

Abstract
COVID-19 outbreak is the recently reported worldwide pandemic threat. As part of our interventions with machine learning and molecular simulation approaches, we report the inhibitory effect of thirty compounds reported from the sacred plant Aerva lanata. The predicted activity of the screened ligands are comparable with the one of the present medication, hydroxy chloroquine (HCQ), on the main protease (PDB:6YB7) of SARS-CoV-2. Our studies pointed out the effectiveness of the plant with twenty seven compounds having potential activity against the main protease compared to the reference HCQ. The robustness of some of the phytochemicals such as ervoside, which is only present in Aerva lanata computed to have very high anticoronavirus activity. The results are indicative of potential natural antivirus source, which subsidizes in thwarting the invasion of coronavirus into the human body. Many phytochemicals which are computed to be effective towards SARS-CoV-2 in this study are used as drugs for various other diseases. Perhaps these compounds could be attractive for the management of COVID-19, but clinical trials must be performed in order to validate this observation.
Keywords: SARS-CoV-2, Aerva lanata, Machine learning, Docking
Introduction
The outbreak of pandemic severe acute respiratory syndrome–coronavirus 2 (SARS-CoV-2) has triggered more than 110 million confirmed cases and around 2.5 deaths globally as of March 2021 [1]. Coronaviruses belong to a large family of viruses that frequently be the reason for common cold in humans. Middle East respiratory syndrome (MERS), severe acute respiratory syndrome (SARS), and currently SARS-CoV-2 are the more severe diseases caused by the coronavirus family [2, 3]. The major drugs under investigation include drugs that have been used to treat malaria and autoimmune diseases; common antiviral drugs, and antibodies from people who have recovered from COVID-19 [4]. Based on the preliminary findings, chloroquine and hydroxychloroquine have been prescribed to patients to recover from the evolution of pneumonia associated with COVID-19 [5]. Even though vaccines may be available, its not sure about the reliability until now. The lack of specific treatments for COVID-19 intended scientists to work hard to develop effective treatments to end up this pandemic. This demands to search for natural and innocuous medications to stop the attack of coronavirus.
As part of our interest in phytochemicals, we selected Aerva lanata, one of the ten sacred herbs of Kerala. It belongs to Amaranthaceae family and is used as traditional medication in snake bites, cough, strangury, headache, urolithiasis and jaundice therapy [6, 7]. The major phytochemicals reported from the plant include alkaloids (ervine, methylervine, ervoside, aervine, methylaervine, aervoside, ervolanine, and aervolanine), flavanoids (kaempferol, quercetin, isorhamnetin, persinol, persinosides A and B), methyl grevillate, lupeol, lupeol acetate benzoic acid, β-sitosteryl acetate and tannic acid. Pharmacological activities such as diuretic, anti-inflammatory, hypoglycemic, anti-diabetic, antiparasitic, antimicrobial, hepoprotective, anti-urolithiasis, antiasthmatic, antifertility and hypolipidemic properties are reported for Aerva lanata [8].
Rational drug design now uses various molecular simulation techniques to reduce the cost and time required for the discovery of a potential drug. Advantages in the field of Computer-Aided Drug Design (CADD) by structure-based (SBDD) and ligand-based drug design (LBDD) suggest that their complementary use, as well as their combination with experimental procedures, has a dominant impact on rational drug design [9]. Machine learning (ML) models are now-a days widely used to predict the druggability of compounds. The availability of effective computational resources and high quality data have made machine learning techniques, a great supporting tool in drug discovery and developement. Molecular docking and dynamics can be used to analyze the binding modes of a small molecule with binding site of a protein at the atomic level, which intern help us to elucidate vital biochemical properties of small organic entities [10]. In the present study, we are trying to explore the SARS-CoV-2 inhibitory activities of phytochemicals from Aerva lanata, compared to hydroxy chloroquine. ML models are built based on the available drug dataset and predicted the IC50 values of thirty phytochemicals already reported from Aerva lanata. The results are cross validated by docking simulations of the screened ligands with SARS-CoV-2 main protease retrieved from RCSB PDB (PDB ID:6YB7). To the best of our knowledge, this is the first report presenting the potential inhibitory activities of phytochemicals from Aerva lanata.
Methodology
The available dataset of 277 drugs with IC50 values screened for SARS-Cov2 were collected from PubChem. The scope of using machine learning to predict the IC50 values of phytochemicals reported from Aerva lanata, were analysed and the best model was selected to predict the values for further trials. The results obtained were confirmed by conventional molecular docking and dynamics simulations. The preparation and optimization of the protein, the ligands and docking simulations to interpret the interaction between these ligands and SARS-CoV-2 main protease, 6YB7 [11] were carried out using AutoDock Vina [12]. UCSF Chimera 1.13 was used for the visualization and analysis of the binding modes [13].
Machine learning
The isomeric SMILES strings of 277 compounds were retrieved using the pubchempy package which provides a way to interact with PubChem from Python. The obtained SMILES were further used to collect the descriptors using PaDELPy package in python which act as Python wrapper for the PaDEL-Descriptor molecular descriptor calculation software. PaDEL Descriptor calculates and returns 1875 descriptors (1444 1D, 2D descriptors and 431 3D Descriptors), which were calculated using The Chemistry Development Kit. Also ADMET properties, calculated using SwissADME were used for building machine learning models. This data along with the IC50 values was used to build models for IC50 prediction of new compounds.
As the number of attributes describing each compound were very much larger than the number of compounds available for constructing the model, the features with low variance were removed to avoid the overfitting of the built model. Also, among two highly correlated attributes only one attribute is retained for constructing the model. The 133 features left after this screening were used in the further process. The most contributing factors were found out using selectkbest method in python. One by third of the collected compounds were randomly selected as the testing data set and the remaining were used to train the models. The training was performed with 183 compounds and testing with 84 compounds.
Various machine learning models—Linear Regression, Support Vector Regression, Lasso Regression, KernelRidge, Random Forest Regression, ElasticNet- were constructed and fitted using Python 3.7.5, in Spyder 3.7 environment using Relevant packages. Among the models created best results were exhibited by Random forest Regressor. The models were evaluated on the basis of r-squared value and means squared error value. The best models constructed were used for predicting the IC50 value of phytochemicals found in the selected plant.
Protein preparation
SARS-CoV-2 main protease with unliganded active site, 6YB7 was retrieved from RCSB PDB [11]. It has a resolution of 1.25 Å, R-value (observed) of 0.172 and contain 306 amino acid residues. Its unit cell edge lengths are a = 112.429, b = 52.798, c = 44.611 and angles are α = 90°, β = 103.03° and γ = 90° respectively. The 3D crystal structure of the target was first optimized using AutoDock Vina [12]. For this, we selected and deleted all nonstandard residues and the protein was minimized using steepest descent step = 200 and size = 0.02 with update interval of 5. Then we added incomplete side chains, hydrogens, charge and deleted the solvent. Assign the charges to the standard residue by AMBER ff145B and to the other residues by AM1-BCC. The minimized structure was further used for docking with the selected ligands.
Ligand preparation and ADME/T
The 3D structures of all the ligands were generated by build structure from SMILES string obtained from PubChem [14]. The physicochemical properties, lipophilicity, aqueous solubility, pharmacokinetics and drug likeness were checked using SwissADME [15–17]. A Bioavailability Radar is demonstrated for a quick evaluation of drug-likeness by taking six physicochemical properties—lipophilicity, size, polarity, solubility, flexibility and saturation. By the specific descriptors adapted [18, 19], physicochemical range on individual axis was well-defined and characterized as a pink area, which represents drug-likeness. The physicochemical parameters like molecular weight (MW), molecular refractivity (MR), count of specific atom types and topological polar surface area (TPSA) were computed with OpenBabel9, version 2.3.0. We calculated partition coefficient between n-octanol and water (log Po/w), a classical descriptor for lipophilicity as the arithmetic mean of the values predicted by the five proposed methods [20–25]. The aqueous solubility, one of the important parameter which influence absorption was predicted by the method suggested by Ali et al. [26] Multiple linear regression is used to predict the skin permeability, Kp, which is linearly correlated with molecular size and lipophilicity. The higher the logKp value, it is more skin permeable [27]. Bioavailability of a drug indicate its “drug-likeness”, that means qualitative chance of a molecule to become an oral drug. PAINS alert gives the number of substructural features which can help to identify promiscuous compounds in many biochemical high throughput screens [28].
Molecular docking
The docking simulations were carried out by AutoDock Vina. After selecting the receptor and ligand, we assign the grid size for binding site. Here we select the grid around the centre (10, 0, 0) and of size 50 × 50 × 50Å for all the ligands. Then we added hydrogens, ignore waters and chains of non-standard residues for the receptor 6YB7 and merge charges, remove lone pairs and non-polar hydrogens for all of the ligands. Then we assigned the number of binding modes as 6, exhaustiveness of search as 8, with maximum energy difference of 3 kcal/mol. Then we analyzed the docking score using ViewDock window. The more negative value with RMSD value of zero is taken as the binding affinity value, which is used for ranking the ligands. After post-dock minimization, the docked ligand and the zone containing atoms and bonds within 2.5–5.0 Å was selected to analyze the active site. Then, we analyzed H-bonded interactions between these residues and the ligand, and create their distances for further interpretation.
Results and discussion
Machine learning for IC50 prediction
In order to predict the IC50 of the phytochemicals from Aerva Lenata plant, we tried building regression models based on machine learning algorithms. Using Python’s sci-kit learn package we built models using Linear Regression, Support Vector Regression, Lasso Regression, KernelRidge, Random Forest Regression and ElasticNet. To validate the model, the dataset was split into training set and testing set in a ratio of 3:1. This was repeated for multiple times after shuffling the dataset before each splitting. Based on this model building and evaluation were performed. An average value of the evaluative measures were considered. The models were constructed using the training set of 193 molecules, which are currently in the clinical trials for COVID19. In order to evaluate the models we used molecules from the data set. The performance of the models built were evaluated on the basis of R-squared value and mean square error value. The R-squared value and mean squared error value of various models are given in Table 1. From the table it is visible that the accuracy of prediction is achieved using random forest regression. The model exhibited a R-squared value closer to 1, which means the data is well fitted to the regression line. The error rate for training and testing data is satisfactory for random forest method, which means the model works well for unseen data too.
Table 1.
Performance of various machine learning models
| Sl. No | Model | R squared Value | Mean squared Error |
|---|---|---|---|
| 1 | Linear Regression | − 0.0005 | 0.0020 |
| 2 | Support Vector Regression | − 1.1671 | 0.0044 |
| 3 | Lasso Regression | − 0.0030 | 0.0020 |
| 4 | Kernel Ridge | 0.1045 | 0.0018 |
| 5 | Random Forest Regression | 0.9516 | 0.0001 |
| 6 | Elastic Net | 0.0001 | 0.0044 |
From the 133 features obtained after the initial feature selection, the number of features to be used for model construction was determined using the SelectKBest method in the Scikit-learn package available in Python. The method selects the k features with the highest scores for building the model. Figure 1 shows the relationship between the number of features and R-squared value. It was observed that the model showed exceptional performance when constructed using 84 features.
Fig. 1.

R-square vs Number of Features
The plot in Fig. 2 for the actual IC50 vs the predicted IC50 demonstrates, that the model predict the IC50 value of the molecules in a satisfactory way, which assures that the random forest model can be used further for predicting the unknown IC50s of the phytochemicals of Aerva Lanata plant.
Fig. 2.

Predicted IC50 vs Actual IC50
The random forest model constructed was used to predict the IC50 of 29 phytochemicals present in the Aerva Lanata plant. The docking score and ADMET properties of the molecules were calculated in order to cross validate the predictions. The relation between docking score and ic50 value were analyzed and found that for most of the molecules, lower the IC50 values, the docking score was negative, which indicates that they have better druggability. The relationship between D-score and IC50 values are shown in the Fig. 3.
Fig. 3.
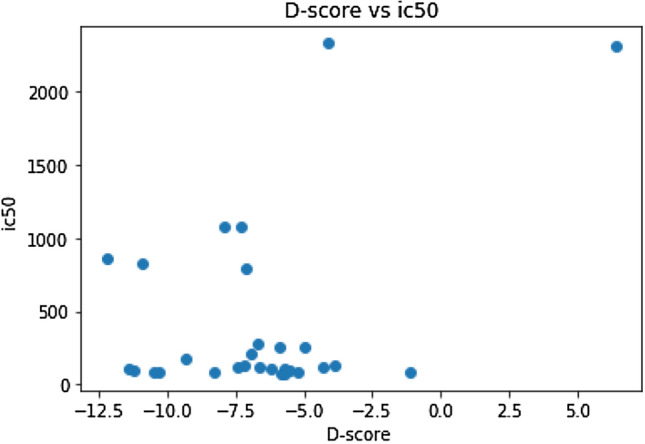
Docking Score vs IC50 for phytochemicals from Aerva lanata
Molecular docking
We have selected 30 compounds (A1-A30) already reported from Aerva lanata and the reference compound hydroxy chloroquine (HCQ) for the present study (Table 2) and compare their activities based on the binding affinity of these ligands with the SARS-CoV-2 main protease, 6YB7. From the ViewDock window analysis, it is very clear that, except methylgrevillate, A13 (−5.4 kcal/mol), hentriacontane, A20 (−4.6 kcal/mol) and syringic acid, A22 (−4.8 kcal/mol), all the other twenty seven ligands show better inhibitory values compared to the refence compound HCQ (−5.5 kcal/mol). The top most compounds are Apigenin-7-O-β-glucoside, A10 (−8.0 kcal/mol), narcissin, A21 (–8.0 kcal/mol) and ervoside A26 (−8.2 kcal/mol). Among these, narcissin, ie, isorhamnetin-3-O-rutinoside is present in many other plants and a patented compound (US6555523) to treat infections [29]. While the compound ervoside with highest docking score is not reported from other sources and its activities are not exploited till now. The compounds ervolanine, A1 (−6.3 kcal/mol), methylervine, A2 (−6.3 kcal/mol), ervoside, A26 (−8.2 kcal/mol) and methylaervine, A28 (−6.3 kcal/mol) are reported mainly from Aerva lanata and exhibiting good binding affinities with SARS-CoV-2 main protease. Among these, A2 and A28 are patented compounds (US2011059977) for the treatment of mycobacteria-linked pathologies [30]. Hence it is very clear that the plant as a whole or by part is beneficial in developing drug candidates for COVID-19.
Table 2.
The ligands, their docking score and interacting residues
| Entry | Molecule | Score | Interacting residues |
|---|---|---|---|
| HCQ | Hydroxychloroquine | − 5.5 | Arg131, Thr199, Tyr237, Leu286, Leu287, Glu288, Asp289 |
| A1 | Ervolanine | − 6.3 | Gln110, Ile200, Val202, Glu240, Ile249, Pro293 |
| A2 | Methylervine | − 6.3 | Pro132, Ile200, Glu240, Thr292, Pro293 |
| A3 | Isorhamnetin | − 6.9 | Arg131, Lys137, Asp197, Tyr237, Leu286, Leu287 |
| A4 | Quercetin | − 7.3 | Tyr25, His41, Phe140, Ser144, His163 |
| A5 | Kaempferol | − 6.8 | Arg131, Lys137, Tyr237, Leu286, Leu287 |
| A6 | Amyrin | − 6.9 | Gln110, Asn151, Asp153, Ser158, Ile249, Phe294, Arg298 |
| A7 | Botulin | − 6.6 | Ile106, Gln107, Gln110, Val202, His246, Ile249, Thr292, Pro293, Phe294 |
| A8 | Campesterol | − 6.3 | Val104, Ile106, Gln107, Gln110, Asn151, Asp153, Ser158, Phe294, Arg298 |
| A9 | 5-hydroxy-3’,4’,6,7-tetramethoxyflavone | − 6.1 | Lys137, Thr199, Tyr239, Leu286, Leu287 |
| A10 | Apigenin-7-O-β-glucoside | − 8.0 | Arg4, Lys5, Arg131, Lys137, Trp207, Leu282, Ser284, Glu288, Phe291 |
| A11 | Chrysin | − 6.7 | Ile200, His246, Ile249, Tyr292, Pro293 |
| A12 | Ecdysone | − 6.7 | Lys55, Gln127, Arg131, Lys137, Thr199, Leu286, Asp289 |
| A13 | Methyl grevillate | − 5.4 | Gln197, Ile200, Val202, Glu240, Thr292, Pro293, Phe294 |
| A14 | Lupeol | − 6.7 | Ile106, Gln107, Gln110, Thr292 |
| A15 | Lupeol acetate | − 6.4 | Val104, Ile106, Gln107, Gln110, Asn151, Ile249, Phe294 |
| A16 | β-sitosteryl acetate | − 5.6 | Val104, Ile106, Gln107, Gln110, Asn151, Asp153, Ser158, Arg298 |
| A17 | Aervitrin | − 7.2 | Ag4, Lys5, Arg131, Lys137, Trp207, Ser284, Glu288, Asp289, Glu290 |
| A18 | β-sitosterol | − 7.0 | Thr199, Tyr239, Leu272, Leu286, Leu287, Glu288, Asp289, Glu290 |
| A19 | Daucosterol | − 6.2 | Val104, Ile106, Gln107, Gln110, Asn151, Ser158, Ile249, Phe294, Arg298 |
| A20 | Hentriacontane | − 4.6 | Ile106, Gln107, Gln110, Asn151, Val202, His246, Ile249, Thr292, Pro293, Phe294, Arg298 |
| A21 | Narcissin | − 8.0 | Lys5, Arg131, Asp197, Asn238, Leu272, Leu286, Glu288, Glu290 |
| A22 | Syringic acid | − 4.8 | Leu220, Asn221, Arg222, Ile259, Asp263, Ser267 |
| A23 | Feruloyltyramine | − 6.3 | Pro108, Pro132, Ile200, Asn203, Glu240, His246, Thr292, Phe294 |
| A24 | Vanilic acid | − 4.7 | Phe3, Arg4, Lys5, Trp207, Glu288 |
| A25 | Ervine | − 6.3 | Asn203, Ile208, Glu240, His246 |
| A26 | Ervoside | − 8.2 | Lys137, Asp197, Thr199, Tyr237, Met276, Leu286, Leu287, Asp289 |
| A27 | Aervine | − 6.3 | Tyr237, Tyr239, Met276, Leu286, Leu287 |
| A28 | Methylaervine | − 6.3 | Pro132, Ile200, Glu240, His246, Thr292, Pro293 |
| A29 | Canthine | − 6.3 | Thr199, Tyr237, Leu272, Met276, Leu286, Leu287 |
| A30 | Aervoside | − 6.4 | Lys5, Ala7, Val125, Gln127 |
The binding site analysis shows that, Leu286 to Arg298 are the major interacting residues found in almost all cases. In the case of topmost ligand, ervoside, the active site includes the amino acid residues Lys137, Asp197, Thr199, Tyr237, Met276, Leu286, Leu287 and Asp289 with two hydrogen bonds (Fig. 4). The H- bonds are formed by oxygen from Asp289, -OH from glucose moiety (2.449 Å), -NH from Lys137 and another -OH from the same glucose unit (3.314 Å), which firmly bind the ligand inside the active site. In the case of narcissin, A21, there are more H-bonds between the ligand moieties and the amino acid residues, which are formed from -OH from glucose moiety with Asp 197(1.623 Å), Glu288(1.541 Å), Lys5(2.082 Å), Glu290 (1.698 Å)and Asn 238(2.490 Å), which stabilizes the ligand inside the active site comprises of Lys5, Arg131, Asp197, Asn238, Leu272, Leu286, Glu288 and Glu290 (Fig. 5).
Fig. 4.

Interaction diagram for ervoside, A28
Fig. 5.

Interaction diagram for narcissin, A21
ADME/T prediction
The pharmacokinetic parameters, ADME/T (absorption, distribution, metabolism, excretion and toxicity) values predicted by SwissADME online tool (Table 3) shows that all the ligands are good drugs with minimum violation from Lipinski’s rule of five (Ro5). All the selected compounds have molecular weight within the range 168.15–624.54 and TPSA (topological polar surface area) is less than 140 Å2 except for A10 and A21. Number of PAINS [31] alert is one only for A5. Octanol/water partition coefficient, LogP is ranging from −0.80 to 11.90 while aqueous solubility, LogS varies between −2.23 and −16.58. Skin permeability, LogKp is negative for all cases and the Abbott bioavailability score is 0.55 or 0.56 in all cases except in A21. Hydrogen bond donors and acceptors (HBD and HBA) varies from 0 to 9 and 0 to 16 respectively. Especially in the case of top most ligands, there are zero violations from Ro5. These values are indicative of the effectiveness of these molecules in developing novel drugs for COVID-19.
Table 3.
The ADME/T properties of the ligands, predicted by SwissADME
| Entry | MW | HBA | HBD | TPSA | LogP | LogS | LogKp | PAINS | BA |
|---|---|---|---|---|---|---|---|---|---|
| HCQ | 335.87 | 3 | 2 | 48.3 | 3.37 | − 4.28 | − 5.81 | 0 | 0.55 |
| A1 | 270.28 | 4 | 2 | 75.2 | 2.16 | − 3.29 | − 6.47 | 0 | 0.56 |
| A2 | 250.25 | 3 | 0 | 43.6 | 2.39 | − 2.95 | − 6.13 | 0 | 0.55 |
| A3 | 316.26 | 7 | 4 | 120.3 | 1.65 | − 4.02 | − 6.90 | 0 | 0.55 |
| A4 | 302.24 | 7 | 5 | 131.3 | 1.23 | − 3.91 | − 7.05 | 1 | 0.55 |
| A5 | 286.24 | 6 | 4 | 111.1 | 1.58 | − 3.86 | − 6.70 | 0 | 0.55 |
| A6 | 426.72 | 1 | 1 | 20.2 | 7.18 | − 9.47 | − 2.41 | 0 | 0.55 |
| A7 | 442.72 | 2 | 2 | 40.4 | 6.39 | − 8.99 | − 3.12 | 0 | 0.55 |
| A8 | 400.68 | 1 | 1 | 20.2 | 6.90 | − 9.11 | − 2.50 | 0 | 0.55 |
| A9 | 358.34 | 7 | 1 | 87.3 | 2.83 | − 4.99 | − 6.02 | 0 | 0.55 |
| A10 | 432.38 | 10 | 6 | 170.0 | 0.55 | − 5.00 | − 7.65 | 0 | 0.55 |
| A11 | 254.24 | 4 | 2 | 70.6 | 2.55 | − 4.69 | − 5.35 | 0 | 0.55 |
| A12 | 464.63 | 6 | 5 | 118.2 | 2.57 | − 3.79 | − 7.93 | 0 | 0.55 |
| A13 | 194.18 | 4 | 2 | 66.7 | 1.31 | − 2.50 | − 6.43 | 0 | 0.55 |
| A14 | 426.72 | 1 | 1 | 20.2 | 7.31 | − 10.22 | − 1.90 | 0 | 0.55 |
| A15 | 468.75 | 2 | 0 | 26.3 | 7.67 | − 10.95 | − 1.74 | 0 | 0.55 |
| A16 | 456.74 | 2 | 0 | 26.3 | 7.63 | − 10.39 | − 2.05 | 0 | 0.55 |
| A17 | 456.74 | 2 | 0 | 26.3 | 7.63 | − 10.39 | − 2.05 | 0 | 0.55 |
| A18 | 414.71 | 1 | 1 | 20.2 | 7.19 | − 9.67 | − 2.20 | 0 | 0.55 |
| A19 | 576.85 | 6 | 4 | 99.3 | 5.51 | − 9.67 | − 4.32 | 0 | 0.55 |
| A20 | 436.84 | 0 | 0 | 0.0 | 11.90 | − 16.58 | 2.69 | 0 | 0.55 |
| A21 | 624.54 | 16 | 9 | 258.4 | − 0.80 | − 4.97 | − 10.12 | 0 | 0.17 |
| A22 | 198.17 | 5 | 2 | 75.9 | 1.02 | − 2.23 | − 6.77 | 0 | 0.56 |
| A23 | 313.35 | 4 | 3 | 78.7 | 2.39 | − 3.38 | − 6.72 | 0 | 0.55 |
| A24 | 168.15 | 4 | 2 | 66.7 | 1.08 | − 2.44 | − 6.31 | 0 | 0.56 |
| A25 | 236.23 | 3 | 1 | 54.6 | 1.99 | − 2.85 | − 6.27 | 0 | 0.55 |
| A26 | 398.37 | 8 | 4 | 133.7 | 0.35 | − 2.63 | − 8.55 | 0 | 0.55 |
| A27 | 236.23 | 3 | 1 | 54.6 | 1.99 | − 2.85 | − 6.27 | 0 | 0.55 |
| A28 | 250.25 | 3 | 0 | 43.6 | 2.39 | − 2.95 | − 6.13 | 0 | 0.55 |
| A29 | 206.24 | 1 | 0 | 17.8 | 2.62 | − 2.80 | − 5.59 | 0 | 0.55 |
| A30 | 220.23 | 2 | 0 | 34.3 | 2.39 | − 2.78 | − 5.93 | 0 | 0.55 |
*Score-AutoDock Vina docking score(kcal/mol), MW-molecular weight(g/mol), HBA-hydrogen bond acceptor, HBD-hydrogen bond donor, TPSA-topological polar surface area(Å2), LogP-lipophilicity, octanol/water partition coefficient, LogS-water solubility (Ali), LogKp-skin permeation (cm/s), PAINS-PAN assay interference structures, BA-Abbott bioavailabilty score
As a representative, drug-likeness of ervoside, A26, based on six physicochemical descriptors—lipophilicity, size, polarity, solubility, flexibility and saturation are represented by Bioavailability Radar (Fig. 6). The physicochemical range on each axis is represented as a pink area, in which the radar plot of the molecule has to fall entirely to be considered drug-like. The coloured zone is suitable physicochemical space for oral availability with values, LIPO (Lipophilicity): −0.7 < XLOGP3 < + 5.0, SIZE: 150 g/mol < MV < 500 g/mol, POLAR (Polarity): 20 Å2 < TPSA < 130Å2, INSOLU (Insolubility): 0 < LogS(ESOL) < 6, INSATU (Insaturation): 0.25 < Fraction Csp3 < 1, FLEX (Flexibility): 0 < Number of rotatable bonds < 9. The BOILED-Egg model, based on the parameters WLogP and TPSA, for A26, an effective interpretation of passive human gastrointestinal absorption (HIA) and blood–brain barrier (BBB) permeation is represented in Fig. 6. This consists, primarily an intuitive way of predicting the two key ADME parameters – HIA and BBB. As shown in the figure, the egg-shaped classification plot includes the yolk and the white which represents the physicochemical space for highly probable BBB permeation and HIA absorption respectively. The grey region stands for molecules with low absorption and limited brain penetration. In addition, the red colored point in the case of all drugs predict that all of them are non-substrates of P-gp (PGP −). That means these drugs are non-inhibitors of cytochrome P450 (CYP), which is essential for the metabolism of many medications.
Fig. 6.

Bioavailability Radar and BOILED-Egg for ervoside, A26
Conclusions
Aerva lanata is well known for its anti-microbial activity and to the best of our knowledge, this is the first report regarding the comparative study of phytochemicals from Aerva lanata and hydroxychloroquine in effective treatment of COVID-19, a pandemic condition reported recently worldwide. Many of the alkaloids and flavonoids from this herb computed to be highly effective towards SARS-CoV-2. Molecules such as narcissin, methylervine, methylaervine etc. are patented medicines for various other diseases. Moreover, we found that some of the phytochemicals such as ervoside, which is reported to be present only from Aerva lanata can be developed as a potential drug in treating SARS-CoV-2 infections. Probably these compounds could be attractive for the management of COVID-19, but clinical trials must be performed in order to validate this observation. The study is expected to pave the way to eradicate this pandemic condition in its initial stage in near future.
Acknowledgements
The authors thanks Planning board, Govt. of Kerala for financial assistance for this project.
Funding
The author thanks Govt. of Kerala for financial assistance for this project.
Declaration
Conflict of interest
There are no conflicts of interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
D. R. Sherin, Email: sherindr84@gmail.com
T. K. Manojkumar, Email: manojtk@iiitmk.ac.in
References
- 1.World Health Organization, Coronavirus Disease 2019 (COVID-19) Dashboard ; https://covid19.who.int/ [PubMed]
- 2.Chen J. Pathogenicity and transmissibility of 2019-nCoV—a quick overview and comparison with other emerging viruses. Microbes Infect. 2020;22:69–71. doi: 10.1016/j.micinf.2020.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275:33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 4.Treatments for COVID-19, What helps, what doesn't, and what's in the pipeline,
- 5.Harvard Health Publishing, Harvard medical school, https://www.health.harvard.edu/diseases-and-conditions/treatments-for-covid-19
- 6.Fabio, S. T.; Julie G.: Vincent, J. L. Hydroxychloroquine in the management of critically ill patients with COVID-19: the need for an evidence base. The Lancet Respiratory Medicine.10.1016/S2213-2600(20)30172-7 [DOI] [PMC free article] [PubMed]
- 7.Medicinal plants used for snake treatment. ToxicologyCentre.com. Archived from the original on 2013–12–13. Retrieved 2013–12–10.
- 8.Tewari D, Mocan A, Parvanov ED, Sah AN, Nabavi SM, Huminiecki L, Ma ZF, Lee YY, Horbańczuk JO, Atanasov AG. Ethnopharmacological approaches for therapy of jaundice: part I. Front Pharmacol. 2017;8:518. doi: 10.3389/fphar.2017.00518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manoj G, Anil P, Nagori BP, Sasmal D. Aerva lanata: A review on phytochemistry and pharmacological aspects. Pharmacogn Rev. 2011;5:195–198. doi: 10.4103/0973-7847.91120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Macalino SJ, Gosu V, Hong S, Choi S. Role of computer-aided drug design in modern drug discovery. Arch Pharm Res. 2015;389:1686–1701. doi: 10.1007/s12272-015-0640-5. [DOI] [PubMed] [Google Scholar]
- 11.Fournet, A. R. Francois, M. Lagoutte, D. Poupon, E. Soriano, F. Use of canthin-6-one and its analogs in the treatment of mycobacteria-linked pathologies, US2011059977.
- 12.Owen, C. D. Lukacik, P. Strain-Damerell, C. M. Douangamath, A. Powell, A. J. Fearon, D. Brandao-Neto, J. Crawshaw, A. D. Aragao, D. Williams, M. Flaig, R. Hall, D. R. McAuley, K. E. Mazzorana, M. Stuart, D. I. von Delft, F. Walsh, M. A. https://www.rcsb.org/structure/6YB7(To be published), 10.2210/pdb6YB7/pdb
- 13.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eric F, Pettersen TD, Goddard C, Huang C, Huang C, Thomas EF. UCSF chimera–a visualization system for exploratory research and analysis. J Comp Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 15.https://pubchem.ncbi.nlm.nih.gov/
- 16.Antoine D, Olivier M, Vincent Z. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Antoine D, Olivier M, Vincent Z. iLOGP: a simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J Chem Inf Model. 2014;54:3284–3301. doi: 10.1021/ci500467k. [DOI] [PubMed] [Google Scholar]
- 18.Antoine D, Vincent Z. A BOILED-Egg to predict gastrointestinal absorption and brain penetration of small molecules. Chem Med Chem. 2016;11:1117–1121. doi: 10.1002/cmdc.201600182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ritchie TJ, Ertl P, Lewis R. The graphical representation of ADME-related molecule properties for medicinal chemists. Drug Discov Today. 2011;16:65–72. doi: 10.1016/j.drudis.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 20.Lovering F, Bikker J, Humblet C. Escape from flatland: increasing saturation as an approach to improving clinical success. J Med Chem. 2009;52:6752–6756. doi: 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- 21.http://silicos-it.be.s3-website-eu-west-1.amazonaws.com/software/filter-it/1.0.2/filter-it.html.
- 22.Daina A, Michielin O, Zoete V. iLOGP: a simple, robust, and efficient description of n-Octanol/water partition coefficient for drug design using the GB/SA approach. J Chem Inf Model. 2014;54:3284–3301. doi: 10.1021/ci500467k. [DOI] [PubMed] [Google Scholar]
- 23.Cheng T, Zhao Y, Li X, Lin F, Xu Y, Zhang X, Li Y, Wang R, Lai L. Computation of octanol−water partition coefficients by guiding an additive model with knowledge. J Chem Inf Model. 2007;47:2140–2148. doi: 10.1021/ci700257y. [DOI] [PubMed] [Google Scholar]
- 24.Wildman SA, Crippen GM. Prediction of physicochemical parameters by atomic contributions. J Chem Inf Model. 1999;39:868–873. [Google Scholar]
- 25.Moriguchi I, Shuichi H, Liu Q, Nakagome I, Matsushita Y. Simple method of calculating octanol/water partition coefficient. Chem Pharm Bull. 1992;40:127–130. doi: 10.1248/cpb.40.127. [DOI] [Google Scholar]
- 26.Moriguchi I, Shuichi H, Nakagome I, Hirano H. Comparison of reliability of log P values for drugs calculated by several methods. Chem Pharm Bull. 1994;42:976–978. doi: 10.1248/cpb.42.976. [DOI] [Google Scholar]
- 27.Ali J, Camilleri P, Brown MB, Hutt AJ, Kirton SB. Revisiting the general solubility equation: in silico prediction of aqueous solubility incorporating the effect of topographical polar surface area. J Chem Inf Model. 2012;52:420–428. doi: 10.1021/ci200387c. [DOI] [PubMed] [Google Scholar]
- 28.Potts RO, Guy RH. Predicting skin permeability. Pharm Res. 1992;9:663–669. doi: 10.1023/A:1015810312465. [DOI] [PubMed] [Google Scholar]
- 29.Xuan YM, Hong XZ, Mihaly M, Meng C. Molecular Docking: A powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des. 2011;7:146–157. doi: 10.2174/157340911795677602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Use of cirsiliol and derivatives to treat infections, US 6555523.
- 31.Jonathan BB, Georgina AH. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]