

Abstract
The RAG1 and RAG2 proteins initiate the process of V(D)J recombination and therefore play an essential role in adaptive immunity. While null mutations in the RAG genes cause severe combined immune deficiency with lack of T and B cells (T−B− SCID) and susceptibility to life-threatening, early-onset infections, studies in humans and mice have demonstrated that hypomorphic RAG mutations are associated with defects of central and peripheral tolerance resulting in immune dysregulation. In this review, we provide an overview of the extended spectrum of RAG deficiencies and their associated clinical and immunological phenotypes in humans. We discuss recent advances in the mechanisms that control RAG expression and function, the effects of perturbed RAG activity on lymphoid development and immune homeostasis, and propose novel approaches to correct this group of disorders.
Keywords: Genotype-phenotype correlation, Immune tolerance, RAG, VDJ recombination
Introduction
Adaptive immune responses require expression of receptors capable of specific antigen recognition. In jawed vertebrates, this function is accomplished through V(D)J recombination, in which the joining of variable (V), diversity (D), and joining (J) coding elements of the immunoglobulin and TCR genes, results in the generation of a diversified repertoire of antigen-specific TCRs and BCRs. The RAG1 and RAG2 proteins initiate the V(D)J recombination process [1–3]. Two molecules of RAG1 and two molecules of RAG2 form a heterotetramer that binds to recombination signal sequences (RSSs) flanking the V, D, and J genes and introduces double-strand breaks in the DNA, which are subsequentially repaired by the nonhomologous end-joining DNA repair pathway by joining the gene segments to form coding joints and the cleaved RSSs and intervening sequences to form signal joints [3]. RAG1 and RAG2 have distinct roles. In particular, RAG1 contains the RNaseH fold catalytic domain and regions that make direct contact with the RSS, and is responsible for the enzymatic activity of the RAG complex [4, 5], while RAG2 promotes DNA binding by scanning the genome for epigenetic signatures characterized by histone 3 trimethylated at lysine 4 (H3K4me3) and facilitates cleavage functions of RAG1 [6]. The lack of either RAG1 or RAG2 protein causes a very similar phenotype with complete lack of mature T and B cells resulting in a nonleaky severe combined immunodeficient (T−B−SCID) phenotype, both in mice and humans [7–9]. However, the phenotype associated with RAG mutations is much broader, with hypomorphic variants allowing various degrees of T- and B-cell development. Furthermore, while susceptibility to severe infections is the main manifestation of SCID, immune dysregulation is a prominent feature in patients with hypomorphic RAG variants [10]. Altogether, these data demonstrate that RAG1 and RAG2 are essential not only to control T- and B-cell development, but that their function is also required to maintain immune tolerance.
Role of RAG proteins in V(D)J recombination: Recent developments
RAG1 and RAG2 expression is tightly controlled in a cell type and developmental stage-specific manner [11]. There are two waves of RAG gene expression in developing T and B lymphocytes [12]. In mice, the first wave of Rag expression in developing thymocytes occurs at the CD4−CD8− double-negative (DN) stage to catalyze Tcrg, Tcrb, and Tcrd gene rearrangement. Thymocytes that successfully rearrange Tcrg and Tcrd to become γδ T cells will then permanently shutdown Rag expression, while cells that successfully rearrange Tcrb transiently downregulate Rag expression and differentiate along the αβ T-cell pathway. The second wave of Rag expression occurs at the CD4+CD8+ double-positive (DP) stage, and promotes rearrangements at the Tcra locus. If an αβ TCR heterodimer is successfully expressed, and DP thymocytes receives sufficient TCR signaling allowing positive selection, Rag gene expression is permanently downregulated [13–15]. Single-cell RNA sequencing studies in human thymic samples have confirmed that RAG gene expression is restricted to DN cells and DP cells; moreover, they have shown that RAG expression starts increasing from the late proliferative phase of each cell stage and peaks during quiescence, demonstrating that cell proliferation and V(D)J recombination need to be uncoupled [16].
Similarly to thymocytes, Rag gene expression in mice occurs at two distinct stages also during B-cell development, with expression in pro-B cells supporting Igh rearrangement and expression in pre-B cells supporting Igk and Igl rearrangement [17].
The mechanisms controlling Rag gene expression have been the object of extensive investigation. The Rag1 and Rag2 genes are juxtaposed on chromosome 11p12 and 2p in humans and mice, respectively, are separated by only approximately 8 kb and their promoters are distant approximately 25 kb. Rag1 and Rag2 are convergently transcribed, and for both the entire protein is encoded by a single exon. Transcriptional regulation of the Rag genes involves cis-regulatory elements that differ between B and T cells [11]; some of these elements have been recently identified in a mouse atlas of open chromatin regions [18]. In mice, Rag expression at DN stage of T-cell development is induced by sequences within 10 kb upstream of Rag2 [19], while in DP thymocytes, when Rag expression reaches the highest levels, it is controlled by a distal antisilencer element (ASE), placed 73 kb upstream of Rag2 [20], which acts as an enhancer through direct interaction with the Rag1 and Rag2 promoters [21]. In B cells, Rag promoters are controlled by proximal, distal, and Erag enhancers upstream of Rag2 [22, 23]. Various transcription factors are also involved in Rag gene expression. In particular, by interacting with the Erag enhancer, Foxo1 serves as a positive regulator [24], while Gfi1b, Ebf1, and c-Myb act as negative regulators [25–28]. Other transcription factors binding directly to the Rag2 promoter include PAX5, MYB, SP1, LEF1, NF-Y, C/EBP GATA3, and NFATc1 [29], whereas NF-Y and NFAT bind directly to the Rag1 promoter [30, 31]. Furthermore, GATA3 and E2A are critical regulators of the ASE, while Runx1 and E2A regulate the Rag1 promoter [32]. The hierarchical assembly of a transcriptionally active chromatin hub containing the ASE and Rag promoters, with Rag2 recruitment and expression secondary to the assembly of a functional ASE-Rag1 framework, has been demonstrated in DP thymocytes [32]. Additionally, it has been shown that downregulation of Rag expression in DP thymocytes depends on IKAROS and occurs with disassembly of the chromatin hub in the Rag locus [32]. Negative regulation of RAG1 expression by IKAROS has been confirmed also in human cells [33].
The broad phenotype of RAG deficiency and genotype–phenotype correlation in humans
Severe combined immune deficiency
RAG deficiency may manifest with a broad range of phenotypes that reflect the degree of adaptive immunity compromise which in turn depends, at least in part, on the residual recombination activity of the mutant RAG protein(s). Null mutations that abrogate recombination activity are responsible for SCID, with lack or severe reduction of T and B cells and agammaglobulinemia; however, development of NK cells is not affected. SCID patients are prone to life-threatening and opportunistic infections since early in life [34, 35]. Immunization with live agents may lead to severe infection and should be strictly avoided [36]. SCID is fatal within the first few years of life unless immune reconstitution is achieved with hematopoietic stem cell transplantation (HSCT) [37, 38]. As compared to patients with typical SCID, those with atypical (or “leaky”) forms of SCID have partially preserved T-cell count and/or function, although markedly lower than age-matched referenced values [39]. In these patients, CMV infection is often associated with expansion of γδ T cells and autoimmune cytopenias [40, 41].
Omenn syndrome
Omenn syndrome (OS) represents another severe presentation of RAG deficiency [34, 35]. It is characterized by prominent immune dysregulation, with erythroderma, lymphadenopathy, hepatosplenomegaly, eosinophilia, and elevated IgE (but otherwise low IgG, IgA, and IgM). T-lymphocytic infiltrates are present in target organs, contributing to tissue damage. Similar features may also be seen in patients with SCID and with maternal T-cell engraftment [42]. However, the T cells of OS patients are autologous and oligoclonal [43]. Both atypical SCID and OS are sustained by hypomorphic RAG variants with minimal levels of recombination activity [44–46] permitting a low number of successful recombination events. Frameshift mutations in the N-terminus of RAG1 are a common cause of atypical SCID and OS [47]. However, it should be noted that these variants with very low recombination activity may also cause typical SCID [48], in particular, when the stochastic process of V(D)J recombination does not yield productive rearrangements. Both OS and atypical SCID are fatal conditions, unless treated with HSCT. Availability of universal newborn screening for SCID has permitted to define that in the United States RAG deficiency (including both RAG1 and RAG2 defects) accounted for 11% of all cases of SCID and as many as 41.2% of cases of atypical SCID and OS observed in the period 2010-2018 [49].
Combined and common variable immunodeficiencies
The extended phenotype associated with hypomorphic RAG mutations includes also combined immune deficiency (CID), which is often associated with granulomas and/or autoimmunity (CID-G/AI) [34, 50–52]. At variance with SCID and OS, CID may have a delayed clinical onset, and may allow survival into adulthood, reflecting milder impairment of adaptive immunity [34]. In particular, T- and B-cell counts as well as immunoglobulin serum levels are often partially preserved [34], consistent with significantly higher levels of recombination activity supported by the RAG mutations, as compared to SCID and OS [34, 45, 46]. In some patients, the disease may manifest as CD4 lymphopenia [53]. Progressive development of hypogammaglobulinemia associated with normal or even increased B-cell count but reduced proportion of switched memory B cells has been also reported [54, 55]. These forms of late-onset CID may mimic common variable immunodeficiency (CVID) [55, 56]. However, at variance with CVID, there is a consistent numerical defect of naïve CD4+ T cells. As in patients with atypical forms of SCID, viral infections, in particular due to CMV or EBV may act as modifiers of the immunological phenotype, inducing lymphoproliferation. Clonotypic T-cell expansions in patients with hypomorphic RAG mutations may also reflect immune tolerance breakdown, with self-reactive T cells infiltrating target organs. Whatever the mechanism, this chronic lymphoproliferation may occasionally progress to clonal disease [57, 58].
Treatment-refractory autoimmune cytopenias are a common manifestation of RAG-dependent CID [51]. Vasculitis and organ-specific autoimmunity have been also frequently reported [34, 51, 59]. Noncaseating granulomas involving the skin or any other organ often develop starting in late childhood or young adulthood. Presence of the rubella virus vaccine (RVV) strain has been documented in several RAG-deficient patients with CID [60]. Immunohistochemical analysis of the granulomatous lesions has revealed that the RV is harbored by M2 macrophages [61]. While immunization with live viral vaccines is contraindicated in patients with immunodeficiency, patients with RAG-dependent CID may not be diagnosed early enough to avoid exposure to the RVV.
Although there are significant immunological differences among SCID, OS, and CID, all conditions are characterized by a reduced proportion of naïve T cells as a result of impaired V(D)J recombination [34]. Furthermore, high-throughput sequencing of the TCR repertoire reveals restrictions and clonotypic expansions [43], and molecular signatures of self-reactivity are often present in the TCR CDR3 [62]. Finally, a unique feature of V(D)J recombination at the TCRA locus is that it happens in waves, with initial rearrangements involving the most downstream TRAV and most upstream TRAJ genes. If such rearrangements are nonproductive, thymocytes proceed with further rearrangements that ultimately involve the most upstream TRAV and most downstream TRAJ genes. In patients with hypomorphic RAG mutations, there is reduced frequency of rearrangements involving these distal TCRA elements, including TRAV1-2, which is expressed as invariant TCR-α chain by mucosa-associated invariant T cells. The low number of mucosa-associated invariant T cell detected in patients with hypomorphic RAG mutations further contributes to the immune dysregulation of this condition [63].
Animal models of RAG deficiency
Genetically engineered mouse models have been used to define the effects of individual gene disruption observed in patients with RAG deficiencies. Because of the inability of developing lymphocytes to initiate V(D)J recombination, Rag1 and Rag2 knockout mice show a block in B- and T-cell development at B220+CD43+ pro-B-cell stage (Fig. 1) and DN stage (Fig. 2), respectively, and complete absence of mature T and B cells [7, 8]. To confirm that this phenotype is not influenced by homologous recombination with introduction of a Neo cassette in embryonic stem cells, Rag1−/− and Rag2−/− pigs and Rag2−/− mice have been generated by TALEN and CRISPR/Cas9 gene editing, respectively [64, 65], confirming that RAG deficiency does not impact viability and development, but causes loss of mature T and B cells with preserved generation of NK cells.
Figure 1.
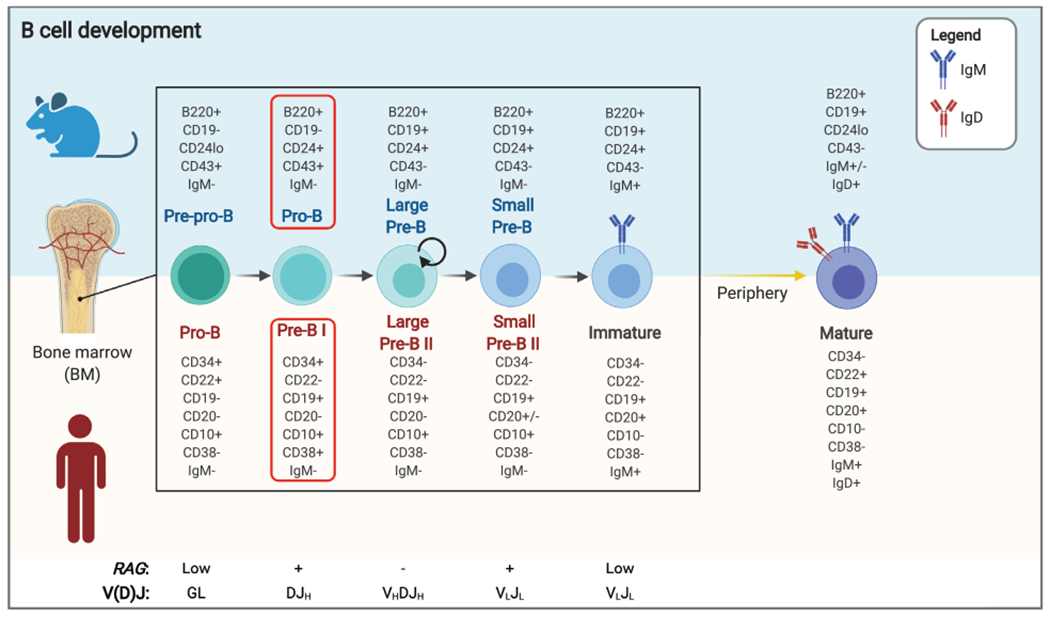
B-cell development block in RAG-deficient patients and mice. Schematic representation of B-cell development in mice and humans, with the list of surface markers that can be used to identify various stages of maturation. Stages at which RAG genes are expressed, and the concurrent nature of VDJ recombination products detected at each stage, are indicated at the bottom. The red boxes indicate the stage (pro-B and pre-B I) at which development is blocked in RAG-deficient mice and patients. Proliferating cells are indicated by a circular arrow. GL, germline.
Figure 2.
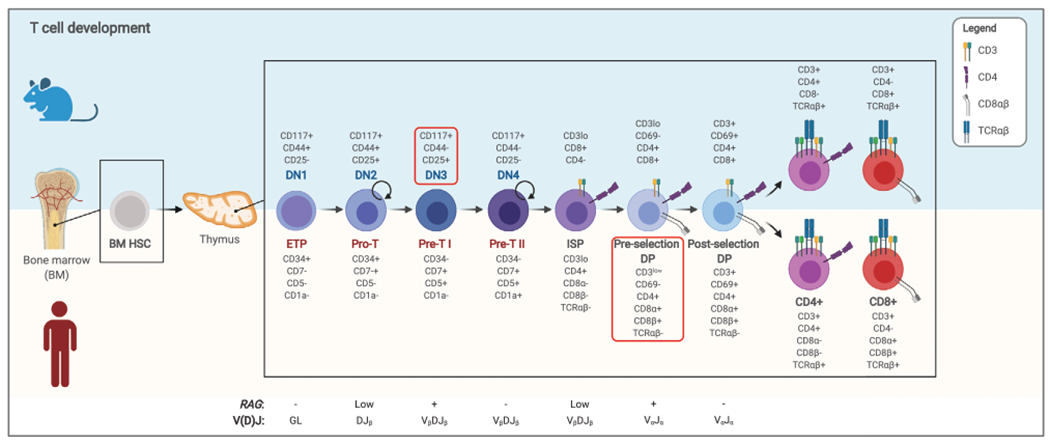
T-cell differentiation blocks in RAG-deficient patients and mice. Representation of T-cell development in mice and humans, with the list of surface markers that can be used to identify the different stages of maturation. Stages at which RAG genes are expressed, and the concurrent nature of VDJ recombination products detected at each stage, are indicated at the bottom. Proliferating cells are indicated by a circular arrow. The red boxes indicate the stage at which development is blocked in RAG deficiency. Of note, use of artificial thymic organoids (ATOs) allowed to establish that T-cell development in RAG-mutated patients occurs at the preselection double-positive (DP) stage, unlike what was found in mice, where the block is present at the double-negative 3 (DN3) stage. DN, double negative; DP, double positive; ETP, early thymic progenitor; GL, germline; HSC, hematopoietic stem cell.
Murine models for Omenn syndrome
The identification of patients with “leaky” forms of disease associated with hypomorphic RAG mutations has prompted the development of knockin mouse models, with the aim of better defining the pathophysiology of these conditions. Two mouse models of OS due to Rag mutations were reported at the same time. The first model, homozygous for a hypomorphic Rag1 variant (R972Q), was the result of a spontaneous mutation and was identified because of the presence of a high proportion of memory CD8+ T cells in the absence of any infection, and for this reason called the memory mutant (MM) mouse [66]. The R972Q Rag1 mutant protein was shown to have decreased recombination activity (about 15% of WT protein). Consistently, MM mice manifested a partial block in T- and B-cell development, reduced numbers of mature T and B cells and abnormal architecture of lymphoid tissues. Additionally, MM mice showed skin redness, hepatosplenomegaly, eosinophilia, oligoclonal T cells, and elevated serum IgE level. Their peripheral T cells showed an activated/memory phenotype and increased production of Th2 cytokines. Because of these features, the MM mouse was proposed to represent a model of OS. However, when another mouse model carrying the R972Q Rag1 variant was generated by CRISPR/Cas gene editing [67], it became clear that only a proportion of these mice have the typical changes of OS. Additionally, RaglR972Q/R972Q mice have detectable B cells, produce IgM and IgG and a broad range of autoantibodies. In this regard, the R972Q Rag1 mutant mice are more a model of CID than of OS. The second mouse model of OS recapitulates more faithfully the patients’ phenotype, and was generated by introducing the R229Q Rag2 mutation previously found in patients with OS [68]. Rag2R229Q/R229Q mice present oligoclonal T cells, absence of circulating B cells and eosinophilia. In addition, they show infiltration of T cells and eosinophils in gut and skin, causing diarrhea, alopecia, and erythroderma in a significant proportion of the animals. These cell infiltrates contrast with the severe lymphopenia of thymus, spleen, and lymph nodes. In the thymus, the cortex/medulla demarcation is lost, and the medullary compartment is severely reduced, with lack of autoimmune regulator (Aire) expression and low number of Treg and NKT cells [68–70]. A detailed analysis of the B-cell compartment revealed that, despite a severe B-cell developmental block, the Rag2R229Q/R229Q mice present a normal or even enlarged compartment of immunoglobulin-secreting cells (ISC), sustained by elevated levels of T cell-derived homeostatic and effector cytokines [71]. Defects of receptor editing and elevated levels of BAFF were found to contribute to the B-cell tolerance breakdown and the development of B cells producing high affinity autoantibodies against target organs, highlighting a previously unidentified role for B cells in the pathogenesis of immune dysregulation of OS [71]. Similar to OS patients, Rag2R229Q/R229Q mice have prominent signs of gut inflammation, with CD4+ Th1/Th17 cells infiltrates in the lamina propria [72]. T cells are directly responsible for these inflammatory changes, since their adoptive transfer in immunodeficient hosts is sufficient to cause colitis, and conversely depletion of CD4+ cells ameliorates bowel inflammation. In these mice, the lack of B cells and consequent reduced amounts of fecal IgA and IgM results in mucosal abnormalities, with enhanced absorption of microbial products and altered composition of the microbiota, which in turn sustain the Th1/Th17 response [72]. Accordingly, use of broad-spectrum antibiotics reduced T-cell infiltrates and improved gut inflammation. Very recently, the study of Rag2R229Q/R229Q mice has provided support for an interplay between gut and skin in promoting cutaneous inflammation, a hallmark of OS [73]. In particular, a high proportion of peripheral T cells from Rag2R229Q/R229Q mice express skin-homing receptors, and the skin of these mice is infiltrated by Th1 cells. Similar abnormalities have been also detected in OS patients. Disruption of the epidermal barrier in Rag2R229Q/R229Q mice is associated with increased microbial challenge. Finally, induction of more severe colitis precipitates enhanced skin infiltration with Th1 cells [73].
Murine models for atypical SCID and combined immune deficiency
Another knockin mouse model was generated to investigate the functional consequences of a Rag1 point mutation (S723C) that does not affect proficiency of DNA double-strand break formation but rather causes defects in postcleavage complex formation and end-joining in vitro [74]. This mutant manifested impaired T- and B-cell development, reminiscent of atypical SCID in humans. Analysis of thymic architecture and composition revealed severe reduction in size and cellularity affecting in particular medullary thymic epithelial cells, markedly reduced expression of Aire and several tissue restricted antigens (TRAs) and reduced number of Treg cells [75]. In the BM, there was a severe but incomplete block in development at the pro-B-cell stage, with virtually absent immature and mature recirculating B cells. In the periphery, profound B-cell lymphopenia was contrasted by residual levels of immunoglobulins and accumulation of ISCs [76]. While responses to T-independent and T-dependent antigens and production of high affinity antibodies were severely impaired in these mice, high amounts of low-affinity self-reactive antibodies were detected. These signs of autoimmunity were associated with defects of receptor editing in the BM and increased serum BAFF levels. Along with significant lymphocytic infiltrates in peripheral tissues, these data support the notion that hypomorphic Rag mutations are often associated with immune dysregulation, as a result of defects of central and peripheral T- and B-cell tolerance. In addition, Rag1-S723C mice presented aberrant DNA double-strand breaks within rearranging loci that, on a p53 mutant background, resulted in predisposition to development of thymic lymphomas [74].
More recently, CRISPR/Cas gene editing has been used to introduce in mice hypomorphic mutations in the C-terminus domain (CTD) of RAG1 (F971L, R972Q, R972W) corresponding to those (F974L, R975Q, R975W) identified in patients with variable phenotypic severity, ranging from atypical SCID to CID-G/AI [67]. All three mouse models showed an incomplete block of T- and B-cell development. Thymic defects were more pronounced in R972W mice, with a marked reduction in size and cellularity and a nearly complete block at the CD25+CD44− DN3 stage. By contrast, a significant proportion of DP and of CD4+ and CD8+ single-positive (SP) cells were detected in R972Q and F971L mice. In the BM, B-cell development was partially blocked at the pro-B and pre-B stages, with increased leakiness in particular in the R972Q mouse [67]. Reduced number of T and B cells were present in the periphery. In particular, T cells mainly expressed an activated/memory phenotype, although a significant fraction of naive cells was present in both F971L and R972Q mice. Partial preservation of immunoglobulin levels and response in particular to T-independent antigen were documented in R972Q and F971L mice, while profound hypogammaglobulinemia but increased serum IgE were present in R972W mice [67]. Broad-spectrum autoantibodies were documented, in particular in the R972Q mouse model, similarly to patients with hypomorphic mutations in the RAG1 CTD domain [52]. However, anticytokine antibodies often found in patients were not observed in these mice. Finally, high-throughput sequencing revealed marked skewing of Ighv and Trbv gene usage in early lymphoid progenitors, with an increased frequency of productive rearrangements. Along with increased apoptosis of B-cell progenitors, these data suggest preferential survival of lymphoid progenitors that successfully rearrange TCR and immunoglobulin genes on the first allele. More recent analysis of the Trb repertoire in splenic T cells isolated from R972W and F971L mutant mice has revealed an enrichment in hydrophobic aminoacid doublets at the apex of the CDR3 in the TCR-β chain [62], a biomarker of self-reactivity [77], similarly to what found in patients with hypomorphic RAG mutations [43, 62].
Impact of RAG gene defects on lymphoid development and immune tolerance: Recent developments
B- and T-cell development in patients with RAG deficiencies
The rarity of the condition, difficulty in obtaining BM and thymus samples, and lack of reliable techniques to model human T-cell development in vitro have hampered the fine characterization of T- and B-cell developmental defects in patients with RAG deficiencies.
Noordzij et al. performed flow cytometric analysis of BM samples obtained from 5 RAG-deficient SCID pediatric patients and demonstrated a block at pro-B and pre-BI cell stage [78] (Fig. 1). However, the relative frequencies of pro-B and pre-BI cells varied, even for patients carrying the same mutation. These results were confirmed by Cassani et al. [71], who analyzed four patients with OS and reported that on average 92% of the B cells in the BM of these patients were represented by pro-B and pre-BI cells. Importantly, some ISCs escape developmental blocks as indicated by ELISpot analysis showing the presence of cells producing either IgM or IgG/A in patients with OS [71]. Ijspert et al. analyzed the BM of 22 patients with similar truncating mutations in the N-terminus of RAG1 [48]. In spite of the variable phenotype (SCID, OS, CID), these patients manifested a similar early block in B-cell development (Fig. 1), with a limited number of pre-BII cells being present only in patients with CID and in one of the OS patients. These data indicate that the nature and functional activity of the mutant alleles are not the only determinants of RAG deficiency phenotypic heterogeneity.
The study of early human T-cell development in patients with RAG deficiency has proven to be even more challenging, because of the very limited access to thymic samples from patients. Taking advantage of the development of methods allowing to model human T-cell development from induced pluripotent stem cells (iPSCs) [79], Braueret al. studied T-cell differentiation of human iPSCs generated from three RAG1-mutated patients with different clinical and immunologic phenotypes (SCID and OS) [80]. In all cases, T-cell development progressed up to immature CD4+CD8− (immature single positive) and DP stages, but only transiently, as these cells were quickly lost over time, due to the accumulation of double-strand breaks and decreased cell survival [80]. The analysis of sorted ISP and DP cells revealed an overall restriction of TCR rearrangements, preferential usage of certain V and J genes, and skewing of CDR3 length. Some of these features had been previously noted in peripheral T cells from patients with OS [81]. Similar results were obtained by Themeli et al. who studied T-cell differentiation of iPSCs generated from a patient carrying a RAG2 null mutation, with only few cells reaching the DP stage [82]. In this study, the impaired T-cell differentiation was accompanied by an increase in CD7−CD56+CD33+ NK-like cells.
More recently, a very efficient protocol for in vitro T-cell differentiation of CD34+ cells in a 3D artificial thymic organoid (ATO) system was devised by Seet et al. [83]. Using this technique, we analyzed the T-cell differentiation of BM or mobilized peripheral blood CD34+ cells from six patients with RAG mutations, manifesting with a wide range of phenotypes [84]. Surprisingly, we observed generation of DP cells in all cases, including samples isolated from T−B−SCID patients, in contrast with what predicted by Rag knockout mouse models in which T-cell development is blocked at DN stage [7, 8] (Fig. 2). However, we noticed that severity of the disease inversely correlated with the persistence of DP cells in the ATOs, with more severe presentations associated with loss of these cells in culture over time [84]. Additionally, in vitro production of CD3−CD56+ NK cells was markedly increased in patients with severe RAG mutations, suggesting a more pronounced unbalance versus NK cell lineage development in T−B−SCID patients [84].
In parallel to our study, Bifsha et al. published a report proposing an alternative approach to study in vitro T-cell differentiation [85]. In this method, CD34+ cells isolated from limited amount of peripheral blood are cocultured in a 3D system with OP9-DLL4 cells used in earlier methods to support T-cell differentiation [86]. In their report, they compared the T-cell differentiation potential of CD34+ cells from a patient with a null RAG2 mutation (the same used in [84]) and an OS patient carrying a hypomorphic RAG1 mutation, and observed a different ability of the cells from these two patients to reach the DP stage, with the patient carrying the null mutation showing a more severe defect. Overall, these data indicate that RAG function is not strictly required to reach the DP stage of T-cell differentiation in humans; however, in the absence of normal RAG, survival of these cells is not sustained, presumably due also to illegitimate rearrangements.
RAG mutations impact establishment of central tolerance in humans
Besides directly impacting on T-cell development, RAG mutations have important consequences also on the composition and maturation of TECs and on the establishment of central tolerance. Cavadini et al. were the first ones to demonstrate lack of AIRE and TRA expression in the thymi of two patients with OS [87]. These results have been subsequently confirmed in other patients with hypomorphic RAG mutations including CID-G/AI [88, 89]. Abnormalities of TEC development and maturation and impairment of AIRE expression in the thymus of patients with RAG deficiency arise from defects of cross-talk between developing T cells and stromal cells. For patients with hypomorphic mutations permitting residual development of T cells, defective AIRE expression in the thymus translates into escape of self-reactive T cells from negative selection and impaired generation of Treg cells, thereby favoring autoimmune manifestations. Similar to patients with Autoimmune Polyendocrinopathy-Enteropathy-Candidiasis-Ectodermal Dystrophy (APECED), a condition due to germline AIRE mutations, patients with hypomorphic RAG variants often produce autoantibodies targeting IFN-α, IFN-ω, IL-17, and IL-22, and some lung antigens such as KCNRG and BPIFB1 [52, 89]. In patients with RAG defects, the occurrence of neutralizing antitype I IFN antibodies is associated with a high risk of severe varicella infection [52, 55], whereas anti-KCNRG and anti-BPIFB1 antibodies may induce or aggravate development of chronic lung disease [90].
Treatment of RAG deficiency: Current status and perspectives
Allogeneic HSCT represents the only curative option currently available for RAG deficiency. For patients with SCID or OS, optimal results (>80% 10-year survival) are achieved with HSCT from HLA-matched related donors; transplantation from haploidentical donors is associated with a very high rate of graft rejection (up to 75%) if no conditioning regimen is used [37]. Furthermore, HSCT from donors other than HLA-matched siblings is often associated with poorer T- and B-cell reconstitution especially when no conditioning or nonmyeloablative regimens are used. These results may reflect competition between mutant and donor-derived cells which in humans may extend up to DP cells in the thymus and pre-B cells in the BM. On the other hand, it is well established that use of myeloablative regimens for HSCT carries the risk of increased toxicity and reduced survival. An alternative strategy would be to use nongenotoxic agents to deplete the endogenous HSCs. A clinical trial using humanized anti-CD117 monoclonal antibody is currently underway, and early data show safety and efficacy [91]. Alternatively, immunotoxins depleting CD45+ cells would have the advantage of purging also lymphoid progenitor and mature cells, which can mediate immune dysregulation especially in patients with CID-G/AI. In this condition, HSCT with conventional conditioning regimens has led to suboptimal results, with 38% of the patients being deceased at a median age of 8.4 years [51]. Preclinical data using anti-CD45-Saporin in mice with null or hypomorphic Rag1 mutations indicate that this agent can efficiently deplete the stem cell and lymphoid compartment, and promote robust and durable engraftment of donor-derived cells [92].
Gene therapy represents an alternative approach to correct RAG deficiency. Initial attempts with retroviral vectors have revealed the risk of lymphoma [93]. Depending on the nature of the promoter used, variable efficiency of T- and B-cell reconstitution has been achieved in Rag1−/− mice reconstituted with lentiviral vectors [94]. Partial immune reconstitution has been associated with development of lymphocytic infiltrates in target organs and autoantibodies [95]. More favorable results have been obtained in Rag2−/− mice [96]; however, when a similar approach was attempted in Rag2R229Q/R229Q mice, the T- and B-cell count of treated animals remained lower than normal [97].
Gene editing represents an alternative strategy, with the advantage of preserving endogenous regulation of gene expression. The size of the RAG1 cDNA may represent a potential limitation, because of difficulties in packaging the entire donor template in adenoassociated viral vectors; however, because of the smaller length of RAG2 cDNA, RAG2 deficiency is an attractive target for gene editing, and proof of concept has been provided in patient-derived iPSCs, with restoration of in vitro T-cell development and generation of a diverse TCR repertoire in gene edited cells [82, 98].
Conclusions
In recent years, important advances have been made in the characterization of the molecular mechanisms that control RAG1 and RAG2 gene expression. With the progressive implementation of whole genome sequencing, it is likely that improved knowledge of the molecular mechanisms controlling RAG expression may help interpret the significance of genetic variants affecting regulatory regions of the RAG genes in patients with defects of lymphoid development. Definition of the crystal structure of the RAG1/RAG2 complex may help predict the functional consequences of naturally occurring RAG variants [4, 5], which can be confirmed using in vitro recombination assays [45, 46]. However, observations in patients have shown that genotype–phenotype correlation in RAG deficiency is not absolute [48], possibly reflecting both the semistochastic nature of V(D)J recombination and the impact of epigenetic factors. An intriguing hypothesis that has yet to be tested is that because of the heterotetrameric nature of the RAG complex, double heterozygosity for hypomorphic variants in the RAG genes may limit the number of fully functional RAG complexes and interfere with normal T- and B-cell development.
Novel cellular and animal models, and more powerful methods to study human T-cell development have offered important insights into the broad immunological and clinical phenotype associated with RAG mutations (Fig. 3). In particular, studies in patients and mice have shown that hypomorphic RAG mutations that allow for subnormal levels of T- and B-cell development have important consequences on lymphostromal cross-talk in the thymus and on mechanisms of T-cell tolerance [75, 87], as well as on the efficiency of receptor editing in the BM and on survival of self-reactive peripheral B cells [71, 76]. Altogether, these abnormalities explain the high frequency of autoimmune manifestations observed in humans and mice with hypomorphic RAG mutations [51, 52, 71]. Use of novel in vitro methods to investigate human T-cell development has revealed important differences between humans and mice with RAG mutations [84], emphasizing the importance of performing such studies directly on human tissues and/or cells whenever possible. Finally, there is hope that novel therapeutic approaches based on use of nongenotoxic conditioning regimens and gene editing (Fig. 3) may improve outcome for patients with RAG deficiency including those with severe immune dysregulation. If successful, these strategies may inform similar developments also for patients with other forms of inborn errors of immunity.
Figure 3.
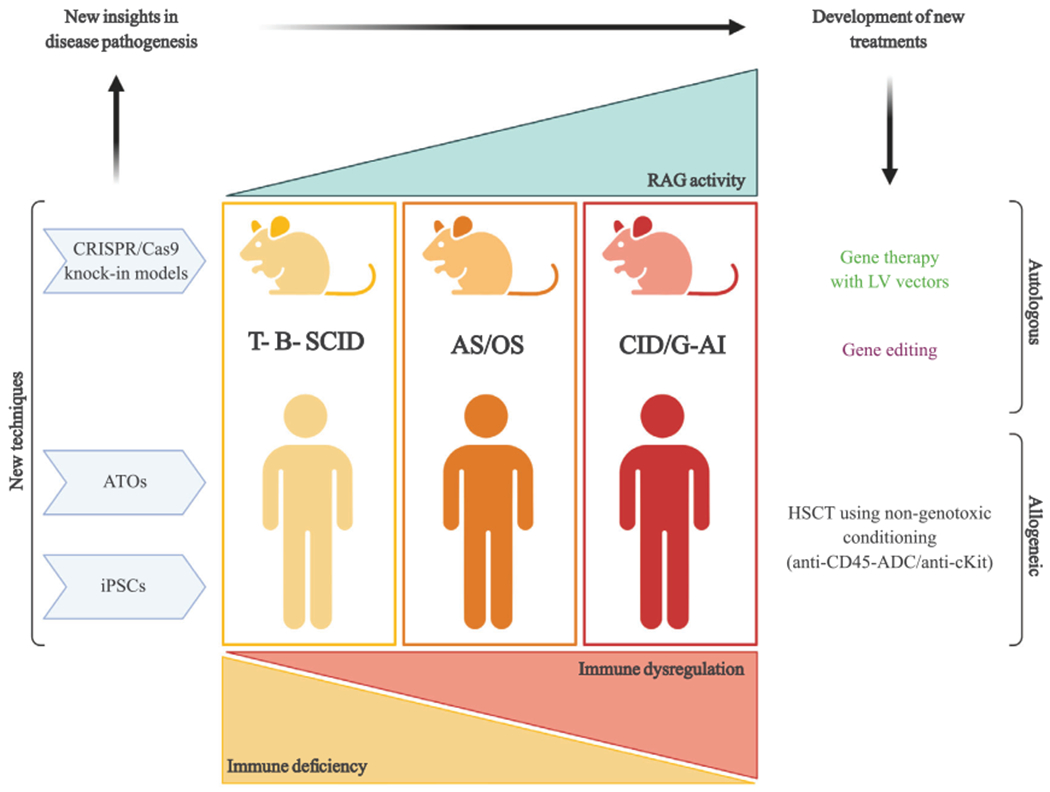
Novel investigational techniques offer new insights into pathophysiology of RAG deficiency and open novel therapeutic perspectives. Depicted here are some of the new experimental techniques that have provided novel insights into RAG deficiencies. In particular, CRISPR/Cas9 technique has been used to generate novel knockin mouse models carrying RAG mutations found in patients, while artificial thymic organoids (ATOs) and induced pluripotent stem cells (iPSCs) have allowed in vitro modeling of faulty T-cell development using cells isolated from RAG-deficient patients. These novel tools have also made possible the development and testing of novel prospective therapeutic approaches such as lentiviral vector-mediated gene therapy, gene editing, and use of nongenotoxic conditioning regimens for hematopoietic stem cell transplantation (HSCT).
Acknowledgements:
This work was supported by The Division of Intramural Research, National Institute of Allergy and Infectious Diseases (Grant ZIA AI001222-04 to LDN) and by The Bench to Bedside grant “RAG deficiency: From pathophysiology to precise gene correction” (to LDN).
Abbreviations:
- AIRE
autoimmune regulator
- ASE
antisilencer element
- ATO
artificial thymic organoid
- CID
combined immune deficiency
- CID-G/AI
combined immune deficiency with granulomas and/or autoimmunity
- CTD
C-terminus domain
- CVID
common variable immune deficiency
- DN
double negative
- DP
double positive
- HSCT
hematopoietic stem cell transplantation
- iPSC
induced pluripotent stem cells
- ISC
immunoglobulin-secreting cell
- ISP
immature single-positive
- MM
memory mutant
- OS
Omenn syndrome
- RAG
Recombinase Activating Gene
- RSS
recombination signal sequence
- RVV
rubella virus vaccine
- SCID
severe combined immune deficiency
- TRAs
tissue restricted antigens
Footnotes
Conflict of interest: The authors declare no commercial or financial conflict of interest.
References
- 1.Schatz DG, Oettinger MA and Baltimore D, The V(D)J recombination activating gene, RAG-1. Cell 1989, 59: 1035–1048. [DOI] [PubMed] [Google Scholar]
- 2.Oettinger MA, Schatz DG, Gorka C and Baltimore D, RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science 1990, 248: 1517–1523. [DOI] [PubMed] [Google Scholar]
- 3.Notarangelo LD, Kim MS, Walter JE and Lee YN, Human RAG mutations: biochemistry and clinical implications. Nat Rev Immunol 2016, 16: 234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ru H, Chambers MG, Fu TM, Tong AB, Liao M andWu H, Molecular mechanism of V(D)J recombination from synaptic RAG1-RAG2 complex structures. Cell 2015, 163: 1138–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim MS, Chuenchor W, Chen X, Cui Y, Zhang X, Zhou ZH, Gellert M et al. , Cracking the DNA code for V(D)J recombination. Mol Cell 2018, 70: 358–370 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodgers W, Byrum JN, Simpson DA, Hoolehan W and Rodgers KK, RAG2 localization and dynamics in the pre-B cell nucleus. PloS one 2019, 14: e0216137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S and Papaioannou VE, RAG-1-deficient mice have no mature B and T lymphocytes. Cell 1992, 68: 869–877. [DOI] [PubMed] [Google Scholar]
- 8.Shinkai Y, Rathbun G, Lam KP, Oltz EM, Stewart V, Mendelsohn M, Charron J et al. , RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell 1992, 68: 855–867. [DOI] [PubMed] [Google Scholar]
- 9.Schwarz K, Gauss GH, Ludwig L, Pannicke U, Li Z, Lindner D, Friedrich W et al. , RAG mutations in human B cell-negative SCID. Science 1996, 274: 97–99. [DOI] [PubMed] [Google Scholar]
- 10.Villa A and Notarangelo LD, RAG gene defects at the verge of immunodeficiency and immune dysregulation. Immunol Rev 2019, 287: 73–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuo TC and Schlissel MS, Mechanisms controlling expression of the RAG locus during lymphocyte development. Curr Opin Immunol 2009. 21: 173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson A, Held W and MacDonald HR, Two waves of recombinase gene expression in developing thymocytes.J Exp Med 1994, 179: 1355–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turka LA, Schatz DG, Oettinger MA, Chun JJ, Gorka C, Lee K, McCormack WT et al. , Thymocyte expression of RAG-1 and RAG-2: termination by T cell receptor cross-linking. Science 1991, 253: 778–781. [DOI] [PubMed] [Google Scholar]
- 14.Borgulya P, Kishi H, Uematsu Y and von Boehmer H, Exclusion and inclusion of alpha and beta T cell receptor alleles. Cell 1992, 69: 529–537. [DOI] [PubMed] [Google Scholar]
- 15.Takahama Y and Singer A, Post-transcriptional regulation of early T cell development by T cell receptor signals. Science 1992, 258: 1456–1462. [DOI] [PubMed] [Google Scholar]
- 16.Park JE, Botting RA, Dominguez Conde C, Popescu DM, Lavaert M, Kunz DJ, Goh I et al. , A cell atlas of human thymic development defines T cell repertoire formation. Science 2020, 367: eaay3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grawunder U, Leu TM, Schatz DG, Werner A, Rolink AG, Melchers F, Winkler TH, Down-regulation of RAG1 and RAG2 gene expression in preB cells after functional immunoglobulin heavy chain rearrangement. Immunity 1995, 3: 601–608. [DOI] [PubMed] [Google Scholar]
- 18.Yoshida H, Lareau CA, Ramirez RN, Rose SA, Maier B, Wroblewska A, Desland F et al. , The cis-regulatory atlas of the mouse immune system. Cell 2019, 176: 897–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu W, Misulovin Z, Suh H, Hardy RR, Jankovic M, Yannoutsos N, Nussenzweig MC, Coordinate regulation of RAG1 and RAG2 by cell type-specific DNA elements 5’ of RAG2. Science 1999, 285: 1080–1084. [DOI] [PubMed] [Google Scholar]
- 20.Yannoutsos N, Barreto V, Misulovin Z, Gazumyan A, Yu W, Rajewsky N, Peixoto BR et al. , A cis element in the recombination activating gene locus regulates gene expression by counteracting a distant silencer. Nat Immunol 2004, 5: 443–450. [DOI] [PubMed] [Google Scholar]
- 21.Hao B, Naik AK, Watanabe A, Tanaka H, Chen L, Richards HW, Kondo M et al. , An anti-silencer- and SATB1-dependent chromatin hub regulates Rag1 and Rag2 gene expression during thymocyte development. J Exp Med 2015, 212: 809–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monroe RJ, Chen F, Ferrini R, Davidson L and Alt FW, RAG2 is regulated differentially in B and T cells by elements 5’ of the promoter. Proc Natl Acad Sci USA 1999, 96: 12713–12718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsu LY, Lauring J, Liang HE, Greenbaum S, Cado D, Zhuang Y and Schlissel MS, A conserved transcriptional enhancer regulates RAG gene expression in developing B cells. Immunity 2003, 19: 105–117. [DOI] [PubMed] [Google Scholar]
- 24.Amin RH and Schlissel MS, Foxo1 directly regulates the transcription of recombination-activating genes during B cell development. Nat Immunol 2008, 9: 613–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schulz D, Vassen L, Chow KT, McWhirter SM, Amin RH, Moroy T and Schlissel MS, Gfi1b negatively regulates Rag expression directly and via the repression of FoxO1. J Exp Med 2012, 209: 187–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Timblin GA and Schlissel MS, Ebf1 and c-Myb repress rag transcription downstream of Stat5 during early B cell development. J Immunol 2013, 191: 4676–4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee BS, Lee BK, Iyer VR, Sleckman BP, Shaffer AL 3rd, Ippolito GC, Tucker HO et al. , Corrected and republished from: BCL11A is a critical component of a transcriptional network that activates RAG expression and V(D)J recombination. Mol Cell Biol 2018, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Timblin GA, Xie L, Tjian R and Schlissel MS, Dual mechanism of Rag gene repression by c-Myb during pre-B cell proliferation. Mol Cell Biol 2017, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schlissel MS, Regulating antigen-receptor gene assembly. Nat Rev Immunol 2003, 3: 890–899. [DOI] [PubMed] [Google Scholar]
- 30.Brown ST, Miranda GA, Galic Z, Hartman IZ, Lyon CJ and Aguilera RJ, Regulation of the RAG-1 promoter by the NF-Y transcription factor. J Immunol 1997, 158: 5071–5074. [PubMed] [Google Scholar]
- 31.Patra AK, Drewes T, Engelmann S, Chuvpilo S, Kishi H, Hunig T, Serfling E et al. , PKB rescues calcineurin/NFAT-induced arrest of Rag expression and pre-T cell differentiation. J Immunol 2006, 177: 4567–4576. [DOI] [PubMed] [Google Scholar]
- 32.Naik AK, Byrd AT, Lucander ACK and Krangel MS, Hierarchical assembly and disassembly of a transcriptionally active RAG locus in CD4(+)CD8(+) thymocytes. J Exp Med 2019, 216: 231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han Q, Ma J, Gu Y, Song H, Kapadia M, Kawasawa YI, Dovat S et al. , RAG1 high expression associated with IKZF1 dysfunction in adult B-cell acute lymphoblastic leukemia. J Cancer 2019, 10: 3842–3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Delmonte OM, Schuetz C and Notarangelo LD, RAG deficiency: two genes, many diseases. J Clin Immunol 2018, 38: 646–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gennery A, Recent advances in understanding RAG deficiencies. F1000Res 2019, 8: F1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Greenberg-Kushnir N, Lee YN, Simon AJ, Lev A, Marcus N, Abuzaitoun O, Somech R et al. , A large cohort of RAG1/2-deficient SCID patients-clinical, immunological, and prognostic analysis. J Clin Immunol 2020, 40: 211–222. [DOI] [PubMed] [Google Scholar]
- 37.Schuetz C, Neven B, Dvorak CC, Leroy S, Ege MJ, Pannicke U, Schwarz K et al. , SCID patients with ARTEMIS vs RAG deficiencies following HCT: increased risk of late toxicity in ARTEMIS-deficient SCID. Blood 2014, 123: 281–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haddad EL BR, Griffith LM, Buckley RH, Parrott RE and Prockop SE, SCID genotype and 6-month post-transplant CD4 count predict survival and immune recovery: a PIDTC Retrospective Study. Blood 2018, 132: 1737–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shearer WT, Dunn E, Notarangelo LD, Dvorak CC, Puck JM, Logan BR, Griffith LM et al. , Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn syndrome: the Primary Immune Deficiency Treatment Consortium experience. J Allergy Clin Immunol 2014, 133: 1092–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ehl S, Schwarz K, Enders A, Duffner U, Pannicke U, Kuhr J, Mascart F et al. , A variant of SCID with specific immune responses and predominance of gamma delta T cells. J Clin Invest 2005, 115: 3140–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Villartay JP, Lim A, Al-Mousa H, Dupont S, Dechanet-Merville J, Coumau-Gatbois E, Gougeon ML et al. , Le Deist F: a novel immunodeficiency associated with hypomorphic RAG1 mutations and CMV infection. J Clin Invest 2005, 115: 3291–3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muller SM, Ege M, Pottharst A, Schulz AS, Schwarz K and Friedrich W, Transplacentally acquired maternal T lymphocytes in severe combined immunodeficiency: a study of 121 patients. Blood 2001, 98: 1847–1851. [DOI] [PubMed] [Google Scholar]
- 43.Rowe JH, Stadinski BD, Henderson LA, de Bruin LO, Delmonte O, Lee YN, de la Morena MT et al. , Abnormalities of T-cell receptor repertoire in CD4(+) regulatory and conventional T cells in patients with RAG mutations: implications for autoimmunity. J Allergy Clin Immunol 2017, 140: 1739–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Villa A, Santagata S, Bozzi F, Giliani S, Frattini A, Imberti L, Gatta LB et al. , Partial V(D)J recombination activity leads to Omenn syndrome. Cell 1998, 93: 885–896. [DOI] [PubMed] [Google Scholar]
- 45.Lee YN, Frugoni F, Dobbs K, Walter JE, Giliani S, Gennery AR, Al-Herz W et al. , A systematic analysis of recombination activity and genotype-phenotype correlation in human recombination-activating gene 1 deficiency. J Allergy Clin Immunol 2014, 133: 1099–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tirosh I, Yamazaki Y, Frugoni F, Ververs FA, Allenspach EJ, Zhang Y, Burns S et al. , Recombination activity of human recombination activating gene 2 (RAG2) mutations and correlation with clinical phenotype. J Allergy Clin Immunol 2019, 143: 726–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sharapova SO, Skomska-Pawliszak M, Rodina YA, Wolska-Kusnierz B, Dabrowska-Leonik N, Mikoluc B, Pashchenko OE et al. , The clinical and genetic spectrum of 82 patients with RAG deficiency including a c.256_257delAA founder variant in slavic countries. Front Immunol 2020, 11: 900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.IJspeert H, Driessen GJ, Moorhouse MJ, Hartwig NG, Wolska-Kusnierz B, Kalwak K, Pituch-Noworolska A et al. , Similar recombination-activating gene (RAG) mutations result in similar immunobiological effects but in different clinical phenotypes. J Allergy Clin Immunol 2014, 133: 1124–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dvorak CC, Haddad E, Buckley RH, Cowan MJ, Logan B, Griffith LM, Kohn DB et al. , The genetic landscape of severe combined immunodeficiency in the United States and Canada in the current era (2010–2018). J Allergy Clin Immunol 2019, 143: 405–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schuetz C, Huck K, Gudowius S, Megahed M, Feyen O, Hubner B, Schneider DT et al. , An immunodeficiency disease with RAG mutations and granulomas. N Engl J Med 2008, 358: 2030–2038. [DOI] [PubMed] [Google Scholar]
- 51.Farmer JR, Foldvari Z, Ujhazi B, De Ravin SS, Chen K, Bleesing JJH, Schuetz C et al. , Outcomes and treatment strategies for autoimmunity and hyperinflammation in patients with RAG deficiency. J Allerg Clin Immunol Pract 2019, 7: 1970–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walter JE, Rosen LB, Csomos K, Rosenberg JM, Mathew D, Keszei M, Ujhazi B et al. , Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J Clin Invest 2016, 126: 4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kuijpers TW, Ijspeert H, van Leeuwen EM, Jansen MH, Hazenberg MD, Weijer KC, van Lier RA et al. , Idiopathic CD4+ T lymphopenia without autoimmunity or granulomatous disease in the slipstream of RAG mutations. Blood 2011, 117: 5892–5896. [DOI] [PubMed] [Google Scholar]
- 54.Dorna MB, Barbosa PFA, Rangel-Santos A, Csomos K, Ujhazi B, Dasso JF, Thwaites D et al. , Combined immunodeficiency with late-onset progressive hypogammaglobulinemia and normal B cell count in a patient with RAG2 deficiency. Front Pediatr 2019, 7: 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buchbinder D, Baker R, Lee YN, Ravell J, Zhang Y, McElwee J, Nugent D et al. , Identification of patients with RAG mutations previously diagnosed with common variable immunodeficiency disorders. J Clin Immunol 2015, 35: 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abolhassani H, Wang N, Aghamohammadi A, Rezaei N, Lee YN, Frugoni F, Notarangelo LD et al. , A hypomorphic recombination-activating gene 1 (RAG1) mutation resulting in a phenotype resembling common variable immunodeficiency. J Allergy Clin Immunol 2014, 134: 1375–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schuetz C, Niehues T, Friedrich W and Schwarz K, Autoimmunity, autoinflammation and lymphoma in combined immunodeficiency (CID). Autoimmun Rev 2010, 9: 477–482. [DOI] [PubMed] [Google Scholar]
- 58.Avitan-Hersh E, Stepensky P, Zaidman I, Nevet MJ, Hanna S and Bergman R, Primary cutaneous clonal CD8+ T-cell lymphoproliferative disorder associated with immunodeficiency due to RAG1 mutation. Am J Dermatopathol 2020, 42: e11–e5. [DOI] [PubMed] [Google Scholar]
- 59.Geier CB, Farmer JR, Foldvari Z, Ujhazi B, Steininger J, Sleasman JW, Parikh S et al. , Vasculitis as a major morbidity factor in patients with partial RAG deficiency. Front Immunol 2020, 11: 574738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Perelygina L, Icenogle J and Sullivan KE, Rubella virus-associated chronic inflammation in primary immunodeficiency diseases. Curr Opin Allergy Clin Immunol 2020, 20: 574–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Perelygina L, Plotkin S, Russo P, Hautala T, Bonilla F, Ochs HD, Joshi A et al. , Rubella persistence in epidermal keratinocytes and granuloma M2 macrophages in patients with primary immunodeficiencies. J Allergy Clin Immunol 2016, 138: 1436–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Daley SR, Koay HF, Dobbs K, Bosticardo M, Wirasinha RC, Pala F, Castagnoli R et al. , Cysteine and hydrophobic residues in CDR3 serve as distinct T-cell self-reactivity indices. J Allergy Clin Immunol 2019, 144: 333–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Berland A, Rosain J, Kaltenbach S, Allain V, Mahlaoui N, Melki I, Fievet A et al. , PROMIDISalpha: a T-cell receptor alpha signature associated with immunodeficiencies caused by V(D)J recombination defects. J Allergy Clin Immunol 2019; 143: 325–334. [DOI] [PubMed] [Google Scholar]
- 64.Huang J, Guo X, Fan N, Song J, Zhao B, Ouyang Z, Liu Z et al. , RAG1/2 knockout pigs with severe combined immunodeficiency. J Immunol 2014, 193: 1496–1503. [DOI] [PubMed] [Google Scholar]
- 65.Kim JI, Park JS, Kim H, Ryu SK, Kwak J, Kwon E, Yun JW et al. , CRISPR/Cas9-mediated knockout of Rag-2 causes systemic lymphopenia with hypoplastic lymphoid organs in FVB mice. Lab Anim Res 2018, 34: 166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Khiong K, Murakami M, Kitabayashi C, Ueda N, Sawa S, Sakamoto A, Kotzin BL et al. , Homeostatically proliferating CD4 T cells are involved in the pathogenesis of an Omenn syndrome murine model. J Clin Invest 2007, 117: 1270–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ott de Bruin LM, Bosticardo M, Barbieri A, Lin SG, Rowe JH, Poliani PL, Ching K et al. , Hypomorphic Rag1 mutations alter the preimmune repertoire at early stages of lymphoid development. Blood 2018, 132: 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marrella V, Poliani PL, Casati A, Rucci F, Frascoli L, Gougeon ML, Lemercier B et al. , A hypomorphic R229Q Rag2 mouse mutant recapitulates human Omenn syndrome. J Clin Invest 2007, 117: 1260–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marrella V, Poliani PL, Notarangelo LD, Grassi F and Villa A, Rag defects and thymic stroma: lessons from animal models. Front Immunol 2014, 5: 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Marrella V, Poliani PL, Fontana E, Casati A, Maina V, Cassani B, Ficara F et al. , Anti-CD3epsilon mAb improves thymic architecture and prevents autoimmune manifestations in a mouse model of Omenn syndrome: therapeutic implications. Blood 2012, 120: 1005–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cassani B, Poliani PL, Marrella V, Schena F, Sauer AV, Ravanini M, Strina D et al. , Homeostatic expansion of autoreactive immunoglobulin-secreting cells in the Rag2 mouse model of Omenn syndrome. J Exp Med 2010, 207: 1525–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rigoni R, Fontana E, Guglielmetti S, Fosso B, D’Erchia AM, Maina V, Taverniti V et al. , Intestinal microbiota sustains inflammation and autoimmunity induced by hypomorphic RAG defects. J Exp Med 2016, 213: 355–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rigoni R, Fontana E, Dobbs K, Marrella V, Taverniti V, Maina V, Facoetti A et al. , Cutaneous barrier leakage and gut inflammation drive skin disease in Omenn syndrome. J Allergy Clin Immunol 2020, 146: 1165–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Giblin W, Chatterji M, Westfield G, Masud T, Theisen B, Cheng HL, DeVido J et al. , Leaky severe combined immunodeficiency and aberrant DNA rearrangements due to a hypomorphic RAG1 mutation. Blood 2009, 113: 2965–2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rucci F, Poliani PL, Caraffi S, Paganini T, Fontana E, Giliani S, Alt FW et al. , Abnormalities of thymic stroma may contribute to immune dysregulation in murine models of leaky severe combined immunodeficiency. Front Immunol 2011, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Walter JE, Rucci F, Patrizi L, Recher M, Regenass S, Paganini T, Keszei M et al. , Expansion of immunoglobulin-secreting cells and defects in B cell tolerance in Rag-dependent immunodeficiency. J Exp Med 2010, 207: 1541–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stadinski BD, Shekhar K, Gomez-Tourino I, Jung J, Sasaki K, Sewell AK, Peakman M et al. , Hydrophobic CDR3 residues promote the development of self-reactive T cells. Nat Immunol 2016, 17: 946–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Noordzij JG, de Bruin-Versteeg S, Verkaik NS, Vossen JM, de Groot R, Bernatowska E, Langerak AW et al. , The immunophenotypic and immunogenotypic B-cell differentiation arrest in bone marrow of RAG-deficient SCID patients corresponds to residual recombination activities of mutated RAG proteins. Blood 2002, 100: 2145–2152. [PubMed] [Google Scholar]
- 79.Kennedy M, Awong G, Sturgeon CM, Ditadi A, LaMotte-Mohs R, Zuniga-Pflucker JC and Keller G, T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Rep 2012, 2: 1722–1735. [DOI] [PubMed] [Google Scholar]
- 80.Brauer PM, Pessach IM, Clarke E, Rowe JH, Ott de Bruin L, Lee YN, Dominguez-Brauer C et al. , Modeling altered T-cell development with human induced pluripotent stem cells from patients with RAG1 mutations and distinct immunological phenotypes. Blood 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yu X, Almeida JR, Darko S, van der Burg M, DeRavin SS, Malech H, Gennery A et al. , Human syndromes of immunodeficiency and dysregulation are characterized by distinct defects in T-cell receptor repertoire development. J Allergy Clin Immunol 2014, 133: 1109–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Themeli M, Chhatta A, Boersma H, Prins HJ, Cordes M, de Wilt E, Farahani AS et al. , Mikkers HMM: iPSC-based modeling of RAG2 Severe Combined Immunodeficiency reveals multiple T cell developmental arrests. Stem Cell Reports 2020, 14: 300–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Seet CS, He C, Bethune MT, Li S, Chick B, Gschweng EH, Zhu Y et al. , Generation of mature T cells from human hematopoietic stem and progenitor cells in artificial thymic organoids. Nat Method 2017, 14: 521–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bosticardo M, Pala F, Calzoni E, Delmonte OM, Dobbs K, Gardner CL, Sacchetti N et al. , Artificial thymic organoids represent a reliable tool to study T-cell differentiation in patients with severe T-cell lymphopenia. Blood Adv 2020, 4: 2611–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bifsha P, Leiding JW, Pai S-Y, Colamartino ABL, Hartog N, Church JA, Oshrine BR et al. , Diagnostic assay to assist clinical decisions for unclassified severe combined immune deficiency. Blood Advances. 2020, 4(12): 2606–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.La Motte-Mohs RN, Herer E and Zuniga-Pflucker JC, Induction of T-cell development from human cord blood hematopoietic stem cells by Delta-like 1 in vitro. Blood 2005, 105: 1431–1439. [DOI] [PubMed] [Google Scholar]
- 87.Cavadini P, Vermi W, Facchetti F, Fontana S, Nagafuchi S, Mazzolari E, Sediva A et al. , AIRE deficiency in thymus of 2 patients with Omenn syndrome. J Clin Invest 2005, 115: 728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Poliani PL, Facchetti F, Ravanini M, Gennery AR, Villa A, Roifman CM and Notarangelo LD, Early defects in human T-cell development severely affect distribution and maturation of thymic stromal cells: possible implications for the pathophysiology of Omenn syndrome. Blood 2009, 114: 105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.De Ravin SS, Cowen EW, Zarember KA, Whiting-Theobald NL, Kuhns DB, Sandler NG, Douek DC et al. , Hypomorphic Rag mutations can cause destructive midline granulomatous disease. Blood 2010, 116: 1263–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ferre EMN, Break TJ, Burbelo PD, Allgauer M, Kleiner DE, Jin D, Xu Z et al. , Lymphocyte-driven regional immunopathology in pneumonitis caused by impaired central immune tolerance. Sci Transl Med 2019, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shaw P, Shizuru J, Hoenig M, Veys P and Iewp E, Conditioning perspectives for primary immunodeficiency stem cell transplants. Front Pediatr 2019, 7: 434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Castiello MC, Bosticardo M, Sacchetti N, Calzoni E, Fontana E, Yamazaki Y, Draghici E et al. , Efficacy and safety of anti-CD45-saporin as conditioning agent for RAG deficiency. J Allergy Clin Immunol 2021, 147:309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lagresle-Peyrou C, Yates F, Malassis-Seris M, Hue C, Morillon E, Garrigue A, Liu A et al. , Cavazzana-Calvo M: long-term immune reconstitution in RAG-1-deficient mice treated by retroviral gene therapy: a balance between efficiency and toxicity. Blood 2006, 107: 63–72. [DOI] [PubMed] [Google Scholar]
- 94.Pike-Overzet K, Rodijk M, Ng YY, Baert MR, Lagresle-Peyrou C, Schambach A, Zhang F et al. , Correction of murine Rag1 deficiency by self-inactivating lentiviral vector-mediated gene transfer. Leukemia 2011, 25: 1471–1483. [DOI] [PubMed] [Google Scholar]
- 95.van Til NP, Sarwari R, Visser TP, Hauer J, Lagresle-Peyrou C, van der Velden G, Malshetty V et al. , Recombination-activating gene 1 (Rag1)-deficient mice with severe combined immunodeficiency treated with lentiviral gene therapy demonstrate autoimmune Omenn-like syndrome. J Allergy Clin Immunol 2014, 133: 1116–1123. [DOI] [PubMed] [Google Scholar]
- 96.van Til NP, de Boer H, Mashamba N, Wabik A, Huston M, Visser TP, Fontana E et al. , Correction of murine Rag2 severe combined immunodeficiency by lentiviral gene therapy using a codon-optimized RAG2 therapeutic transgene. Mol Ther 2012, 20: 1968–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Capo V, Castiello MC, Fontana E, Penna S, Bosticardo M, Draghici E, Poliani LP et al. , Efficacy of lentivirus-mediated gene therapy in an Omenn syndrome recombination-activating gene 2 mouse model is not hindered by inflammation and immune dysregulation. J Allergy Clin Immunol 2018, 142: 928–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gardner CL, Pavel-Dinu M, Dobbs K, Bosticardo M, Reardon PK, Lack J, DeRavin SS et al. , Gene Editing Rescues In vitro T Cell Development of RAG2-Deficient Induced Pluripotent Stem Cells in an Artificial Thymic Organoid System. J Clin Immunol. 2021. 10.1007/s10875-021-00989-6. [DOI] [PMC free article] [PubMed] [Google Scholar]