

Keywords: E/I balance, GABA, inhibition, NMDA receptor, SRR
Abstract
There is substantial evidence that both N-methyl-D-aspartate receptor (NMDAR) hypofunction and dysfunction of GABAergic neurotransmission contribute to schizophrenia, though the relationship between these pathophysiological processes remains largely unknown. Although models using cell-type-specific genetic deletion of NMDARs have been informative, they display overly pronounced phenotypes extending beyond those of schizophrenia. Here, we used the serine racemase knockout (SRKO) mice, a model of reduced NMDAR activity rather than complete receptor elimination, to examine the link between NMDAR hypofunction and decreased GABAergic inhibition. The SRKO mice, in which there is a >90% reduction in the NMDAR coagonist d-serine, exhibit many of the neurochemical and behavioral abnormalities observed in schizophrenia. We found a significant reduction in inhibitory synapses onto CA1 pyramidal neurons in the SRKO mice. This reduction increases the excitation/inhibition balance resulting in enhanced synaptically driven neuronal excitability without changes in intrinsic excitability. Consistently, significant reductions in inhibitory synapse density in CA1 were observed by immunohistochemistry. We further show, using a single-neuron genetic deletion approach, that the loss of GABAergic synapses onto pyramidal neurons observed in the SRKO mice is driven in a cell-autonomous manner following the deletion of SR in individual CA1 pyramidal cells. These results support a model whereby NMDAR hypofunction in pyramidal cells disrupts GABAergic synapses leading to disrupted feedback inhibition and impaired neuronal synchrony.
NEW & NOTEWORTHY Recently, disruption of excitation/inhibition (E/I) balance has become an area of considerable interest for psychiatric research. Here, we report a reduction in inhibition in the serine racemase knockout mouse model of schizophrenia that increases E/I balance and enhances synaptically driven neuronal excitability. This reduced inhibition was driven cell-autonomously in pyramidal cells lacking serine racemase, suggesting a novel mechanism for how chronic NMDA receptor hypofunction can disrupt information processing in schizophrenia.
INTRODUCTION
Schizophrenia is a devastating psychiatric disease characterized by psychosis along with profound cognitive and social impairments. One prominent and enduring model implicates hypofunction of N-methyl-d-aspartate receptors (NMDARs) in the broad symptomatology of schizophrenia (1–4). For example, open channel NMDAR inhibitors, such as phencyclidine (PCP) and ketamine, induce schizophrenia-like symptoms in healthy subjects (5, 6), and exacerbate both positive and negative symptoms in patients with schizophrenia (6–8), supporting a shared mechanism between NMDAR dysfunction and schizophrenia pathophysiology. In addition, mice with low levels of the obligatory GluN1 subunit of NMDA receptor, so-called GluN1 hypomorphs, display behaviors and endophenotypes consistent with schizophrenia (9–19).
Another well-supported hypothesis states that schizophrenia arises from changes in the ratio of excitatory to inhibitory activity in the brain (E/I imbalance), specifically through downregulation of GABAergic inhibition, and may represent a point of overlap between schizophrenia and autism (20, 21). Decreases in GABAergic markers in schizophrenia have been consistently observed in postmortem tissue (22–27). Furthermore, decreased GABAergic signaling disrupts oscillatory activity in the brain, particularly γ oscillations (28), that may be important for a variety of cognitive processes (29) including perceptual binding (30), cognitive flexibility (31), and attention (32, 33).
In the present study, we evaluated E/I balance in a mouse model of NMDAR hypofunction associated with the knockout of serine racemase (SR), the biosynthetic enzyme for the NMDAR coagonist d-serine (34, 35). In contrast to mouse models using broad genetic deletion of NMDARs which have phenotypes extending beyond the bounds of schizophrenia phenomenology (36), similar to the NMDAR hypomorph mice which have a severe reduction in NMDAR expression (37–39), the SR knockout (SRKO) mice provide a more subtle and potentially physiologically relevant model of NMDAR hypofunction (35). Indeed, deficiency of d-serine and the subsequent hypofunction of NMDARs has been implicated in the pathophysiology of schizophrenia (40). Genetic studies have suggested that SR, as well as the degradation enzyme d-amino acid oxidase (DAAO) and G72, an activator of DAAO, are putative risk genes for schizophrenia (41–45). In addition, d-serine levels in the CSF and serum are decreased in individuals with schizophrenia (46, 47) and supplementation of antipsychotics with d-serine improves negative and cognitive symptoms in patients with schizophrenia (48–50). Consistent with well-characterized hallmarks of schizophrenia, the SRKO mice have reductions in cortical dendritic complexity and spine density, reduced hippocampal volume (51, 52), and impaired performance on cognitive tasks that can be improved with exogenous d-serine administration (51, 53–55).
Here, we show that SRKO mice also have a significant reduction in GABAergic synapses onto the soma and apical dendrites of CA1 pyramidal neurons. This reduction in inhibition increases the E/I ratio resulting in enhanced synaptically driven neuronal excitability. Single neuron deletion of SR revealed that the loss of inhibitory synapses is driven cell-autonomously by the loss of SR in the pyramidal neurons, consistent with recent evidence that NMDARs on pyramidal neurons regulate GABAergic synapse development (56–58). These results support a model of pyramidal cell NMDAR hypofunction directly leading to GABAergic dysfunction.
MATERIALS AND METHODS
Animals
The SRKO mice are derived from the floxed SR mice (SRfl), in which the first coding exon (exon 3) is flanked by loxP sites as described (53, 59) and are maintained on a C57Bl/6J background. Mice were group-housed in polycarbonate cages and maintained on a 12-h light/dark cycle. Animals were given access to food and water ad libitum. The University of California Davis Institutional Animal Care and Use Committee approved all animal procedures.
Slice Preparation
Male SRfl (labeled as WT) and SRKO mice (2- to 3-mo-old) were deeply anesthetized with isoflurane, followed by cervical dislocation and decapitation. The brain was rapidly removed and submerged in ice-cold, oxygenated (95% O2/5% CO2) artificial cerebrospinal fluid (ACSF) containing (in mm) as follows: 124 NaCl, 4 KCl, 25 NaHCO3, 1 NaH2PO4, 2 CaCl2, 1.2 MgSO4, and 10 glucose (Sigma-Aldrich). On a cold plate, the brain hemispheres were separated, blocked, and the hippocampi removed. For extracellular recordings, 400-μm-thick slices of dorsal hippocampus were cut using a McIlwain tissue chopper (Brinkman, Westbury, NY). For whole cell recordings, a modified transverse 300 µm slices of dorsal hippocampus were prepared by performing a ∼10° angle blocking cut of the dorsal portion of each cerebral hemisphere (60) then mounting the cut side down on a Leica VT1200 vibratome in ice-cold, oxygenated (95% O2/5% CO2) ACSF. Slices were incubated (at 32°C) for 20 min and then maintained in submerged-type chambers that were continuously perfused (2–3 mL/min) with oxygenated (95% O2/5% CO2) ACSF at room temperature and allowed to recover for at least 1.5–2 h before recordings. Just before the start of experiments, slices were transferred to a submersion chamber on an upright Olympus microscope, perfused with warmed to 30.4°C using a temperature controller (Medical System Corp.) normal ACSF saturated with 95% O2/5% CO2. For intracellular experiments, the slices were bathed in a modified ACSF containing 2.4 mm KCl.
Extracellular Recordings
A bipolar, nichrome wire stimulating electrode (MicroProbes) was placed in stratum radiatum of the CA1 region and used to activate Schaffer collateral (SC)-CA1 synapses. For extracellular recordings, evoked field excitatory postsynaptic potentials (fEPSPs) (basal stimulation rate = 0.033 Hz) were recorded in stratum radiatum using borosilicate pipettes (Sutter Instruments, Novato, CA) filled with ACSF (resistance ranged from 5–10 MΩ). To determine response parameters of excitatory synapses, basal synaptic strength was determined by comparing the amplitudes of presynaptic fiber volleys and postsynaptic fEPSP slopes for responses elicited by different intensities of SC fiber stimulation. Presynaptic neurotransmitter release probability was compared by paired-pulse ratio (PPR) experiments, performed at 25, 50, 100, and 200 ms stimulation intervals. Long-term potentiation (LTP) was induced by high-frequency stimulation (HFS) using a 1× tetanus (1 s train of 100 Hz stimulation). At the start of each experiment, the maximal fEPSP amplitude was determined and the intensity of presynaptic fiber stimulation was adjusted to evoke fEPSPs with an amplitude ∼30%–40% of the maximal amplitude. The mean slope of excitatory postsynaptic potentials (EPSPs) elicited 55–60 min after HFS (normalized to baseline) was used for statistical comparisons. For experiments performed in picrotoxin (PTX, Sigma-Aldrich; 50 µM) the CA3 region was removed. Analyses were performed with the Clampex 10.6 software suite (Molecular Devices, San Jose, CA) and Prism 9.1 software (GraphPad Software, San Diego, CA).
Whole Cell Current Clamp Recordings
CA1 pyramidal neurons were visualized by infrared differential interference contrast microscopy, and current clamp recordings were performed using borosilicate recording electrodes (3–5 MΩ) filled with a K+-based electrode-filling solution containing (in mM) 135 K-gluconate, 5 NaCl, 10 HEPES, 2 MgCl, 0.2 EGTA, 10 Na2-phosphocreatine, 4 Na-ATP, 0.4 Na-GTP (pH = 7.3, 290 osmol/kgH2O). Passive and active membrane properties of CA1 pyramidal cells were determined using three 500 ms current pulses 10 s apart. Current injections were first recorded in increasing order (i.e., 0, 25, 50, 75, 100, 125, 150, and 200 pA) and then in decreasing order. Values obtained from the responses elicited by the same current injection were averaged. For input resistance, 500 ms current steps of 0 to −200 pA were injected in −20 pA increments. Steady-state responses were measured as the average change in voltage in the last 100 ms of the pulse. The slope of a regression line fitted to the voltage versus current data was used to calculate input resistance. Sag currents were measured during the 100 pA hyperpolarizing steps and calculated as the initial voltage trough minus the steady-state voltage change. Firing frequency versus injected current was measured as the number of spikes per 500 ms step in 25 pA increments from 0 to 200 pA. Rheobase was determined by injecting 0.5 ms square pulses in 2 pA steps and recording the strength of the first pulse to elicit an action potential. Spike firing threshold and action potential height were calculated by injecting a 2 ms square pulse of 1.8 nA. To measure the E/I ratio from CA1 pyramidal neurons, current clamp recordings at holding potential of −60 mV were made in the absence of synaptic blockers. E/I ratio was calculated from averaged baseline subtracted traces as the maximum depolarization amplitude (in mV) divided by the maximum hyperpolarization amplitude in the 300 ms after the stimulus. Synaptically mediated excitability was determined with short trains of synaptic stimulation (5 pulses at 100 Hz SC fiber stimulation) with the CA1 pyramidal neurons at holding potential of −60 mV in the absence of synaptic blockers. For both the E/I ratio and stimulation trains, the stimulus strength was adjusted so that the initial PSP depolarization ∼5 mV.
Whole Cell Voltage Clamp Recordings
CA1 pyramidal neurons were visualized by infrared differential interference contrast microscopy, and voltage-clamp recordings were performed using borosilicate glass recording pipettes (3–5 MΩ) filled with a Cs+-based electrode-filling solution containing (in mM): 135 Cs-methanesulfonate, 8 NaCl, 5 QX314 (Sigma-Aldrich), 0.3 EGTA, 4 Mg-ATP, 0.3 Na-GTP, and 10 HEPES (pH = 7.3, 290 osmol/kgH2O). Evoked inhibitory postsynaptic currents (IPSCs; eIPSCs), spontaneous IPSCs (sIPSCs), and miniature IPSCs (mIPSCs) were recorded in presence of DL-2-amino-5-phosphonovaleric acid (APV) and 2,3-dioxo-6-nitro-7-sulfamoyl-benzo[f]quinoxaline (NBQX) (Tocris; 50 µM and 10 µM respectively) to block AMPAR and NMDAR currents. Miniature excitatory postsynaptic currents (EPSCs; mEPSCs) were recorded in the presence of 100 µM PTX and 1 µM tetrodotoxin (TTX; Alomone Laboratories, Jerusalem, Israel) to block action potential-dependent neurotransmitter release, whereas mIPSCs were recorded in presence of 1 µM TTX alone. The outward IPSCs were completely blocked by PTX (50 µM). For the input/output (I/O) curves of eIPSCs, the stimulus intensity of the threshold evoked response was first determined and then stimulation was increased to develop the I/O curves. Recordings where series resistance was ≥25 MΩ or unstable were discarded. Series resistance compensation was used in all voltage-clamp recordings except in experiments examining miniature postsynaptic currents. All recordings were obtained with a MultiClamp 700B amplifier (Molecular Devices), filtered at 2 kHz, and digitized at 10 Hz. Analysis was performed with the Clampex 10.6 software suite and GraphPad Prism 9.1.
Single Neuron SR Deletion Experiments
Neonatal [P0] SRfl mice of both sexes were stereotaxically injected with a low-titer rAAV1-Cre:GFP viral stock (∼1 × 1012 vg/mL) targeting hippocampal CA1 as previously described (61, 62), resulting in very sparse transduction of CA1 pyramidal cells. At 2–3 mo, the injected mice were anesthetized with isoflurane and transcardially perfused with ice-cold artificial cerebrospinal fluid (ACSF), containing (in mM) 119 NaCl, 26.2 NaHCO3, 11 glucose, 2.5 KCl, 1 NaH2PO4, 2.5 CaCl2, and 1.3 MgSO4. Modified transverse 300 µm slices of dorsal hippocampus were prepared by performing a ∼10° angle blocking cut of the dorsal portion of each cerebral hemisphere (60) then mounting the cut side down on a Leica VT1200 vibratome in ice-cold cutting buffer. Slices were incubated in 32°C NMDG solution containing (in mM) 93 NMDG, 93 HCl, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 sodium ascorbate, 2 thiourea, 3 sodium pyruvate, 10 MgSO4, and 0.5 CaCl2 (63) for 15 min, which we have previously used to increase cell health in slices from older animals (64). Slices were transferred to room temperature ACSF and held for at least 1 h before recording. All solutions were vigorously perfused with 95% O2 and 5% CO2. Slices were transferred to a submersion chamber on an upright Olympus microscope, perfused in room temperature ACSF, and saturated with 95% O2 and 5% CO2. CA1 neurons were visualized by infrared differential interference contrast microscopy, and GFP+ cells were identified by epifluorescence microscopy. Cre expression was generally limited to the hippocampus within a sparse population of CA1 pyramidal neurons. Cells were patched with 3–5 MΩ borosilicate pipettes filled with intracellular solution containing (in mM) 135 cesium methanesulfonate, 8 NaCl, 10 HEPES, 0.3 Na-GTP, 4 Mg-ATP, 0.3 EGTA, and 5 QX-314 (Sigma, St. Louis, MO), and mIPSCs were recorded at 0 mV in the presence of 50 µM APV, 10 µM NBQX, and 0.5 µM TTX. Series resistance was monitored and not compensated, and cells were discarded if series resistance varied more than 25%. Recordings were obtained with a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA), filtered at 2 kHz, and digitized at 10 Hz. Analysis was performed with the Clampex 10.6, MiniAnalysis, and GraphPad Prism 9.1 (GraphPad Software, San Diego, CA).
Immunohistochemistry
Male C57Bl/6J, SRfl (labeled as WT) and SRKO mice (2- to 3-mo-old) were deeply anesthetized with isoflurane and injected with a lethal dose of Fatal Plus (Vortech Pharmaceuticals) pentobarbital solution. The mice were then perfused transcardially with 1×PBS followed by 4% paraformaldehyde (PFA; Electron Microscopy Sciences) in 1×PBS. Brains were removed and postfixed for 3 h in 4% PFA in 1×PBS. The fixed brains were then cryoprotected stepwise, first in 10% sucrose in 1×PBS overnight, then in 30% sucrose in 1×PBS overnight. Brains were then mounted and frozen in O.C.T. compound (Tissue-Tek). Coronal sections through the dorsal hippocampus were cut on a Leica CM3050 S cryostat at 10 µm and collected onto Superfrost Plus slides (Thermo Fisher Scientific). Sections were outlined with a hydrophobic barrier pen and all subsequent incubation steps were performed in a humidified chamber. The sections were blocked with 10% normal donkey serum in 1×PBS-T (0.5% Triton X-100) for 1 h at room temperature and then probed overnight with rabbit anti-VGAT antibody (Synaptic Systems, Cat. No. 131 003, 1:500) in blocking solution at 4°C. The next day sections were rinsed 3 times with 1×PBS-T and then incubated with secondary antibody (donkey anti-rabbit 647, Jackson, Cat. No. 711-605-152, 1:400) in 1×PBS-T for 90 min at room temperature. The sections were then rinsed 3 times with 1×PBS-T and counterstained with DAPI. The sections were then mounted with Mowoil mounting medium and covered with a glass coverslip. Wild-type and SRKO slices were prepared and stained in parallel. After drying, a series of images covering the hippocampus were collected on a Nikon C2 LSM with a Nikon CFI Apo Lambda 60 × 1.4 NA oil objective. Laser and PMT settings remained constant between individuals and genotypes. Single images covering the regions of interest were stitched in together in Nikon Elements software. Regions of interest of the stratum pyramidale or stratum radiatum of hippocampal CA1 were analyzed using custom-written journals (65) in Metamorph software (v7.5, Molecular Devices) to identify and quantify VGAT puncta density and intensity. Constraints for puncta identification is semiautomated with the output visually inspected and calibrated to capture the majority of punctal signal while removing artifacts. Two regions of interest for both stratum pyramidale or stratum radiatum were analyzed for each of three individual animals per genotype. Data were graphed and analyzed using GraphPad Prism 9.1 (GraphPad Software, San Diego, CA). Unpaired Student’s t tests were used to test for statistically significant differences between genotypes.
Statistical Analysis
Statistical comparisons were made with Student’s unpaired t test or two-way ANOVA with Bonferroni’s multiple comparisons test as specified and appropriate, using GraphPad Prism 9.1 (GraphPad Software, San Diego, CA). Spontaneous and miniature inhibitory synaptic events were analyzed using Mini Analysis software (Synaptosoft, Fort Lee, NJ). Peaks of events were first automatically detected by the software according to a set threshold amplitude of 6 pA. To generate cumulative probability plots for both amplitude and interevent time interval, the same number of events (50–200 events acquired after an initial 3 min of recording) from each CA1 pyramidal neuron was pooled for each group, and input into the Mini Analysis program. The Kolmogorov–Smirnov two-sample statistical test (KS test) was used to compare the distribution of spontaneous and miniature events between WT and SRKO mice.
RESULTS
Increased E/I Balance in CA1 Pyramidal Cells in SRKO Mice
To investigate the properties of excitatory synaptic transmission in the SRKO mice, we first conducted extracellular field recordings of SC-CA1 synapses in the SRKO mice. Consistent with previous studies (52), the basal excitatory synaptic strength, determined by comparing the amplitudes of presynaptic fiber volleys and fEPSP slopes for responses elicited by different intensities of SC fiber stimulation (input-output curve), was unaltered in SRKO compared with WT slices (Fig. 1A, P = 0.49, two-way ANOVA, F(1,122) = 0.467). We next examined the input-output (I/O) function of evoked monosynaptic IPSCs through stimulation in the stratum radiatum in the presence of 10 µM NBQX and 50 µM AP5. We found a significant decrease in monosynaptic inhibition onto CA1 pyramidal neurons in SRKO mice compared with WT (Fig. 1B, P = 0.0001, two-way ANOVA, F(1,224) = 58.90; Bonferroni’s multiple comparisons test, *P < 0.05). This difference was characterized by a downward shift in the I/O curve showing the relationship between eIPSC amplitude and stimulus intensity. There was also no change in paired-pulse ratio (PPR) of the fEPSPs in the SRKO mice compared with WT mice (Fig. 1C, P = 0.91, two-way ANOVA, F(1,23) = 0.012), which together with unaltered change in basal excitatory synaptic strength suggests that there is no alteration of excitatory neurotransmission or presynaptic glutamate release probability in the SRKO mice. PPR of eIPSCs was also unchanged in SRKO mice compared with WT mice (Fig. 1D, P = 0.82, unpaired t test, t(34) = 0.230), suggesting that the reduction in inhibitory currents is not due to a change in the probability of GABA release. Using whole cell current clamp recordings, we next examined the impact of the reduction in synaptic inhibition in the SRKO on the E/I ratio in CA1 pyramidal neurons by recording compound EPSP/inhibitory excitatory postsynaptic potentials (IPSPs) at holding potential of −60 mV using current injection upon SC stimulation. For this experiment, the peak depolarization of the PSP was set to ∼5 mV (WT: 5.2 ± 0.1 mV, n = 15; SRKO: 5.1 ± 0.1, n = 20, P = 0.343, unpaired t test, t(33) = 0.96) to draw out the inhibitory component of compound EPSP/IPSPs (Fig. 1E). We found a significant reduction in the IPSP component of the compound EPSP/IPSP (Fig. 1E, peak IPSP amplitude, P = 0.0008, unpaired t test, t(33) = 3.71). This decrease in IPSP amplitude results in an increased E/I ratio (Fig. 1E, E/I ratio, P = 0.0026, unpaired t test, t(33) = 3.26). Together, these results suggest that a selective GABAergic impairment in the SRKO mice leads to an increase in the E/I balance.
Figure 1.
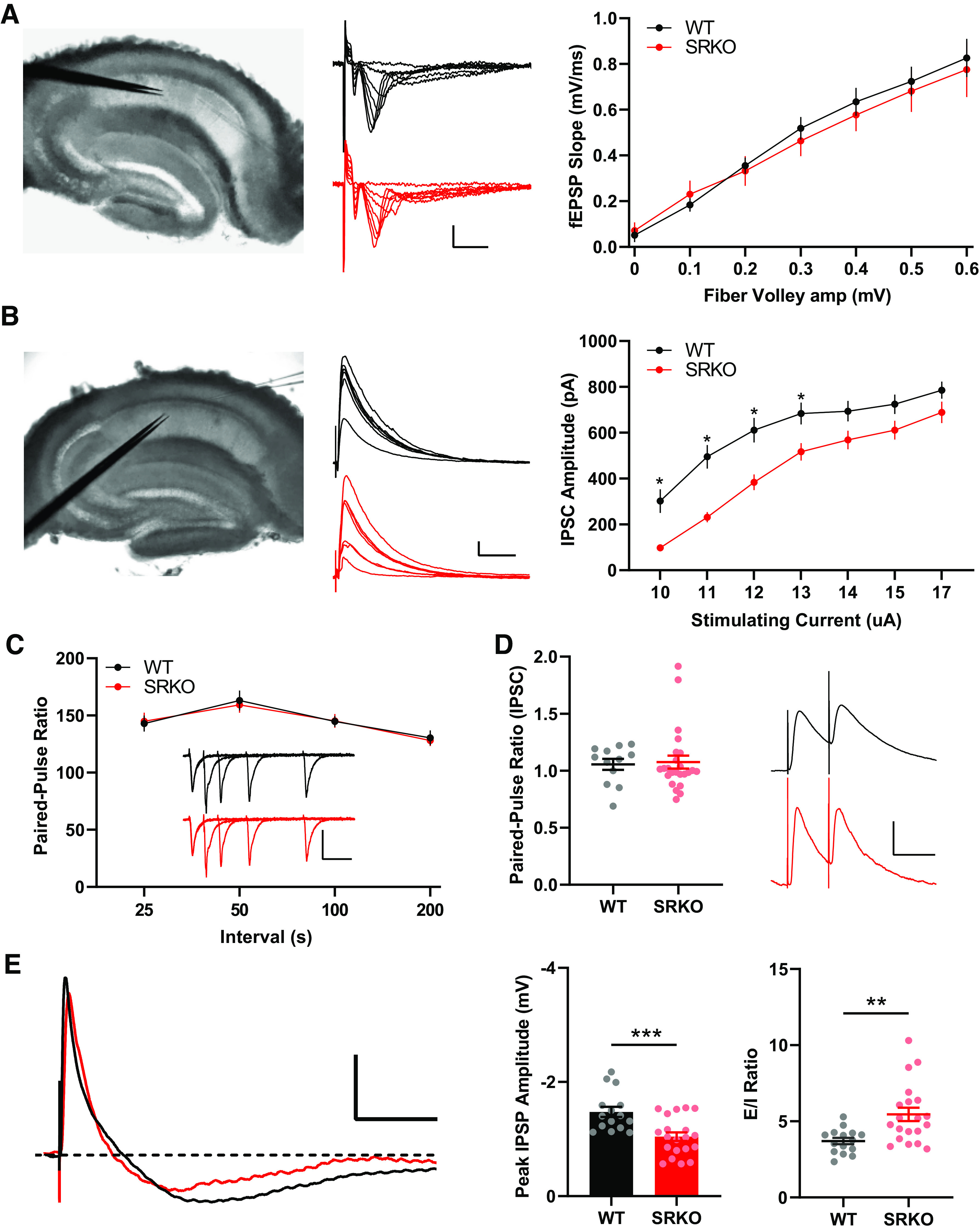
Increased excitation/inhibition (E/I) ratio in SRKO mice. A: left, view through the upright Olympus microscope of hippocampal slice with stimulating electrode (left) and recording electrode (right) in stratum radiatum. Middle, representative sample traces of extracellular field recordings for WT and SRKO; scale bars: 0.5 mV, 5 ms. Normal basal synaptic transmission as measured by presynaptic fiber volley amplitudes and postsynaptic field EPSP (fEPSP) slopes for responses elicited by different intensities of Schaffer collateral (SC) fiber stimulation in WT (n = 10) and SRKO (n = 10) hippocampal slices (P = 0.49, two-way ANOVA, F(1,122) = 0.467). B: Left, view through the upright Olympus microscope of hippocampal slice with stimulating electrode (left) in stratum radiatum and patch-clamp recording electrode (right) in the CA1 pyramidal cell layer. Middle, representative sample traces of evoked IPSC from WT and SRKO CA1 pyramidal cells at holding potential of 0 mV: scale bars: 100 pA, 200 ms. Input-output function of evoked IPSC amplitude vs. stimulating current strength show a significant decrease in inhibition in SRKO mice (P = 0.0001, two-way ANOVA, F(1,224) = 58.90; Bonferroni’s multiple comparisons test; WT n = 17, SRKO n = 17). C: paired-pulse ratio is unchanged at SRKO SC-CA1 synapses compared with WT (P = 0.91, two-way ANOVA, F(1,23) = 0.012; WT: n = 12, SRKO: n = 13). Inset, traces represent fEPSPs evoked by stimulation pulses delivered with a 25-, 50-, 100-, and 200-ms interpulse interval; scale bars: 0.5 mV, 50 ms. D: paired pulse ratio of IPSCs at a 50-ms interpulse interval (WT: 1.056 ± 0.049, n = 12; SRKO: 1.076 ± 0.057, n = 24) indicating that there is no change in the probability of inhibitory neurotransmitter release from presynaptic terminals. Right, representative traces of evoked IPSCs from WT and SRKO CA1 pyramidal cells; scale bars: 50 pA, 50 ms. E: Left, overlaid traces of compound excitatory (EPSP) and inhibitory (IPSP) postsynaptic potentials evoked by SC stimulation in absence of synaptic blockers at holding potential of −60 mV from SRKO (red) and WT (black) mice; dashed line indicates the baseline; scale bars: 2 mV, 100 ms. Peak PSP depolarization was set to approximately 5 mV for each cell. Peak IPSP amplitude is significantly decreased in SRKO mice compared with WT mice (P = 0.0008, unpaired t test, t(33)=3.71, WT: 1.5 ± 0.1 mV, n = 15; SRKO: 1.0 ± 0.1, n = 20). The E/I ratio in CA1 pyramidal cells calculated from EPSP and IPSP peak amplitudes is greater in SRKO mice compared with WT (P = 0.0026, unpaired t test, t(33)=3.26, WT: 3.7 ± 0.2, n = 16; SRKO: 5.5 ± 0.4, n = 20). Data represent means ± SE. EPSP, excitatory postsynaptic potential; IPSC, inhibitory postsynaptic current; IPSP, inhibitory postsynaptic potential; SRKO, serine racemase knockout; WT, wild type. *P < 0.05, **P < 0.01, ***P < 0.001.
Enhanced Pyramidal Cell Excitability to Synaptic Stimulation in SRKO Mice
Synaptic inhibition plays a key role in synaptic integration and spike initiation in neurons (66). Indeed, at hippocampal SC-CA1 synapses, EPSP-spike potentiation, an enhancement of spike probability in response to a synaptic input of a fixed slope, is dependent on changes in GABAergic inhibition (67). Thus, in the SRKO mice, we examined EPSP-spike coupling using short trains of SC stimulation (5 pulses at 100 Hz). Stimulation intensity was adjusted for each neuron to normalize the initial subthreshold EPSP to ∼5 mV. We found a significantly increased probability of spiking in SRKO CA1 pyramidal cells compared to WT (Fig. 2A, P < 0.0001, two-way ANOVA, F(1,150) = 31.4), especially for the second, fourth and fifth stimulus (***P = 0.0001, Bonferroni’s multiple comparisons test, F(150) = 4.34; **P = 0.004, Bonferroni’s multiple comparisons test, F(150) = 3.41; *P = 0.013, Bonferroni’s multiple comparisons test, F(150) = 3.07, respectively). Post hoc analysis of the data in Fig. 2A showed a correlated increase in temporal summation (Fig. 2B). Here, the peak PSP amplitude was measured after each stimulus, excluding data after the cell fired its first action potential. The differing number of data points precluded statistical analyses but this qualitative analysis supports an increase in temporal summation from the reduction in inhibition in the SRKO CA1 pyramidal cells. Importantly, there were no differences in the intrinsic excitability of CA1 pyramidal cells between SRKO and WT mice (Fig. 2C, Table 1). We analyzed the number of spikes elicited during 500 ms steps of somatically injected current and found no significant differences in the number of spikes between WT and SRKO neurons at steps of any intensity (Fig. 2C, P = 0.759, two-way ANOVA, F(8,207) = 0.621). There were also no significant differences in the resting membrane potential, input resistance (Fig. 2D), rheobase, action potential threshold or height, or sag amplitude between the CA1 neurons of WT and SRKO mice (Table 1). Together, these data suggest that a reduction in inhibitory input onto CA1 pyramidal neurons in the SRKO mice increases the E/I balance resulting in enhanced synaptically driven neuronal excitability without changes in intrinsic excitability.
Figure 2.
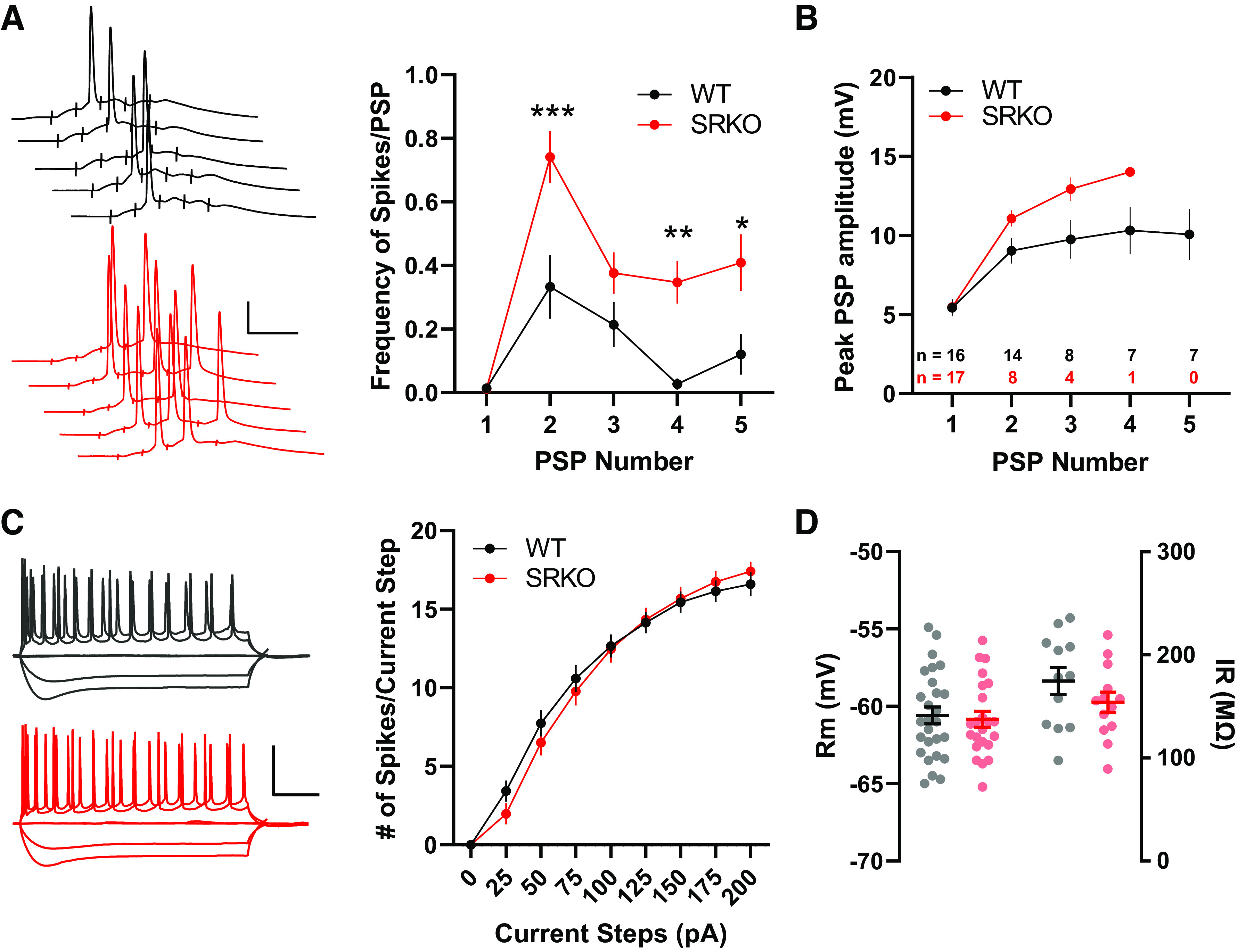
Increased synaptic excitability in SRKO mice. A: left, sample traces of APs/PSPs evoked by 5 pulses at 100 Hz Schaffer collateral fiber stimulation; scale bars: 25 mV, 20 ms. Right, short trains of synaptic stimulation leads to significantly more APs/PSP in SRKO compared with WT (PSP 1: P = 0.0001, two-way ANOVA, F(150) = 4.34, PSP 4: P = 0.004, two-way ANOVA, F(150) = 3.41, PSP 5: P = 0.013, two-way ANOVA, F(150) = 3.07; Bonferroni’s multiple comparisons test); WT: n = 15, SRKO: n = 17). B: temporal summation of PSPs measured from the 100 Hz stimulation in A until the first action potential for each cell (final n for each PSP shown in inset). C: left, sample traces for 0, −100, −200, +100, and +200 pA current steps; scale bars: 50 mV, 100 ms. Right, intrinsic excitability is unchanged in SRKO CA1 pyramidal neurons. Depolarization induced by somatic current injection elicits similar numbers of APs in WT and SRKO cells (P = 0.759, two-way ANOVA, F(8,207) = 0.6212; WT: n = 12, SRKO: n = 13) suggesting basal synaptic transmission is unaffected. D: summary graph of resting membrane potential (Rm) (right) and input resistance (IR) (left) showing no significance difference between WT and SRKO CA1 pyramidal cells. Data represent means ± SE. AP, action potential; PSP, postsynaptic potential; SRKO, serine racemase knockout; WT, wild-type. *P < 0.05, **P < 0.01, ***P < 0.001.
Table 1.
Intrinsic excitability in wild-type and SRKO CA1 pyramidal neurons
| Property | Wild Type (n = 12) | SRKO (n = 13) | Student’s t Test (unpaired) | P Value |
|---|---|---|---|---|
| RMP, mVa | 60.6 ± 0.5 [−65 to −55] |
−60.8 ± 0.5 [−65 to −55] |
t(48) = 0.34 | 0.736 |
| Rinput, MΩ | 174.6 ± 13.0 [97.7 to 235.8] |
154.0 ± 9.9 [89.4 to 219.3] |
t(23) = 1.27 | 0.216 |
| Sag, mVb | −0.20 ± 0.001 [−0.17 to −0.24] |
−0.22 ± 0.01 [−0.14 to −0.26] |
t(23) = 1.27 | 0.219 |
| Rheobase, pA | 15.1 ± 5.1 [2.7 to 59.3] |
25.2 ± 4.8 [2.7 to 70.2] |
t(23) = 1.45 | 0.159 |
| AP threshold, mVb | 49.2 ± 0.8 [−44.3 to −52.7] |
−50.4 ± 0.6 [−44.8 to −53.1] |
t(23) = 1.19 | 0.247 |
| AP height, mV | 120.9 ± 2.3 [105 to 140] |
120.9 ± 1.7 [110 to 134] |
t(23) = 0.01 | 0.989 |
| AHP peak, mV | −2.79 ± 0.4 [−5.75 to −0.845] |
−1.60 ± 0.3 [−3.74 to −0.46] |
t(23) = 2.35 | 0.028* |
Means ± SE [range].
For RMP: wild-type n = 27, SRKO n = 23.
bJunction potential not adjusted. AHP, afterhyperpolarization; AP, action potential; Rinput, input resistance; RMP, resting membrane potential; SRKO, serine racemase knockout; *P < 0.05.
Loss of Picrotoxin-Induced Disinhibition during LTP in SRKO Mice
In hippocampal SC-CA1 field LTP experiments induced with a HFS (e.g., 100 Hz tetanus), the addition of a GABAA inhibitor (e.g., PTX) causes a disinhibition that enhances LTP (Fig. 3A, P = 0.0002, unpaired t test, t(26) = 4.38) (68). Because of the reduced inhibition observed in the SRKO mice, we hypothesized that PTX-induced disinhibition might be disrupted. Consistently, we found that, in hippocampal slices from the SRKO mice, the addition of PTX (50 µM) did not affect the magnitude of LTP induced with a single 100 Hz tetanus (Fig. 3B, P = 0.394, unpaired t test, t(21) = 0.871). Interestingly, comparing data between WT and SRKO slices, we only observed significantly different LTP in the presence of PTX (P = 0.046, unpaired t test, t(22) = 2.11). In the absence of PTX, there was no difference in LTP between WT and SRKO slices (P = 0.623, unpaired t test, t(25) = 0.498), likely due to baseline disinhibition in the SRKO slices. Thus, by removing the impact of the reduced inhibition in the SRKO slices, the addition of PTX provides a more direct measure of the impact of synaptic NMDAR hypofunction in LTP, consistent with previous studies (52, 53, 59, 69, 70).
Figure 3.
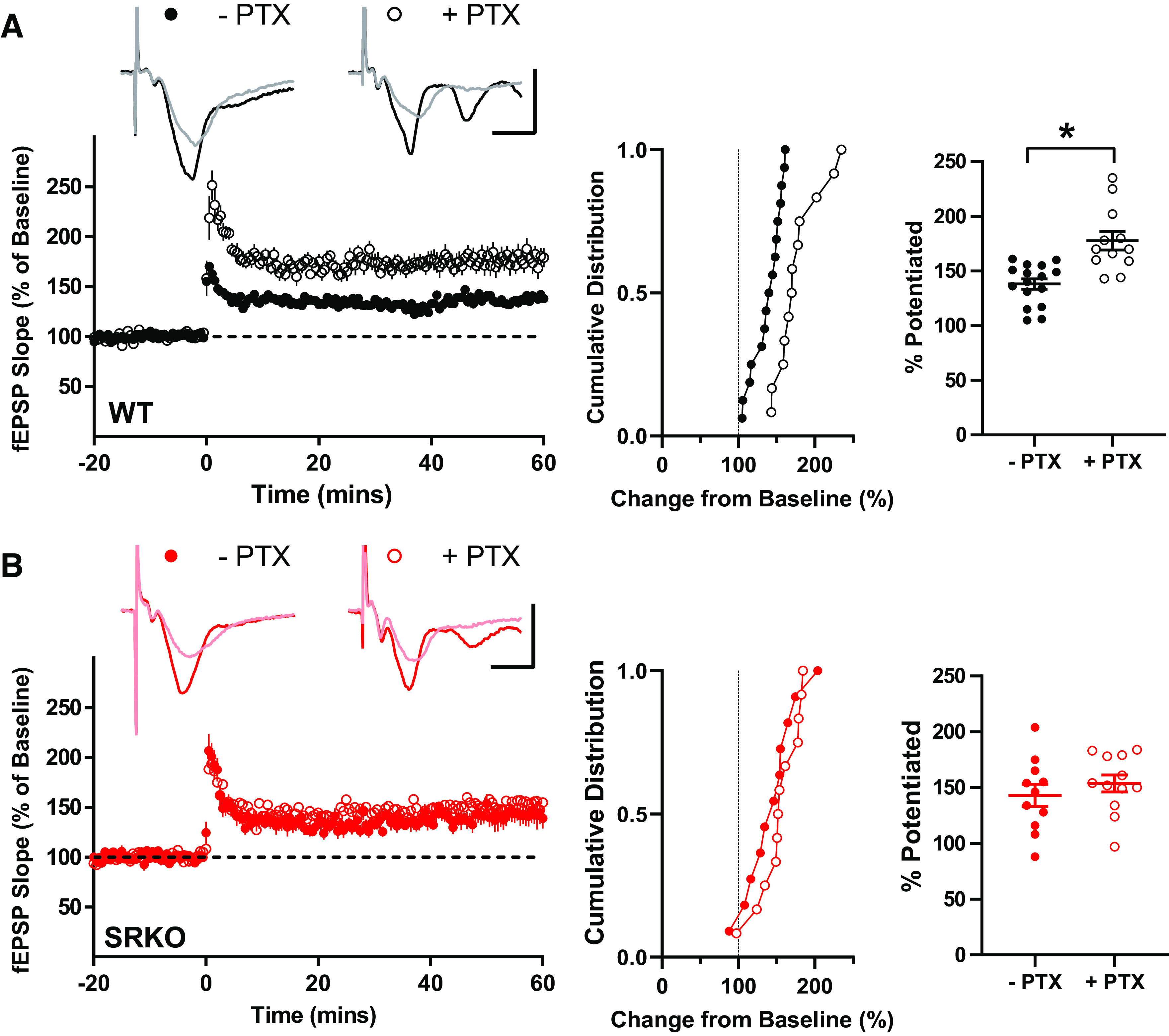
Loss of picrotoxin-induced enhancement of long-term potentiation (LTP) in SRKO mice. A: traces represent superimposed fEPSPs recorded from WT slices during baseline and 60 min after high-frequency stimulation (HFS) in the presence (+) and absence (−) of 50 µM picrotoxin (PTX); scale bars: 1 mV, 20 ms. In slices from WT mice, PTX enhances LTP. Middle, the cumulative distribution of experiments. Right, summary graph of mean percentage potentiation relative to baseline demonstrating that PTX results in significantly enhanced LTP (−PTX: 138 ± 5% of baseline, n = 16; +PTX: 178 ± 7% of baseline, n = 12; P = 0.0002). B: traces represent superimposed fEPSPs recorded from SRKO slices during baseline and 60 min after HFS in the presence (+) and absence (−) of 50 µM PTX; scale bars: 1 mV, 20 ms. In slices from SRKO mice, PTX does not enhance LTP. Middle, the cumulative distribution of experiments. Right, summary graph of mean percentage potentiation relative to baseline showing no effect of PTX on LTP in slices from SRKO mice (−PTX: 143 ± 10% of baseline, n = 11; +PTX: 154 ± 8% of baseline, n = 11; p = 0.394). Data represent means ± SE. fEPSPs, field excitatory postsynaptic potentials; SRKO, serine racemase knockout; WT, wild type. *P < 0.05.
Reduced Inhibitory Synapses Onto CA1 Pyramidal Neurons of SRKO Mice
To examine the source of the reduced GABAergic inhibition in the SRKO mice, we recorded spontaneous IPSCs (sIPSC) from CA1 pyramidal cells (Fig. 4, A–C). There were no significant differences in sIPSC amplitude between SRKO and WT mice (Fig. 4A, P = 0.138, unpaired t test, t(34) = 1.42), though sIPSC frequency was significantly reduced (Fig. 4B, P = 0.006, unpaired t test, t(34) = 2.96). Similarly, mIPSC (Fig. 4, D–F) frequency was significantly reduced in CA1 pyramidal cells from the SRKO mice compared with WT (Fig. 4E, P = 0.0003, unpaired t test, t(23)=4.29). There was also a small decrease in mIPSC amplitude in the SRKO neurons (Fig. 4D, P = 0.042, unpaired t test, t(23) = 2.15). These results suggest that there is a significant reduction of inhibitory synapses onto CA1 pyramidal neurons in the SRKO mice. Though there were no apparent differences in the I/O of excitatory responses at SC-CA1 synapses (Fig. 1A), evoked and spontaneous neurotransmission may be distinct (71). Thus, we also examined sEPSCs and mEPSCs from CA1 pyramidal neurons (Fig. 5). We found no significant differences between cells from WT and SRKO mice in sEPSC amplitude (Fig. 5A, P = 0.79, unpaired t test, t(22) = 0.259), sEPSC frequency (Fig. 5B, P = 0.47, unpaired t test, t(22) = 0.732), or mEPSC frequency (Fig. 5D, P = 0.70, unpaired t test, t(26) = 0.383). There was a small, significant increase in mEPSC amplitude in the SRKO cells (Fig. 5E, P = 0.016, unpaired t test, t(26) = 2.57) that appeared to be most at larger amplitude synapses. Overall, these results, combined with Fig. 1, suggest that fast excitatory neurotransmission is largely normal in CA1 pyramidal cells from the SRKO mice.
Figure 4.
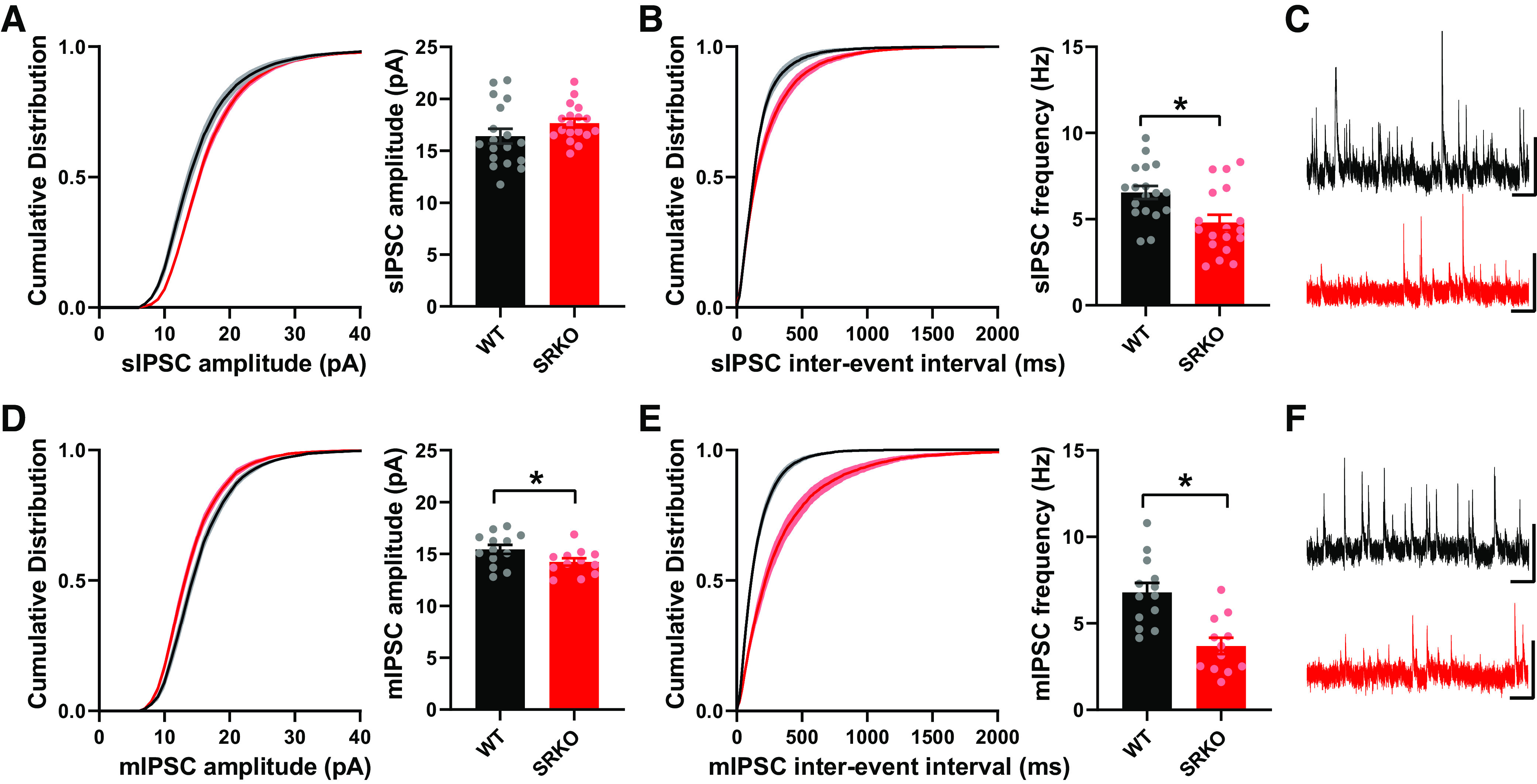
Reduced spontaneous GABAergic synaptic transmission in SRKO mice. A–C: spontaneous IPSCs (sIPSCs) from CA1 pyramidal cells. A: the cumulative distribution of sIPSC amplitude indicated larger amplitudes in SRKO compared with WT (KS test, P < 0.0001), though the mean amplitude of sIPSCs are unchanged between slices from WT and SRKO mice (WT: 16.41 ± 0.708, n = 18; SRKO: 17.66 ± 0.414, n = 18; P = 0.138). B: the cumulative probability (KS test, P < 0.0001) of interevent intervals reveals a shift toward longer intervals and the mean frequency of sIPSCs was significantly decreased in SRKO compared to WT cells (WT: 6.55 ± 0.38 Hz, n = 19; SRKO: 4.80 ± 0.45 Hz, n = 18; P = 0.006). C: sample sIPSC traces from WT (black) and SRKO (red) mice; scale bars: 25 pA and 0.5 s. D–F: miniature IPSCs (mIPSCs) from CA1 pyramidal cells. D: the cumulative distribution (KS test, P < 0.0001) and mean amplitude of mIPSC were significantly reduced in SRKO compared with WT mice (WT: 15.45 ± 0.43 pA, n = 13; SRKO: 14.23 ± 0.36 pA, n = 12; P = 0.042). E: the cumulative distribution (KS Test, P < 0.0001) of interevent intervals and the mean frequency of mIPSCs are significantly decreased in SRKO compared with WT cells (WT: 6.79 ± 0.54 Hz, n = 13; SRKO: 3.69 ± 0.47 Hz, n = 12; P = 0.0003). F: sample mIPSC traces from WT (black) and SRKO (red) mice; scale bars: 25 pA and 0.5 s. Data represent means ± SE. IPSC, inhibitory postsynaptic current; KS test, Kolmogorov–Smirnov test; SRKO, serine racemase knockout; WT, wild type. *P < 0.05.
Figure 5.
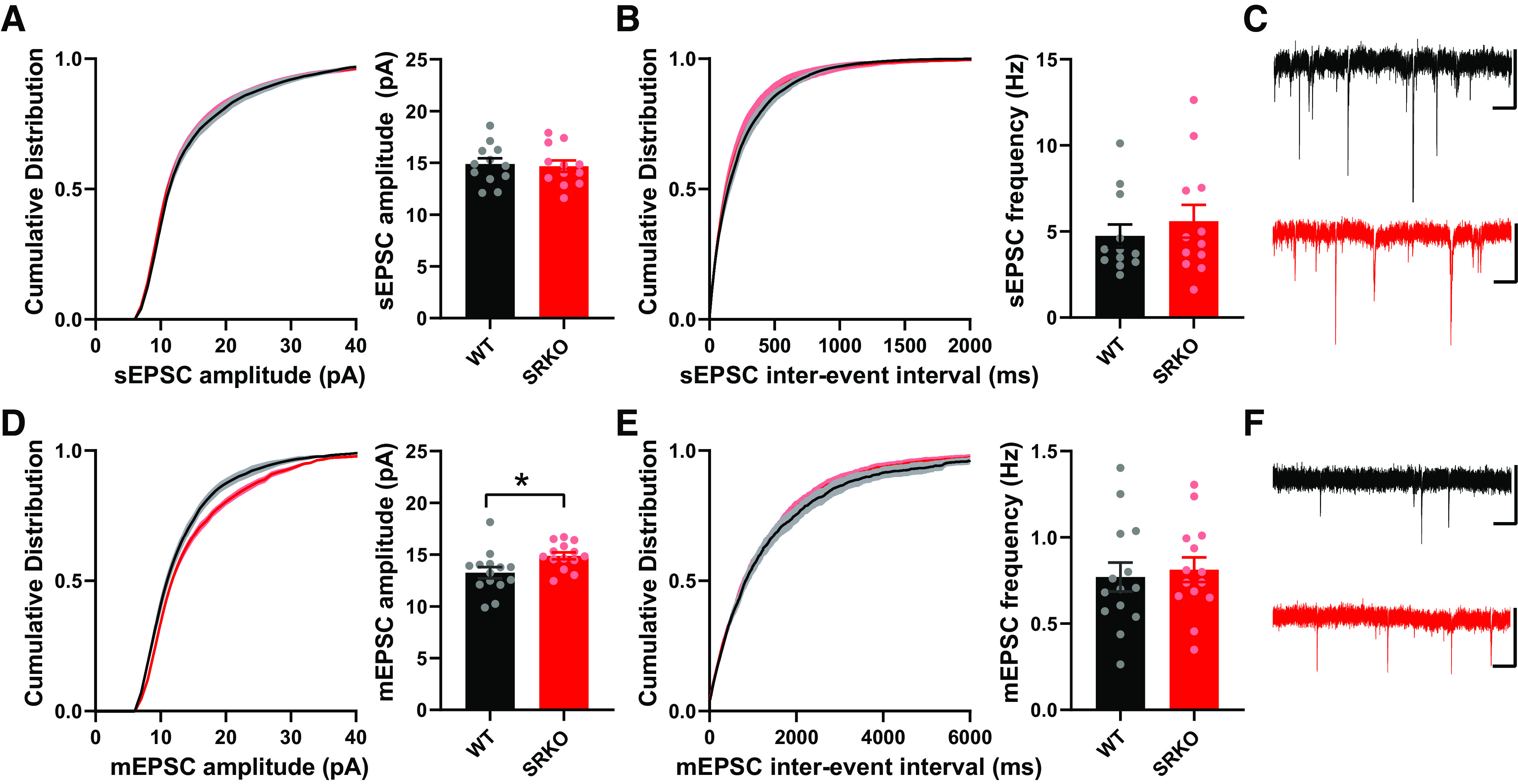
Normal spontaneous excitatory synaptic transmission in SRKO mice. A–C: spontaneous EPSCs from CA1 pyramidal cells. A: the cumulative probability and mean of sEPSC amplitudes were not significantly different between SRKO and WT mice (KS Test, P > 0.05; WT: 14.88 ± 0.56 pA, n = 12; SRKO: 14.68 ± 0.56 pA, n = 12; P = 0.798). B: the cumulative probability (KS test, P > 0.05) of interevent intervals and mean frequency of sEPSCs were also unchanged (WT: 4.74 ± 0.68 Hz, n = 12; SRKO: 5.59 ± 0.95 Hz, n = 12; P = 0.472). C: sample sEPSC traces from WT (black) and SRKO (red) mice; scale bars: 25 pA and 0.5 s. D–F: miniature EPSCs from CA1 pyramidal cells. D: cumulative probability (KS Test, P < 0.0001) and mean amplitude of mEPSCs were significantly changed between SRKO and WT mice (WT: 13.24 ± 0.54 pA, n = 14; SRKO: 14.89 ± 0.34 pA, n = 14; P = 0.016). E: the cumulative probability (KS test, P > 0.05) of interevent intervals and mean frequency of mEPSCs were not significantly different between SRKO and WT mice (WT: 0.770 ± 0.084 Hz, n = 14; SRKO: 0.812 ± 0.072 Hz, n = 14; P = 0.705). F: sample mEPSC traces from WT (black) and SRKO (red) mice; scale bars: 25 pA and 0.5 s. Data represent means ± SE. EPSC, excitatory postsynaptic current; KS test, Kolmogorov–Smirnov test; SRKO, serine racemase knockout; WT, wild type. *P < 0.05.
The reduced frequency of mIPSCs (Fig. 4E), in the absence of apparent changes in presynaptic release probability (Fig. 1D), suggests a reduction in the number of GABAergic synapses onto CA1 pyramidal neurons in the SRKO mice. We then confirmed this synaptic reduction using immunohistochemistry (Fig. 6) by staining for the vesicular GABA transporter (VGAT) in hippocampal slices. Consistent with a reduction of synapses from PV+ interneurons, which form perisomatic synapses onto CA1 pyramidal cells, there was a significant reduction of VGAT density (Fig. 6, A and B, left, P = 0.028, unpaired t test, t(4) = 3.36) and intensity (Fig. 6, A and B, right, P = 0.042, unpaired t test, t(4) = 2.95) in the CA1 pyramidal cell layer in the SRKO mice compared with WT. Similarly, in the stratum radiatum, there was a nonsignificant reduction in VGAT density (Fig. 6, C and D, left, P = 0.092, unpaired t test, t(4) = 2.21) and a significant decrease in VGAT intensity (Fig. 6, C and D, right, P = 0.024, unpaired t test, t(4) = 3.53), that was evenly distributed throughout the stratum radiatum (Fig. 6E) suggesting a broader GABAergic synapse deficit. Taken together with the significant reduction in mIPSC frequency, these results suggest that a loss of GABAergic synapse density in the hippocampus underlies the increased E/I ratio in the SRKO mice.
Figure 6.

Reduced GABAergic synapses onto CA1 pyramidal neurons in SRKO mice. A: representative images of VGAT labeling in the stratum pyramidale of CA1 hippocampus show a reduction in VGAT antibody labeling in SRKO mice; scale bar indicates 5 µm. B: both normalized mean VGAT puncta density (WT: 1.000 ± 0.069, n = 3; SRKO: 0.654 ± 0.076, n = 3; P = 0.028) and normalized mean VGAT puncta intensity in CA1 stratum pyramidale (WT: 1.000 ± 0.156, n = 3; SRKO: 0.453 ± 0.101, n = 3; P = 0.042) are significantly lower in SRKO mice. C: representative images of VGAT labeling in the stratum radiatum of CA1 hippocampus show a reduction in VGAT labeling in SRKO mice; scale bar indicates 5 µm. D: there is a nonsignificant reduction in the normalized mean VGAT puncta density in s. radiatum of CA1 of SRKO mice (WT: 1.000 ± 0.028, n = 3; SRKO: 0.609 ± 0.175, n = 3; P = 0.092), whereas the normalized mean VGAT puncta intensity in s. radiatum is significantly reduced in the SRKO mice (WT: 1.000 ± 0.128, n = 3; SRKO: 0.452 ± 0.088, n = 3; P = 0.024). E: representative images of hippocampal CA1 show that the reduction in VGAT signal is consistent across strata of CA1 in SRKO mice; scale bar indicates 20 µm. Data represent means ± SE. SRKO, serine racemase knockout; VGAT, vesicular GABA transporter; WT, wild type. *P < 0.05.
Deletion of SR from CA1 Pyramidal Neurons Results in a Cell-Autonomous Reduction in GABAergic Synapses
Early studies suggested that d-serine is exclusively synthesized and released by astrocytes (34, 72, 73), leading to the classification of d-serine as a gliotransmitter (74–76). More recent studies, using the SRKO mice as controls, have strongly supported a predominantly neuronal localization (53, 77–84). Furthermore, in agreement with previous studies in cultured neurons (85, 86), we recently reported that SR localizes to the apical dendrites and the post-synaptic density in situ in hippocampal CA1 pyramidal neurons and regulates postsynaptic NMDARs (64). Importantly, while conditional knockout (cKO) of SR from astrocytes has minimal impact on SR levels, cKO from CaMKIIα-expressing forebrain glutamatergic neurons results in ∼65% reduction of SR expression in the cortex and hippocampus (59). The remainder of SR expression is thought to be from GABAergic interneurons. As such, we sought to determine if the decrease in GABAergic synapses onto CA1 pyramidal neurons in the SRKO mice was due to the loss of SR in the pyramidal cells themselves. We utilized a single-neuron genetic approach in the SRfl mice in which SR was removed in a sparse subset of CA1 pyramidal neurons by neonatal stereotaxic injection of adeno-associated virus, serotype 1 expressing a Cre recombinase GFP fusion protein (AAV1-Cre:GFP) (Fig. 7A). This mosaic transduction allows for whole-cell recordings from Cre-expressing (Cre) and untransduced neurons (Ctrl) (Fig. 7B) providing a measurement of the cell-autonomous effects of SR deletion. Similar to the SRKO mice (Fig. 4), we found no differences in mIPSC amplitude (Fig. 7C, P = 0.939, unpaired t test, t(19) = 2.022), but significantly reduced mIPSC frequency (Fig. 7D, P = 0.039, unpaired t test, t(19) = 2.218) in Cre-expressing CA1 pyramidal neurons compared to control neurons. These results suggest that cKO of SR from CA1 pyramidal neurons results in a cell-autonomous reduction in GABAergic synapses.
Figure 7.

Cell-autonomous reductions in spontaneous GABAergic synaptic transmission onto CA1 pyramidal cells following single-neuron SR deletion. A: representative image of the sparse transduction of CA1 pyramidal cells by AAV1-Cre:GFP counterstained by DAPI. Scale bar indicates 100 μm. B: schematic of the experimental setup. Whole cell mIPSC recordings were made from transduced (Cre+) and control CA1 pyramidal cells. C: cumulative probability and mean mIPSC amplitude. Although cumulative probability (KS test, P < 0.0001) of mIPSC amplitude was significantly changed between Cre and Cre+ neurons, the mean mIPSC amplitude from Cre+ neurons was not significantly different than those from control cells (WT: 12.37 ± 0.32 pA, n = 11; SRKO: 11.42 ± 0.34 pA, n = 10; P = 0.939). D: cumulative probability of interevent intervals and mean frequency of mIPSCs. Cumulative probability (KS test, P < 0.0001) and mean frequency from Cre+ neurons were significantly decreased compared to control cells (WT: 1.74 ± 0.27 Hz, n = 11; SRKO: 0.88 ± 0.28 Hz, n = 10; P = 0.039. E: sample mIPSC traces from control (black, top) and Cre+ (green, bottom) pyramidal neurons; scale bars: 25 pA and 0.5 s; inset, 25 pA and 100 ms. Data represent means ± SE. mIPSC, miniature inhibitory postsynaptic current; SRKO, serine racemase knockout; WT, wild type. *P < 0.05.
DISCUSSION
Broad NMDAR deletion causes overly pronounced phenotypes that do not adequately model schizophrenia (36). Germline deletion of NMDARs from mice is perinatally lethal (87–89) and embryonic deletion from only forebrain pyramidal neurons results in death within the first month (90–92). Similarly, mice with a homozygous embryonic deletion of NMDARs from migrating forebrain GABAergic neurons expressing the Dlx5/6 promoter (93), are reportedly nonviable (36). Moreover, broad and regional deletion of NMDARs severely disrupts cortical patterning during development (88, 90). The NMDAR hypomorph mice (9), which have only 5%–10% of wild-type NMDAR expression, have been hailed as a major transgenic model of the NMDAR hypofunction in schizophrenia (94), though they have also been highly criticized for having more global cognitive impairments with earlier onset than what is seen in schizophrenia (37–39). Interestingly, decreases in NMDAR protein is not a consistent finding in schizophrenia (95), suggesting that the hypofunction may be more functional (e.g., downstream signaling) than structural (96). Indeed, NMDARs are macromolecular machines (97) involved in a plethora of signaling processes in neurons and complete loss of NMDARs could lead to a broad range of allostatic changes. In this study, we used a mouse model of NMDAR hypofunction that involves a functional rather than structural reduction in NMDAR activity, the SRKO mice (53). In the SRKO mice, there is a >90% decrease in the levels of d-serine, the primary coagonist for synaptic NMDARs in the forebrain (53, 98). Indeed, deficiency of d-serine and the subsequent hypofunction of NMDARs has been implicated in the pathophysiology of schizophrenia (40) and the SRKO mice display many well-characterized hallmarks of schizophrenia, including reductions in dendritic complexity and spine density (51, 52, 99) and impaired performance on various cognitive tasks (52, 53).
Using the SRKO mice, we have explored the relationship between NMDAR hypofunction and GABAergic inhibition. Because interneurons expressing the calcium-binding protein parvalbumin (PV+) are particularly affected in schizophrenia (46, 100, 101), previous studies have examined PV expression in the SRKO mice. Although one study reported a 26% reduction in PV+ cells in the anterior cingulate cortex of the SRKO mice (102), another found no change in PV immunoreactivity in the hippocampus, prelimbic and infralimbic cortices (103). However, using electrophysiological approaches in ex vivo hippocampal slices we found a significant reduction of GABAergic synapses onto CA1 pyramidal neurons in the SRKO mice. This reduction of GABAergic synaptic inhibition onto pyramidal cells increases the E/I balance resulting in enhanced synaptically driven neuronal excitability.
Consistent with previous studies, baseline excitatory transmission and presynaptic release probability were largely preserved in the SRKO mice (52, 53). Surprisingly, we found normal levels of LTP in the SRKO mice, which initially seemed to be counter to previous studies (52, 53, 59, 69, 70). In each of those studies, however, inhibition was blocked with picrotoxin. Indeed, in the presence of picrotoxin, we also observed a clear reduction in LTP due to the isolation of the NMDAR hypofunction in the SRKO mice. These results also suggested a loss of picrotoxin-induced disinhibition in the SRKO mice which we show is due to a reduction in GABAergic synapses onto CA1 pyramidal neurons in the SRKO mice. We speculate that this reduction of inhibitory synapses and the resulting increase in E/I ratio in the SRKO mice represents a homeostatic compensation to normalize synaptic plasticity. This is similar to recent work in four autism models where the increases in E/I ratio were demonstrated to be homeostatic changes (104), though in that study there was a stabilization of synaptic drive and spiking by a coordinated decrease in excitatory conductance (104). In contrast, we observed increased synaptically-driven spiking in ex vivo slices from the SRKO mice along with generally normal excitatory responses. These differences may represent disparate compensatory demands and homeostatic mechanisms in the cortical layer 2/3 neurons examined in the autism mutants (104) compared with the CA1 pyramidal cells studied here. Importantly, even with the increase in E/I ratio, no epileptiform activity has been reported in the SRKO mice during in vivo electrophysiology nor reported or observed seizure activity (105–107), and one study reported that the SRKO mice had a reduced susceptibility to seizures (108). The lack of apparent seizure activity with the increase E/I ratio further suggests concurrent homeostatic processes, though we cannot rule out covert temporal lobe epileptiform bursting in the SRKO mice. Furthermore, other compensatory mechanisms could contribute to the normalization of LTP in the SRKO mice, including an increase in hippocampal glycine levels (109), and an increased in synaptic GluN2B (53, 64). Overall, these homeostatic changes suggest that there is a prioritization of synaptic and cellular functions over network function resulting in a disruption of the signal-to-noise ratio and impairing cognition. Indeed, SRKO mice display impairments in task-elicited gamma power, enhanced background broadband gamma activity, sensory gating impairments, working memory deficits (106), and disruptions in the auditory steady-state response (107), together supporting an aberrant signal-to-noise ratio impairing cognitive function.
We further show, using a single-neuron genetic deletion approach, that the loss of GABAergic synapses onto pyramidal neurons observed in the SRKO mice is driven in a cell-autonomous manner following the deletion of SR in individual CA1 pyramidal cells. Indeed, recent studies have shown a critical role for NMDARs on pyramidal neurons in regulating GABAergic synapse development (56–58). Specifically, deletion of the obligatory GluN1 subunit of NMDARs from single CA1 pyramidal cells in early development leads to a significant reduction in mIPSC frequency and a loss of GABAergic synapses (57). Importantly, a similar loss of GABAergic synapses upon GluN1 deletion was observed in layer 2/3 pyramidal neurons in the motor cortex and midbrain dopaminergic neurons in the ventral tegmental area (58), suggesting a more generalizable mechanism. This work builds upon older pharmacological studies showing that NMDAR activity can accelerate GABAergic synapse development (110–113). Interestingly, NMDARs have been found to co-localize with GABAA receptors at GABAergic synapses in the developing brain (114–116), though the function of this localization remains unclear. Here, the Cre-expressing virus was injected within 24 h after birth and the stochastic loss of the gene is thought to be complete by 4–5 days (117), followed by loss of the mRNA and protein. This time course overlaps with inhibitory synapse formation, so it remains to be determined if there is disrupted synaptogenesis or a loss of formed or maturing inhibitory synapses. However, these results together support a model whereby NMDAR hypofunction on pyramidal neurons can lead to GABAergic dysfunction through a loss of GABAergic synapses.
The cellular location of the NMDAR hypofunction in schizophrenia has been intensely studied yet remains poorly understood. A large body of pharmacological studies using uncompetitive NMDAR antagonists support a locus of NMDAR hypofunction on cortical GABAergic interneurons, particularly PV positive cells (46, 100, 101). Notably, acute systemic administration of NMDAR antagonists results in the increased activity of cortical pyramidal neurons (118, 119), spillover of cortical glutamate (120, 121), and increases in cortical γ power (122, 123), indicative of increased E/I balance and pyramidal cell disinhibition. Similar evidence for increased cortical excitability following administration of NMDAR antagonists have been found in human studies (7, 124–126). These findings are consistent with the increase in E/I balance and disinhibition we observe here in the SRKO mice and in another recent study (109); however, NMDAR antagonists are thought to preferentially inhibit receptors on fast-spiking PV-positive interneurons (127).
Cell-type-specific knockouts of GluN1 from either pyramidal neurons or PV+ interneurons have provided additional insights into the locus of NMDAR hypofunction in schizophrenia. For example, deletion of GluN1 from PV+ interneurons leads to cortical and hippocampal disinhibition and an increase in the baseline gamma power in the hippocampus (128–131). In addition, acute MK801-induced behaviors were not detected in these mice (129), providing decisive evidence for PV+ interneurons being the locus of NMDAR hypofunction upon systemic NMDAR antagonist administration in adult rodents. Behaviorally, these mice have selective impairments in working memory, habituation, and sociability, but display normal pre-pulse inhibition (PPI) (128, 129, 132). Importantly, because PV-selective promoter expression, and thus NMDAR removal, begins at 2–4 wk of age (129, 130, 132, 133), these mice may not fully model the neurodevelopmental changes occurring in schizophrenia.
Similarly, mice with a deletion of GluN1 from forebrain pyramidal neurons using the CaMKII promoter display a variety of schizophrenia-related phenotypes, including reductions in social interaction, nest-building, and spatial working memory (134, 135). Interestingly, there was also an increase in locomotor activity in the CaMKII-Cre/GluN1 KO mice consistent with dopaminergic models of psychosis (135, 136). Similar to our results, CA1 pyramidal cell excitability was increased along with increased broadband local field potential power in the CaMKII-Cre/GluN1 KO mice (135); however, this was an increase in intrinsic excitability attributable to a reduction in GIRK2 channel activity, rather than due to the loss of synaptic inhibition seen here. Furthermore, no changes in mRNA levels were found in the hippocampus for the GABAergic markers GAD67, PV, cholecystokinin, and somatostatin (135), suggesting a lack of effects on inhibition. Importantly, the CaMKII promoter drives GluN1 deletion in these mice beginning at 3–4 wk of age in CA1 pyramidal neurons which then spreads more broadly throughout the forebrain by 4 mo (137). Thus, as with the deletion of GluN1 from PV+ interneurons, these mice may not recapitulate the developmental aspects of NMDAR hypofunction.
Consistent with a reduction in synapses from PV+ basket cells, we found a significant reduction in perisomatic VGAT puncta density and intensity in the CA1 pyramidal cell layer. However, the density and intensity of VGAT puncta were also decreased in the stratum radiatum with no apparent proximal-distal differences along the apical dendrites of CA1 pyramidal neurons, supporting a broad reduction of GABAergic synapses. Indeed, while PV+ interneurons are particularly affected in schizophrenia (46, 100, 101), multiple interneuron subtypes have been implicated (100, 138–140) and hippocampal inhibitory networks appear especially sensitive to NMDAR hypofunction (141, 142). Interestingly, the decreases in VGAT puncta density and intensity were more extensive than the reductions in mIPSC frequency and amplitude. This difference may be methodological or a sampling bias, but may also represent changes in VGAT expression that are not linearly correlated with postsynaptic responsiveness.
Overall, our data suggest that a pyramidal cell locus of synaptic NMDAR hypofunction could lead to GABAergic deficits through the impaired development of feedback inhibitory synapses. Additional studies will be needed to elucidate the molecular mechanisms underlying the role of NMDARs in GABAergic synapse development and to ascertain the relationship between inhibitory synapses on pyramidal neurons and endophenotypes in schizophrenia.
GRANTS
This work was supported by R21MH116315 and R01MH117130 to J.A.G.; and R01NS060125 to A.K.M.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.A.J., S.C., J.M.W., and J.A.G. conceived and designed research; S.A.J., S.C., J.M.W., and E.R.D. performed experiments; S.A.J., S.C., J.M.W., E.R.D., and J.A.G. analyzed data; S.A.J., S.C., J.M.W., A.K.M., and J.A.G. interpreted results of experiments; S.A.J., S.C., J.M.W., and J.A.G. prepared figures; S.A.J. and J.A.G. drafted manuscript; S.C. and J.M.W. edited and revised manuscript; A.K.M. and J.A.G. approved final version of manuscript.
ACKNOWLEDGMENTS
We would like to thank Joseph Coyle for the SR floxed and knockout mice, and Haley Martin, Zaiyang “Sunny” Zhang, and Casey Martin for assistance in mouse breeding and genotyping.
REFERENCES
- 1.Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry 148: 1301–1308, 1991. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- 2.Jentsch JD, Redmond DE Jr, Elsworth JD, Taylor JR, Youngren KD, Roth RH. Enduring cognitive deficits and cortical dopamine dysfunction in monkeys after long-term administration of phencyclidine. Science 277: 953–955, 1997. doi: 10.1126/science.277.5328.953. [DOI] [PubMed] [Google Scholar]
- 3.Kirihara K, Rissling AJ, Swerdlow NR, Braff DL, Light GA. Hierarchical organization of gamma and theta oscillatory dynamics in schizophrenia. Biol Psychiatry 71: 873–880, 2012. doi: 10.1016/j.biopsych.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakazawa K, Sapkota K. The origin of NMDA receptor hypofunction in schizophrenia. Pharmacol Ther 205: 107426, 2020. doi: 10.1016/j.pharmthera.2019.107426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB Jr, Charney DS. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 51: 199–214, 1994. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- 6.Lahti AC, Weiler MA, Tamara MB, Parwani A, Tamminga CA. Effects of ketamine in normal and schizophrenic volunteers. Neuropsychopharmacology 25: 455–467, 2001. doi: 10.1016/S0893-133X(01)00243-3. [DOI] [PubMed] [Google Scholar]
- 7.Lahti AC, Koffel B, LaPorte D, Tamminga CA. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology 13: 9–19, 1995. doi: 10.1016/0893-133X(94)00131-I. [DOI] [PubMed] [Google Scholar]
- 8.Malhotra AK, Pinals DA, Adler CM, Elman I, Clifton A, Pickar D, Breier A. Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology 17: 141–150, 1997. doi: 10.1016/S0893-133X(97)00036-5. [DOI] [PubMed] [Google Scholar]
- 9.Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell 98: 427–436, 1999. doi: 10.1016/s0092-8674(00)81972-8. [DOI] [PubMed] [Google Scholar]
- 10.Duncan G, Miyamoto S, Gu H, Lieberman J, Koller B, Snouwaert J. Alterations in regional brain metabolism in genetic and pharmacological models of reduced NMDA receptor function. Brain Res 951: 166–176, 2002. doi: 10.1016/s0006-8993(02)03156-6. [DOI] [PubMed] [Google Scholar]
- 11.Duncan GE, Moy SS, Perez A, Eddy DM, Zinzow WM, Lieberman JA, Snouwaert JN, Koller BH. Deficits in sensorimotor gating and tests of social behavior in a genetic model of reduced NMDA receptor function. Behav Brain Res 153: 507–519, 2004. doi: 10.1016/j.bbr.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 12.Fradley RL, O'Meara GF, Newman RJ, Andrieux A, Job D, Reynolds DS. STOP knockout and NMDA NR1 hypomorphic mice exhibit deficits in sensorimotor gating. Behav Brain Res 163: 257–264, 2005. doi: 10.1016/j.bbr.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 13.Duncan GE, Moy SS, Lieberman JA, Koller BH. Typical and atypical antipsychotic drug effects on locomotor hyperactivity and deficits in sensorimotor gating in a genetic model of NMDA receptor hypofunction. Pharmacol Biochem Behav 85: 481–491, 2006. doi: 10.1016/j.pbb.2006.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moy SS, Perez A, Koller BH, Duncan GE. Amphetamine-induced disruption of prepulse inhibition in mice with reduced NMDA receptor function. Brain Res 1089: 186–194, 2006. doi: 10.1016/j.brainres.2006.03.073. [DOI] [PubMed] [Google Scholar]
- 15.Bickel S, Lipp HP, Umbricht D. Early auditory sensory processing deficits in mouse mutants with reduced NMDA receptor function. Neuropsychopharmacology 33: 1680–1689, 2008. doi: 10.1038/sj.npp.1301536. [DOI] [PubMed] [Google Scholar]
- 16.Dzirasa K, Ramsey AJ, Takahashi DY, Stapleton J, Potes JM, Williams JK, Gainetdinov RR, Sameshima K, Caron MG, Nicolelis MA. Hyperdopaminergia and NMDA receptor hypofunction disrupt neural phase signaling. J Neurosci 29: 8215–8224, 2009. doi: 10.1523/JNEUROSCI.1773-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halene TB, Ehrlichman RS, Liang Y, Christian EP, Jonak GJ, Gur TL, Blendy JA, Dow HC, Brodkin ES, Schneider F, Gur RC, Siegel SJ. Assessment of NMDA receptor NR1 subunit hypofunction in mice as a model for schizophrenia. Genes Brain Behav 8: 661–675, 2009. doi: 10.1111/j.1601-183X.2009.00504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramsey AJ. NR1 knockdown mice as a representative model of the glutamate hypothesis of schizophrenia. Prog Brain Res 179: 51–58, 2009. doi: 10.1016/S0079-6123(09)17906-2. [DOI] [PubMed] [Google Scholar]
- 19.Saunders JA, Gandal MJ, Siegel SJ. NMDA antagonists recreate signal-to-noise ratio and timing perturbations present in schizophrenia. Neurobiol Dis 46: 93–100, 2012. doi: 10.1016/j.nbd.2011.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci 6: 312–324, 2005. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- 21.Sohal VS, Rubenstein JLR. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol Psychiatry 24: 1248–1257, 2019. doi: 10.1038/s41380-019-0426-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewis DA, Pierri JN, Volk DW, Melchitzky DS, Woo TU. Altered GABA neurotransmission and prefrontal cortical dysfunction in schizophrenia. Biol Psychiatry 46: 616–626, 1999. doi: 10.1016/s0006-3223(99)00061-x. [DOI] [PubMed] [Google Scholar]
- 23.Lewis DA, Volk DW, Hashimoto T. Selective alterations in prefrontal cortical GABA neurotransmission in schizophrenia: a novel target for the treatment of working memory dysfunction. Psychopharmacology (Berl) 174: 143–150, 2004. doi: 10.1007/s00213-003-1673-x. [DOI] [PubMed] [Google Scholar]
- 24.Lewis DA, Hashimoto T, Morris HM. Cell and receptor type-specific alterations in markers of GABA neurotransmission in the prefrontal cortex of subjects with schizophrenia. Neurotox Res 14: 237–248, 2008. doi: 10.1007/BF03033813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonzalez-Burgos G, Fish KN, Lewis DA. GABA neuron alterations, cortical circuit dysfunction and cognitive deficits in schizophrenia. Neural Plast 2011: 723184, 2011. doi: 10.1155/2011/723184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stan AD, Lewis DA. Altered cortical GABA neurotransmission in schizophrenia: insights into novel therapeutic strategies. Curr Pharm Biotechnol 13: 1557–1562, 2012. doi: 10.2174/138920112800784925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glausier JR, Lewis DA. GABA and schizophrenia: where we stand and where we need to go. Schizophr Res 181: 2–3, 2017. doi: 10.1016/j.schres.2017.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lodge DJ, Behrens MM, Grace AA. A loss of parvalbumin-containing interneurons is associated with diminished oscillatory activity in an animal model of schizophrenia. J Neurosci 29: 2344–2354, 2009. doi: 10.1523/JNEUROSCI.5419-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sohal VS. How close are we to understanding what (if anything) gamma oscillations do in cortical circuits? J Neurosci 36: 10489–10495, 2016. doi: 10.1523/JNEUROSCI.0990-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singer W, Gray CM. Visual feature integration and the temporal correlation hypothesis. Annu Rev Neurosci 18: 555–586, 1995.doi: 10.1146/annurev.ne.18.030195.003011. [DOI] [PubMed] [Google Scholar]
- 31.Cho KK, Hoch R, Lee AT, Patel T, Rubenstein JL, Sohal VS. Gamma rhythms link prefrontal interneuron dysfunction with cognitive inflexibility in Dlx5/6(+/−) mice. Neuron 85: 1332–1343, 2015. doi: 10.1016/j.neuron.2015.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tiesinga PH, Fellous JM, Salinas E, Jose JV, Sejnowski TJ. Inhibitory synchrony as a mechanism for attentional gain modulation. J Physiol Paris 98: 296–314, 2004. doi: 10.1016/j.jphysparis.2005.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim H, Ahrlund-Richter S, Wang X, Deisseroth K, Carlen M. Prefrontal parvalbumin neurons in control of attention. Cell 164: 208–218, 2016. doi: 10.1016/j.cell.2015.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wolosker H, Blackshaw S, Snyder SH. Serine racemase: a glial enzyme synthesizing D-serine to regulate glutamate-N-methyl-D-aspartate neurotransmission. Proc Natl Acad Sci USA 96: 13409–13414, 1999. doi: 10.1073/pnas.96.23.13409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coyle JT, Balu DT. The role of serine racemase in the pathophysiology of brain disorders. Adv Pharmacol 82: 35–56, 2018. doi: 10.1016/bs.apha.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakazawa K, Jeevakumar V, Nakao K. Spatial and temporal boundaries of NMDA receptor hypofunction leading to schizophrenia. NPJ Schizophr 3: 7, 2017. doi: 10.1038/s41537-016-0003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barkus C, Dawson LA, Sharp T, Bannerman DM. GluN1 hypomorph mice exhibit wide-ranging behavioral alterations. Genes Brain Behav 11: 342–351, 2012. doi: 10.1111/j.1601-183X.2012.00767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gandal MJ, Anderson RL, Billingslea EN, Carlson GC, Roberts TP, Siegel SJ. Mice with reduced NMDA receptor expression: more consistent with autism than schizophrenia? Genes Brain Behav 11: 740–750, 2012. doi: 10.1111/j.1601-183X.2012.00816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moy SS, Nikolova VD, Riddick NV, Baker LK, Koller BH. Preweaning sensorimotor deficits and adolescent hypersociability in Grin1 knockdown mice. Dev Neurosci 34: 159–173, 2012. doi: 10.1159/000337984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coyle JT. NMDA receptor and schizophrenia: a brief history. Schizophr Bull 38: 920–926, 2012. doi: 10.1093/schbul/sbs076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chumakov I, Blumenfeld M, Guerassimenko O, Cavarec L, Palicio M, Abderrahim H, et al. Genetic and physiological data implicating the new human gene G72 and the gene for D-amino acid oxidase in schizophrenia. Proc Natl Acad Sci USA 99: 13675–13680, 2002[Erratum inProc Natl Acad Sci USA99: 17221, 2002]. doi: 10.1073/pnas.182412499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Detera-Wadleigh SD, McMahon FJ. G72/G30 in schizophrenia and bipolar disorder: review and meta-analysis. Biol Psychiatry 60: 106–114, 2006. doi: 10.1016/j.biopsych.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 43.Goltsov AY, Loseva JG, Andreeva TV, Grigorenko AP, Abramova LI, Kaleda VG, Orlova VA, Moliaka YK, Rogaev EI. Polymorphism in the 5'-promoter region of serine racemase gene in schizophrenia. Mol Psychiatry 11: 325–326, 2006. doi: 10.1038/sj.mp.4001801. [DOI] [PubMed] [Google Scholar]
- 44.Morita Y, Ujike H, Tanaka Y, Otani K, Kishimoto M, Morio A, Kotaka T, Okahisa Y, Matsushita M, Morikawa A, Hamase K, Zaitsu K, Kuroda S. A genetic variant of the serine racemase gene is associated with schizophrenia. Biol Psychiatry 61: 1200–1203, 2007. doi: 10.1016/j.biopsych.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 45.Shi J, Badner JA, Gershon ES, Liu C. Allelic association of G72/G30 with schizophrenia and bipolar disorder: a comprehensive meta-analysis. Schizophr Res 98: 89–97, 2008. doi: 10.1016/j.schres.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, Sampson AR, Lewis DA. Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci 23: 6315–6326, 2003. doi: 10.1523/JNEUROSCI.23-15-06315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bendikov I, Nadri C, Amar S, Panizzutti R, De Miranda J, Wolosker H, Agam G. A CSF and postmortem brain study of D-serine metabolic parameters in schizophrenia. Schizophr Res 90: 41–51, 2007. doi: 10.1016/j.schres.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 48.Tsai G, Yang P, Chung LC, Lange N, Coyle JT. D-serine added to antipsychotics for the treatment of schizophrenia. Biol Psychiatry 44: 1081–1089, 1998. doi: 10.1016/s0006-3223(98)00279-0. [DOI] [PubMed] [Google Scholar]
- 49.Heresco-Levy U, Javitt DC, Ebstein R, Vass A, Lichtenberg P, Bar G, Catinari S, Ermilov M. D-serine efficacy as add-on pharmacotherapy to risperidone and olanzapine for treatment-refractory schizophrenia. Biol Psychiatry 57: 577–585, 2005. doi: 10.1016/j.biopsych.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 50.Lane HY, Chang YC, Liu YC, Chiu CC, Tsai GE. Sarcosine or D-serine add-on treatment for acute exacerbation of schizophrenia: a randomized, double-blind, placebo-controlled study. Arch Gen Psychiatry 62: 1196–1204, 2005. doi: 10.1001/archpsyc.62.11.1196. [DOI] [PubMed] [Google Scholar]
- 51.Balu DT, Basu AC, Corradi JP, Cacace AM, Coyle JT. The NMDA receptor co-agonists, D-serine and glycine, regulate neuronal dendritic architecture in the somatosensory cortex. Neurobiol Dis 45: 671–682, 2012. doi: 10.1016/j.nbd.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Balu DT, Li Y, Puhl MD, Benneyworth MA, Basu AC, Takagi S, Bolshakov VY, Coyle JT. Multiple risk pathways for schizophrenia converge in serine racemase knockout mice, a mouse model of NMDA receptor hypofunction. Proc Natl Acad Sci USA 110: E2400–E2409, 2013. doi: 10.1073/pnas.1304308110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Basu AC, Tsai GE, Ma CL, Ehmsen JT, Mustafa AK, Han L, Jiang ZI, Benneyworth MA, Froimowitz MP, Lange N, Snyder SH, Bergeron R, Coyle JT. Targeted disruption of serine racemase affects glutamatergic neurotransmission and behavior. Mol Psychiatry 14: 719–727, 2009[Erratum inMol Psychiatry15: 1122, 2010]. doi: 10.1038/mp.2008.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.DeVito LM, Balu DT, Kanter BR, Lykken C, Basu AC, Coyle JT, Eichenbaum H. Serine racemase deletion disrupts memory for order and alters cortical dendritic morphology. Genes Brain Behav 10: 210–222, 2011. doi: 10.1111/j.1601-183X.2010.00656.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Balu DT, Coyle JT. Chronic D-serine reverses arc expression and partially rescues dendritic abnormalities in a mouse model of NMDA receptor hypofunction. Neurochem Int 75: 76–78, 2014. doi: 10.1016/j.neuint.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lu W, Bushong EA, Shih TP, Ellisman MH, Nicoll RA. The cell-autonomous role of excitatory synaptic transmission in the regulation of neuronal structure and function. Neuron 78: 433–439, 2013. doi: 10.1016/j.neuron.2013.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gu X, Zhou L, Lu W. An NMDA receptor-dependent mechanism underlies inhibitory synapse development. Cell Rep 14: 471–478, 2016. doi: 10.1016/j.celrep.2015.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gu X, Lu W. Genetic deletion of NMDA receptors suppresses GABAergic synaptic transmission in two distinct types of central neurons. Neurosci Lett 668: 147–153, 2018. doi: 10.1016/j.neulet.2018.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Benneyworth MA, Li Y, Basu AC, Bolshakov VY, Coyle JT. Cell selective conditional null mutations of serine racemase demonstrate a predominate localization in cortical glutamatergic neurons. Cell Mol Neurobiol 32: 613–624, 2012. doi: 10.1007/s10571-012-9808-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bischofberger J, Engel D, Li L, Geiger JR, Jonas P. Patch-clamp recording from mossy fiber terminals in hippocampal slices. Nat Protoc 1: 2075–2081, 2006. doi: 10.1038/nprot.2006.312. [DOI] [PubMed] [Google Scholar]
- 61.Gray JA, Shi Y, Usui H, During MJ, Sakimura K, Nicoll RA. Distinct modes of AMPA receptor suppression at developing synapses by GluN2A and GluN2B: single-cell NMDA receptor subunit deletion in vivo. Neuron 71: 1085–1101, 2011. doi: 10.1016/j.neuron.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wong JM, Gray JA. Long-term depression is independent of GluN2 subunit composition. J Neurosci 38: 4462–4470, 2018. doi: 10.1523/JNEUROSCI.0394-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ting JT, Lee Br Chong P, Soler-Llavina G, Cobbs C, Koch C, Zeng H, Lein E. Preparation of acute brain slices using an optimized N-methyl-D-glucamine protective recovery method. J Vis Exp: 53825, 2018. doi: 10.3791/53825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wong JM, Folorunso OO, Barragan EV, Berciu C, Harvey TL, Coyle JT, Balu DT, Gray JA. Postsynaptic serine racemase regulates NMDA receptor function. J Neurosci 40: 9564–9575, 2020. doi: 10.1523/JNEUROSCI.1525-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Elmer BM, Estes ML, Barrow SL, McAllister AK. MHCI requires MEF2 transcription factors to negatively regulate synapse density during development and in disease. J Neurosci 33: 13791–13804, 2013. doi: 10.1523/JNEUROSCI.2366-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gulledge AT, Kampa BM, Stuart GJ. Synaptic integration in dendritic trees. J Neurobiol 64: 75–90, 2005[Erratum inJ Neurobiol65: 205-6, 2005]. doi: 10.1002/neu.20144. [DOI] [PubMed] [Google Scholar]
- 67.Marder CP, Buonomano DV. Timing and balance of inhibition enhance the effect of long-term potentiation on cell firing. J Neurosci 24: 8873–8884, 2004. doi: 10.1523/JNEUROSCI.2661-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wigstrom H, Gustafsson B. Facilitated induction of hippocampal long-lasting potentiation during blockade of inhibition. Nature 301: 603–604, 1983. doi: 10.1038/301603a0. [DOI] [PubMed] [Google Scholar]
- 69.Henneberger C, Papouin T, Oliet SH, Rusakov DA. Long-term potentiation depends on release of D-serine from astrocytes. Nature 463: 232–236, 2010. doi: 10.1038/nature08673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Balu DT, Li Y, Takagi S, Presti KT, Ramikie TS, Rook JM, Jones CK, Lindsley CW, Conn PJ, Bolshakov VY, Coyle JT. An mGlu5-Positive Allosteric modulator rescues the neuroplasticity deficits in a genetic model of NMDA receptor hypofunction in schizophrenia. Neuropsychopharmacology 41: 2052–2061, 2016. doi: 10.1038/npp.2016.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kavalali ET. The mechanisms and functions of spontaneous neurotransmitter release. Nat Rev Neurosci 16: 5–16, 2015. doi: 10.1038/nrn3875. [DOI] [PubMed] [Google Scholar]
- 72.Schell MJ, Molliver ME, Snyder SH. D-serine, an endogenous synaptic modulator: localization to astrocytes and glutamate-stimulated release. Proc Natl Acad Sci USA 92: 3948–3952, 1995. doi: 10.1073/pnas.92.9.3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schell MJ, Brady RO Jr, Molliver ME, Snyder SH. D-serine as a neuromodulator: regional and developmental localizations in rat brain glia resemble NMDA receptors. J Neurosci 17: 1604–1615, 1997. doi: 10.1523/JNEUROSCI.17-05-01604.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wolosker H, Panizzutti R, De Miranda J. Neurobiology through the looking-glass: D-serine as a new glial-derived transmitter. Neurochem Int 41: 327–332, 2002. doi: 10.1016/s0197-0186(02)00055-4. [DOI] [PubMed] [Google Scholar]
- 75.Miller RF. D-Serine as a glial modulator of nerve cells. Glia 47: 275–283, 2004. doi: 10.1002/glia.20073. [DOI] [PubMed] [Google Scholar]
- 76.Panatier A, Theodosis DT, Mothet JP, Touquet B, Pollegioni L, Poulain DA, Oliet SH. Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell 125: 775–784, 2006. doi: 10.1016/j.cell.2006.02.051. [DOI] [PubMed] [Google Scholar]
- 77.Kartvelishvily E, Shleper M, Balan L, Dumin E, Wolosker H. Neuron-derived D-serine release provides a novel means to activate N-methyl-D-aspartate receptors. J Biol Chem 281: 14151–14162, 2006. doi: 10.1074/jbc.M512927200. [DOI] [PubMed] [Google Scholar]
- 78.Yoshikawa M, Takayasu N, Hashimoto A, Sato Y, Tamaki R, Tsukamoto H, Kobayashi H, Noda S. The serine racemase mRNA is predominantly expressed in rat brain neurons. Arch Histol Cytol 70: 127–134, 2007. doi: 10.1679/aohc.70.127. [DOI] [PubMed] [Google Scholar]
- 79.Miya K, Inoue R, Takata Y, Abe M, Natsume R, Sakimura K, Hongou K, Miyawaki T, Mori H. Serine racemase is predominantly localized in neurons in mouse brain. J Comp Neurol 510: 641–654, 2008. doi: 10.1002/cne.21822. [DOI] [PubMed] [Google Scholar]
- 80.Ding X, Ma N, Nagahama M, Yamada K, Semba R. Localization of D-serine and serine racemase in neurons and neuroglias in mouse brain. Neurol Sci 32: 263–267, 2011. doi: 10.1007/s10072-010-0422-2. [DOI] [PubMed] [Google Scholar]
- 81.Ehmsen JT, Ma TM, Sason H, Rosenberg D, Ogo T, Furuya S, Snyder SH, Wolosker H. D-serine in glia and neurons derives from 3-phosphoglycerate dehydrogenase. J Neurosci 33: 12464–12469, 2013. doi: 10.1523/JNEUROSCI.4914-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Balu DT, Takagi S, Puhl MD, Benneyworth MA, Coyle JT. D-serine and serine racemase are localized to neurons in the adult mouse and human forebrain. Cell Mol Neurobiol 34: 419–435, 2014. doi: 10.1007/s10571-014-0027-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wolosker H, Balu DT, Coyle JT. The rise and fall of the d-serine-mediated gliotransmission hypothesis. Trends Neurosci 39: 712–721, 2016. doi: 10.1016/j.tins.2016.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Balu DT, Presti KT, Huang CCY, Muszynski K, Radzishevsky I, Wolosker H, Guffanti G, Ressler KJ, Coyle JT. Serine racemase and D-serine in the amygdala are dynamically involved in fear learning. Biol Psychiatry 83: 273–283, 2018. doi: 10.1016/j.biopsych.2017.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ma TM, Paul BD, Fu C, Hu S, Zhu H, Blackshaw S, Wolosker H, Snyder SH. Serine racemase regulated by binding to stargazin and PSD-95: potential N-methyl-D-aspartate-alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (NMDA-AMPA) glutamate neurotransmission cross-talk. J Biol Chem 289: 29631–29641, 2014. doi: 10.1074/jbc.M114.571604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lin H, Jacobi AA, Anderson SA, Lynch DR. D-Serine and serine racemase are associated with PSD-95 and glutamatergic synapse stability. Front Cell Neurosci 10: 34, 2016. doi: 10.3389/fncel.2016.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Forrest D, Yuzaki M, Soares HD, Ng L, Luk DC, Sheng M, Stewart CL, Morgan JI, Connor JA, Curran T. Targeted disruption of NMDA receptor 1 gene abolishes NMDA response and results in neonatal death. Neuron 13: 325–338, 1994. doi: 10.1016/0896-6273(94)90350-6. [DOI] [PubMed] [Google Scholar]
- 88.Li Y, Erzurumlu RS, Chen C, Jhaveri S, Tonegawa S. Whisker-related neuronal patterns fail to develop in the trigeminal brainstem nuclei of NMDAR1 knockout mice. Cell 76: 427–437, 1994. doi: 10.1016/0092-8674(94)90108-2. [DOI] [PubMed] [Google Scholar]
- 89.Kutsuwada T, Sakimura K, Manabe T, Takayama C, Katakura N, Kushiya E, Natsume R, Watanabe M, Inoue Y, Yagi T, Aizawa S, Arakawa M, Takahashi T, Nakamura Y, Mori H, Mishina M. Impairment of suckling response, trigeminal neuronal pattern formation, and hippocampal LTD in NMDA receptor epsilon 2 subunit mutant mice. Neuron 16: 333–344, 1996. doi: 10.1016/s0896-6273(00)80051-3. [DOI] [PubMed] [Google Scholar]
- 90.Iwasato T, Datwani A, Wolf AM, Nishiyama H, Taguchi Y, Tonegawa S, Knopfel T, Erzurumlu RS, Itohara S. Cortex-restricted disruption of NMDAR1 impairs neuronal patterns in the barrel cortex. Nature 406: 726–731, 2000. doi: 10.1038/35021059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ultanir SK, Kim JE, Hall BJ, Deerinck T, Ellisman M, Ghosh A. Regulation of spine morphology and spine density by NMDA receptor signaling in vivo. Proc Natl Acad Sci USA 104: 19553–19558, 2007. doi: 10.1073/pnas.0704031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Quintero GC, Erzurumlu RS, Vaccarino AL. Evaluation of morphine analgesia and motor coordination in mice following cortex-specific knockout of the N-methyl-D-aspartate NR1-subunit. Neurosci Lett 437: 55–58, 2008. doi: 10.1016/j.neulet.2008.03.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zerucha T, Stuhmer T, Hatch G, Park BK, Long Q, Yu G, Gambarotta A, Schultz JR, Rubenstein JL, Ekker M. A highly conserved enhancer in the Dlx5/Dlx6 intergenic region is the site of cross-regulatory interactions between Dlx genes in the embryonic forebrain. J Neurosci 20: 709–721, 2000. doi: 10.1523/JNEUROSCI.20-02-00709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gainetdinov RR, Mohn AR, Caron MG. Genetic animal models: focus on schizophrenia. Trends Neurosci 24: 527–533, 2001. doi: 10.1016/s0166-2236(00)01886-5. [DOI] [PubMed] [Google Scholar]
- 95.Catts VS, Lai YL, Weickert CS, Weickert TW, Catts SV. A quantitative review of the postmortem evidence for decreased cortical N-methyl-D-aspartate receptor expression levels in schizophrenia: how can we link molecular abnormalities to mismatch negativity deficits? Biol Psychol 116: 57–67, 2016. doi: 10.1016/j.biopsycho.2015.10.013. [DOI] [PubMed] [Google Scholar]
- 96.Banerjee A, Wang HY, Borgmann-Winter KE, MacDonald ML, Kaprielian H, Stucky A, Kvasic J, Egbujo C, Ray R, Talbot K, Hemby SE, Siegel SJ, Arnold SE, Sleiman P, Chang X, Hakonarson H, Gur RE, Hahn CG. Src kinase as a mediator of convergent molecular abnormalities leading to NMDAR hypoactivity in schizophrenia. Mol Psychiatry 20: 1091–1100, 2015. doi: 10.1038/mp.2014.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fan X, Jin WY, Wang YT. The NMDA receptor complex: a multifunctional machine at the glutamatergic synapse. Front Cell Neurosci 8: 160, 2014. doi: 10.3389/fncel.2014.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mothet JP, Parent AT, Wolosker H, Brady RO Jr, Linden DJ, Ferris CD, Rogawski MA, Snyder SH. D-serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc Natl Acad Sci USA 97: 4926–4931, 2000. doi: 10.1073/pnas.97.9.4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rosoklija G, Toomayan G, Ellis SP, Keilp J, Mann JJ, Latov N, Hays AP, Dwork AJ. Structural abnormalities of subicular dendrites in subjects with schizophrenia and mood disorders: preliminary findings. Arch Gen Psychiatry 57: 349–356, 2000. doi: 10.1001/archpsyc.57.4.349. [DOI] [PubMed] [Google Scholar]
- 100.Hashimoto T, Arion D, Unger T, Maldonado-Aviles JG, Morris HM, Volk DW, Mirnics K, Lewis DA. Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol Psychiatry 13: 147–161, 2008. doi: 10.1038/sj.mp.4002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mellios N, Huang HS, Baker SP, Galdzicka M, Ginns E, Akbarian S. Molecular determinants of dysregulated GABAergic gene expression in the prefrontal cortex of subjects with schizophrenia. Biol Psychiatry 65: 1006–1014, 2009. doi: 10.1016/j.biopsych.2008.11.019. [DOI] [PubMed] [Google Scholar]
- 102.Steullet P, Cabungcal JH, Coyle J, Didriksen M, Gill K, Grace AA, Hensch TK, LaMantia AS, Lindemann L, Maynard TM, Meyer U, Morishita H, O'Donnell P, Puhl M, Cuenod M, Do KQ. Oxidative stress-driven parvalbumin interneuron impairment as a common mechanism in models of schizophrenia. Mol Psychiatry 22: 936–943, 2017. doi: 10.1038/mp.2017.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Benneyworth MA, Roseman AS, Basu AC, Coyle JT. Failure of NMDA receptor hypofunction to induce a pathological reduction in PV-positive GABAergic cell markers. Neurosci Lett 488: 267–271, 2011. doi: 10.1016/j.neulet.2010.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Antoine MW, Langberg T, Schnepel P, Feldman DE. Increased excitation-inhibition ratio stabilizes synapse and circuit excitability in four autism mouse models. Neuron 101: 648–661,2019. doi: 10.1016/j.neuron.2018.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Perez EJ, Tapanes SA, Loris ZB, Balu DT, Sick TJ, Coyle JT, Liebl DJ. Enhanced astrocytic d-serine underlies synaptic damage after traumatic brain injury. J Clin Invest 127: 3114–3125, 2017. doi: 10.1172/JCI92300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Aguilar DD, Radzik LK, Schiffino FL, Folorunso O, Zielinski MR, Coyle JT, Balu DT, McNally JM. Altered neural oscillations and behavior in a genetic mouse model of NMDA receptor hypofunction. Sci Rep 11: 9031, 2021. doi: 10.1038/s41598-021-88428-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Balla A, Ginsberg SD, Abbas AI, Sershen H, Javitt DC. Translational neurophysiological biomarkers of N-methyl-d-aspartate receptor dysfunction in serine racemase knockout mice. Biomark Neuropsychiatry 2: 100019, 2020. doi: 10.1016/j.bionps.2020.100019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Harai T, Inoue R, Fujita Y, Tanaka A, Horio M, Hashimoto K, Hongou K, Miyawaki T, Mori H. Decreased susceptibility to seizures induced by pentylenetetrazole in serine racemase knockout mice. Epilepsy Res 102: 180–187, 2012. doi: 10.1016/j.eplepsyres.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 109.Ploux E, Bouet V, Radzishevsky I, Wolosker H, Freret T, Billard JM. Serine racemase deletion affects the excitatory/inhibitory balance of the hippocampal CA1 network. Int J Mol Sci 21: 9447, 2020. doi: 10.3390/ijms21249447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Harris BT, Costa E, Grayson DR. Exposure of neuronal cultures to K+ depolarization or to N-methyl-D-aspartate increases the transcription of genes encoding the alpha 1 and alpha 5 GABAA receptor subunits. Brain Res Mol Brain Res 28: 338–342, 1995. doi: 10.1016/0169-328x(94)00240-f. [DOI] [PubMed] [Google Scholar]
- 111.Aamodt SM, Shi J, Colonnese MT, Veras W, Constantine-Paton M. Chronic NMDA exposure accelerates development of GABAergic inhibition in the superior colliculus. J Neurophysiol 83: 1580–1591, 2000. doi: 10.1152/jn.2000.83.3.1580. [DOI] [PubMed] [Google Scholar]
- 112.Henneberger C, Juttner R, Schmidt SA, Walter J, Meier JC, Rothe T, Grantyn R. GluR- and TrkB-mediated maturation of GABA receptor function during the period of eye opening. Eur J Neurosci 21: 431–440, 2005. doi: 10.1111/j.1460-9568.2005.03869.x. [DOI] [PubMed] [Google Scholar]
- 113.Lin Y, Bloodgood BL, Hauser JL, Lapan AD, Koon AC, Kim TK, Hu LS, Malik AN, Greenberg ME. Activity-dependent regulation of inhibitory synapse development by Npas4. Nature 455: 1198–1204, 2008. doi: 10.1038/nature07319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gundersen V, Holten AT, Storm-Mathisen J. GABAergic synapses in hippocampus exocytose aspartate on to NMDA receptors: quantitative immunogold evidence for co-transmission. Mol Cell Neurosci 26: 156–165, 2004. doi: 10.1016/j.mcn.2004.01.017. [DOI] [PubMed] [Google Scholar]
- 115.Szabadits E, Cserep C, Szonyi A, Fukazawa Y, Shigemoto R, Watanabe M, Itohara S, Freund TF, Nyiri G. NMDA receptors in hippocampal GABAergic synapses and their role in nitric oxide signaling. J Neurosci 31: 5893–5904, 2011. doi: 10.1523/JNEUROSCI.5938-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cserép C, Szabadits E, Szőnyi A, Watanabe M, Freund TF, Nyiri G. NMDA receptors in GABAergic synapses during postnatal development. PLoS One 7: e37753, 2012. doi: 10.1371/journal.pone.0037753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kaspar BK, Vissel B, Bengoechea T, Crone S, Randolph-Moore L, Muller R, Brandon EP, Schaffer D, Verma IM, Lee KF, Heinemann SF, Gage FH. Adeno-associated virus effectively mediates conditional gene modification in the brain. Proc Natl Acad Sci USA 99: 2320–2325, 2002. doi: 10.1073/pnas.042678699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Suzuki Y, Jodo E, Takeuchi S, Niwa S, Kayama Y. Acute administration of phencyclidine induces tonic activation of medial prefrontal cortex neurons in freely moving rats. Neuroscience 114: 769–779, 2002. doi: 10.1016/s0306-4522(02)00298-1. [DOI] [PubMed] [Google Scholar]
- 119.Jackson ME, Homayoun H, Moghaddam B. NMDA receptor hypofunction produces concomitant firing rate potentiation and burst activity reduction in the prefrontal cortex. Proc Natl Acad Sci USA 101: 8467–8472, 2004. doi: 10.1073/pnas.0308455101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci 17: 2921–2927, 1997. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lorrain DS, Baccei CS, Bristow LJ, Anderson JJ, Varney MA. Effects of ketamine and N-methyl-D-aspartate on glutamate and dopamine release in the rat prefrontal cortex: modulation by a group II selective metabotropic glutamate receptor agonist LY379268. Neuroscience 117: 697–706, 2003. doi: 10.1016/s0306-4522(02)00652-8. [DOI] [PubMed] [Google Scholar]
- 122.Driesen NR, McCarthy G, Bhagwagar Z, Bloch M, Calhoun V, D'Souza DC, Gueorguieva R, He G, Ramachandran R, Suckow RF, Anticevic A, Morgan PT, Krystal JH. Relationship of resting brain hyperconnectivity and schizophrenia-like symptoms produced by the NMDA receptor antagonist ketamine in humans. Mol Psychiatry 18: 1199–1204, 2013. doi: 10.1038/mp.2012.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hunt MJ, Kasicki S. A systematic review of the effects of NMDA receptor antagonists on oscillatory activity recorded in vivo. J Psychopharmacol 27: 972–986, 2013. doi: 10.1177/0269881113495117. [DOI] [PubMed] [Google Scholar]
- 124.Lahti AC, Holcomb HH, Medoff DR, Tamminga CA. Ketamine activates psychosis and alters limbic blood flow in schizophrenia. Neuroreport 6: 869–872, 1995. doi: 10.1097/00001756-199504190-00011. [DOI] [PubMed] [Google Scholar]
- 125.Breier A, Malhotra AK, Pinals DA, Weisenfeld NI, Pickar D. Association of ketamine-induced psychosis with focal activation of the prefrontal cortex in healthy volunteers. Am J Psychiatry 154: 805–811, 1997. doi: 10.1176/ajp.154.6.805. [DOI] [PubMed] [Google Scholar]
- 126.Vollenweider FX, Leenders KL, Scharfetter C, Antonini A, Maguire P, Missimer J, Angst J. Metabolic hyperfrontality and psychopathology in the ketamine model of psychosis using positron emission tomography (PET) and [18F]fluorodeoxyglucose (FDG). Eur Neuropsychopharmacol 7: 9–24, 1997. doi: 10.1016/S0924-977X(96)00039-9. [DOI] [PubMed] [Google Scholar]
- 127.Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci 27: 11496–11500, 2007. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Korotkova T, Fuchs EC, Ponomarenko A, von Engelhardt J, Monyer H. NMDA receptor ablation on parvalbumin-positive interneurons impairs hippocampal synchrony, spatial representations, and working memory. Neuron 68: 557–569, 2010. doi: 10.1016/j.neuron.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 129.Carlen M, Meletis K, Siegle JH, Cardin JA, Futai K, Vierling-Claassen D, Ruhlmann C, Jones SR, Deisseroth K, Sheng M, Moore CI, Tsai LH. A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol Psychiatry 17: 537–548, 2012. doi: 10.1038/mp.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Alvarez RJ, Pafundo DE, Zold CL, Belforte JE. Interneuron NMDA receptor ablation induces hippocampus-prefrontal cortex functional hypoconnectivity after adolescence in a mouse model of schizophrenia. J Neurosci 40: 3304–3317, 2020. doi: 10.1523/JNEUROSCI.1897-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pafundo DE, Pretell Annan CA, Fulginiti NM, Belforte JE. Early NMDA receptor ablation in interneurons causes an activity-dependent E/I imbalance in vivo in prefrontal cortex pyramidal neurons of a mouse model useful for the study of schizophrenia. Schizophr Bull : sbab030, 2021. doi: 10.1093/schbul/sbab030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Saunders JA, Tatard-Leitman VM, Suh J, Billingslea EN, Roberts TP, Siegel SJ. Knockout of NMDA receptors in parvalbumin interneurons recreates autism-like phenotypes. Autism Res 6: 69–77, 2013. doi: 10.1002/aur.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, Kvitsiani D, Kvitsani D, Fu Y, Lu J, Lin Y, Miyoshi G, Shima Y, Fishell G, Nelson SB, Huang ZJ. A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron 71: 995–1013, 2011[Erratum inNeuron72: 1091, 2011]. doi: 10.1016/j.neuron.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.McHugh TJ, Blum KI, Tsien JZ, Tonegawa S, Wilson MA. Impaired hippocampal representation of space in CA1-specific NMDAR1 knockout mice. Cell 87: 1339–1349, 1996. doi: 10.1016/s0092-8674(00)81828-0. [DOI] [PubMed] [Google Scholar]
- 135.Tatard-Leitman VM, Jutzeler CR, Suh J, Saunders JA, Billingslea EN, Morita S, White R, Featherstone RE, Ray R, Ortinski PI, Banerjee A, Gandal MJ, Lin R, Alexandrescu A, Liang Y, Gur RE, Borgmann-Winter KE, Carlson GC, Hahn CG, Siegel SJ. Pyramidal cell selective ablation of N-methyl-D-aspartate receptor 1 causes increase in cellular and network excitability. Biol Psychiatry 77: 556–568, 2015. doi: 10.1016/j.biopsych.2014.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.van den Buuse M. Modeling the positive symptoms of schizophrenia in genetically modified mice: pharmacology and methodology aspects. Schizophr Bull 36: 246–270, 2010. doi: 10.1093/schbul/sbp132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell 87: 1327–1338, 1996. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- 138.Benes FM, Lim B, Matzilevich D, Subburaju S, Walsh JP. Circuitry-based gene expression profiles in GABA cells of the trisynaptic pathway in schizophrenics versus bipolars. Proc Natl Acad Sci USA 105: 20935–20940, 2008. doi: 10.1073/pnas.0810153105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Morris HM, Hashimoto T, Lewis DA. Alterations in somatostatin mRNA expression in the dorsolateral prefrontal cortex of subjects with schizophrenia or schizoaffective disorder. Cereb Cortex 18: 1575–1587, 2008. doi: 10.1093/cercor/bhm186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Beneyto M, Morris HM, Rovensky KC, Lewis DA. Lamina- and cell-specific alterations in cortical somatostatin receptor 2 mRNA expression in schizophrenia. Neuropharmacology 62: 1598–1605, 2012. doi: 10.1016/j.neuropharm.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ling DS, Benardo LS. Recruitment of GABAA inhibition in rat neocortex is limited and not NMDA dependent. J Neurophysiol 74: 2329–2335, 1995. doi: 10.1152/jn.1995.74.6.2329. [DOI] [PubMed] [Google Scholar]
- 142.Grunze HC, Rainnie DG, Hasselmo ME, Barkai E, Hearn EF, McCarley RW, Greene RW. NMDA-dependent modulation of CA1 local circuit inhibition. J Neurosci 16: 2034–2043, 1996. doi: 10.1523/JNEUROSCI.16-06-02034.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]