

Abstract
Fungi in the genus Metarhizium are soil-borne plant–root endophytes and rhizosphere colonizers, but also potent insect pathogens with highly variable host ranges. These ascomycete fungi are predominantly asexually reproducing and ancestrally haploid, but two independent origins of persistent diploidy within the Coleoptera-infecting Metarhizium majus species complex are known and has been attributed to incomplete chromosomal segregation following meiosis during the sexual cycle. There is also evidence for infrequent sexual cycles in the locust-specific pathogenic fungus Metarhizium acridum (Hypocreales: Clavicipitaceae), which is an important entomopathogenic biocontrol agent used for the control of grasshoppers in agricultural systems as an alternative to chemical control. Here, we show that the genome of the M. acridum isolate ARSEF 324, which is formulated and commercially utilized is functionally diploid. We used single-molecule real-time sequencing technology to complete a high-quality assembly of ARSEF 324. K-mer frequencies, intragenomic collinearity between contigs and single nucleotide variant read depths across the genome revealed the first incidence of diploidy described within the species M. acridum. The haploid assembly of 44.7 Mb consisted of 20.8% repetitive elements, which is the highest proportion described of any Metarhizium species. The long-read diploid genome assembly sheds light on past research on this strain, such as unusual high UVB tolerance. The data presented here could fuel future investigation into the fitness landscape of fungi with infrequent sexual reproduction and aberrant ploidy levels, not least in the context of biocontrol agents.
Keywords: genome duplication, ploidy, parasexual, Metarhizium, entomopathogen, ascomycete
Introduction
Significance
Increasing evidence suggests that chromosome copy number variation is an adaptive trait of many fungi and present at low prevalence in natural populations. Here, we describe a rare case of diploidy and the first case within the species Metarhizium acridum, which in contrast to more intensely studied haploid Metarhizium species with broad host ranges such as Metarhizium robertsii and Metarhizium anisopliae, is a locust-specific pathogen with infrequent sexual reproduction. Using long-read sequencing, we show that the diploid genome of M. acridum is markedly different in repeat content and signature of the fungus-specific repeat-induced point mutation defensive machinery compared with generalist Metarhizium species. This is relevant for studying evolutionary patterns of genome structure and ploidy levels within this genus of important biological control agents.
The duplication of single genes, genomic segments, chromosomes, and whole-genome duplications (WGD) has played a vital role in eukaryotic evolution by providing genetic material for adaptation. Throughout evolution, recurrent WGD events have been linked to the emergence, diversification, and survival of species (Crow et al. 2006). For example, WGD has been coupled with the emergence of angiosperms within plants (De Bodt et al. 2005), and with the emergence of vertebrates, gnathostomes, and teleosts within metazoan chordate evolution (Sacerdot et al. 2018).
A well-described ancient WGD event in the fungal kingdom is the ancient diploidization that led to the formation of the Saccharomyces genus (Wolfe and Shields 1997; Dietrich 2004; Kellis et al. 2004). Although some WGD events lead to stable polyploids, as in the case of Saccharomyces where an ancient hybridization event is thought to have provided stability and fertility to the new diploid (Marcet-Houben and Gabaldón 2015), duplications are usually perceived as transient. Duplicated genes undergo subsequent reductions by loss or mutational decay leading to pseudogenization or neofunctionalization (Lynch and Conery 2000; Levasseur and Pontarotti 2011). It should be noted that it can be difficult to distinguish ancient WGD events from cases of multiple segmental duplications caused by transposon activity (Roelofs et al. 2020). Regardless of origin, duplications have long been recognized as a driver of evolution by providing the material for natural selection to work on (Bridges 1919; Metz 1947; Ohno 1970; Crow et al. 2006).
WGD arise from abnormal cell cycles post chromosome duplication, either by the absence of mitotic division or cytokinesis. The duplication itself can occur by auto- or allopolyploidization. The latter involves the fusion of individual cells and subsequent fusion of nuclei bringing together the variants accumulated between the genetically distinct individuals. In contrast, autopolyploidization, or endoreplication, only involves a single cell and has been associated with environmental stress. Within plants, several studies have shown that endoreplication can be induced by abiotic stress, such as heat (Monjardino et al. 2006), drought (Cookson et al. 2006), elevated salinity (Barkla et al. 2018), and toxins (Biskup and Izmaiłow 2004). Duplications are considered to mediate an acute increase in the transcription of metabolic and stress-mitigating genes (Scholes and Paige 2015). The human pathogenic fungi Candida albicans and Cryptococcus neoformans have been shown to repeatedly gain resistance to antifungal drugs such as fluconazole through chromosomal duplications (Sionov et al. 2010; Kronstad et al. 2011). The fungus C. neoformans can also evade phagocytosis within the lungs by creating titan cells (Okagaki et al. 2010; Zaragoza et al. 2010). The increased cell size is induced by multiple rounds of endoreplication and is observed in up to one-fifth of the C. neoformans cells residing in infected lung tissue (Okagaki et al. 2010). Transient somatic endoreplication has also been recognized as a driver of developmental change of some cell types in plants, insects and mammals (Fox and Duronio 2013). Examples of somatic endoreplication include the giant cells of the mammalian trophoblast that develop into a significant part of the placenta, and the >1,000× sister chromatids observed in the salivary glands of Drosophila melanogaster (Sher et al. 2013).
Within fungi, increasing evidence suggests widespread cryptic population-level ploidy variations. Whole-genome sequencing of an extensive collection of 794 natural (wild) isolates of Saccharomyces cerevisiae found alternative ploidy levels within 13% of the otherwise predominately diploid yeast (Peter et al. 2018). The same study found chromosomal copy number variation (CCNV or aneuploidy) in 19% of the isolates (193 of the 1,011), reiterating the importance of variation in chromosome numbers for organismal evolution (Mayrose and Lysak 2020). Although at least some fungi readily undergo endoreplication, such levels of genome plasticity are not without constraints. A long-term evolutionary experiment with S. cerevisiae showed a convergence from alternative ploidies to the ancestral diploid form (Gerstein et al. 2006). The chytrid fungus Batrachochytrium dendrobatidis threatening amphibians worldwide has a high rate of CCNV in nature (Rosenblum et al. 2013). This plasticity has been linked to pathogenicity through lab experiments, which showed a gain of chromosomal copies upon induced stress by a characteristic host antimicrobial peptide (Farrer et al. 2013).
Although the majority of known fungi are dikaryotic at some stage, diploidy is the exception. Filamentous ascomycetes (Pezizomycotina) are only dikaryotic in specific life stages, namely the ascogenous hyphae and ascocarp, whereas the bulk of the mycelium remains monokaryotic, and in most species, haploid. Exceptions to haploidy within this important group of fungi are found in the Phyllactinia, Stephensia, Xylaria, Botrytis, and Zygosaccharomyces genera (Albertin and Marullo 2012), and within the genus Metarhizium (Kepler et al. 2016). When these fungi contain two different idiomorphs of the mating-type genes within the same diploid, it indicates mating events that failed to complete meiosis, leading to the observed allopolyploidization. Diploidy may also arise from the parasexual cycle of fungi, which allows for nuclei fusion (karyogamy) following anastomosis of strains from somatically compatible groups. Contrary to the sexual cycle, fused nuclei of the parasexual cycle do not undergo meiosis, but continue to divide mitotically. The original ploidy level is restored by random chromosome loss, through a series of aneuploidy intermediates (Moore et al. 2011), which is known from the “asexual” human pathogen Candida albicans. Reversion is a gradual process with generations of aneuploidy. Sexual reproduction requires different mating-types, whereas the parasexual cycle normally requires compatible identities. The reproductive isolation of the parasexual cycle is enforced by heterokaryon incompatibility protein domains that abort incompatible hyphal fusion attempts (Glass and Dementhon 2006). This entails that any parasexual fusion of nuclei will be of closely related and highly similar genomes.
Although sexual reproduction facilitates the recombination of genomes, it also opens a door for disseminating “selfish DNA” such as transposons and other mobile sequences throughout the genome. Selfish DNA elements can have detrimental effects on host fitness why Ascomycete genomes have different modes of defence to silence transposons and ensure genome integrity. Some ascomycetes have a repeat-induced point (RIP) mutation pathway (Selker and Garrett 1988) that induce cytosine to thymine mutations (Cambareri et al. 1991; Watters et al. 1999) in any kind of repetitive element, which are thereby degraded and deactivated. Because the RIP pathway is considered only to be active during the dikaryotic phase between fertilization and nuclear fusion within the sexual cycle (Selker and Garrett 1988), it is also considered exclusively available to sexually reproducing fungi (Galagan and Selker 2004; Gladyshev 2017). The genomic footprint of RIP in the form of cytosine to thymine transitions that preferentially affect the frequency of CpA di-nucleotides (Hane and Oliver 2008), can therefore be used to indicate a sexual cycle within fungal species where sex otherwise is not known. It is important not to associate the presence of an active RIP pathway with the absence of recombination, because recombination also occurs during the parasexual cycle (Arnau and Oliver 1993). However, although the RIP pathway should not be associated with recombination per se, an active RIP pathway is considered linked to the sexual cycle (Galagan and Selker 2004; Gladyshev 2017).
For entomopathogenic fungi, infected insects can be hot spots of fungal parasexuality, and the formation of diploid conidia (Riba et al. 1980; Leal-Bertioli et al. 2000; Wang et al. 2011). Investigating the conidia harvested from an insect coinfected with two fluorescently labeled isolates of the same Metarhiziumrobertsii strain revealed that 24% of the conidia investigated were diploid (Wang et al. 2011) and 13% of conidia when coinfected with different M. robertsii isolates (Li et al. 2021). Similar observations have been reported from experiments with Metarhiziumanisopliae and Metarhiziummajus (Riba et al. 1980; Leal-Bertioli et al. 2000).
The entomopathogenic fungus Metarhiziumacridum (Hypocreales: Clavicipitaceae) is a specialist with a narrow host range of insects of the order Orthoptera. Host specificity is enforced by the requirement of Orthopteran cuticle signals facilitating spore germination and appressorial formation (Wang and St. Leger 2005). The M. acridum strain ARSEF 324 is the active ingredient in the biocontrol agent “Green Guard” targeting grasshoppers (Acrididae), which is commercially deployed in Australia where these insects are agricultural pests of concern. Within the genus Metarhizium, M. acridum, as well as the specialist Metarhiziumalbum, are considered to be cryptic sexual species (Hu et al. 2014; St. Leger and Wang 2020). No teleomorphs are known, but genomic analyses have found signatures of an active RIP pathway as an indirect sign of meiosis and sexual recombination (Galagan and Selker 2004; Gladyshev 2017). This is in contrast to the co-generic generalists with wide host ranges, Metarhiziumpingshaense, M. anisopliae, M. robertsii, and Metarhiziumbrunneum (the PARB clade, sensu Bischoff et al. 2009), where no RIP footprint is present, consequently causing these species to be considered primarily asexual.
The ascomycete genus Metarhizium is generally haploid, but Kepler et al. (2016) found two independent origins of stable diploid clades interspersed among lineages comprised entirely of haploid individuals within the larger Metarhizium guizhouense/majus/taii clade (MGT clade; sensu Bischoff et al. 2009). All isolates within these diploid taxa were consistently heterozygotic for the analyzed microsatellite markers and contained two different idiomorphs of the mating-type genes (MAT1 and MAT2). The presence of both mating-types was interpreted as a mating event that failed to complete meiosis, which lead to the observed allopolyploidization. The locust-specific M. acridum has so far been regarded as entirely haploid, and previously the only available genome for this species was from the haploid strain CQMa 102. This strain was sequenced and assembled into 241 scaffolds (>1 kb; N50, 329.5 kb) containing 1,609 contigs with a total genome size of 38.0 Mb (Gao et al. 2011).
In this study, we describe another instance of diploidy within the genus Metarhizium and the first within M. acridum. We present a de-duplicated haploid genome assembly of M. acridum strain ARSEF 324, consisting of 35 contigs representing a haploid genome of 44.7 Mb. Ploidy is established through k-mer analysis on unassembled sequencing reads, and intragenomic collinearity analysis by all-versus-all mapping of contigs. Euploidy was established through sequencing read depth comparison across each contig, and through estimation of heterozygosity ratios of single nucleotide variants (SNV), both in 10-kb nonoverlapping sliding windows. Finally, using DAPI fluorescence staining we demonstrate that conidia are uninucleate.
Results
K-mer Analysis
The k-mer analysis on the PacBio subreads predicted that the sequenced genome was diploid with a haploid length of 45 Mb (fig. 1A). The profile in figure 1A shows two peaks in the frequency of observed unique 21-mers within the sequencing data. The smaller peak has half the read coverage of the taller peak indicating the presence of heterozygous loci. The genome is estimated to have a heterozygosity rate of 0.5%. Analyzing the sum of k-mer pair coverages to coverage ratios confirmed the inferred diploidy of the data in figure 1B (for full GenomeScope results see supplementary table S1, Supplementary Material online).
Fig. 1.

The k-mer analysis for Metarhizium acridum ARSEF 324 PacBio subread data revealed a diploid structure of heterozygous k-mer pairs. (A) Based on the number of unique 21-mers the genome is estimated to have a haploid length of 45 Mb, with 18.2% repeats. The two peaks of the k-mer frequency profile of the observed data, where the lesser have half the read coverage of the higher peak, indicate heterozygocity, consistent with diploidy. The low frequency of k-mers from heterozygous loci indicate that most 21-mers were heterozygous (0.5%). (B) The diploidy of the genome was confirmed by Smudgeplot analysis, comparing the sum of k-mer pair coverages (CovA + CovB) to their relative coverage (CovB/(CovA + CovB)). No higher-level ploidy was observed. Max and min estimates from the GenomeScope analysis are available in supplementary table S1, Supplementary Material online.
Genome Assembly
A total of 478,156 filtered PacBio reads with an average length of 12.7 kb was used to assemble the genome into 35 primary contigs representing a haploid genome of 44.71 Mb. The primary assembly H1 has an L50 of 6, with a N50 of 23.37 Mb, and 4,276 complete BUSCO genes, out of the 4,494 BUSCO genes sought, or 95.1% (Scores in BUSCO format: C: 95.1% [S: 94.2%, D: 0.9%], F: 1.4%, M: 3.5%, n: 4494). Out of the 44.7 Mb in H1, 9.3 Mb (20.8%) was identified to be from repetitive elements, 74.2% of which had a strong GC-bias (<33.2%). The alternative haplotig (H2) is represented by 565 contigs with a collective size of 39.24 Mb, that is, 88% of the assumed haploid length. This assembly has an L50 of 144, with an N50 of 0.08 Mb. This set of contigs only contains loci sufficiently divergent from H1 to be forked out during the genome assembly. This implies that conserved loci between H1 and H2 will be present in the H1 primary assembly but largely absent from the H2 haplotigs. This is reflected in the associated BUSCO scores where 43.7% of the highly conserved BUSCO genes are missing from H2 (C: 54.1% [S: 51.7%, D: 2.4%], F: 2.2%, M: 43.7%, n: 4,494), likely because the genomic regions are highly conserved between the two haplotypes and therefore collapsed and only present in the H1 assembly. The outer band of the outer tracks of figure 2 depict the size of the 35 primary contigs representing the complete haploid genome. The second band depicts the mapping of the 565 H2 contigs to H1, indicating their redundancy (fig. 2). The mitochondrion genome was assembled to a length of 96,363 bp and contained 17 genes. The number and order of genes correspond with the conserved structure of mito-genomes within Hypocreales (Aguileta et al. 2014).
Fig. 2.

Genome-wide statistics of sequencing data, assembly and traits of the 35 primary haploid contigs of Metarhizium acridum strain ARSEF 324 genome. The plot consists of five major tracks; from the outside: (i) 35 contigs placed in the outer band (H1) represent the haploid genome. The remaining 565 contigs (H2) map to H1. H2 contigs are given in light and dark blue, where dark blue indicates the fraction of each contig mapped to H1. The following tracks, show (ii) the read depth of long reads (PacBio) and short reads (BGI DNBseq) mapped to H1 (mean depth within 20-kb windows), (iii) Allele frequencies calculated from short reads (BGI DNBseq) mapped to H1 (mean within 10-kb windows), (iv) Repeat and gene count within 20-kb windows shown in black and red respectively, (v) The GC levels within 10-kb windows. The contig length of the 35 primary contigs is indicated by the scale bar legend.
Ideally, the sequencing of an euploid genome should result in an equal read depth across the genome. We mapped both long and short-read data to the H1 assembly, to assess CCNV. The genome wide mean read depth was 54× and 103× (excluding >500× depth, at repetitive elements), for long and short-read data respectively (fig. 2). Smaller contigs (<400 kb) tended to have lower read depth, likely due to poor mapping in repeat dense regions. Contig 34 had a noticeable lower coverage than other contigs with a mean long-read depth of 52×. Contig 34 has an H2 haplotig that uniquely maps to it why it is unlikely that contig 34 constitutes a small haploid chromosome and more likely a haploid segment in the otherwise diploid chromosome. The equal depth across the genome is a strong indication of euploidy across all chromosomes represented in the assembled contigs.
The ploidy of each contig was also assessed by mean ratios of SNV read depth in 10-kb nonoverlapping windows. The expectation is that chromosomes with an even ploidy will tend toward a 50:50 distribution across each SNV, whereas chromosomes with odd ploidy will tend toward a 33:66 or 33:33:33 ratio. All contigs have distributions of ratios with means of 0.5 (50:50) (fig. 2), confirming the diploidy of all contigs. Deviations from 0.5 correlate with read depth of mapped short-read DBNseq data on which the SNV data is based. Mapping of short-reads likewise correlates with the repeat density (fig. 2), because high repeat densities impede the mapping.
The mean GC content across the 44.7 Mb of the H1 contigs is 45% (fig. 2, track 5). The GC-content analyzed in 100-bp windows shows a bimodal distribution with peaks at 17.6% and 51.3% (fig. 3A). In the following, we delimitate the low and high GC peaks at 33.2%. Repetitive elements explain 81.5% of the low GC peak, 40.5% are unknown repetitive elements, simple repeats 28.7%, retroelements 6.4%, and DNA transposons explain 5.9%. Within low GC windows, 17% were without annotations (fig. 3A). Analyzing the dinucleotide composition across the 100-bp windows using the RIP composite index, showed that 33.1% of the genome have a composite index value above zero and are thereby interpreted as affected by RIP. Only 19.3% of the windows with GC content above 33.2% are RIP affected, whereas 95.7% of the low GC peak are affected by RIP (fig. 3B). Across GC levels, 82% of transposable elements are affected by RIP. Similarly, 83% of unknown repeats are RIP affected.
Fig. 3.
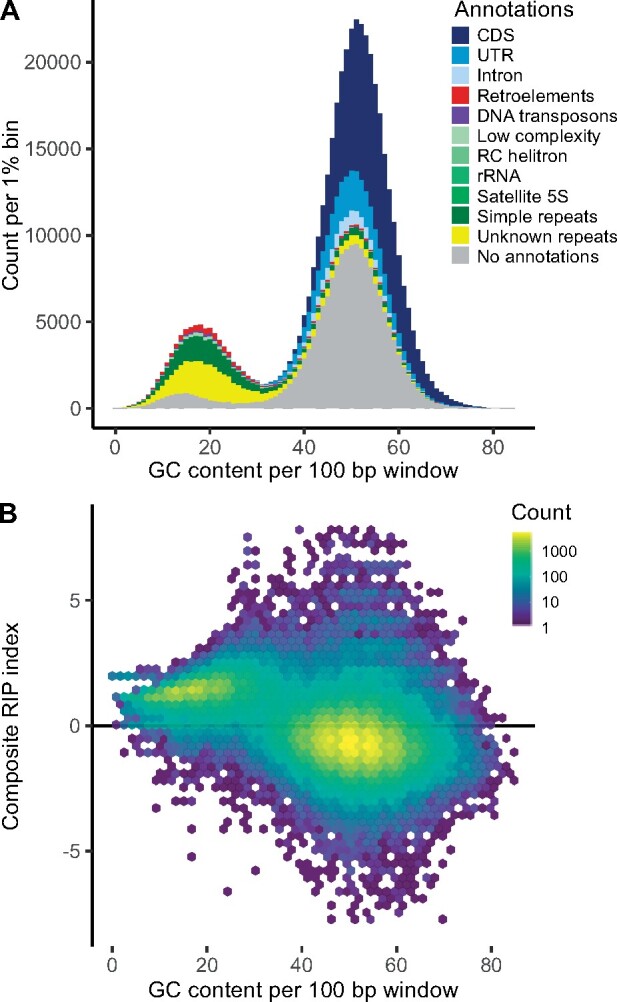
GC content calculated in 100-bp windows across the 44.7 Mb H1 haploid genome. (A) If a window overlapped with more than half of its length with an annotated feature this were signed to the window. The bimodal distribution has peak GC-levels of 17.6% and 51.3%. Each peak approximates normal distributions with standard deviations of 5.6 and 6.5, respectively. The fitted normal distributions intersect at 33.2%; this level is set to split high and low GC values. Genomic windows with high GC values are largely comprised CDS (37.3%), UTR (10.9%), introns (3.3%) and sequence that were without annotations (42.3%). Genomic windows with low GC values are dominated by repetitive elements. Of these repetitive elements 40.5% are unknown, 28.7% are simple repeats, 6.4% are retroelements, 5.9% are DNA transposons. Of the low GC windows 17% were without annotations. (B) Composite RIP index as a function of GC content. Regions with a composite RIP index >0 are considered RIP affected, which comprise 33.1% of the total genome, or 95.7% of the windows within the low GC peak. Combining different indices of RIP will provide a more conservative estimate of the proportion of the genome that is affected by RIP (17.9%, fig. 5).
Metarhizium acridum ARSEF 324 is homozygous for the MAT1-2 mating-type idiomorph (fig. 4). The two mating-type alleles of M. acridum ARSEF 324 and the mating-type allele of M. acridum CQMa 102 have notable differences in insertions. H1 contains a Mutator-like element (MULE) DNA transposon downstream of MAT1-2-3 not present in H2. In contrast, H2 harbors a LAGLIDADG homing endonuclease not present in H1. Metarhizium acridum CQMa 102 contains a WD-repeat domain that is not found in either of the two ARSEF 324 alleles.
Fig. 4.

Synteny between mating-type loci of the two alleles Metarhizium acridum strain ARSEF 324 and M. arcridum strain CQMa 102. The M. acridum strain ARSEF 324 is homozygous for the MAT1-2 mating-type idiomorph. Down-stream of the ARSEF H1 MAT1-2-3 gene, a 3 kb DNA transposon of the “MULE” (Mutator-like elements) superfamily is found, this is not present in ARSEF H2. Mutator transposable elements are among the most mutagenic transposons known (Lisch 2015; Dupeyron et al. 2019). Conversely, H2 harbors a LAGLIDADG endonuclease fragment (red) together with a fragment of the mitochondrial COX3 gene (orange), not present in H1. Neither of these insertions is present in the CQMa 102 strain, which contains a WD domain missing in ARSEF 324.
Analysis of single-copy homolog gene pairs within the M. acridum ARSEF 324 diploid strain showed that homologous gene pairs on average were as divergent when compared with homologs of the Chinese M. acridum strain CQMa 102 (supplementary fig. S3, Supplementary Material online). The divergence observed between homologous genes within strain ARSEF 324 were likewise comparable to the intraspecific divergence between isolates of M. bruneum or M. robertsii (supplementary fig. S3, Supplementary Material online). Among the divergent single-copy homologs within ARSEF 324, no Gene Ontology functional annotations were notably enriched for synonymous substitutions (supplementary fig. S4B, Supplementary Material online) or correlated to the composite RIP index (supplementary fig. S4C, Supplementary Material online).
Genome Comparison
To assess if our haplotig assembly differed from other species within the genus Metarhizium, we compared the assembly of strain ARSEF 324 with genomes of eight Metarhizium species (fig. 5). For the phylogenetic analysis, two strains of the closely related Pochonia chlamydosporia was selected as outgroups. The Metarhizium genomes presented range in haploid size from 30.8 to 44.7 Mb. The 12.536 predicted genes within ARSEF 324 (H1), fall within the range predicted for other Metarhizium isolates, that is, 8,472−13,646. A notable difference can be seen in the proportion of repetitive elements between genomes. The median repeat level across the Metarhizium isolates is 6.1%, M. anisopliae strain JEF-290 has twice this, and the diploid M. acridum strain ARSEF 324 has three times this level, with 20.8% of the genome comprised repetitive elements. The proportion of the genome of M. acridum strain ARSEF 324 that is affected by RIP is likewise higher than observed for any other Metarhizium genome analyzed (fig. 5). A conservative estimate of the RIP affected region can be given by only including regions that are assessed as RIP affected with all three RIP calculating methods: RIP product, RIP substrate, and RIP composite as used above. With this conservative estimate, 17.9% of the M. acridum strain ARSEF 324 genome is affected by RIP (calculated in 1,000-bp window, in 500 bp steps). Evaluating the genomes ordered in descending order of RIP content, the diploid M. acridum strain is followed by M. acridum CQMa 102 and M. rileyi RCEF 4,871 with 3.8% and 3.6%, respectively (fig. 5). The effect of window size on total RIP proportions is shown in supplementary table S3, Supplementary Material online. DAPI staining of individual conidia supported that cells are homokaryotic diploid and could rule out that the strain was heterkaryotic by showing a single nucleus within each conidia (fig. 6).
Fig. 5.

A consensus maximum-likelihood phylogeny based on 444 known single-copy orthologous genes for sequenced Metarhizium species and the related Pochonia chlamydosporia. All splits within the phylogeny had 100% bootstrap support, except the artificial split between H1 and H2. From the left: ML phylogeny, tip icons indicating the host adaptation, species names and strain id. Genome size and number of genes is retrieved from NCBI Genbank and the protein file available under the accession found in supplementary table S2, Supplementary Material online. The proportion of repetitive elements within each isolate was found as described in the method section. The figure highlights that the genome assembly of M. acridum ARSEF 324 generated in this study is the longest genome assembly of any Metarhizium isolate, and the one with the highest proportion of repetitive elements.
Fig. 6.

DAPI stained nuclei in Metarhizium acridum strain ARSEF 324 conidia. All inspected spores contained monokaryons.
Discussion
Metarhizium species are predominately haploid, with only two independent lineages within the M. majus complex described as stable diploids. Here we present a highly improved genome assembly of M. acridum based on long-read sequencing data assembled across repeat regions. This reveals a genome enriched in repetitive elements relative to other Metarhizium isolates or species studied. All presented analyses indicate that the sequences M. acridum strain ARSEF 324 show the genomic characteristics of a diploid. All 35 contigs of the representative assembled haplotype were subject to the mapping of smaller contigs, and all were heterozygous with mapped read allele frequencies around 0.5. The only exceptions are two minor contigs, contig 17 and contig 34, which could be examples of misassembly, or segmental aneuploidy.
A growing body of evidence indicates that CCNV is a common adaptive trait of many fungi and has been observed at low prevalence in natural populations (Farrer et al. 2013). The effect on fitness induced by CCNVs has been shown to vary, and can be life-stage specific, and how often these variants are reproductively stable is unknown. We did not see any signs of chromosomal loss to indicate ongoing haploidization within this diploid strain of M. acridum. The absence of chromosome loss suggests that the polyploidization is recent, or that the strain constitutes a stable diploid. Kepler et al. (2016) found monophyletic clades of diploid isolates in a population genetic study of the M. majus species complex (the MGT clade, sensu Bischoff et al 2009). A similar population study could shed light on the prevalence of diploidy or CCNV within M. acridum. Alternatively, the diploid stability can be assessed by long-term evolution experiment focusing on possible reversions to the ancestral haploid state. Future research is needed to investigate gene evolution within this strain and identify concerted gene loss levels and rates of pseudo- and neofunctionalization. If the diploidization of this strain is not recent, it should be possible to trace divergence between homolog genes, where gene duplication did not result in increased fitness and purifying selection.
The highly continuous genome assembly ARSEF 324 includes the highest proportion of repetitive elements of any Metharhizium species reported so far. Part of the reason for this is likely the fragmented character of many short read based assemblies and the inherent bias that spring from difficulties in assembling across repeats using these technologies. Our comparative analysis includes, besides the genome assembly of M. acridum ARSEF 324, three assemblies based on long-read technologies, that is, M. anisopliae JEF-290, M. brunneum ARSEF 4556, and P. chlamydosporia 170 (supplementary table S2, Supplementary Material online). Assuming that these assemblies give credible indications of their species’ general repeat content, it is noticeable that the long-read assemblies of M. anisopliae and M. brunneum, both belonging to the presumable asexual PARB clade and have considerable less repetitive elements compared with ARSEF 324. This difference between the primarily asexual M. anisopliae and M. brunneum and the occasional sexual M. acridum corroborates the theory that sexual reproduction allows for the proliferation of transposable elements within populations like “a sexually transmitted nuclear parasite” (Hickey 1982). Another difference between asexual and sexual fungal species is in their defense against such selfish DNA elements. The fungal RIP pathway that mutates TE’s is only active during the sexual cycle and should therefore only be observed in species that sexually recombine and be almost absent in primarily asexually reproducing species. Comparing the repeat content to the proportion of the genome affected by RIP across the Metarhizium species show that the two well-assembled genomes of M. anisopliae JEF-290 and M. brunneum ARSEF 4556 both have a higher ratio of “repeats %” to “RIP %” compared with M. acridum ARSEF 324, indicating a lack of an active RIP control of repeats. Both the high repeat content and the active RIP defence supports that M. acridum is at least occasionally sexually reproducing, even though no teleomorph is known (Mongkolsamrit et al. 2020). The impact of RIP defence on the genome of M. acridum has changed the nucleotide composition of approximately one-fifth of the genome. RIP affected regions have a reduced GC content that is less than half that of the background GC frequency (51%).
Cambareri et al. (1991) showed that both copies of duplicated gene-sized sequences within Neurospora crassa are affected by RIP in a single sexual cycle at frequencies of 50 − 100%. The effectiveness of the RIP machinery varies between species, but it generally affects homologous sequences longer than 400 bp within a nucleus, including selfish mobile elements, viruses and genes. The diploid nature of this M. acridum genome is therefore incompatible with an active RIP pathway and sexual reproduction because all duplicated gene regions would have been mutated. The activation of the RIP pathway must therefore have preceded the described genome duplication.
The presence of two identical mating-type ideomorphs within ARSEF 324 makes it unlikely that the diploidization results from a mating event as hypothesized of the diploid strains within the M. majus species complex. This supports a model where either endoreplication, or allopolyploidization through parasexuality are responsible for diploidization. If the former were the case, the duplication event should be old enough for the measurable sequence divergence to have occurred between homologous genes. The phylogenetic distances between homologous genes within the diploid were comparable to the distance observed between this diploid strain and the conspecific strain QCMa 102, as well as the distances observed between haploid strains of M. bruneum and M. robertsii (supplementary fig. S3, Supplementary Material online). This indicates that the observed phylogenetic distance between ARSEF 324 homologs is consistent with allopolyploidization. The high prevalence of parasexuality observed in M. robertsii (Wang et al. 2011) renders alloploidization a likely mode of forming the observed diploid M. acridum isolate.
CCNV has thoroughly been linked with both stress and enhanced fitness (Hu et al. 2011; Farrer et al. 2013). In Metarhizium the high frequency of parasexuality observed in coinfected insects (Riba et al. 1980; Leal-Bertioli et al. 2000; Wang et al. 2011) supports that coinfected insects provides an opportunity for recombination between pathogenic isolates. If the diploidization of ARSEF 324 arose from a parasexual fusion, the question remains how stable or long-lived this is. The diploid strain ARSEF 324 is likely to be phenotypically different from haploid M. acridum strains because higher ploidy can enable increased transcription of virulence factors or effectors, which could make the strain a more potent biocontrol agent. Diploidy can also work as a shield against detrimental mutation through functional redundancy (Haldane 1932, p. 110; Orr 1995) This could explain the strain-specific high tolerance to UV-B radiation of ARSEF 324 compared with other M. acridum isolates reported by Braga et al. (2001).
Although the diploidy reported here for an isolate within M. acridum is independent from the cases of diploidy in M. majus (Kepler et al. 2016), it is noteworthy that there are several instances of ploidy variation in the genus Metarhizium. It is tempting to speculate whether the complex species associations where many isolates are soil-dwelling plant–root endophytes and rhizosphere colonizers as well as potent insect pathogens influence the tendency for ploidy-level variation (St. Leger and Wang 2020). It certainly provides the potential for these fungi to experience several stressful microhabitats (Lovett and St. Leger 2015), which could be triggering ploidy-level variation among Metarhizium strains.
Materials and Methods
Strain and Culturing
The Metarhizium acridum strain ARSEF 324 was isolated in 1979 from a spur-throated locust (Austracris guttulosa) in Australia and obtained from the ARSEF collection (ARS Collection of Entomopathogenic Fungal Cultures, Ithaca, New York). A single-spore isolate was obtained by plating serial-dilutions of conidia from the original culture. Conidia from the single-spore culture were grown in liquid Sabouraud dextrose broth with yeast extract (40 g/l dextrose, 10 g/l peptone, 10 g/l yeast extract, pH = 6.5) while being stirred at 170 rpm on a shaking table for 24 h before DNA extraction. Fluorescence staining with DAPI (blue, 4′,6-diamidino-2-phenylindole) was used to visualize the number of nuclei within mature conidia, as described in Kepler et al. (2016). Imaging was done on a Zeiss Axioscope microscope (×100 objective) with an AxioCam ICm1, and spore sizes measured using ImageJ v1.53e (Schneider et al. 2012).
DNA and RNA Extraction and Sequencing
Liquid culture filtrate was ground with liquid nitrogen in a mortar and approximately 500 mg finely ground powder transferred to 50-ml Falcon tube. DNA was extracted using a CTAB method by adding 17.5 ml CTAB buffer with 0.1% 2-Mercaptoethanol and 125 µl Proteinase K and incubated for 1 h at 60 °C. After centrifugation for 20 min at 5.000 g at 4 °C, the supernatant was removed and washed with 1 v of Phenol/chloroform/isoamyl alcohol (25:24:1) followed by two rounds of 1 v chloroform/isoamyl alcohol (24:1) and centrifuged for 10 min at 11,000 rpm at each step. To remove RNA, 150 µl RNAse was added to the aqueous phase and incubated for 120 min at 37 °C. To precipitate DNA, 0.6 v 2-propanol was added and incubated at −20 °C overnight, before centrifugation at 11,000 rpm at 4 °C for 30 min. The resulting pellet was washed twice with 2 ml 70% ethanol and centrifuged for 10 min at 11,000 rpm at 4 °C before being suspended in 500 µl TE-buffer. Purity was checked using NanoDrop reading and DNA quantity using a Qubit Broad-Range analysis kit.
A single SMART Cell library was sequenced on a PacBio Sequel platform using the Sequel Sequencing Kit 2.1 v2 (Sequencing Kit par number 101-309-500). Each continuous long read had one passage of sequencing; subsequently, subreads were generated by the removal of adapters and bases were called with the basecaller v5.0.0.6236. Sequencing was performed by Genewiz (Takeley, United Kingdom) with a yield from the single SMRT Cell equaling a coverage of 134×, assuming a 45 Mb genome, with a mean subread length of 11.2 kb. Short read sequence data were obtained using the PCR-free DBNseq platform at BGI-tech Copenhagen. Sequencing adapters were removed by the sequencing company, delivering a total of 15,111,206 reads of 150 bp, equaling a coverage of 50×, assuming a 45 Mb genome.
To obtain RNAseq samples to aid in the genome prediction, four replicate flasks with 20 ml media were grown for four days as described above but with 100 rpm agitation. After filtration through a Whatman filter paper (5 − 13 µm), the fungal material was collected and flash-frozen in liquid nitrogen before being pulverized in a tissue lyzer and extracted using a QIAGEN plant RNeasy kit following manufacturers specifications. The four samples were sequenced separately using the DBNseq platform at BGI-tech Copenhagen yielding approximately 16,000,000 paired-end 150 bp reads per sample.
K-mer Analysis and Genome Assembly
K-mers frequencies within PacBio subread data were obtained with KMC 3 (Kokot et al. 2017), and genome size and ploidy were inferred using GenomeScope v2.0 (Ranallo-Benavidez et al. 2020). Ploidy was further investigated comparing the sum of k-mer pair coverages using Smudgeplot implemented in GenomeScope. Sequence reads were both error-corrected and assembled with Canu v. 2.0, using the following parameters to co-assemble haplotypes: genomeSize = 45m correctedErrorRate = 0.03 corOutCoverage = 200 batOptions=-dg 3 -db 3 -dr 1 -ca 500 -cp 50. Two rounds of polishing were conducted to improve the assembly, each consisting of mapping raw-reads to the assembly using Minimap2 v.2.6 (Li 2018), and reducing remaining insertion-deletion and base substitution errors by polishing the consensus sequence using Arrow v2.3.3 (https://github.com/PacificBiosciences/GenomicConsensus, last accessed April 30, 2021). Mirror-reads (putatively an effect of missing adapters during sequencing) were detected in the PacBio subreads set; therefore, reads longer than 40 kb were filtered out prior to assembly. Different levels of the correctedErrorRate parameter were tested (i.e., 0.020, 0.025, 0.030, 0.035, 0.040). Each of the five assemblies was assessed based on the complementarity of phased contigs. The value 0.030 were chosen for the final assembly (see supplementary fig. S1, Supplementary Material online).
Contigs were ordered according to length and grouped into primary contigs, forming a haploid representative genome assembly [Haplotig 1 (H1)] and shorter contigs (H2) that mapped to the primary contigs. Primary contigs were identified by an all-versus-all contig mapping on the repeat masked assembly with minimap2 v.2.17. The best match of each contig to a longer contig was assessed from the accumulated length of alignment between each pair of contigs. Primary contigs were manually curated; three contigs were not only subjects to mapping, but also query to other primary contigs. These contigs, and the regions they mapped to had approximately half the read depth of the mean read depth of the other primary contigs. Because the read depth of the subject regions could be raised to the mean read depth, if the query contigs were removed before mapping, these three contigs were assumed to be haplotigs and moved to the H2 group.
The mitochondrion was assembled from 2,806 reads mapping to the mitochondrion of Metarhizium rileyi strain RCEF 4871 (NCBI accession number: NC_047289.1; Zhang et al. 2020). The alignment was done with Minimap2 v2.17(r941) (Li 2018). Only reads above 8 kb, which mapped with more than 70% of their length was kept. The mitochondrion was assembled using Canu v. 2.0 (Koren et al. 2017) with the settings: “genomeSize = 62k” and “corOutCoverage = 999.” The mitochondrion was attempted circularized using circlator v1.5.5 (Hunt et al. 2015). To facilitate synteny analysis, the mt-genome fasta file was rearranged to start with rnl gene and end with nad6 gene.
Transcriptome Assembly
Adapter and low-quality sequences were trimmed from RNA-seq reads with Trimmomatic v.0.36 (Bolger et al. 2014) and poly-A sequences were clipped from transcripts using SeqClean (available at http://compbio.dfci.harvard.edu/tgi/software/, last accessed April 30, 2021). Reads were aligned to the complete assembled genome (H1 + H2) of M. acridum ARSEF 324 using HISAT2 (Kim et al. 2019), allowing for a maximum intron length of 3 kb. This was followed by clustering of aligned reads using Trinity v2.8.5 (Grabherr et al. 2011) in a genome-guided de novo transcriptomic assembly, using the jaccard clip parameter to reduce transcript fusion. The Trinity assembled transcripts were input to gene prediction training to support genome annotation.
Genome Annotation
A custom repeat library was created by adding de novo identified repeats from RepeatModeler (Hubley et al. 2016) to the repeat databases, Dfam_Consensus-20170127 (Hubley et al. 2016), and RepBase-20170127 (Bao et al. 2015). RepeatModeler was run on the genome masked by these two public databases, and the iteratively growing custom library, using RepeatMasker v4.0.7 (Smit et al. 2015) with the option [-species] set to fungi. RepeatModeler relies on the three de novo repeat finding programs RECON (Bao and Eddy 2002), RepeatScout (Price et al. 2005), and LtrHarvester/Ltr_retriever (Ellinghaus et al. 2008). Repeats were identified using RepeatMasker [option: sensitive] and the generated custom repeat library. The custom library was built on repeat data from the following four Metarhizium strains: M. acridum CQMa 102, M. anisopliae JEF-290, M. brunneum ARSEF 4556, M. rileyi RCEF 4871.
Gene prediction and functional annotation of the polished assembly was conducted using the Funannotate pipeline v1.8.4 (https://github.com/nextgenusfs/funannotate, last accessed April 30, 2021). Repeats were identified with RepeatModeler and soft masked using RepeatMasker. Protein evidence from a UniProtKB/Swiss-Prot-curated database (v2021_01) (Bateman et al. 2021) was aligned to the genomes using TBlastN and Exonerate (Slater and Birney 2005). Two gene prediction tools were used: AUGUSTUS v3.3.3 (Stanke and Morgenstern 2005) and GeneMark-ES v4.33 (Besemer and Borodovsky 2005), with Fusarium graminearum as the model for the AUGUSTUS gene predicter and BRAKER1 (Hoff et al. 2016) for the training of GeneMark-ES. tRNAs were predicted with tRNAscan-SE (Lowe and Eddy 1997). Consensus gene models were found with EvidenceModeler (Haas et al. 2008). Gene models were further updated with the “update” step in funannotate, which uses the tool PASA (Haas et al. 2008) to further extend untranslated regions and identify alternatively spliced isoforms based on alignments of RNA-Seq assembled transcripts. The completeness of the assembled genome was evaluated through comparison to the 4,494 single-copy ortholog genes of the hypocreales_odb10 data set (Creation date: 2020-08-05, https://busco-data.ezlab.org/v5/data/lineages/, last accessed April 30, 2021), using BUSCO v5 (Simão et al. 2015).
Functional annotations for the predicted proteins were obtained using BlastP to search the UniProt/SwissProt protein database (v2021_01). Protein families (Pfam) and Gene Ontology (GO) terms were assigned with InterProScan v5.48-83.0 (Jones et al. 2014). Functional predictions were also inferred by alignments to the eggNOG 5 orthology database (Huerta-Cepas et al. 2019) using emapper v2.0.1b-2-g816e190 (Huerta-Cepas et al. 2017). The secretome was predicted using SignalP v5.0 (Almagro Armenteros et al. 2019) and Phobius v1.01 (Käll et al. 2007), identifying proteins carrying a signal peptide. To further characterize the secretome, putative CAZymes were identified using HMMER v3.3 (Eddy 2011) and family-specific hidden Markov model profiles of dbCAN2 meta server (Lombard et al. 2014). Putative proteases and protease inhibitors were predicted using the MEROPS database (October 04, 2017) (Rawlings et al. 2016). Biosynthetic gene clusters were annotated using strict parameters of the antibiotics and Secondary Metabolites Analysis Shell v5 (antiSMASH) (Blin et al. 2019). All functional predictions and annotations were executed through the Funannotate pipeline.
The guanine-cytosine (GC) content was determined in nonoverlapping windows across the 35 H1 contigs, using seqkit v0.13.2 (seqkit sliding -s 100 -W 100 | seqkit fx2tab –name –gc). Each window was annotated with the gene or repeat annotation (or no annotation), which it primarily overlapped. This was achieved with bedtools v.2.28.0 option [intersect -wa -wb -f 0.51] (Quinlan 2014).
Variant Calling and Chromosome CCNV
The 150 bp PE DNBseq reads were mapped with the Burrow−Wheeler Aligner, BWA-MEM v.0.7.16a (Li 2013) to the primary haplotig (H1) of the assembled nuclear genome, and PCR duplicates were removed from the bam file using samtools v1.10 (markdup –r) (Li et al. 2009). Coverage and mean depth to reference was assessed using samtools (coverage). Allele frequency distribution across the genome was calculated based on SNV called using Bcftools v1.10.2 (Li 2011a). Variants were called using a combination of BCFtools “mpileup” and “call” using a mapping quality filter of 30, a base quality filter of 20, and a minimum depth of 10, together with default parameters including BAQ (Li 2011b). Mean SNV allele ratios were summarized in nonoverlapping 10-kb windows using a custom R script (supplementary script 1, Supplementary Material online). Genome annotations and repeat annotations were combined for the mating-type loci including the flanking APN2 and SLA2 genes, for the two genome assemblies of M. acridum ARSEF 324 and M. acridum CQMa 102 (GenBank accession no. GCA_000187405.1). Synteny between loci was visualized using the R package genoPlotR v0.8.10 (Guy et al. 2010).
Genome Comparison
Single-copy homologous (SCH) genes were identified between H1 and H2, and 11 Metarhizium isolates representing eight species, along with two isolates of Pochonia chlamydosporia and two isolates used as outgroup, that is, Epichloe festucae strain Fl1, and Villosiclava virens strain UV-8b. This was done using OrthoFinder v2.2.7 (Emms and Kelly 2019). The proteomes of the 16 isolates were obtained from NCBI GenBank; accession numbers are available in supplementary table S2, Supplementary Material online. Protein sequence alignment was generated with MAFFT v 7.453 as implemented in OrthoFinder. Substitution models for each gene were predicted using ModelFinder (Kalyaanamoorthy et al. 2017) as implemented in IQtree v2.1.2. A subset of 444 known genes with less than 10% gaps within the alignment was concatenated and a maximum likelihood phylogeny with gene-specific substitution models using IQtree (Nguyen et al. 2015; Chernomor et al. 2016). Per gene maximum likelihood distances were likewise calculated using IQtree and utilized in supplementary figure S3, Supplementary Material online, to summarize the distance between homologous genes.
SCH identified between H1 and H2 had their synonymous point substitution rates estimated with the pairwise alignment tool in subopt-kaks (Stajich 2019) (available at: https://github.com/hyphaltip/subopt-kaks, last accessed April 30, 2021), which implements yn00 pairwise dN/dS estimations (Yang and Nielsen 2000). Estimations are displayed in supplementary figure S4, Supplementary Material online, per gene, along with respective gene ontology annotations. RIP mutations indices were calculated using “The RIPper” (http://theripper.hawk.rocks). Three different indices were calculated, based on dinucleotide frequencies: RIP substrate [(CpA+TpG)/(ApC+GpT)]: 0.75 ≥ x, RIP product [TpA/ApT]: x ≥ 1.1 and RIP composite [(TpA/ApT) – ((CpA + TpG)/(ApC + GpT))]: x ≥ 0, where x are values indicating RIP on a given sequence. Conservative estimates of the RIP affected proportion of the genome are given as regions where all the above indices indicate RIP (fig. 5).
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgements
This work was supported by the Independent Research Fund Denmark through a Sapere Aude Grant to HHDFL. J.E.S. is a CIFAR Fellow in the program Fungal Kingdom: Threats and Opportunities and was supported by the National Science Foundation (NSF DEB 1441715, NSF DEB 1557110), and USDA-NIFA Hatch project (CA-R-PPA-5062-H).
Author Contributions
H.H.D.F.L. initiated the project. M.N. cultured fungi, performed DNA extraction, and initial data QC. K.N.N. and T.K. performed genome assembly, J.F.M.S. and J.S. carried out genome annotation, K.N.N. and J.F.M.S. carried out all other data analyses with input from H.H.D.F.L. and J.S., K.N.N. drafted the manuscript and all authors were involved in the finalization of the manuscript.
Data Availability
Sequencing data and genome assemblies of Metarhizium acridum strain ARSEF 324 are available at DDBJ/ENA/GenBank under the BioProject accession numbers PRJNA714114 and PRJNA714115, and genome accession numbers JAGRQK000000000 and JAGRQL000000000, for the primary genome assembly H1 and the secondary haplotig assembly H2, respectively. Gene expression data used to support genome annotation are available in the Short Read Archive under BioProject PRJNA708346. The DNA for the genome assembly is associated with BioSample accession number: SAMN18235592. Genbank accession numbers of all genomes used in this study are given in supplementary table S2, Supplementary Material online.
Literature Cited
- Aguileta G, et al. 2014. High variability of mitochondrial gene order among fungi. Genome Biol Evol. 6(2):451–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertin W, Marullo P.. 2012. Polyploidy in fungi%: evolution after whole-genome duplication. Proc Biol Sci. 279(1738):2497–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almagro Armenteros JJ, et al. 2019. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol. 37(4):420–423. [DOI] [PubMed] [Google Scholar]
- Arnau J, Oliver RP.. 1993. Inheritance and alteration of transforming DNA during an induced parasexual cycle in the imperfect fungus Cladosporium fulvum. Curr Genet. 23(5 − 6):508–511. [DOI] [PubMed] [Google Scholar]
- Bao W, Kojima K, Kohany O.. 2015. Repbase update, a database of repetitive elements in eukaryotic genomes. Mob DNA 6: 1–11. doi: 10.1186/s13100-015-0041-9. Available from: https://www.girinst.org/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Z, Eddy SR.. 2002. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12:1269–1276. doi: 10.1101/gr.88502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkla BJ, et al. 2018. Making epidermal bladder cells bigger: developmental- and salinity-induced endopolyploidy in a model halophyte. Plant Physiol. 177(2):615–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, et al. ; UniProt Consortium. 2021. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49(D1):D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besemer J, Borodovsky M.. 2005. GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 33:451–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff JF, Rehner SA, Humber RA.. 2009. A multilocus phylogeny of the Metarhizium anisopliae lineage. Mycologia 101(4):512–530. [DOI] [PubMed] [Google Scholar]
- Biskup A, Izmaiłow R.. 2004. Endosperm development in seeds of Echium vulgare L. (Boraginaceae) from polluted sites. Acta Biol Cracoviensia Ser Bot. 46:39–44. [Google Scholar]
- Blin K, et al. 2019. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 47(W1):W81–W87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bodt S, Maere S, Van de Peer Y.. 2005. Genome duplication and the origin of angiosperms. Trends Ecol Evol. 20(11):591–597. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga GUL, Flint SD, Miller CD, Anderson AJ, Roberts DW.. 2001. Variability in response to UV-B among species and strains of Metarhizium isolated from sites at latitudes from 61°N to 54°S. J Invertebr Pathol. 78(2):98–108. [DOI] [PubMed] [Google Scholar]
- Bridges CB. 1919. Duplication. Anat Rec. 15:357–358. [Google Scholar]
- Cambareri E, Singer M, Selker E.. 1991. Recurrence of repeat-induced point mutation (RIP) in Neurospora crassa. Genetics. 127(4):699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomor O, von Haeseler A, Minh BQ.. 2016. Terrace Aware Data Structure for Phylogenomic Inference from Supermatrices. Syst Biol. 65(6):997–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson SJ, Radziejwoski A, Granier C.. 2006. Cell and leaf size plasticity in Arabidopsis: what is the role of endoreduplication? Plant Cell Environ. 29(7):1273–1283. [DOI] [PubMed] [Google Scholar]
- Crow KD, Wagner GP, SMBE Tri-National Young Investigators. 2006. What is the role of genome duplication in the evolution of complexity and diversity? Mol Biol Evol. 23(5):887–892. [DOI] [PubMed] [Google Scholar]
- Dietrich FS, et al. 2004. The Ashbya gossypii Genome as a tool for mapping the ancient Saccharomyces cerevisiae Genome. Science 304(5668):304–307. [DOI] [PubMed] [Google Scholar]
- Dupeyron M, Singh KS, Bass C, Hayward A.. 2019. Evolution of Mutator transposable elements across eukaryotic diversity. Mob DNA 10: 1–12. doi:10.1186/s13100-019-0153-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR. 2011. Accelerated profile HMM searches. PLoS Comput Biol. 7(10):e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellinghaus D, Kurtz S, Willhoeft U.. 2008. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms DM, Kelly S.. 2019. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20(1):238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer RA, et al. 2013. Chromosomal copy number variation, selection and uneven rates of recombination reveal cryptic genome diversity linked to pathogenicity. PLoS Genet. 9(8):e1003703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox DT, Duronio RJ.. 2013. Endoreplication and polyploidy: insights into development and disease. Development 140(1):3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galagan JE, Selker EU.. 2004. RIP: the evolutionary cost of genome defense. Trends Genet. 20(9):417–423. [DOI] [PubMed] [Google Scholar]
- Gao Q, et al. 2011. Genome sequencing and comparative transcriptomics of the model entomopathogenic fungi Metarhizium anisopliae and M. acridum. PLoS Genet. 7(1):e1001264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein AC, Chun HE, Grant A, Otto SP.. 2006. Genomic convergence toward diploidy in Saccharomyces cerevisiae. PLoS Genet. 2(9):e145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladyshev E. 2017. Repeat-induced point mutation and other genome defense mechanisms in fungi. In: Heitman J, et al., editors. The fungal kingdom. Washington (DC): ASM Press. p. 687–699. doi: 10.1128/9781555819583.ch33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass NL, Dementhon K.. 2006. Non-self recognition and programmed cell death in filamentous fungi. Curr Opin Microbiol. 9(6):553–558. [DOI] [PubMed] [Google Scholar]
- Grabherr MG, et al. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29:644–652. doi:10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy L, Roat Kultima J, Andersson SGE.. 2010. genoPlotR: comparative gene and genome visualisation in R. Bioinformatics 26(18):2334–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hane JK, Oliver RP.. 2008. RIPCAL: a tool for alignment-based analysis of repeat-induced point mutations in fungal genomic sequences. BMC Bioinformatics 9:478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, et al. 2008. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9(1):R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldane JBS. 1932. The causes of evolution. London/New York/Toronto (Canada): Longmans, Green [Google Scholar]
- Hickey DA. 1982. Selfish DNA: a sexually transmitted nuclear parasite. Genetics 101(3 − 4):519–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff KJ, Lange S, Lomsadze A, Borodovsky M, Stanke M.. 2016. BRAKER1: unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 32(5):767–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, et al. 2011. Variation in chromosome copy number influences the virulence of Cryptococcus neoformans and occurs in isolates from AIDS patients. BMC Genomics 12:526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, et al. 2014. Trajectory and genomic determinants of fungal-pathogen speciation and host adaptation. Proc Natl Acad Sci U S A. 111(47):16796–16801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubley R, et al. 2016. The Dfam database of repetitive DNA families. Nucleic Acids Res. 44:D81–D89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J, et al. 2019. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47(D1):D309–D314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J, et al. 2017. Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol Biol Evol. 34(8):2115–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt M, et al. 2015. Circlator: automated circularisation of genome assemblies using long sequencing reads. Genome Biol. 16(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Yi W, Chen S, Wang C.. 2021. Empirical support for the pattern of competitive exclusion between insect parasitic fungi. J Fungi. 7(5):385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P, et al. 2014. InterProScan 5: genome-scale protein function classification. Bioinformatics 30(9):1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käll L, Krogh A, Sonnhammer ELL.. 2007. Advantages of combined transmembrane topology and signal peptide prediction-the Phobius web server. Nucleic Acids Res. doi: 10.1093/nar/gkm256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS.. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis M, Birren BW, Lander ES.. 2004. Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature 428(6983):617–624. [DOI] [PubMed] [Google Scholar]
- Kepler RM, Chen Y, Kilcrease J, Shao J, Rehner SA.. 2016. Independent origins of diploidy in Metarhizium. Mycologia 108(6):1091–1103. [DOI] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL.. 2019. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokot M, Długosz M, Deorowicz S.. 2017. KMC 3: counting and manipulating k-mer statistics. Bioinformatics 33(17):2759–2761. [DOI] [PubMed] [Google Scholar]
- Koren S, et al. 2017. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27(5):722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronstad JW, et al. 2011. Expanding fungal pathogenesis: cryptococcus breaks out of the opportunistic box. Nat Rev Microbiol. 9(3):193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal-Bertioli SCM, Butt TM, Peberdy JF, Bertioli DJ.. 2000. Genetic exchange in Metarhizium anisopliae strains coinfecting Phaedon cochleariae is revealed by molecular markers. Mycol Res. 104(4):409–414. [Google Scholar]
- St. Leger RJ, Wang JB.. 2020. Metarhizium: jack of all trades, master of many. Open Biol. 10(12):200307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levasseur A, Pontarotti P.. 2011. The role of duplications in the evolution of genomes highlights the need for evolutionary-based approaches in comparative genomics. Biol Direct. 6:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. ; 1000 Genome Project Data Processing Subgroup. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25(16):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2011a. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27(21):2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2011b. Improving SNP discovery by base alignment quality. Bioinformatics 27(8):1157–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Available from: http://arxiv.org/abs/1303.3997. Accessed April 30, 2021.
- Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34(18):3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisch D. 2015. Mutator and MULE transposons. Microbiol Spectr. 3(2):MDNA3-0032-2014. [DOI] [PubMed] [Google Scholar]
- Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B.. 2014. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42:490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett B, St. Leger RJ.. 2015. Stress is the rule rather than the exception for Metarhizium. Curr Genet. 61(3):253–261. [DOI] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR.. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25(5):955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Conery JS.. 2000. The evolutionary fate and consequences of duplicate genes. Science 290(5494):1151–1155. [DOI] [PubMed] [Google Scholar]
- Marcet-Houben M, Gabaldón T.. 2015. Beyond the whole-genome duplication: phylogenetic evidence for an ancient interspecies hybridisation in the Baker's yeast lineage. PLoS Biol. 13:e1002220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayrose I, Lysak MA.. 2020. The evolution of chromosome numbers: mechanistic models and experimental approaches. Genome Biol. Evol. 13:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz CW. 1947. Duplication of chromosome parts as a factor in evolution. Am Nat. 81(797):81–103. [DOI] [PubMed] [Google Scholar]
- Mongkolsamrit S, et al. 2020. Revisiting Metarhizium and the description of new species from Thailand. Stud Mycol. 95:171–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monjardino P, Smith AG, Jones RJ.. 2006. Zein transcription and endoreduplication in Maise endosperm are differentially affected by heat stress. Crop Sci. 46(6):2581–2589. [Google Scholar]
- Moore D, Robson GD, Trinci APJ.. 2011. 21st century guidebook to fungi. Cambridge: Cambridge University Press. [Google Scholar]
- Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ.. 2015. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol Biol Evol. 32(1):268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S. 1970. Evolution by gene duplication. Berlin, Heidelberg (Germany): Springer. [Google Scholar]
- Okagaki LH, et al. 2010. Cryptococcal cell morphology affects host cell interactions and pathogenicity. PLoS Pathog. 6(6):e1000953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HA. 1995. Somatic mutation favors the evolution of diploidy. Genetics 139(3):1441–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter J, et al. 2018. Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 556(7701):339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Jones NC, Pevzner PA.. 2005. De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl 1):i351–i358. [DOI] [PubMed] [Google Scholar]
- Quinlan AR. 2014. BEDTools: the Swiss-army tool for genome feature analysis. Curr Protoc Bioinformatics 47:11.12.1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranallo-Benavidez TR, Jaron KS, Schatz MC.. 2020. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun. 11(1):1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings ND, Barrett AJ, Finn R.. 2016. Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 44(D1):D343–D350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riba G, Glandard A, Ravelojoana AM, Ferron P.. 1980. Isolement de recombines mitotiques stables de type intermediaire chez Metarhizium anisopliae (Metschnikoff) par hybridation de biotypes sauvages. Comptes Rendus Des Seances l ’Academie Sci. Paris D. 29:657–660. [Google Scholar]
- Roelofs D, et al. 2020. Multi-faceted analysis provides little evidence for recurrent whole-genome duplications during hexapod evolution. BMC Biol. 18(1):57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum EB, et al. 2013. Complex history of the amphibian-killing chytrid fungus revealed with genome resequencing data. Proc Natl Acad Sci U S A. 110(23):9385–9390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacerdot C, Louis A, Bon C, Berthelot C, Roest Crollius H.. 2018. Chromosome evolution at the origin of the ancestral vertebrate genome. Genome Biol. 19(1):166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW.. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 9(7):671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholes DR, Paige KN.. 2015. Plasticity in ploidy: a generalised response to stress. Trends Plant Sci. 20(3):165–175. [DOI] [PubMed] [Google Scholar]
- Selker EU, Garrett PW.. 1988. DNA sequence duplications trigger gene inactivation in Neurospora crassa. Proc Natl Acad Sci U S A. 85(18):6870–6874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sher N, et al. 2013. Fundamental differences in endoreplication in mammals and Drosophila revealed by analysis of endocycling and endomitotic cells. Proc Natl Acad Sci U S A. 110(23):9368–9373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM.. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31(19):3210–3212. [DOI] [PubMed] [Google Scholar]
- Sionov E, Lee H, Chang YC, Kwon-Chung KJ.. 2010. Cryptococcus neoformans overcomes stress of azole drugs by formation of disomy in specific multiple chromosomes. PLoS Pathog. 6(4):e1000848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater GSC, Birney E.. 2005. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6(1):31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit A, Hubley R, Green P.. 2015. RepeatMasker. Available from: http://www.repeatmasker.org.
- Stajich JE. 2019. subopt-kaks v1.0.0. Zenodo. doi:10.5281/zenodo.2548016. Available from: https://github.com/hyphaltip/subopt-kaks. Accessed April 30, 2021.
- Stanke M, Morgenstern B.. 2005. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33:465–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watters MK, Randall TA, Margolin BS, Selker EU, Stadler DR.. 1999. Action of repeat-induced point mutation on both strands of a duplex and on tandem duplications of various sizes in Neurospora. Genetics 153(2):705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, St. Leger RJ.. 2005. Developmental and transcriptional responses to host and nonhost cuticles by the specific locust pathogen Metarhizium anisopliae var. acridum. Eukaryot Cell 4(5):937–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, O'Brien TR, Pava-Ripoll M, St. Leger RJ.. 2011. Local adaptation of an introduced transgenic insect fungal pathogen due to new beneficial mutations. Proc Natl Acad Sci U S A. 108(51):20449–20454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe KH, Shields DC.. 1997. Molecular evidence for an ancient duplication of the entire yeast genome. Nature 387(6634):708–713. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R.. 2000. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol. 17(1):32–43. [DOI] [PubMed] [Google Scholar]
- Zaragoza O, et al. 2010. Fungal cell gigantism during mammalian infection. PLoS Pathog. 6(6):e1000945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Ren LY, Zhang YJ.. 2020. Complete mitogenome of the entomopathogenic fungus Metarhizium rileyi. Mitochondrial DNA Part B Resour. 5(2):1494–1495. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data and genome assemblies of Metarhizium acridum strain ARSEF 324 are available at DDBJ/ENA/GenBank under the BioProject accession numbers PRJNA714114 and PRJNA714115, and genome accession numbers JAGRQK000000000 and JAGRQL000000000, for the primary genome assembly H1 and the secondary haplotig assembly H2, respectively. Gene expression data used to support genome annotation are available in the Short Read Archive under BioProject PRJNA708346. The DNA for the genome assembly is associated with BioSample accession number: SAMN18235592. Genbank accession numbers of all genomes used in this study are given in supplementary table S2, Supplementary Material online.