

Abstract
Systemic lupus erythematosus (SLE) is an autoimmune-mediated diffuse connective tissue disease characterized by immune inflammation with an unclear aetiology and pathogenesis. This work profiled the intestinal flora and faecal metabolome of patients with SLE using 16S RNA sequencing and gas chromatography-mass spectrometry (GC-MS). We identified unchanged alpha diversity and partially altered beta diversity of the intestinal flora. Another important finding was the increase in Proteobacteria and Enterobacteriales and the decrease in Ruminococcaceae among SLE patients. For metabolites, amino acids and short-chain fatty acids were enriched when long-chain fatty acids were downregulated in SLE faecal samples. KEGG analysis showed the significance of the protein digestion and absorption pathway, and association analysis revealed the key role of 3-phenylpropanoic acid and Sphingomonas. Sphingomonas were reported to be less abundant in healthy periodontal sites of SLE patients than in those of HCs, indicating transmission of oral species to the gut. This study contributes to the understanding of the pathogenesis of SLE disease from the perspective of intestinal microorganisms, explains the pathogenesis of SLE, and serves as a basis for exploring potential treatments for the disease.
1. Introduction
Systemic lupus erythematosus (SLE) is an autoimmune-mediated diffuse connective tissue disease characterized by immune inflammation [1]. The presence of antinuclear antibodies in the serum and multisystem involvement are the two main clinical features of SLE, and the incidence rate of SLE in women is significantly higher than that in men [2]. Lupus nephritis (LN) occurs when SLE is complicated by renal damage. Approximately 60% of SLE patients suffer from LN complications that cause a higher mortality than SLE patients without LN [3]. At present, the aetiology and pathogenesis of SLE and LN are still unclear [4]. Studies have shown that the occurrence and development of some autoimmune diseases, such as inflammatory bowel disease [5, 6], rheumatoid arthritis [7, 8], and SLE [9, 10], are correlated with changes in intestinal microecology. Some studies found that the intestinal microbes of patients or animals with SLE were different from those of the control group through experimental models and clinical models [11–13], and the reconstruction of intestinal microecology through diet and drug regulation could help improve the severity of the disease [14]. There are many hypotheses on the interaction mechanism between intestinal microbes and the host and its relationship with SLE, but there are no mature theories yet. Currently, an increasing number of studies have shown that metabolites of the intestinal flora, such as short-chain fatty acids (SCFAs), free fatty acids (FFAs), amino acids, and arachidonic acid, correlate with autoimmune reactions [14–16]. However, the relationship between microbial metabolites and SLE remains unknown, and previously published studies are limited to adult patients. In this study, we collected the intestinal flora and metabolite data of 33 SLE children and 28 healthy controls (HCs) to clarify the pathogenesis of the disease from the perspective of intestinal microorganisms as much as possible, explain the pathogenesis of SLE, and serve as a basis for exploring potential treatments for the disease.
2. Material and Methods
2.1. Ethical Statement
All enrolled patients and healthy controls understood the sampling process and study plan in detail before sampling and signed informed consent forms. Ethical approval for this study was obtained from the Ethics Committee of the Second Xiangya Hospital of Central South University.
2.2. Study Subjects
A total of 28 healthy controls and 33 SLE patients were enrolled from the Second Xiangya Hospital of Central South University between December 2018 and April 2019. The diagnostic criteria of SLE were consistent with SLE classification criteria 2 revised by American College Rheumatology (ACR) in 1997 [17]. SLE patients were given hormone and/or immunosuppressive therapy. None of the selected subjects had applied or taken antibiotics, probiotics, symbiotic, or other microecological preparations for at least 3 months before sampling.
2.3. 16S rRNA Microbiota Analysis
Total genomic DNA from samples was extracted using the CTAB/SDS method. 16S/18S rRNA genes were amplified using primers including 16S V4-V5 : 515F-907R, 18S V9 : 1380F-1510R, and ITS1 : ITS1F-ITS2R. All PCRs were carried out in 30 μl reactions with 15 μl of Phusion® High-Fidelity PCR Master Mix (New England Biolabs), 0.2 μM forward and reverse primers, and approximately 10 ng of template DNA. Thermal cycling consisted of initial denaturation at 98°C for 1 min, 30 cycles of denaturation at 98°C for 10 s, annealing at 50°C for 30 s, and elongation at 72°C for 60 s then 5 min. Electrophoresis was performed on a 2% agarose gel. PCR products were purified with GeneJET Gel Extraction Kit (Thermo Scientific). Sequencing libraries were generated using the NEBNext® Ultra™ DNA Library Prep Kit for Illumina (NEB, USA), and the library quality was assessed on the Qubit@ 2.0 Fluorometer (Thermo Scientific) and Agilent Bioanalyzer 2100 system. The library was sequenced on an Illumina MiSeq platform.
A total of 5507169 sequences were used for analysis. Sequence analysis was performed by the UPARSE software package using the UPARSE-OTU and UPARSE-OTUref algorithms. In-house Perl scripts were used to analyse the alpha diversity and beta diversity. Cluster analysis was preceded by principal component analysis (PCA). QIIME calculates both weighted and unweighted UniFrac distances. We used unweighted UniFrac distance for principal coordinate analysis (PCoA) and unweighted pair group method with arithmetic mean (UPGMA) clustering. Metastats software was utilized to confirm differences in the abundances of individual taxa between the two groups, and LEfSe was used for the quantitative analysis of biomarkers within different groups. To identify differences in microbial communities between the two groups, ANOSIM [18] and MRPP (multiresponse permutation procedure) [19] were performed based on Bray-Curtis dissimilarity distance matrices.
2.4. GC-MS Analysis
The instrumental status and balance as well as the stability of the gas chromatography-mass spectrometry system were evaluated using QC samples. Samples (60 mg) were mixed with 200 μg water and vortexed for 60 s before being added to 800 μl methanol acetonitrile (Merck, 1499230-935) solution (1 : 1, v/v), followed by vortexing for 60 s and low-temperature ultrasound for 30 min twice. Samples were stored at -20°C for 1 h to precipitate protein. Finally, the samples were centrifuged for 20 min at 14000 RCF at 4°C, and the supernatant liquor was collected for freeze drying and stored at -80°C.
For GC analysis, the samples were separated by an Agilent 1290 Infinity LC ultra-high-performance liquid chromatography (UHPLC) HILIC column at 25°C with a flow rate of 0.3 ml/min. The mobile phase was the composite of A (water, 25 mm ammonium acetate, 25 mm ammonium) and B (acetonitrile). Gradient elution procedures were as follows: B was 95% in 0-1 min, then changed linearly from 95% to 65% in 1-14 min and 65% to 40% in 14-16 min, and maintained at 40% in 16-18 min before changing linearly from 40% to 95% in 18-18.1 min, maintaining at 95% in 18.1-23 min. The samples were placed in an automatic sampler at 4°C throughout the analysis. A random sequence was used for the continuous analysis. QC samples were inserted into the sample queue for monitoring and evaluation of the system.
For Q-TOF MS analysis, electrospray ionized positive ions and negative ions were detected. The samples were separated by UHPLC and analysed by a Triple TOF 5600 mass spectrometer (AB SCIEX). The ESI source conditions after HILIC chromatographic separation were as follows: ion source gas 1 (Gas1): 60, ion source gas 2 (Gas2): 60, curtain gas (CUR): 30, source temperature: 600°C, ion spray voltage floating (ISVF) ±5500 V (positive and negative modes), TOF MS scan m/zrange: 60-1000 Da, product ion scan m/zrange: 25-1000 Da, TOF MS scan accumulation time 0.20 s/spectra, and product ion scan accumulation time 0.05 s/spectra. The second-order mass spectrum was obtained by information-dependent acquisition (IDA) with a high sensitivity mode, a declustering potential (DP): of ±60 V (positive and negative modes), collision energy of 35 ± 15 eV, and IDA excluded isotopes within 4 Da with 6 candidate ions to monitor per cycle.
The original data were converted into the mzXML format by ProteoWizard, and then, the XCMS program was used for peak alignment, retention time correction, and peak area extraction. Accurate mass number matching (<25 ppm) and secondary spectrogram matching were used to identify the metabolite structure, and the laboratory database was retrieved. After pretreatment by Pareto scaling, multidimensional statistical analysis was performed, including principal component analysis (PCA), supervised partial least square discriminant analysis (PLS-DA), and orthogonal partial least square discriminant analysis (OPLS-DA). One-dimensional statistical analysis included Student's t-test and multiple analysis of variation.
2.5. Association Analysis
The correlation analysis is mainly based on statistical algorithms to find the correlation between significantly different flora (p < 0.05) and the significantly different metabolites obtained through nontarget metabolomics analysis. Principal component analysis (PCA) and multivariate statistical analysis were performed with SIMCA version 14.1. KEGG pathway analysis was based on the online Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.kegg.jp/). Differentially abundant proteins/modified peptides/genes/and metabolites/lipids were log2 scaled (TMT/iTRAQ) or Z-score scaled (label free) and concatenated into one matrix. Then, the correlation coefficients among all the molecules in the matrix were calculated with the Pearson algorithm in R version 3.5.1. Cytoscape version 3.5.1 was used to calculate the correlation network.
3. Results
3.1. Clinical Features of SLE Patients
The basic characteristics of the participants at the time point of sample collection are listed in Table 1, including their sex, age, disease activity parameters, autoantibody status, and treatment. According to two-tailed unpaired Student's t-test, BMI was not significantly different between the SLE group and HC group (p = 0.234). All the children in the SLE group were also diagnosed with LN.
Table 1.
Characteristics of the study cohorts.
| SLE | HC | |
|---|---|---|
| Fecal samples | 33 | 28 |
| Female | 26 (78.79%) | 14 (50%) |
| Age, years, mean ± SD | 12.39 ± 2.40 | 10.61 ± 3.67 |
| BMI, kg/m2, mean ± SD | 18.79 ± 2.30 | 18.57 ± 4.51 |
| DISEASE ACTIVITY PARAMETERS IN SERUM | ||
| Elevated ESR, mm/h | 11 (33.33%) | |
| Elevated CRP, mg/L | 1 (0.30%) | |
| Reduced complement C3 | 18 (54.55%) | |
| Reduced complement C4 | 17 (51.52%) | |
| Positive ANA | 21 (63.64%) | |
| Positive ANCA | 2 (6.06%) | |
| Positive C1q | 16 (48.48%) | |
| SLEDAI SCORE | ||
| No or mild activity (0-4) | 22 (66.67%) | |
| Moderate activity (5-9) | 2 (6.06%) | |
| High activity (10-14) | 9 (27.27%) | |
| Very high activity (≥15) | 0 | |
| TREATMENT | ||
| GCs + HCQ | 3 (9.09%) | |
| GCs + HCQ + CTX | 12 (36.36%) | |
| GCs + HCQ + CsA + CTX | 2 (6.06%) | |
| GCs + HCQ + MMF | 15 (45.45%) | |
| GCs + HCQ + CTX + MMF | 1 (3.03%) | |
SD: standard deviation, ESR: erythrocyte sedimentation rate, CRP: C-reactive protein, ANA: antinuclear antibody, ANCA: antineutrophil cytoplasmic antibody, C1q: anti-C1q antibody. GCs: glucocorticoids, HCQ: hydroxychloroquine, CTX: cyclophosphamide, CsA: cyclosporin A, MMF: mycophenolate mofetil.
3.2. Intestinal Flora Is Different between SLE Patients and Healthy Groups
After data optimization, the average length of 99.87% of the sequence bars was within the range of 401-450 bp, and a total of 2,159 operational taxonomic units (OTUs) were retained for subsequent analysis.
Using a t-test, we compared the Chao1 index (p = 0.327) of the two groups and found no significant difference, suggesting that the species abundance does not change in SLE patients. Additionally, Shannon (p = 0.214), coverage (p = 0.988), and Simpson (p = 0.414) indexes that estimate community diversity also showed similar results. Taken together, these results show that the richness of the intestinal flora did not change among SLE patients.
As we can see in Figure 1, the sample distance of the control vs. control was significantly shorter than SLE vs. SLE group and control vs. SLE group (p < 0.05). Interestingly, although the alpha diversity of SLE patients remained unchanged compared with that of healthy controls, the sample distance of the former was obviously longer, suggesting healthy children possess microbiota with more similar species composition and the heterogeneity of the microbiota among SLE patients.
Figure 1.
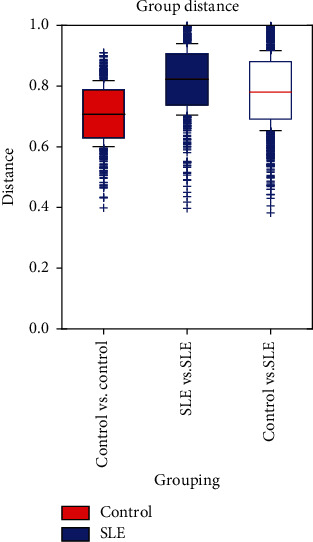
Bray-Curtis statistical algorithm was used to calculate the distance between each sample and obtain the distance matrix. The distance between the SLE group and the HC group with quartiles was calculated, and a box diagram of the distance between the two groups was drawn to compare the distance distribution differences. Student's two-sample t-test method was used to compare the distance between each group.
A scatter plot based on principal coordinates analysis (PCoA) revealed a clear separation between the SLE and HC groups (Figure 2). It should be highlighted that a principal component analysis (PCA) of SLE and HC subjects, based on 16 rRNA microbiota profiles, did not show distinct clustering patterns. We witnessed the association between gastrointestinal microbiota and immune factors at the level of bacterial composition but not at the level of the metabolite landscape of gut microbiota, which suggests that SLE can influence the heterogeneous species inhabiting the gut but not the metabolic pattern [20].
Figure 2.

PCoA analysis and PCA analysis were conducted by R version 3.5.1. The red dots represent health controls, and the blue dots represent systemic lupus erythematosus patient samples. PCoA analysis sorts a series of eigenvalues and eigenvectors. It selects the most important eigenvalues ranked in the first few and shows them in the coordinate system. The result can be regarded as a rotation of the distance matrix. The red dots and the blue dots distribute in different clusters on the coordinate axis, revealing a clear separation between the SLE and HC groups. PCA analysis uses variance decomposition to reflect the differences of multiple groups of data on the two-dimensional coordinate graph. The PCA analysis of our study did not show clear separation.
Twenty phyla were detected, among which Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria, Verrucomicrobia, Fusobacteria, and Synergistetes were the most abundant, with Firmicutes being the most dominant. At the genus level, a total of 380 genera were detected in all the samples, and the top 10 genera with the largest abundance were Faecalibacterium (average abundance was 17.26%), Agathobacter (6.23%), Roseburia (5.45%), Megamonas (5.37%), Lachnoclostridium (3.45%), Subdoligranululum (2.58%), Bacteroides of Bacteroidetes (7.14%), Prevotella 9 (5.46%), Escherichia-Shigella (Proteobacteria, average abundance was 3.97%), and Bifidobacterium (Actinomycetes, average abundance was 2.79%).
Differently abundant species in both groups are listed in Table 2. There were a total of 54 bacteria, including 1 phylum, 3 classes, 5 orders, 8 families, and 37 genera. Proteobacteria was the only different bacterium at the phylum level, increasing in SLE patients. Gammaproteobacteria, Bacilli, Enterobacteriaceae, Escherichia_Shigella, Ruminococcus_gnavus_group, Lachnoclostridium, and Kluyvera were also more abundant in SLE patients, while Ruminococcaceae, Agathobacter, Ruminococcus_2, Coprococcus_2, Eubacterium_coprostanoligenes_group, Dialister, Faecalibacterium, and Subdoligranulum were distinctly richer among the healthy controls.
Table 2.
Different species.
| Flora category | Log value of abundance | Group | LDA value | p value | Level |
|---|---|---|---|---|---|
| Proteobacteria | 4.967 | SLE | 4.49 | 0.007 | Phylum |
| Alphaproteobacteria | 2.883 | SLE | 3.255 | 0 | Class |
| Gammaproteobacteria | 4.95061 | SLE | 4.486 | 0.007 | Class |
| Bacilli | 4.154298 | SLE | 3.656 | 0.006 | Class |
| Enterobacteriales | 4.92 | SLE | 4.495 | 0.006 | Order |
| Xanthomonadales | 2.178 | SLE | 3.563 | 0 | Order |
| Caulobacterales | 1.81 | SLE | 3.61 | 0.001 | Order |
| Sphingomonadales | 2.497641 | SLE | 3.013 | 0 | Order |
| Lactobacillales | 4.152046 | SLE | 3.655 | 0.008 | Order |
| Ruminococcaceae | 5.556 | HC | 4.821 | 0.005 | Family |
| Streptococcaceae | 3.838 | SLE | 3.284 | 0.017 | Family |
| Christensenellaceae | 3.909 | HC | 3.377 | 0.04 | Family |
| Caulobacteraceae | 1.788 | SLE | 3.589 | 0.001 | Family |
| Enterobacteriaceae | 4.920289 | SLE | 4.495 | 0.006 | Family |
| Family_XIII | 2.853446 | HC | 2.797 | 0.036 | Family |
| Sphingomonadaceae | 2.497641 | SLE | 3.021 | 0 | Family |
| Rhodanobacteraceae | 1.816683 | SLE | 3.679 | 0 | Family |
| Erysipelotrichaceae_UCG_003 | 3.358 | HC | 2.768 | 0.003 | Genus |
| Lachnospiraceae_UCG_008 | 1.772055 | HC | 3.586 | 0.043 | Genus |
| Lachnospiraceae_UCG_004 | 3.600504 | HC | 2.964 | 0.025 | Genus |
| Ruminococcaceae_NK4A214_group | 3.618 | HC | 3.006 | 0.006 | Genus |
| Lachnospiraceae_FCS020_group | 2.815 | HC | 2.738 | 0.033 | Genus |
| Family_XIII.Family_XIII_UCG_001 | 2.302 | HC | 3.209 | 0.01 | Genus |
| Ruminococcaceae_UCG_013 | 3.423 | HC | 2.885 | 0.002 | Genus |
| Ruminococcaceae_UCG_010 | 3.021 | HC | 2.775 | 0.03 | Genus |
| Lachnospiraceae_NC2004_group | 2.246211 | HC | 3.241 | 0.034 | Genus |
| Christensenellaceae_R_7_group | 3.89737 | HC | 3.376 | 0.021 | Genus |
| Family_XIII_AD3011_group | 2.580169 | HC | 3.234 | 0.037 | Genus |
| Lachnospiraceae_NK4A136_group | 3.869956 | HC | 3.367 | 0.001 | Genus |
| Escherichia_Shigella | 4.809 | SLE | 4.393 | 0.019 | Genus |
| Agathobacter | 4.932 | HC | 4.313 | 0.004 | Genus |
| Akkermansiaceae | 3.328 | HC | 2.979 | 0.007 | Genus |
| Ruminococcus_2 | 4.314 | HC | 3.767 | 0.049 | Genus |
| Clostridium_innocuum_group | 2.778 | SLE | 2.938 | 0.037 | Genus |
| Fusicatenibacter | 3.892 | HC | 3.342 | 0.019 | Genus |
| Streptococcus | 3.829 | SLE | 3.271 | 0.032 | Genus |
| Dorea | 3.792 | HC | 3.122 | 0 | Genus |
| Coprococcus_2 | 4.047 | HC | 3.686 | 0 | Genus |
| Hungatella | 3.387 | SLE | 3.087 | 0.033 | Genus |
| Erysipelatoclostridium | 3.226 | SLE | 2.98 | 0.017 | Genus |
| Eubacterium_coprostanoligenes_group | 4.435 | HC | 3.809 | 0.008 | Genus |
| Ruminococcus_gnavus_group | 4.308 | SLE | 3.958 | 0.003 | Genus |
| Dialister | 4.603 | HC | 4.101 | 0.001 | Genus |
| Klebsiella | 3.669467 | SLE | 3.274 | 0 | Genus |
| Lachnoclostridium | 4.698468 | SLE | 4.244 | 0.027 | Genus |
| Faecalibacterium | 5.329871 | HC | 4.598 | 0.015 | Genus |
| Ruminiclostridium_5 | 3.464955 | HC | 2.995 | 0.014 | Genus |
| Rudaea | 1.816683 | SLE | 3.68 | 0 | Genus |
| Sphingomonas | 2.425476 | SLE | 3.099 | 0 | Genus |
| Kluyvera | 4.006708 | SLE | 3.48 | 0.001 | Genus |
| Subdoligranulum | 4.57793 | HC | 4.024 | 0.012 | Genus |
| Odoribacter | 2.434041 | HC | 3.256 | 0.048 | Genus |
| Akkermansia | 3.327618 | HC | 2.973 | 0.007 | Genus |
| Eubacterium_eligens_group | 3.929 | HC | 3.478 | 0.022 | Genus |
SLE: systemic lupus erythematosus group; HC: health control group; LDA: linear discriminant analysis.
Using LEfSe software, a nonparametric factorial Kruskal-Wallis (KW) sum-rank test was conducted to identify species with distinct abundance differences before undertaking linear discriminant analysis (LDA) to estimate the influence of each component (species) on the difference. The difference in microflora from phylum to genus was determined by an LDA value greater than 2 and a p value less than 0.05 (Table 2). We can see that for SLE patients, Proteobacteria at the phylum level, Gammaproteobacteria at the class level, Enterobacteriales at the order level, and Enterobacteriaceae at the family level are the most relevant components, and they possess a subordinate relationship. On the other hand, Ruminococcaceae, Christensenellaceae, and Family_XIII at the family level and Akkermansiaceae at the genus level were abundant in the healthy controls (Figure 3).
Figure 3.
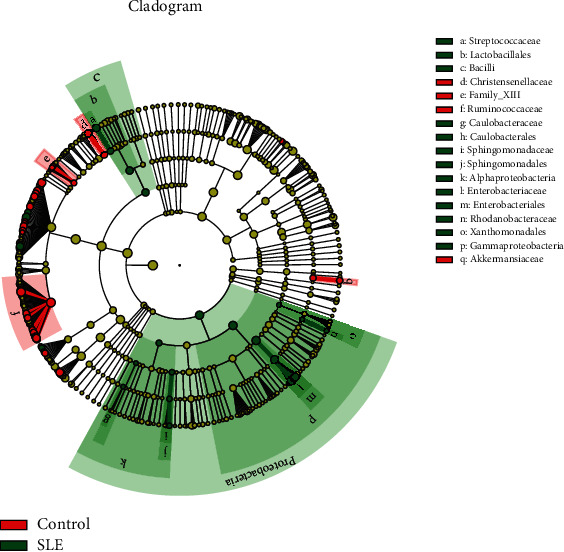
Differential analysis using LEfSe software. Red nodes represent the microbial group that plays an important role in the HC group, green nodes represent the microbial group that plays an important role in the SLE group, and yellow nodes represent the microbial group that does not play an important role in both groups.
The nonparametric factorial Kruskal-Wallis sum-rank test was used for calculating the difference of abundance. Log value of abundance reflects the abundance of the different species. LDA value estimates the influence of the abundance of each component (species) on the overall abundance differences. p < 0.05 and LDA > 2 represent significantly different species.
3.3. Different Metabolites' Analysis by GC-MS
Variable importance for the projection (VIP) obtained from the OPLS-DA model was used to measure the influence intensity and explanatory ability of the expression pattern of each metabolite on the classification and discrimination of samples of each group, and the different metabolites with biological significance were excavated.
This study defined metabolites with VIP > 1 in the multidimensional statistical analysis and p < 0.05 in univariate statistical analysis as significantly different metabolites, including 24 in positive ion mode and 37 in anion mode. Ruling out 6 overlapping data, there were a total of 55 significantly different metabolites (Table 3), including mainly long-chain fatty acids and amino acids. Among them, cyclohexylsulfamate had the highest VIP value, indicating that its decline had a greater impact on the occurrence of SLE diseases.
Table 3.
Significantly different metabolites.
| Description | VIP | Fold change (SLE/HC) | p value | m/z | rt (s) |
|---|---|---|---|---|---|
| (S)-2-Aminobutyric acid | 1.134457 | 7.300207 | 0.027901 | 102.0559 | 714.909 |
| (Z)-6-Octadecenoic acid | 1.460343 | 0.604532 | 0.00674 | 282.2502 | 91.72 |
| 16-Hydroxypalmitic acid | 1.874055 | 0.499351 | 0.009079 | 314.2681 | 92.142 |
| 1-Oleoyl-sn-glycero-3-phosphocholine | 1.904087 | 0.331859 | 0.032946 | 522.3537 | 354.483 |
| 1-Palmitoyl-2-hydroxy-sn-glycero-3-phosphoethanolamine | 2.796585 | 0.225656 | 7.95E − 06 | 454.2913 | 372.123 |
| 2-Hydroxy-3-methylbutyric acid | 4.457651 | 4.059644 | 0.024633 | 117.0556 | 283.2115 |
| 2-Hydroxy-butanoic acid | 1.365271 | 2.252586 | 0.000967 | 103.0398 | 364.888 |
| 3-(3-Hydroxyphenyl)propanoic acid | 7.504876 | 3.292347 | 0.042029 | 165.0556 | 213.446 |
| 3-Phenylpropanoic acid | 8.186158 | 0.482206 | 0.018599 | 149.0607 | 197.716 |
| 5,10-Methylene-THF | 2.02514 | 5.177854 | 0.038319 | 456.1695 | 81.174 |
| 9,10-DiHOME | 1.759624 | 0.622241 | 0.018751 | 313.2384 | 197.908 |
| Acetyl-DL-leucine | 2.436625 | 3.258067 | 0.020322 | 172.0978 | 372.417 |
| all cis-(6,9,12)-Linolenic acid | 1.996995 | 0.552529 | 0.003271 | 279.2309 | 91.4225 |
| Alpha-linolenic acid | 1.125754 | 0.495219 | 0.027657 | 296.2578 | 85.836 |
| Alpha-N-phenylacetyl-L-glutamine | 1.032981 | 6.993584 | 0.03739 | 263.1035 | 434.981 |
| Bisindolylmaleimide I | 1.251916 | 0.533825 | 0.034091 | 411.1837 | 60.485 |
| cis-9-Palmitoleic acid | 2.112812 | 0.530253 | 0.010487 | 315.2523 | 157.6505 |
| Cyclohexylsulfamate | 18.63932 | 0.399938 | 0.01068 | 178.0541 | 134.362 |
| Daidzein | 1.860198 | 0.37232 | 0.049911 | 253.0506 | 90.079 |
| DL-Indole-3-lactic acid | 1.389089 | 1.868281 | 2.75E − 05 | 188.0698 | 482.9055 |
| DL-Methionine sulfoxide | 1.188165 | 1.570971 | 0.013233 | 164.0387 | 705.022 |
| Enalapril | 2.770035 | 3.292851 | 0.02267 | 375.1845 | 335.5975 |
| Formononetin | 1.159497 | 12.16737 | 0.031198 | 269.08 | 75.902 |
| Formylanthranilic acid | 1.108129 | 1.780982 | 0.030492 | 164.035 | 124.961 |
| Gamma-L-glutamyl-L-valine | 1.260303 | 1.995892 | 0.001099 | 245.1144 | 738.78 |
| Glycyl-L-leucine | 1.714816 | 1.468498 | 0.009308 | 187.1089 | 549.388 |
| Hydrocortisone (cortisol) | 2.126813 | 6.47518 | 0.003054 | 421.2224 | 94.857 |
| Hydroxyisocaproic acid | 10.89455 | 4.366298 | 0.014686 | 131.0711 | 244.439 |
| L-Alanine | 1.917025 | 1.859043 | 0.000362 | 88.04036 | 663.2525 |
| L-Glutamine | 1.81976 | 2.137778 | 0.00051 | 145.0619 | 715.39 |
| Linoleic acid | 3.644522 | 0.377719 | 4.77E − 06 | 298.2736 | 84.196 |
| L-Isoleucine | 1.163985 | 4.459251 | 0.006748 | 261.182 | 497.738 |
| L-Leucine | 7.61484 | 1.835031 | 9.53E − 05 | 130.0874 | 497.981 |
| L-Methionine | 2.024436 | 2.05734 | 0.005488 | 148.0437 | 536.5665 |
| L-Norleucine | 1.193737 | 5.606042 | 0.047861 | 263.1959 | 493.5235 |
| L-Phenylalanine | 6.291687 | 1.785839 | 0.000231 | 164.072 | 482.6735 |
| L-Tryptophan | 2.780279 | 1.785305 | 2.45E − 05 | 203.0828 | 482.5715 |
| L-Tyrosine | 3.001494 | 1.658265 | 0.006866 | 180.0666 | 569.619 |
| L-Valine | 3.438313 | 3.006204 | 0.000931 | 116.0716 | 574.471 |
| Methoprene (S) | 1.236084 | 0.334761 | 0.004683 | 328.2844 | 64.9405 |
| MG(18 : 2(9Z,12Z)/0 : 0/0 : 0)[rac] | 3.617466 | 0.313071 | 7.55E − 05 | 355.2834 | 69.542 |
| N-Acetyl-L-aspartic acid | 1.020191 | 0.48076 | 0.016099 | 174.0408 | 773.394 |
| N-Acetylneuraminic acid | 1.083761 | 0.539489 | 0.027489 | 310.1126 | 716.366 |
| Nicotinate | 1.873086 | 0.543751 | 0.001525 | 124.0383 | 412.64 |
| Oleic acid | 1.514581 | 0.359758 | 0.001028 | 265.2517 | 120.4355 |
| Oxindole | 1.01039 | 0.407327 | 0.033063 | 134.0594 | 80.262 |
| Palmitic acid | 4.166731 | 0.485449 | 0.035091 | 255.2329 | 88.202 |
| p-Hydroxyphenylacetic acid | 1.625958 | 2.672582 | 0.032862 | 151.0399 | 386.525 |
| Pregnenolone sulfate | 1.984757 | 0.31761 | 0.008648 | 395.1888 | 55.708 |
| Stearidonic acid | 2.760427 | 23.20345 | 0.040155 | 337.2366 | 372.7625 |
| Sunitinib | 3.289336 | 0.283469 | 0.002701 | 397.2046 | 61.461 |
| trans-Cinnamate | 1.407338 | 1.766262 | 0.000204 | 147.045 | 482.794 |
| trans-Vaccenic acid | 3.069738 | 0.496621 | 0.012385 | 283.2626 | 93.176 |
| Tridecanoic acid (tridecylic acid) | 1.351245 | 0.338885 | 0.032456 | 213.186 | 91.512 |
| Tyramine | 2.011381 | 1.73279 | 0.001232 | 120.0798 | 479.944 |
| Vanillin | 1.308743 | 1.345909 | 0.016492 | 151.0398 | 75.058 |
VIP: variable importance for the projection; m/z: mass-to-charge ratio; rt (s): retention time. The VIP value was calculated based on orthogonal partial least squares discriminant analysis to measure the influence and explanatory ability of each metabolite expression pattern on the classification discrimination of each group of samples, and to excavate the different metabolites with biological significance. Fold change describes the ratio of metabolite content (SLE/HC). p value < 0.05 represents that metabolites have significant difference in one-way ANOVA or two-way ANOVA analysis.
Of the 55 SLE-altered metabolites submitted to the KEGG (Kyoto Encyclopedia of Genes and Genomes) website (http://www.kegg.jp/), we were able to match 27 of them with 66 pathways. The p value represents the significance of enrichment of the KEGG pathway, and the number of differentially expressed metabolites contained in the KEGG pathway reflects the degree of influence of SLE on each pathway to some extent (Figure 4). Taking two factors into consideration, protein digestion and absorption (p = 1.75E − 9) were the most significant among all these pathways, involving L-tryptophan, tyramine, L-phenylalanine, L-leucine, L-methionine, L-alanine, L-glutamine, L-valine, L-isoleucine, and L-tyrosine.
Figure 4.
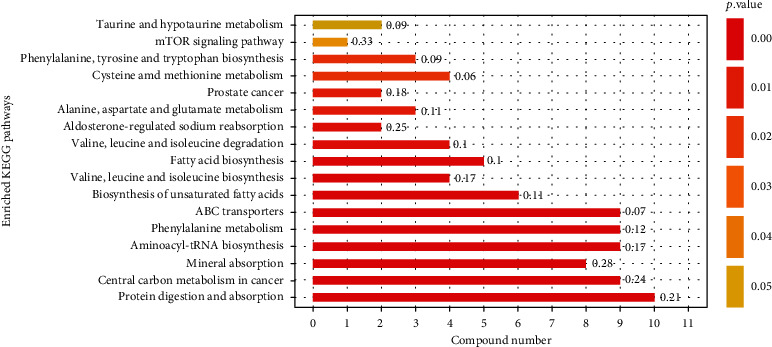
KEGG pathways. Numbers on the right of the bar are rich factor, reflecting the degree of enrichment (rich factor = the number of genes enriched in this pathway/the number of all genes in this pathway).
The network of metabolites and intestinal flora using Cytoscape version 3.5.1 is shown in Figure 5. We can see that the most central location belongs to 3-phenylpropanoic acid, which exhibits a strong positive correlation with Family XIII UCG-001 and a negative correlation with all 5 differentially expressed genera of Proteobacteria. Sphingomonas of the Proteobacteria phylum plays the most important role as a bacterium, and it is obviously positively associated with all 10 metabolites in the protein digestion and absorption pathway mentioned above.
Figure 5.
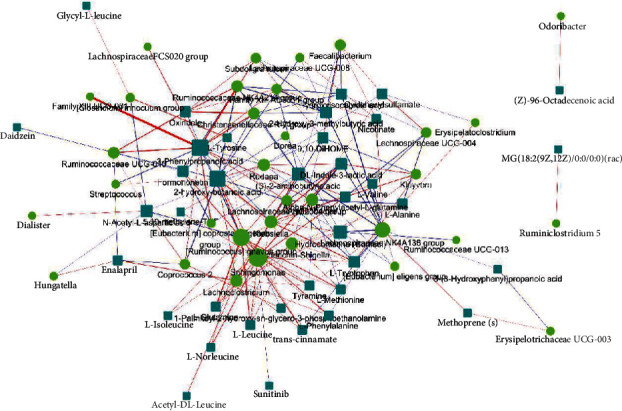
Circles represent significantly different genera, and rectangles represent significantly different metabolites. The color of the line represents the positive and negative value of correlation coefficient (blue represents a negative correlation and red represents a positive correlation), and the thickness of the line is proportional to the absolute value of the correlation coefficient. The node size is positively correlated with its degree (the greater the degree, the larger the node size).
4. Discussion
The current study found that alpha diversity of the SLE group is not different when compared to healthy controls, and the beta diversity was partially altered, which was in accordance with a previous study among adult SLE patients [20]. The fact that the alpha diversity is not statistically different between the healthy control group and the group affected by the pathology could be related to age-dependent factors and also to the small number of children in the analysis. To be precise, PCoA successfully identified 2 separated clusters of SLE and HC subjects when PCA did not, indicating that the changes induced by SLE became marked at the functional level, i.e., the level of microbial population structure (or 16S rRNA), regardless of the heterogeneities that appeared above at the highest level of the functional hierarchy, i.e., the metabolite level (Figure 1). In contrast to our outcomes on the alpha and beta diversity, the outcomes of a study by Zhu revealed upregulated alpha diversity in the SLE group; in addition, Rojo et al. reported beta diversity in PCA but not PCoA [16, 21]. This inconsistency may be due to age and sex differences since previous papers contained only adult patients and fewer or no male patients.
Another important finding was the increase in Proteobacteria and Enterobacteriales at the phylum and family levels among SLE patients in our project in southern China, and this finding was also reported by Wei et al., He et al., and Hevia and Milani from northern China. However, results from studies conducted in Spain and southern China do not support our results [22–24]. In Spain, the intestinal flora of SLE patients was characterized by a significantly lower Firmicutes/Bacteroidetes ratio and showed a depletion of Lachnospiraceae and Ruminococcaceae and an enrichment of Bacteroidaceae and Prevotellaceae [24]. In southern China, the intestinal flora of SLE patients was characterized by a significant increase in Actinobacteria [22]. It can therefore be assumed that the flora composition is irrelevant to the geographical location. On the other hand, consistent with the literature, this research found that Ruminococcaceae levels were higher in healthy controls [21, 23, 24]. Moreover, there was a list of other genus assigned to the Lachnospiraceae family that are widely descripted as beneficial bacteria in literature elevated in health controls, including Lachnospiraceae_UCG_008 and Lachnospiraceae_UCG_004 [25, 26]. Additionally, previous research has established that the increase in Proteobacteria and the decrease in Ruminococcaceae were associated with lupus nephritis with gastrointestinal damage [27]. Na et al. proposed that an increased prevalence of the bacterial phylum Proteobacteria is a marker for an unstable microbial community (dysbiosis) and a potential diagnostic criterion for disease [28]. Shang et al. identified Ruminococcaceae to be responsible for the degradation of diverse polysaccharides and fibres [29].
An obvious finding to emerge from the analysis is that amino acids were enriched in SLE faecal samples compared to HCs (Table 3), which was in accordance with previous research that found that their plasma concentration decreased [30–33]. Using gas chromatography-mass spectrometry, Yan et al. were able to detect the serum depletion of 12 amino acids, including tryptophan, alanine, proline, glycine, serine, threonine, aspartate, glutamine, asparagine, lysine, histidine, and tyrosine [30]. Similarly, Ouyang et al. found that L-alanine, L-isoleucine, L-lysine, L-phenylalanine, L-tyrosine, and L-valine were downregulated in the plasma of SLE patients [31]. Notably, four essential amino acids, L-valine, L-leucine, L-phenylalanine, and L-tryptophan, were listed among the metabolites depleted in plasma but enriched in faeces. These four amino acids have important physiological functions, such as the regulation of immunity, metabolism, and neural activity. Choi et al. reported that low dietary tryptophan prevented autoimmune pathology in lupus-prone mice, whereas high dietary tryptophan exacerbated disease [32]. Moreover, glucogenic amino acids, such as proline and L-methionine, and glucogenic and ketogenic amino acids, such as L-tyrosine, were increased in the faeces of SLE patients, indicating that there might be disorders in glucose metabolism and energy metabolism, as these amino acids could emerge as potential energy sources [33, 34]. Regarding the fatty acids, we found the abundance varied, as most long-chain fatty acids decreased while short-chain fatty acids increased in SLE children. This finding is contrary to a recent study that reported enriched fatty acids in the faeces of the SLE group [35]. Another study also established that long-chain fatty acids were less abundant in SLE patient serum [36]. At least half of SLE patients have gastrointestinal symptoms, including nausea, vomiting, anorexia, abdominal pain, diarrhoea, and abdominal distension, which may lead to an imbalance of amino acids and fatty acids [37].
The results of this study show that SLE-enriched faecal amino acids were significantly located in the protein digestion and absorption, ABC transporters, and aminoacyl-tRNA biosynthesis pathways. Amino acids are mainly absorbed in human intestinal mucosal cells through carrier proteins and the γ-glutamine cycle. Through type I ABC transporters in the third subgroup, intestinal prokaryotes and archaea obtain amino acids, which can be used for the synthesis of microbial proteins, including the biosynthesis of aminoacyl-tRNA, microbial energy metabolism, and conversion into a variety of physiologically active substances, such as neurotransmitters and hormones [38]. As a result, the increase in amino acids in faeces can impact the physiological activities of microorganisms and their hosts. For example, SLE-rich faecal branched-chain amino acids, including L-leucine and L-valine, can be used as precursors of membrane fatty acids, as well as key synergistic regulators of pathogen growth and virulence. Low-proline or low-protein diets in germ-free mice parasitized by human intestinal flora led to reduced expansion of wild-type C. difficile after challenge, indicating that the effectiveness of amino acids may be important for C. difficile infection [39]. Tryptophan is essential for the growth of certain pathogens, and severe tryptophan deficiency may hinder normal Chlamydia trachomatis onset and reduce its activation [40].
Of association analysis, this study found that Sphingomonas plays the most important role in the network of metabolites and intestinal flora. We found higher levels of the bacterium in SLE children. It is encouraging to compare this rise with that found by Corrêa et al., who found that Sphingomonas was at lower relative levels in healthy periodontal sites of SLE patients [41]. Chen et al.'s research provided evidence for increased microbial transmission along the gastrointestinal tract of most certain species in SLE patients compared to HCs, implicating a damaged barrier in the upper gastrointestinal tract in SLE patients who possibly cause transmissible oral species to colonize the gut [42].
It is unfortunate that most studies concerning gut microbiota and metabolites in SLE and LN patients include a study cohort of only adults, so our literature search may not be comprehensive enough. There is also a potential confounding effect of medications, as most patients were receiving treatment at the time of sample collection, and a large list of medications is postulated to affect the balance of taxa within the gut microbiome community [43, 44]. Another limitation of this study is that the SLE group and the HC group were not perfectly matched.
5. Conclusion
This study set out to investigate intestinal flora and metabolomics in SLE children. This study has shown that Proteobacteria was increased in SLE patients. Of this phylum, the genus Sphingomonas plays a very important role, as it exerts a significant positive correlation with a series of proteins in the protein digestion and absorption pathway. Sphingomonas were reported to be less abundant in healthy periodontal sites of SLE patients than in those of HCs, indicating the transmission of oral species to the gut. Adjunct therapeutic modalities of adjusting microbiota can be innocuous and worth trying. Considerably, more work will need to be done to determine the pathogenesis and treatment of SLE in children.
Contributor Information
Xiaojie He, Email: hexiaojie@csu.edu.cn.
Qingnan He, Email: heqn2629@csu.edu.cn.
Data Availability
All data are available in our manuscript.
Ethical Approval
Ethical approval for this study was obtained from Ethics Committee of the Second Xiangya Hospital of Central South University.
Conflicts of Interest
All authors declare that they have no conflict of interest.
Authors' Contributions
Min Wen, Taohua Liu, Mingyi Zhao, and Xiqiang Dang participated in the experiments, data analysis, and manuscript writing. Shipin Feng, Xuewei Ding, Zhiquan Xu, Xiaoyan Huang, Qiuyu Lin, Wei Xiang, and Xiaoyan Li contributed to the data collection, management, and analysis. Xiaojie He and Qingnan He designed the experiment. Min Wen and Taohua Liu contributed equally to this work and should be considered co-first authors.
References
- 1.Aringer M., Costenbader K., Daikh D., et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Annals of the Rheumatic Diseases. 2019;78(9):1151–1159. doi: 10.1136/annrheumdis-2018-214819. [DOI] [PubMed] [Google Scholar]
- 2.Hiraki L. T., Feldman C. H., Liu J., et al. Prevalence, incidence, and demographics of systemic lupus erythematosus and lupus nephritis from 2000 to 2004 among children in the US Medicaid beneficiary population. Arthritis and Rheumatism. 2012;64(8):2669–2676. doi: 10.1002/art.34472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim Y., Shim S. C. Wolves trapped in the NETs the pathogenesis of lupus nephritis. J Rheum Dis. 2018;25(2):81–99. doi: 10.4078/jrd.2018.25.2.81. [DOI] [Google Scholar]
- 4.Moulton V. R., Suarez-Fueyo A., Meidan E., Li H., Mizui M., Tsokos G. C. Pathogenesis of human systemic lupus erythematosus: a cellular perspective. Trends in Molecular Medicine. 2017;23(7):615–635. doi: 10.1016/j.molmed.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shamoon M., Martin N. M., O'Brien C. L. Recent advances in gut microbiota mediated therapeutic targets in inflammatory bowel diseases: emerging modalities for future pharmacological implications. Pharmacological Research. 2019;148:p. 104344. doi: 10.1016/j.phrs.2019.104344. [DOI] [PubMed] [Google Scholar]
- 6.Castro F., de Souza H. S. P. Dietary composition and effects in inflammatory bowel disease. Nutrients. 2019;11(6):p. 1398. doi: 10.3390/nu11061398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhong D., Wu C., Zeng X., Wang Q. The role of gut microbiota in the pathogenesis of rheumatic diseases. Clinical Rheumatology. 2018;37(1):25–34. doi: 10.1007/s10067-017-3821-4. [DOI] [PubMed] [Google Scholar]
- 8.Sayers E., MacGregor A., Carding S. R. Drug-microbiota interactions and treatment response: relevance to rheumatoid arthritis. AIMS Microbiology. 2018;4(4):642–654. doi: 10.3934/microbiol.2018.4.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruiz L., López P., Suárez A., Sánchez B., Margolles A. The role of gut microbiota in lupus: what we know in 2018? Expert Review of Clinical Immunology. 2018;14(10):787–792. doi: 10.1080/1744666X.2018.1519395. [DOI] [PubMed] [Google Scholar]
- 10.Neuman H., Koren O. The gut microbiota: a possible factor influencing systemic lupus erythematosus. Current Opinion in Rheumatology. 2017;29(4):374–377. doi: 10.1097/BOR.0000000000000395. [DOI] [PubMed] [Google Scholar]
- 11.Luo X. M., Edwards M. R., Mu Q., et al. Gut microbiota in human systemic lupus erythematosus and a mouse model of lupus. Applied and Environmental Microbiology. 2018;84(4) doi: 10.1128/AEM.02288-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katz-Agranov N., Zandman-Goddard G. The microbiome and systemic lupus erythematosus. Immunologic Research. 2017;65(2):432–437. doi: 10.1007/s12026-017-8906-2. [DOI] [PubMed] [Google Scholar]
- 13.Rosser E. C., Mauri C. A clinical update on the significance of the gut microbiota in systemic autoimmunity. Journal of Autoimmunity. 2016;74:85–93. doi: 10.1016/j.jaut.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 14.Zhang H., Liao X., Sparks J. B., Luo X. M. Dynamics of gut microbiota in autoimmune lupus. Applied and Environmental Microbiology. 2014;80(24):7551–7560. doi: 10.1128/AEM.02676-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo X. M., Edwards M. R., Reilly C. M. Diet and Microbes in the Pathogenesis of Lupus. Lupus: 2017. [Google Scholar]
- 16.Rojo D., Hevia A., Bargiela R., et al. Ranking the impact of human health disorders on gut metabolism: systemic lupus erythematosus and obesity as study cases. Scientific Reports. 2015;5(1):p. 8310. doi: 10.1038/srep08310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hochberg M. C. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis and Rheumatism. 1997;40(9):p. 1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 18.Philippot L., Spor A., Hénault C., et al. Loss in microbial diversity affects nitrogen cycling in soil. The ISME Journal. 2013;7(8):1609–1619. doi: 10.1038/ismej.2013.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Price M. N., Dehal P. S., Arkin A. P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5(3, article e9490) doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He J., Chan T., Hong X., et al. Microbiome and metabolome analyses reveal the disruption of lipid metabolism in systemic lupus erythematosus. Frontiers in Immunology. 2020;11:p. 1703. doi: 10.3389/fimmu.2020.01703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu W. F. Alteration of Gut Microbiome in Patients with Systemic Lupus Erythematosus. Chinese: Zhejiang: Univ. of Zhejiang; 2018. [Google Scholar]
- 22.Wei F., Xu H., Yan C., Rong C., Liu B., Zhou H. Changes of intestinal flora in patients with systemic lupus erythematosus in northeast China. PLoS One. 2019;14(3, article e0213063) doi: 10.1371/journal.pone.0213063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He Z., Shao T., Li H., Xie Z., Wen C. Alterations of the gut microbiome in Chinese patients with systemic lupus erythematosus. Gut Pathogens. 2016;8(1) doi: 10.1186/s13099-016-0146-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hevia A., Milani C. Intestinal dysbiosis associated with systemic lupus erythematosus. mBio. 2014;5(5) doi: 10.1128/mBio.01548-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vacca M., Celano G., Calabrese F. M., Portincasa P., Gobbetti M., de Angelis M. The controversial role of human gut Lachnospiraceae. Microorganisms. 2020;8(4):p. 573. doi: 10.3390/microorganisms8040573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greco R., Nitti R., Mancini N., et al. Microbiome markers are early predictors of acute GVHD in allogeneic hematopoietic stem cell transplant recipients. Blood. 2021;137(11):1556–1559. doi: 10.1182/blood.2020007158. [DOI] [PubMed] [Google Scholar]
- 27.Li Z. Gastrointestinal System Involvement in Systemic Lupus Erythematosus. Beijing: Peking Union Medical University; 2017. [DOI] [PubMed] [Google Scholar]
- 28.Na R. S., Tae W. W., Jin W. B. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends in Biotechnology. 2015;33(9):496–503. doi: 10.1016/j.tibtech.2015.06.011. [DOI] [PubMed] [Google Scholar]
- 29.Shang Q., Shan X., Cai C., Hao J., Li G., Yu G. Dietary fucoidan modulates the gut microbiota in mice by increasing the abundance of Lactobacillus and Ruminococcaceae. Food & Function. 2016;7(7):3224–3232. doi: 10.1039/C6FO00309E. [DOI] [PubMed] [Google Scholar]
- 30.Yan B., Huang J., Zhang C., et al. Serum metabolomic profiling in patients with systemic lupus erythematosus by GC/MS. Modern Rheumatology. 2016;26(6):914–922. doi: 10.3109/14397595.2016.1158895. [DOI] [PubMed] [Google Scholar]
- 31.Ouyang X., Dai Y., Wen J. L., Wang L. X. 1H NMR-based metabolomic study of metabolic profiling for systemic lupus erythematosus. Lupus. 2011;20(13):1411–1420. doi: 10.1177/0961203311418707. [DOI] [PubMed] [Google Scholar]
- 32.Choi S.-C., Brown J., Gong M., et al. Gut microbiota dysbiosis and altered tryptophan catabolism contribute to autoimmunity in lupus-susceptible mice. Science Translational Medicine. 2020;12(551):p. eaax2220. doi: 10.1126/scitranslmed.aax2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bengtsson A. A., Trygg J., Wuttge D. M., et al. Metabolic profiling of systemic lupus erythematosus and comparison with primary Sjögren’s syndrome and systemic sclerosis. PLoS One. 2016;11(7, article e0159384) doi: 10.1371/journal.pone.0159384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guleria A., Phatak S., Dubey D., et al. NMR-based serum metabolomics reveals reprogramming of lipid dysregulation following cyclophosphamide-based induction therapy in lupus nephritis. Journal of Proteome Research. 2018;17(7):2440–2448. doi: 10.1021/acs.jproteome.8b00192. [DOI] [PubMed] [Google Scholar]
- 35.Yan R., Jiang H., Gu S., et al. Fecal metabolites were altered, identified as biomarkers and correlated with disease activity in patients with systemic lupus erythematosus in a GC-MS-based metabolomics study. Frontiers in Immunology. 2020;11 doi: 10.3389/fimmu.2020.02138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu T., Xie C., Han J., et al. Metabolic disturbances associated with systemic lupus erythematosus. PLoS One. 2012;7(6, article e37210) doi: 10.1371/journal.pone.0037210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Z., Xu D., Wang Z., et al. Gastrointestinal system involvement in systemic lupus erythematosus. Lupus. 2017;26(11):1127–1138. doi: 10.1177/0961203317707825. [DOI] [PubMed] [Google Scholar]
- 38.Lewis V. G., Ween M. P., McDevitt C. A. The role of ATP-binding cassette transporters in bacterial pathogenicity. Protoplasma. 2012;249(4):919–942. doi: 10.1007/s00709-011-0360-8. [DOI] [PubMed] [Google Scholar]
- 39.Battaglioli E. J., Hale V. L., Chen J., et al. Clostridioides difficileuses amino acids associated with gut microbial dysbiosis in a subset of patients with diarrhea. Science Translational Medicine. 2018;10(464):p. eaam7019. doi: 10.1126/scitranslmed.aam7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leonhardt R. M., Lee S. J., Kavathas P. B., Cresswell P. Severe tryptophan starvation blocks onset of conventional persistence and reduces reactivation of Chlamydia trachomatis. Infection and Immunity. 2007;75(11):5105–5117. doi: 10.1128/IAI.00668-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Corrêa J. D., Calderaro D. C., Ferreira G. A., et al. Subgingival microbiota dysbiosis in systemic lupus erythematosus: association with periodontal status. Microbiome. 2017;5(1):p. 34. doi: 10.1186/s40168-017-0252-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen B., Jia X., Xu J., et al. An Autoimmunogenic and Proinflammatory Profile Defined by the Gut Microbiota of Patients With Untreated Systemic Lupus Erythematosus. Arthritis & Rheumatology. 2021;73(2):232–243. doi: 10.1002/art.41511. [DOI] [PubMed] [Google Scholar]
- 43.Gul'neva M., Shilkina N. P. Intestinal microbial biocenosis in patients with systemic lupus erythematosus treated with prednisolone. Klinicheskaia Meditsina. 2009;87(6):42–45. [PubMed] [Google Scholar]
- 44.Maier L., Pruteanu M., Kuhn M., et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature. 2018;555(7698):623–628. doi: 10.1038/nature25979. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are available in our manuscript.