the ligand binding site, although as with all protein-observed NMR experiments that rely on chemical shift perturbations, conformational effects cannot be ruled out. However, we have found that the chemical shift perturbations for bromodomains tend to localize to residues near the binding site.24 This type of analysis is useful if there are multiple binding sites on a protein construct where only one is the desired target. Gee et al. previously showed this in the context of the protein KIX, which possesses two binding sites.21 Additionally, because multiple labels are present which may be outside known binding sites, there is the potential for serendipitously uncovering a cryptic binding site.
In conclusion, in this study we have benchmarked PrOF NMR as having comparable sensitivity to 1H CPMG with 85% assay overlap when an additional competition experiment is employed in the 1H CPMG NMR experiment in the context of bromodomain screening. Without a competitor, the agreement between the two assays dropped to 80%. The similar hit-rate of PrOF NMR with the well-utilized 1H CPMG further validates PrOF NMR as a screening method for ligand discovery for similar proteins. Because the detection abilities of both methods were so similar, the decision of which biophysical screen to use as a primary screen is protein dependent, and the complementary nature of the data supports conducting both ligand- and protein-based experiments. Factors that can influence this decision are the availability of reference compounds, the ability to express protein in sufficient yields, protein size, and the presence of multiple binding sites. One advantage of 1H CPMG over PrOF NMR is the ability to avoid a time-consuming deconvolution step for the fragment mixtures. We found an 85% similarity between PrOF NMR of a representative set of deconvoluted mixtures and 1H CPMG, similar to the mixture data which was 77% similar between methods; as such, no further deconvolution was pursued. Due to the similarities in assay conditions, the possibilities of doing a sequential ligand-observed and PrOF NMR experiment during the same NMR screen may prove beneficial, eliminating the need for deconvolution.
MATERIALS AND METHODS
Preparation of Fragment Library.
The fragments in the screening library were initially analyzed by 1H NMR, and any compounds with low resonance intensity from low solubility or aggregation propensity were removed from the set. The fragments were then combined into mixtures of five in such a way as to maximize the diversity of compounds in each mixture, while avoiding mixing reactive functional groups. Although the fragment library consists of a majority of commercial fragments, several fragments are proprietary compounds of Eli Lilly & Company. Compound 12 was one such compound, whose structure has been omitted.
Expression of 5FW-Brd4 (42–168).
Unlabeled and 5FW-labeled Brd4 were expressed based on established methods23,24 using E. coli Bl21(DE3) + pRARE strains. To express the labeled protein, the secondary culture in LB media was grown until an OD600 of 0.6 was reached followed by harvesting. Cells were resuspended in defined media of Muchmore et al.56 containing 5-fluoroindole (60 mg/L) in place of tryptophan.57 The resuspended E. coli were incubated at 37 °C while shaking for 1.5 h followed by cooling to 20 °C and media temperature equilibration for 30 min. Protein expression was induced with 1 mM IPTG overnight (14–16 h) at 20 °C. The cells were harvested and stored at −20 °C. Cell pellets were thawed at RT followed by the addition of lysis buffer (50 mM phosphate at pH 7.4, 300 mM NaCl) containing protease inhibitor PMSF (5 mM) as well as the Halt protease inhibitor and purified according to methods described in the Supporting Information using Ni-affinity chromatography. Yields following purification are 120 mg/L 5FW-Brd4 (>94% fluorine incorporation assessed by mass spectrometry). Purity of proteins was assessed by SDS-PAGE. Fluorinated amino acid incorporation efficiency in proteins was measured by mass spectrometry as described in the Supporting Information. Concentration was determined via absorbance at 280 nm.58
Protein-Observed Fluorine (PrOF) NMR.
1D 19F NMR Parameters: 19F NMR spectra were acquired at 565 MHz on a Bruker Avance III spectrometer equipped with a quadruple resonance HFCN CryoProbe without proton decoupling, unless otherwise specified. Samples for binding assays contained 50 μM 5FW-Brd4 and five fragments each at 400 μM in 50 mM Tris, 100 mM NaCl, and 5% D2O, at pH 7.4. Spectra were referenced to trifluoroacetate (−76.55 ppm). Measurement parameters included a relaxation delay time of 0.7 s and a 58° pulse flip angle (based on the Ernst angle from T1 determination). An acquisition time of 0.05 s and a spectral width of 10 ppm were used for all experiments. Thirteen-minute screening experiments used 1000 scans, while 2 min screening experiments used 160 scans. Small molecules were titrated into the protein solution from concentrated stock solutions of DMSO (100 mM). Final DMSO concentrations were kept at or below 2%. Proton decoupling was not used because it results in a reduction in signal intensity due to the negative nuclear Overhauser effect with large molecules. Additionally, the increased NMR line width from large biomolecules obscures couplings.
1H CPMG.
1H NMR spectra were first collected on all compounds in the fragment library to be used as reference spectra for deconvoluting screening data. Samples were prepared with 100 μM of each fragment and a 1H CPMG spectrum acquired of each mixture (with a CPMG filter length of 1.2 s and an interpulse delay of 2.5 ms). In a second step, a concentrated protein stock solution was then added to each sample to a final concentration of 10 μM Brd4, and a 1H CPMG spectrum was recorded. Finally, known competitor (+)-JQ140 was added to a concentration of 20 μM, and competition was monitored by recovery of signal intensity.
Fluorescence Anisotropy.
Fluorescence anisotropy was measured from an excitation wavelength of 485 nm and an emission wavelength of 535 nm on a Tecan Infinite 500 plate reader using low volume 384-well plates (Corning 4511). All experiments were carried out in 50 mM Tris, 150 mM NaCl, and 4 mM CHAPS at pH 7.4. The fluorescently labeled tracer, BI-BODIPY, was kindly provided by the laboratory of Prof. Wei Zhang and was synthesized according to published methods.59 For both direct binding and competition experiments, 25 μM stocks of a BI-BODIPY stock solution in DMSO were diluted to a final concentration of 25 nM. For direct binding experiments, Brd4 was serially diluted from micromolar to subnanomolar concentrations. For competition experiments, Brd4 was kept at a constant concentration of 156 nM, equivalent to 80% bound tracer as determined in direct binding experiments, and the concentration of competing ligand was serially diluted from micromolar to subnanomolar concentrations. Data were collected within 30 min after plating to minimize Brd4 binding to the plate surface. All experiments were carried out in triplicate. Acquired data were fit using GraphPad Prism. Kd values were determined by fitting to eq 1, which accounts for ligand depletion. In this equation, b and c are the maximal and minimal anisotropy values, respectively, a is the concentration of fluorescently labeled tracer, x is the protein concentration, and y is the observed anisotropy value.
| (1) |
IC50 values were determined using GraphPad Prism’s log(inhibitor) vs response function. Ki values were obtained using a variant of the Cheng-Prussof equation from Huang.60
Isothermal Titration Calorimetry.
Experiments were carried out on a MicroCal Auto-iITC200 titration from Malvern with a cell volume of 200 μL and a 40 μL microsyringe. Experiments were carried out at 25 °C while stirring at 750 rpm, in ITC buffer (50 mM phosphate buffer at pH 7.4 and 150 mM NaCl). The microsyringe was loaded with a solution of ligands whose concentrations were accurately measured by quantitative NMR (ITC buffer with 0.5% DMSO-d6) and was automatically inserted into the calorimetric cell which was filled with an amount of the protein, Brd4, and 5FW-Brd4 (200 μL, 30 μM in ITC buffer with 0.5% DMSO-d6). The system was first allowed to equilibrate until the cell temperature reached 25 °C, and an additional delay of 60 s was applied. These first titrations were conducted using an initial control injection of 0.5 μL followed by 18 identical injections of 2 μL with a duration of 4 s per injection and a spacing of 150 s between injections. Afterward, a second titration was performed over this first titration without cleaning the cell. This second titration was executed with Continue Injections as Automation Method in MicroCal Auto-iTC200. Second titrations were conducted using 19 identical injections of 2 μL with duration of 4 s per injection and a spacing of 150 s between injections. The titration experiments were designed in such a fashion as to ensure complete saturation of the protein before the final injection. The heat of dilution for the ligands was independent of ligand concentration and corresponded to the heat observed from the last injection, following saturation of protein binding, thus facilitating the estimation of the baseline of each titration from the last injection. The collected data were corrected for ligand heats of dilution and deconvoluted using the MicroCal PEAQ-ITC Analysis Software to yield enthalpy of binding (ΔH) and binding constant (Kd). Thermodynamic parameters were calculated using the basic equation of thermodynamics (ΔG = ΔH − TΔS = −RT ln Kd, where ΔG, ΔH, and ΔS are the changes in free energy, enthalpy, and entropy of binding respectively). A single binding site model was employed by MicroCal PEAQ-ITC Analysis Software.
Thermal Shift Assays.
Thermal shift assay (TSA) measures the thermal stability of the Brd4 wild-type and the comparison between the melting temperature of apoprotein and melting temperatures in the presence of different compounds. The previous exploratory phase was performed to determine optimal experimental conditions for this assay in the same buffer as ITC (50 mM phosphate buffer at pH 7.4 and 150 mM NaCl). In a 96-well PCR plate with a sample of 20 μL in each well (2.5% DMSO), Brd4 was at 26 μM, fragments were assayed at 250 μM, and Protein Thermal Shift Dye was utilized at 2× (Protein Thermal Shift Dye Kit from ThermoFisher Scientific). Fluorescence data were collected on an Applied Biosystems 7500 FAST RealTime PCR System with an excitation range of 580 ± 10 nm. The fluorescence emission signal at 623 ± 14 nm was used for data analysis. Samples were preheated for 2 min at 25 °C. Then the temperature was continuously increased 2 °C/min from 24 to 99 °C, and finally samples were maintained for 2 min at 99 °C.
Supplementary Material
ACKNOWLEDGMENTS
This project was funded in part by the NSF-CAREER Award CHE-1352091 (W.C.K.P.) and the National Institutes of Health Biotechnology training grant 5T32GM008347-23 (A.K.U.). Some NMR spectra were recorded on an instrument purchased with support from the National Institutes of Health Shared Instrumentation Grant program (S10OD011952). We kindly thank Alex Ayoub for assistance with the fluorescence anisotropy experiments.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.6b00730.
Additional experimental procedures, tables, figures, and characterization data (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Warner KD, Homan P, Weeks KM, Smith AG, Abell C, and Ferré-D’Amaré AR (2014) Validating Fragment-Based Drug Discovery for Biological RNAs: Lead Fragments Bind and Remodel the TPP Riboswitch Specifically. Chem. Biol 21, 591–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Fontaine F, Overman J, and François M (2015) Pharmacological manipulation of transcription factor protein-protein interactions: opportunities and obstacles. Cell Regener. 4, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bamborough P, and Chung C (2015) Fragments in bromodomain drug discovery. MedChemComm 6, 1587–1604. [Google Scholar]
- (4).Hajduk PJ, and Greer J (2007) A decade of fragment-based drug design: strategic advances and lessons learned. Nat. Rev. Drug Discovery 6, 211–219. [DOI] [PubMed] [Google Scholar]
- (5).Bollag G, Tsai J, Zhang J, Zhang C, Ibrahim P, Nolop K, and Hirth P (2012) Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discovery 11, 873–886. [DOI] [PubMed] [Google Scholar]
- (6).Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DCS, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park C-M, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang H, Humerickhouse R a, Rosenberg SH, and Elmore SW (2013) ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med 19, 202–8. [DOI] [PubMed] [Google Scholar]
- (7).Niesen FH, Berglund H, and Vedadi M (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc 2, 2212–2221. [DOI] [PubMed] [Google Scholar]
- (8).Shepherd CA, Hopkins AL, and Navratilova I (2014) Fragment screening by SPR and advanced application to GPCRs. Prog. Biophys. Mol. Biol 116, 113–123. [DOI] [PubMed] [Google Scholar]
- (9).Davies TG, and Hyvönen M (2012) Fragment-Based Drug Discovery and X-Ray Crystallography (Davies TG, and Hyvönen M, Eds.), Topics in Current Chemistry, Vol. 317, Springer, Berlin. [Google Scholar]
- (10).Begley DW, Moen SO, Pierce PG, and Zartler ER (2013) Saturation Transfer Difference NMR for Fragment Screening, In Current Protocols in Chemical Biology, pp 251–268, John Wiley & Sons, Inc., Hoboken, NJ. [DOI] [PubMed] [Google Scholar]
- (11).Dalvit C, Fagerness PE, Hadden DT a, Sarver RW, and Stockman BJ (2003) Fluorine-NMR Experiments for High-Throughput Screening: Theoretical Aspects, Practical Considerations, and Range of Applicability. J. Am. Chem. Soc 125, 7696–7703. [DOI] [PubMed] [Google Scholar]
- (12).Dalvit C, Pevarello P, Tatò M, Veronesi M, Vulpetti A, and Sundström M (2000) Identification of compounds with binding affinity to proteins via magnetization transfer from bulk water. J. Biomol. NMR 18, 65–8. [DOI] [PubMed] [Google Scholar]
- (13).Shuker SB, Hajduk PJ, Meadows RP, and Fesik SW (1996) Discovering High-Affinity Ligands for Proteins: SAR by NMR. Science 274, 1531–1534. [DOI] [PubMed] [Google Scholar]
- (14).Schiebel J, Radeva N, Krimmer SG, Wang X, Stieler M, Ehrmann FR, Fu K, Metz A, Huschmann FU, Weiss MS, Mueller U, Heine A, and Klebe G (2016) Six Biophysical Screening Methods Miss a Large Proportion of Crystallographically Discovered Fragment Hits: A Case Study. ACS Chem. Biol 11, 1693. [DOI] [PubMed] [Google Scholar]
- (15).Schiebel J, Radeva N, Köster H, Metz A, Krotzky T, Kuhnert M, Diederich WE, Heine A, Neumann L, Atmanene C, Roecklin D, Vivat-Hannah V, Renaud JP, Meinecke R, Schlinck N, Sitte A, Popp F, Zeeb M, and Klebe G (2015) One Question, Multiple Answers: Biochemical and Biophysical Screening Methods Retrieve Deviating Fragment Hit Lists. ChemMedChem 10, 1511–1521. [DOI] [PubMed] [Google Scholar]
- (16).Kutchukian PS, Wassermann AM, Lindvall MK, Wright SK, Ottl J, Jacob J, Scheufler C, Marzinzik A, Brooijmans N, and Glick M (2015) Large Scale Meta-Analysis of Fragment-Based Screening Campaigns: Privileged Fragments and Complementary Technologies. J. Biomol. Screening 20, 588–596. [DOI] [PubMed] [Google Scholar]
- (17).Keserű GM, Erlanson DA, Ferenczy GG, Hann MM, Murray CW, and Pickett SD (2016) Design Principles for Fragment Libraries: Maximizing the Value of Learnings from Pharma Fragment-Based Drug Discovery (FBDD) Programs for Use in Academia. J. Med. Chem, DOI: 10.1021/acs.jmedchem.6b00197. [DOI] [PubMed] [Google Scholar]
- (18).Rodriguez-Mias R a, and Pellecchia M (2003) Use of Selective Trp Side Chain Labeling To Characterize Protein–Protein and Protein–Ligand Interactions by NMR Spectroscopy. J. Am. Chem. Soc 125, 2892–2893. [DOI] [PubMed] [Google Scholar]
- (19).Arntson KE, and Pomerantz WCK (2016) Protein-Observed Fluorine NMR: A Bioorthogonal Approach for Small Molecule Discovery. J. Med. Chem 59, 5158–5171. [DOI] [PubMed] [Google Scholar]
- (20).Gerig JT Fluorine NMR (http://www.biophysics.org/Portals/1/PDFs/Education/gerig.pdf). Previously published in (2001) Biophysics Textbook Online, pp 1–35.
- (21).Gee CT, Koleski EJ, and Pomerantz WCK (2015) Fragment Screening and Druggability Assessment for the CBP/p300 KIX Domain through Protein-Observed 19F NMR Spectroscopy. Angew. Chem., Int. Ed 54, 3735–3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Pomerantz WC, Wang N, Lipinski AK, Wang R, Cierpicki T, and Mapp AK (2012) Profiling the Dynamic Interfaces of Fluorinated Transcription Complexes for Ligand Discovery and Characterization. ACS Chem. Biol 7, 1345–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Urick AK, Hawk LML, Cassel MK, Mishra NK, Liu S, Adhikari N, Zhang W, dos Santos CO, Hall JL, and Pomerantz WCK (2015) Dual Screening of BPTF and Brd4 Using Protein-Observed Fluorine NMR Uncovers New Bromodomain Probe Molecules. ACS Chem. Biol 10, 2246–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Mishra NK, Urick AK, Ember SWJ, Schönbrunn E, and Pomerantz WC (2014) Fluorinated Aromatic Amino Acids Are Sensitive 19F NMR Probes for Bromodomain-Ligand Interactions. ACS Chem. Biol 9, 2755–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Jordan JB, Whittington DA, Bartberger MD, Sickmier EA, Chen K, Cheng Y, and Judd T (2016) Fragment-Linking Approach Using 19 F NMR Spectroscopy To Obtain Highly Potent and Selective Inhibitors of β-Secretase. J. Med. Chem 59, 3732–3749. [DOI] [PubMed] [Google Scholar]
- (26).Ge X, MacRaild CA, Devine SM, Debono CO, Wang G, Scammells PJ, Scanlon MJ, Anders RF, Foley M, and Norton RS (2014) Ligand-Induced Conformational Change of Plasmodium falciparum AMA1 Detected Using F-19 NMR. J. Med. Chem 57, 6419–6427. [DOI] [PubMed] [Google Scholar]
- (27).Wamhoff E-C, Hanske J, Schnirch L, Aretz J, Grube M, Varón Silva D, and Rademacher C (2016) 19 F NMR-Guided Design of Glycomimetic Langerin Ligands. ACS Chem. Biol 11, 2407. [DOI] [PubMed] [Google Scholar]
- (28).Devine SM, Mulcair MD, Debono CO, Leung EWW, Nissink JWM, Lim SS, Chandrashekaran IR, Vazirani M, Mohanty B, Simpson JS, Baell JB, Scammells PJ, Norton RS, and Scanlon MJ (2015) Promiscuous 2-Aminothiazoles (PrATs): A Frequent Hitting Scaffold. J. Med. Chem 58, 1205. [DOI] [PubMed] [Google Scholar]
- (29).Norton R, Leung E, Chandrashekaran I, and MacRaild C (2016) Applications of 19F-NMR in Fragment-Based Drug Discovery. Molecules 21, 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Gee CT, Arntson KE, Urick AK, Mishra NK, Hawk LML, Wisniewski AJ, and Pomerantz WCK (2016) Protein-observed 19F-NMR for fragment screening, affinity quantification and druggability assessment. Nat. Protoc 11, 1414–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Anderluh G, Razpotnik A, Podlesek Z, Maček P, Separovic F, and Norton RS (2005) Interaction of the Eukaryotic Pore-forming Cytolysin Equinatoxin II with Model Membranes: 19F NMR Studies. J. Mol. Biol 347, 27–39. [DOI] [PubMed] [Google Scholar]
- (32).Harner MJ, Chauder BA, Phan J, and Fesik SW (2014) Fragment-Based Screening of the Bromodomain of ATAD2. J. Med. Chem 57, 9687–9692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Richards KL, Rowe ML, Hudson PB, Williamson RA, and Howard MJ (2016) Combined ligand-observe 19F and protein-observe 15N,1H-HSQC NMR suggests phenylalanine as the key Δ-somatostatin residue recognized by human protein disulfide isomerase. Sci. Rep 6, 19518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Barile E, and Pellecchia M (2014) NMR-Based Approaches for the Identification and Optimization of Inhibitors of Protein–Protein Interactions. Chem. Rev 114, 4749–4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Prinjha RK, Witherington J, and Lee K (2012) Place your BETs: the therapeutic potential of bromodomains. Trends Pharmacol. Sci 33, 146–153. [DOI] [PubMed] [Google Scholar]
- (36).Practical Fragments - NMR Poll Results. http://practicalfragments.blogspot.com/search/label/CPMG.
- (37).Halgren T (2007) New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des 69, 146–148. [DOI] [PubMed] [Google Scholar]
- (38).Halgren T a (2009) Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Model 49, 377–389. [DOI] [PubMed] [Google Scholar]
- (39).Schrödinger Release 2015–3: SiteMap, version 3.6; Schrödinger, LLC: New York, 2016. [Google Scholar]
- (40).Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French C a, Wiest O, Kung AL, Knapp S, and Bradner JE (2010) Selective inhibition of BET bromodomains. Nature 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Ferreira RS, Bryant C, Ang KKH, McKerrow JH, Shoichet BK, and Renslo AR (2009) Divergent Modes of Enzyme Inhibition in a Homologous Structure–Activity Series. J. Med. Chem 52, 5005–5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Bamborough P, Diallo H, Goodacre JD, Gordon L, Lewis A, Seal JT, Wilson DM, Woodrow MD, and Chung CW (2012) Fragment-based discovery of bromodomain inhibitors part 2: Optimization of phenylisoxazole sulfonamides. J. Med. Chem 55, 587–596. [DOI] [PubMed] [Google Scholar]
- (43).Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung C-W, Chandwani R, Marazzi I, Wilson P, Coste H, White J, Kirilovsky J, Rice CM, Lora JM, Prinjha RK, Lee K, and Tarakhovsky A (2010) Suppression of inflammation by a synthetic histone mimic. Nature 468, 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Chung C-W, Dean AW, Woolven JM, and Bamborough P (2012) Fragment-Based Discovery of Bromodomain Inhibitors Part 1: Inhibitor Binding Modes and Implications for Lead Discovery. J. Med. Chem 55, 576–586. [DOI] [PubMed] [Google Scholar]
- (45).Dalvit C, Fogliatto G, Stewart A, Veronesi M, and Stockman B (2001) WaterLOGSY as a method for primary NMR screening: Practical aspects and range of applicability. J. Biomol. NMR 21, 349–359. [DOI] [PubMed] [Google Scholar]
- (46).Ho C, Pratt EA, and Rule GS (1989) Membrane-bound d-lactate dehydrogenase of Escherichia coli: a model for protein interactions in membranes. Biochim. Biophys. Acta, Rev. Biomembr 988, 173–184. [DOI] [PubMed] [Google Scholar]
- (47).Structural Genomics Consortium (SGC): Bromosporine. http://www.thesgc.org/?chemical-probes/?bromosporine.
- (48).Cala O, and Krimm I (2015) Ligand-Orientation Based Fragment Selection in STD NMR Screening. J. Med. Chem 58, 8739–8742. [DOI] [PubMed] [Google Scholar]
- (49).Cozzi F, Ponzini F, Annunziata R, Cinquini M, and Siegel JS (1995) Polar Interactions between Stacked π Systems in Fluorinated 1,8-Diarylnaphthalenes: Importance of Quadrupole Moments in Molecular Recognition. Angew. Chem., Int. Ed. Engl 34, 1019–1020. [Google Scholar]
- (50).Torrice MM, Bower KS, Lester HA, and Dougherty DA (2009) Probing the role of the cation–π interaction in the binding sites of GPCRs using unnatural amino acids. Proc. Natl. Acad. Sci. U. S. A 106, 11919–11924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Zhong W, Gallivan JP, Zhang Y, Li L, Lester H a, and Dougherty D a (1998) From ab initio quantum mechanics to molecular neurobiology: A cation-π binding site in the nicotinic receptor. Proc. Natl. Acad. Sci. U. S. A 95, 12088–12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Bogan AA, and Thorn KS (1998) Anatomy of hot spots in protein interfaces. J. Mol. Biol 280, 1–9. [DOI] [PubMed] [Google Scholar]
- (53).Liu JJ, Horst R, Katritch V, Stevens RC, and Wuthrich K (2012) Biased Signaling Pathways in 2-Adrenergic Receptor Characterized by 19F-NMR. Science 335, 1106–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Manglik A, Kim TH, Masureel M, Altenbach C, Yang Z, Hilger D, Lerch MT, Kobilka TS, Thian FS, Hubbell WL, Prosser RS, and Kobilka BK (2015) Structural Insights into the Dynamic Process of β2-Adrenergic Receptor Signaling. Cell 161, 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Peng C, Frommlet A, Perez M, Cobas C, Blechschmidt A, Dominguez S, and Lingel A (2016) Fast and Efficient Fragment-Based Lead Generation by Fully Automated Processing and Analysis of Ligand-Observed NMR Binding Data. J. Med. Chem 59, 3303–3310. [DOI] [PubMed] [Google Scholar]
- (56).Muchmore DC, McIntosh LP, Russell CB, Anderson DE, and Dahlquist FW (1989) Nuclear Magnetic Resonance Part B Structure and Mechanism, Methods in Enzymology, Vol. 177, Elsevier. [DOI] [PubMed] [Google Scholar]
- (57).Crowley PB, Kyne C, and Monteith WB (2012) Simple and inexpensive incorporation of 19F-Tryptophan for protein NMR spectroscopy. Chem. Commun 48, 10681. [DOI] [PubMed] [Google Scholar]
- (58).Pace CN, Vajdos F, Fee L, Grimsley G, and Gray T (1995) How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 4, 2411–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Zhang ZJ, Kwiatkowski N, Zeng H, Lim SM, Gray NS, Zhang W, and Yang PL (2012) Leveraging kinase inhibitors to develop small molecule tools for imaging kinases by fluorescence microscopy. Mol. BioSyst 8, 2523–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Huang X (2003) Fluorescence Polarization Competition Assay: The Range of Resolvable Inhibitor Potency Is Limited by the Affinity of the Fluorescent Ligand. J. Biomol. Screening 8, 34–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.