

Abstract
Cancer survivors are more susceptible to pathologies such as hypertension, liver disease, depression, and coronary artery disease when compared with individuals who have never been diagnosed with cancer. Therefore, it is important to understand how tumor burden negatively impacts nontumor-bearing tissues that may impact future disease susceptibility. We hypothesized that the energetic costs of a tumor would compromise proteostatic maintenance in other tissues. Therefore, the purpose of this study was to determine if tumor burden changes protein synthesis and proliferation rates in heart, brain, and liver. One million Lewis lung carcinoma (LLC) cells or phosphate-buffered saline (PBS, sham) were injected into the hind flank of female mice at ∼4.5 mo of age, and the tumor developed for 3 wk. Rates of proliferation and protein synthesis were measured in heart, brain, liver, and tumor tissue. Compared with sham, rates of protein synthesis (structural/nuclear, cytosolic, mitochondrial, and collagen) relative to proliferation were lower in the heart and liver of LLC mice, but higher in the brain of LLC mice. In the tumor tissue, the ratio of protein synthesis to DNA synthesis was approximately 1.0 showing that protein synthesis in the tumor was used for proliferation with little proteostatic maintenance. We further provide evidence that the differences in tissue responses may be due to energetic stress. We concluded that the decrease in proteostatic maintenance in liver, heart, and muscle might contribute to the increased risk of disease in cancer survivors.
NEW & NOTEWORTHY We present data showing that simultaneously measuring protein synthesis and cell proliferation can help in the understanding of protein turnover as a proteostatic process in response to tumor burden. In some tissues, like hepatic, cardiac, and skeletal muscle, there was a decrease in the protein to DNA synthesis ratio indicating less proteostatic maintenance. In contrast, the brain maintained or even increased this protein to DNA synthesis ratio indicating more proteostatic maintenance.
Keywords: cancer, organs, proliferation, protein turnover
INTRODUCTION
Cancer survivors are more susceptible to disorders such as hypertension, coronary artery disease, liver disease, and depression (1, 2). Therefore, it is important to understand how tumor burden negatively impacts other nontumor-bearing tissues to increase future disease susceptibility. Cell proliferation is an energy-consuming process, thus the increased proliferation rates in tumors require a tremendous amount of energy. In fact, patient survival rates decrease in cancers with a high rate of proliferation (3, 4), and it is thought that extra energy expenditure from tumor proliferation may contribute to body mass loss that is observed in 80% of patients with cancer (5–7). Protein turnover (the continual synthesis and breakdown of proteins) is also one of the most energetically costly cellular processes with protein synthesis using 20% of basal metabolic rate (8, 9), whereas protein breakdown uses another 5%–15% (10). Protein turnover is often not a priority for cellular processes and can be compromised in the face of other energetic demands. Thus, the energetic demands of tumor proliferation can lead to energy shortages in noncancerous tissues, which may alter rates of protein turnover (5, 11–14).
Maintaining high-quality proteins (proteostasis) is important for cellular function. The loss of proteostasis is a feature of aging and many of the pathologies that cancer survivors are more susceptible to such as liver disease, diabetes, and hypertension (15–22). Protein turnover is a critical process for maintaining proteostasis (15, 23, 24). Our studies have demonstrated that the assessment of the contribution of protein turnover to maintaining proteostasis should include measurements of cell proliferation (24, 25). The rationale for both measurements is that cell proliferation requires a doubling of the proteome and therefore has a large impact on the measured rate of protein synthesis. When there are high rates of proliferation, protein synthesis rates are also high, but in an indiscriminate manner compared with proteostatic maintenance. We have shown that what appears to be a decrease of protein synthesis during treatment with rapamycin or caloric restriction is really the result of decreased proliferation (24, 25). Further, with caloric restriction or treatment with rapamycin a greater proportion of protein synthesis is used to maintain the existing cellular structures (proteostatic maintenance). Although we have made protein synthesis and proliferation measurements in relatively slowly proliferating cell types, we have not confirmed the impact of cell proliferation in a highly proliferative tissue.
Decreased rates of protein synthesis in muscle due to tumor burden is well established (7, 26–32); however, the influence of tumor burden on modulating rates of protein synthesis in other noncancerous tissue types is understudied. Using the puromycin method to measure protein synthesis, it was shown that tumor burden increases protein synthesis rates in liver (33). However, puromycin method has some limitations that affect the interpretation of the resultant data. Because of the 20–30-min period of labeling with puromycin, the method is biased toward rapidly turning over proteins (34). Further, sample preparation usually includes a slow-speed spin that pellets many of the structural and mitochondrial proteins with the soluble proteins in the supernatant used for analysis. An additional paper used 24-h deuterium oxide (D2O) labeling, a more integrated measurement, and showed that the synthesis of new mitochondrial components in cardiac tissue does change in tumor-bearing mice (35). In our recent study using 5 days of D2O labeling, we showed in skeletal muscle that Lewis lung carcinoma (LLC) had lower rates of mitochondrial protein synthesis, whereas myofibrillar protein synthesis did not change (32). However, compared with control animals, mice with LLC had higher rates of myofibrillar and mitochondrial protein breakdown, indicating an energetic stress to skeletal muscle. To our knowledge, the effects of tumor burden on the rates of protein turnover in other tissues such as brain and cardiac have not fully been investigated.
The purpose of this study was to determine if tumor burden changes protein synthesis and proliferation rates in heart, brain, and liver. Second, we used tumor tissue to further validate the importance of measuring cell proliferation when examining protein turnover as a proteostatic mechanism. We hypothesized that there is an energetic stress with cancer that leads to reduced rates of protein synthesis and proliferation in noncancerous tissues. In addition, we hypothesized that in tumor tissue the rate of protein synthesis will be fully reflective of changes in cell proliferation.
MATERIALS AND METHODS
Animals and Interventions
Animal experiments were approved by the Institutional Animal Care and Use Committees and performed at the Oklahoma Medical Research Foundation. In the current study, we have utilized the Lewis lung carcinoma (LLC) preclinical model. C57BL/6J female mice (4.5-mo-old) were group housed, kept on a 12:12-h light-dark cycle, and had access to standard rodent chow and water ad libitum.
LLC Growth and Tumor Implantation
LLC cells were grown and implanted as previously described (32, 36). Briefly, LLC cells (ATCC CRL-1642) were plated in 250-mL culture flasks in DMEM supplemented with 10% fetal bovine serum plus 1% penicillin and streptomycin. Once confluent, cells were trypsinized, counted, and diluted in PBS for implantation. Mice were implanted with 1 × 106 LLC cells (LLC, n = 9) or equal volume saline (sham, n = 9) treated at ∼4.5 mo of age. LLC cells were plated at passages 2–5 (Fig. 1).
Figure 1.

Study design schematic.
Determination of Protein Synthesis and Proliferation
Protein synthesis was determined via deuterium incorporation into alanine according to methods described in Refs 37–41. A subset of the mice used for the study (n = 4/group) was labeled with deuterium oxide. Mice received a bolus intraperitoneal injection (∼20 μL/g body wt) of 99% deuterium oxide (D2O) 5 days before tissue collection (i.e., day 16 after saline or LLC injection; Fig. 1). Drinking water was thereafter supplemented with 8% D2O until euthanasia (37–41). We chose to start labeling 5 days before euthanasia because this period corresponds to the time where tumor is beginning to grow exponentially (26, 36, 42). Euthanasia was performed via CO2 asphyxiation and cervical dislocation followed by blood collection via the inferior vena cava. Blood was then allowed to clot at room temperature for 10 min and spun at 2,000 g for 10 min at 4°C and serum was aliquoted and frozen until analyses. As we previously reported, all animals were euthanized at 21 days post saline (sham) or LLC injection, instead of our planned 28 days, due to early mortality in our genetic knockout mice (32). Following euthanasia, hind limb skeletal muscles, organs, and tumors were quickly excised, trimmed of excess fat or connective tissue, weighed, and flash frozen in liquid nitrogen. Approximately 50 mg of livers, hearts, brains, muscles, and tumors were powdered and fractionated via differential centrifugation to obtain structural/nuclear, cytosolic, mitochondrial, and collagen protein fractions according to our previously published protocols (37–41). We used tibialis anterior (TA) muscle for the current study to have enough tissue to simultaneously capture protein and DNA synthesis, which is sometimes challenging with smaller muscles (e.g., soleus), whereas the gastrocnemius and quadricep muscles were used for other analyses in our previously published study (32).
Cell proliferation was determined via deuterium incorporation in purine deoxyribose in the same tissues analyzed for protein synthesis (37, 43). The use of D2O to measure proliferation rates has been established by our laboratory and others (44–46), and D2O offers several advantages over traditional methods to measure proliferation. Briefly, D2O does not require an antibody for detection and D2O is incorporated into DNA through de novo pathways only (44). Further, D2O is not toxic, does not affect proliferation rates, and is nonmutagenic (44). DNA was isolated from ∼20 mg of powdered tissues using a kit (QiAmp DNA Mini Kit Qiagen, Valencia, CA) following established procedures (37, 44). Isolated DNA was then hydrolyzed overnight at 37°C with nuclease S1 and potato acid phosphatase. Hydrolysates were reacted with pentafluorobenzyl hydroxylamine and acetic acid and then acetylated with acetic anhydride and 1-methylimidazole. Dichloromethane extracts were dried, resuspended in ethyl acetate, and analyzed by GC-MS as previously described (37, 43).
To determine the precursor pool enrichment, serum samples were prepared for analysis of deuterium enrichment on a liquid water isotope analyzer (Los Gatos Research, Los Gatos, CA) using 0%–12% deuterium standards (47). Precursor pool enrichments for alanine and deoxyribose were calculated using the measured serum deuterium enrichments adjusted by mass isotopomer distribution analysis (MIDA) as previously described (37, 40, 43). The protein or the fraction of new DNA synthesized were calculated using the enrichment of protein or DNA hydrolysates (product), respectively, divided by free alanine or deoxyribose enrichment (precursor), respectively. From the fraction new, the rate of protein or DNA synthesis (i.e., fractional synthesis rate, FSR) was calculated using length of deuterium oxide labeling (i.e., 5 days). From the synthesis of protein and DNA, we calculated the ratio of protein synthesis to DNA synthesis (25, 37).
Hydroxyproline Assay
Collagen concentration was determined in the liver using an established hydroxyproline assay in skeletal muscle, with modifications for liver (48, 49). Four mice per group were used for the hydroxyproline assay. Briefly, ∼250 mg of whole powdered liver was hydrolyzed in 6 M HCl overnight at 110°C. Hydrolyzed sample (10 µL) was mixed with 150-µL isopropanol and 75 µL of chloramine-T (EMD Millipore Sigma, St. Louis, MO) in citrate buffer and oxidized for 10 min at room temperature. The oxidized samples were then mixed with 1 mL of a 3:13 solution of Ehrlich reagent [3 g of p-dimethylaminobenzaldehyde (Sigma), 10 mL of ethanol, and 675 µL of sulfuric acid) to isopropanol and incubated for 45 min at 55°C. Quantification was determined by measuring absorbance at 558 nm on a 96-well plate reader in duplicate. Hydroxyproline concentration (µg/mg tissue) was then measured using a 0–1,000 nM standard curve of trans-4-hydroxy-l-proline (Sigma) as proxy of collagen concentration. Total collagen content was then calculated using collagen concentration and total liver wet weight.
Immunoblotting
Immunoblot was performed as previously described (32, 50). Three to 5 mice per group (n = 3–5) were used for immunoblot analysis. Antibodies were used for neutrophil gelatinase-associated lipocalin (NGAL, PA5-79590, Thermo Fisher, Waltham, MA), α-fetoprotein (AFP, PA5-21004, Thermo Fisher, Waltham, MA) phosphorylated AMP-activated protein kinase alphaThr172 (p-AMPK, 40H9, Cell Signaling, Danvers, MA), AMP-activated protein kinase α (AMPK, 2532, Cell Signaling, Danvers, MA), and MYC proto-oncogene, basic Helix-Loop-Helix transcription factor (c-Myc, D3N8F Cell Signaling, Danvers, MA). Primary antibodies were diluted by 1:1,000 in 5% bovine serum albumin in Tris-buffered saline. Appropriate secondary antibody was used and membranes were imaged on a Syngene G Box.
Statistical Analysis
A two-tailed independent samples t test was used for comparisons between LLC and sham treatments. For all experiments, the comparison-wise error rate, α, was set at 0.05 for all statistical tests. Asterisk was used to denote significant differences between saline and LLC implantation groups. We present the tumor data (for the LLC group only) alongside our organ data for qualitative comparisons only, with no statistical comparisons done between the tumor and other tissues. All data were analyzed, and graphs were generated using GraphPad Prism (La Jolla, CA). Data were expressed as means ± SD.
RESULTS
Bodyweights and muscle mass data has already been published in a prior study (32). Briefly, bodyweights were not different between sham-treated and LLC mice (32). Muscle mass of quadricep, TA, and soleus muscles was 10%–15% lower in LLC mice than sham-treated mice (32). There was no change in gastrocnemius muscle mass between groups (32).
Compared with sham, LLC tumor had significantly higher liver wet weights (Fig. 2A) and wet weights normalized to body weights (Fig. 2B). There were no differences in the wet weights of heart between groups (Fig. 2C) or when wet weights of heart were normalized to body weight (Fig. 2D). There were no differences between groups in the wet weights of brains (Fig. 2E) or when wet weights of brains were normalized to body weight (Fig. 2F).
Figure 2.

A: liver wet weight. B: liver wet weight normalized to bodyweight. C: heart wet weight. D: heart wet weight normalized to bodyweight. E: brain wet weight. F: brain wet weight normalized to bodyweight. n = 8–9 mice per group. A Student’s t test was used for statistical analysis. *Statistical significance at α set at P < 0.05. Values are means ± SD.
Protein synthesis rates in the structural/nuclear (Fig. 3A), cytosolic (Fig. 3B), and mitochondrial (Fig. 3C) fractions did not differ between sham-treated and LLC mice for liver, heart, or brain (Fig. 3, A–C). LLC mice had higher liver collagen protein synthesis than sham-treated mice, with no differences in collagen protein synthesis in heart or brain (Fig. 3D).
Figure 3.

A: protein synthesis rates in heart, liver, brain, and tumor in the structural/nuclear fraction. B: protein synthesis rates in heart, liver, brain, and tumor in the cytosolic fraction. C: protein synthesis rates in heart, liver, brain, and tumor in the mitochondrial fraction. D: protein synthesis rates in heart, liver, brain, and tumor in the collagen fraction. n = 3–4 mice per group. A Student’s t-test was used for statistical analysis. *Statistical significance at α set at P < 0.05. Values are means ± SD. FSR, fractional synthesis rate.
Cell proliferation rates were higher for LLC than sham in both liver and heart, but was lower in the brain (Fig. 4A). In our previously published study, we did not include cell proliferation rates for muscle, but here we show that LLC was not different from sham in the TA (Fig. 4A). As expected, rates of proliferation in the tumor were very rapid with rates that were ∼5–1,000 times greater than the other organs. To account for the impact of cell proliferation on rates of protein synthesis we calculated the ratio of protein synthesis to DNA synthesis for every tissue (Fig. 4, B–E). Compared with sham, LLC had a lower protein synthesis to DNA synthesis ratio in all protein fractions of the liver (Fig. 4, B–E). Ratios in the heart followed the same pattern except for the cytosolic, which only had a strong trend (P = 0.078) to be lower (Fig. 4, B–E). In stark contrast, the cytosolic fraction of the brain had a higher ratio of protein to DNA synthesis in LLC than sham, with a trend (P = 0.053) toward a higher ratio in mitochondrial fraction, and no change in the structural/nuclear proteins or collagen proteins (Fig. 4, B, D, and E). Using previously published protein synthesis values for the TA with the DNA synthesis values herein (32), we show that in the TA, the protein synthesis to DNA synthesis ratio was only different for the mitochondrial fraction where LLC was lower than sham (Fig. 4, B, C, and E). Finally, in the tumor, the ratio of the rate or protein synthesis to DNA synthesis approximated 1.0 for all protein fractions, indicating a match of protein synthesis to DNA proliferation.
Figure 4.

A: proliferation rates in heart, liver, brain, tumor, and TA muscle. B: rates of protein synthesis normalized to rates of DNA synthesis in heart, liver, brain, tumor, and TA muscle in the nuclear/structural fraction. C: rates of protein synthesis normalized to rates of DNA synthesis in heart, liver, brain, tumor, and TA muscle in the cytosolic fraction. D: rates of protein synthesis normalized to rates of DNA synthesis in heart, liver, brain, tumor, and TA muscle in the mitochondrial fraction. E: rates of protein synthesis normalized to rates of DNA synthesis in heart, liver, brain, tumor, and TA muscle in the collagen fraction. n = 3–4 mice per group. A Student’s t test was used for statistical analysis. *Statistical significance at α set at P < 0.05. Values are means ± SD. TA, tibialis anterior.
We explored the enlarged liver with cancer treatment by examining collagen amount and a marker of liver hepatocyte proliferation and liver cancer. Cancer treatment did not change liver collagen concentration compared with sham (Fig. 5A), but did result in a significant upregulation in liver collagen content (Fig. 5B). Further, no differences were observed in the content of liver cell proliferation marker AFP in the liver of mice without or with cancer (Fig. 5C). Representative AFP blot can be found in Fig. 5D.
Figure 5.

A: liver collagen concentration. B: liver collagen content. C: Western blot quantification for the liver cell proliferation marker AFP. D: representative AFP Western blot images. n = 4–5 mice per group. A Student’s t test was used for statistical analysis. *Statistical significance at α set at P < 0.05. Values are means ± SD.
We confirmed that c-Myc, a tumor marker, was only present in the tumors of LLC mice and not in any other tissues assessed in sham or LLC mice (Fig. 6A). We then assessed NGAL expression as an inflammatory marker. NGAL protein content was not detected in muscle and brain (Fig. 6B). In contrast, in heart and liver, NGAL expression was higher in LLC than in sham (Fig. 6B). Representative NGAL Western blot can be found in Fig. 6E. We measured phosphorylated AMPK α and total AMPK α as a surrogate marker for energy stress. We observed more phosphorylated AMPK α in liver tissue, but not in brain or heart tissue (Fig. 6C). Total AMPK α did not change in liver, brain, or heart tissues (Fig. 6D). Representative AMPK α blots can be found in Fig. 6F.
Figure 6.
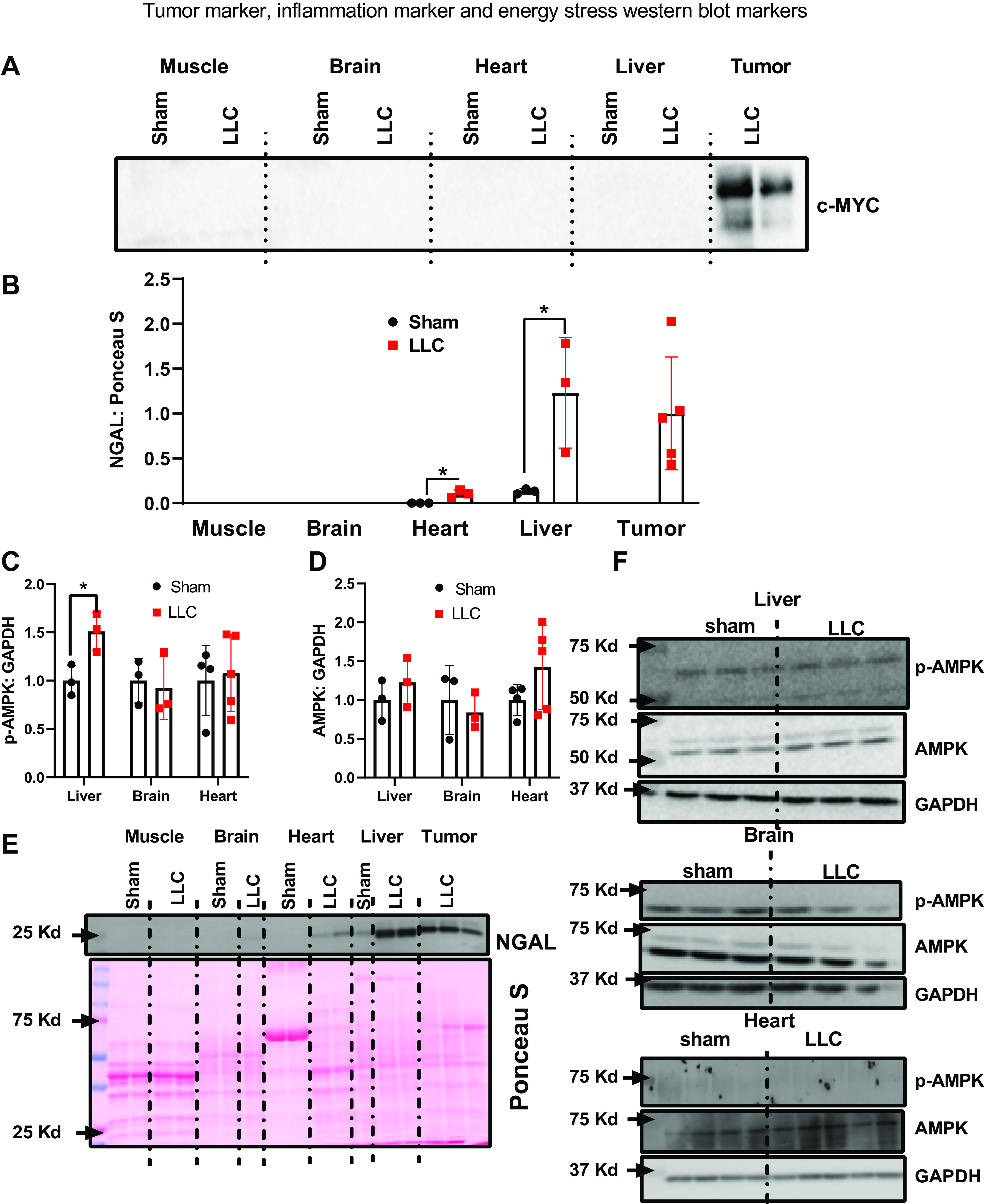
A: representative image of c-myc Western blot to confirm that cancer did not invade heart, liver, brain, and muscle. B: Western blot quantification of the inflammatory immune cell marker NGAL. C: Western blot quantification of phosphorylated AMPK. D: Western blot quantification of AMPK. E: representative NGAL Western blot images. F: representative AMPK Western blot images. n = 3–5 mice per group. A Student’s t test was used for statistical analysis. *Statistical significance at α set at P < 0.05. Values are means ± SD. AMPK, AMP-activated protein kinase α.
DISCUSSION
In this project, we hypothesized that energetic demands of tumor proliferation can lead to energy shortages in noncancerous tissues, which may alter rates of protein turnover. Our primary findings are that in the liver, heart, and skeletal muscle, there were decreased protein synthesis rates for proteostatic processes as measured by rates of protein synthesis normalized to proliferation rates. In contrast, the brain was protected against such decrements and may even have improved protein synthesis for proteostatic maintenance. The influence of the energetic state on these turnover processes was demonstrated by more activation of AMPK α in liver, but not in brain. In sum, these data indicate that tumor burden compromises proteostatic processes in liver and heart that could lead to the higher propensity for chronic diseases after cancer recovery.
Proliferation requires protein synthesis to double the protein content to equip two cells (51). Therefore, to separate the contributions of the doubling of protein concentration (a rather indiscriminate process) and protein turnover to maintain proteostasis, we measure cell proliferation and protein synthesis simultaneously (24, 25). Our laboratory has shown that in treatments that extend life span, there is a suppression of proliferation and increased maintenance of proteins in existing cells, as reflected by an increased protein synthesis to DNA synthesis ratio (24, 38, 39, 46, 52). We hypothesize that this increased ratio is tied to cellular energetics whereby there is a decrease in one energetically costly process (proliferation) to support another energetically costly process (protein turnover). In the current study, as a proof of principle, we measured the proliferation rates in a tumor, which were between 5 and 1,000 times higher than other organ tissues. In the tumor, the ratio of protein synthesis to DNA synthesis approximated 1.0, which indicates that 100% of the protein synthesis was for cell proliferation. Since protein synthesis in tumors is predominately being used for proliferation, synthesis of proteins for the maintenance of existing cellular components is lacking, which may explain why tumors have little proteostatic maintenance (53, 54).
Cancer survivors are more susceptible to liver disease (1, 2). An enlarged liver is often the result of inflammation and liver disease, (55–57) and an enlarged liver has been observed in multiple models of cancer cachexia and in human patients with cancer (33, 58–60). In agreement, the liver mass in LLC mice from our study was ∼20% heavier than control mice. More collagen is also commonly associated with liver disease (61–64) and has been observed in livers of LLC mice (58). In our study, we showed that total liver collagen content was higher in LLC mice, and this was at least partially attributable to higher rates of collagen protein synthesis. When we examined the protein synthesis rates relative to proliferation rates, we found that the ratio is lower in LLC mice than sham, which indicates that in the LLC mice, a greater proportion of the protein synthesis is being used for proliferation rather than proteostatic maintenance. To follow up with this finding, we measured AFP protein content. AFP is a clinical marker of progenitor cell induction in injured, fibrotic, or regenerating liver (65–67). AFP protein content was not different between groups, which showed that the increased proliferation rates were not due to liver regeneration. We hypothesized that immune cells may be infiltrating the liver in LLC mice; therefore, we measured the neutrophil marker NGAL. NGAL was elevated in the livers from tumor-bearing mice, which suggests the increased proliferation rates in liver were due to immune cells.
Prior work has shown that cardiac pathology occurs in between 8% and 42% of patients suffering from cancer (35, 68–71), and cancer-induced muscle loss occurs in up to 80% of cancer cases (72–74). Type II muscle fibers are more susceptible to cancer-induced muscle loss (75). The TA muscles in C57Bl6 mice are predominately composed of type IIB muscle fibers (76). Therefore, the TA muscle is more susceptible to cancer-induced changes in protein synthesis when compared with the more oxidative muscles. Like liver tissue, we observe that protein synthesis relative to proliferation rates is lower in all structural/nuclear, cytosolic, collagen, and mitochondrial fractions in heart tissue of LLC mice compared with sham, whereas only mitochondrial protein synthesis relative to proliferation was lower in skeletal muscle. These data show that like liver tissue, proteostatic maintenance is impaired in both skeletal and cardiac muscles in LLC mice. Both cardiac and skeletal muscles are thought to be postmitotic tissues because of the high abundance of postmitotic myocytes. Importantly though, ∼50% of the nuclei in these tissues are in myocytes while the remaining reside in mononuclear cells that have varying degrees of proliferative capacity (77–80). One potential replicating cell type is inflammatory cells. Our data showed that the inflammation marker NGAL is elevated in cardiac tissue of LLC mice indicating that infiltrating inflammatory cells could be one source of the higher proliferation rates we observed in cardiac tissue. Another potential source of elevated proliferation are resident stem cells. Previous studies have shown that in skeletal muscle, markers of satellite cell proliferation are elevated in LLC mice (81). This increase in stem cell proliferation may be indicative of skeletal muscle damage due to cancer.
Cancer survivors are more likely to have adverse mental health outcomes, but not necessarily neurodegenerative outcomes (82–84). In contrast to heart and liver, brains of LLC mice did not have a lower protein synthesis to DNA synthesis ratio when compared with sham. In fact, our data showed that a greater portion of protein synthesis is being used to maintain existing cellular components in brains of LLC mice. One possible explanation for why brain is protected from reduced proteostatic maintenance in LLC mice is the fact that ATP levels in the brains are protected during conditions of energetic stress (85–87).
To support our hypotheses related to energetic stresses in tissues outside the tumor, we measured AMPK α activity (88). We observed greater AMPK α phosphorylation in the livers of LLC mice compared with sham, which supports that there was an energetic stress in livers. Like livers in LLC mice, we showed that tumors have little proteostatic maintenance. Recent evidence show that AMPK α activation is elevated in tumors to promote tumor cell survival (89), which supports our hypothesis that energetic stress may contribute to reduced proteostatic maintenance. In contrast, the brain did not have a difference in AMPK αactivation between LLC and sham mice, which may reflect why the brain is able to use protein synthesis to maintain current structures in LLC mice. We also did not observe increased AMPK α phosphorylation in cardiac tissue. We observed that LLC reduced proteostatic maintenance in cardiac tissue. However, the reduction in proteostatic maintenance in the liver was greater than cardiac tissue. Although our turnover data indicate a relationship between the energetic status of a tissue and lack of proteostatic maintenance during cancer, more studies are needed to directly demonstrate this relationship, including why some tissues like the brain are protected against these decrements.
We have shown that simultaneously measuring protein synthesis and cell proliferation can help in the understanding of protein turnover as a proteostatic process. We support those data in the present study by demonstrating that in a tumor, all the protein synthesis is directed toward cell proliferation with little proteostatic maintenance. In some tissues, like hepatic, cardiac, and skeletal muscle, there was a decrease in the protein to DNA synthesis ratio, indicating less proteostatic maintenance. In contrast, the brain maintained or even increased this protein to DNA synthesis ratio. The decrease in proteostatic maintenance in liver, heart, and muscle might contribute to the increased risk of disease in cancer survivors. In summary, these data provide insight into why cancer may impact tissue health and future disease risk in nonaffected tissues.
ETHICAL APPROVALS
The authors certify that they comply with the ethical guidelines for authorship and publishing.
GRANTS
Support for this work has been provided by National Institute on Aging P01AG051442 (to H. Van Remmen). Dr. Van Remmen is the recipient of a VA Senior Research Career Scientist award (12F-RCS-011). This work was also partially supported by NIA T32AG052363 (to J. L. Brown and M. M. Lawrence) and an American Physiological Society (APS) Postdoctoral Fellowship (to M. M. Lawrence).
DISCLAIMERS
Contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.L.B., H.V.R., and B.F.M. conceived and designed research; J.L.B., M.M.L., L.O., and F.F.P. performed experiments; J.L.B., M.M.L., A.B., L.O., F.F.P., and B.F.M. analyzed data; J.L.B., M.M.L., A.B., H.V.R., and B.F.M. interpreted results of experiments; J.L.B. and M.M.L. prepared figures; J.L.B., M.M.L., A.B., and B.F.M. drafted manuscript; J.L.B., A.B., L.O., F.F.P., H.V.R., and B.F.M. edited and revised manuscript; J.L.B., M.M.L., A.B., L.O., F.F.P., H.V.R., and B.F.M. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank the members of the Holly Van Remmen Laboratory for contributions to the experiments presented here. We extend our gratitude to the numerous other faculties, staff, and other researchers at the Oklahoma Medical Research Foundation and Oklahoma University Health Science Center for helpful discussions.
REFERENCES
- 1.Meacham LR, Sklar CA, Li S, Liu Q, Gimpel N, Yasui Y, Whitton JA, Stovall M, Robison LL, Oeffinger KC. Diabetes mellitus in long-term survivors of childhood cancer. Increased risk associated with radiation therapy: a report for the childhood cancer survivor study. Arch Intern Med 169: 1381–1388, 2009. doi: 10.1001/archinternmed.2009.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oeffinger KC, Mertens AC, Sklar CA, Kawashima T, Hudson MM, Meadows AT, Friedman DL, Marina N, Hobbie W, Kadan-Lottick NS, Schwartz CL, Leisenring W, Robison LL. ; Childhood Cancer Survivor Study. Chronic health conditions in adult survivors of childhood cancer. N Engl J Med 355: 1572–1582, 2006. doi: 10.1056/NEJMsa060185. [DOI] [PubMed] [Google Scholar]
- 3.Rosenwald A, Wright G, Wiestner A, Chan WC, Connors JM, Campo E, Gascoyne RD, Grogan TM, Muller-Hermelink HK, Smeland EB, Chiorazzi M, Giltnane JM, Hurt EM, Zhao H, Averett L, Henrickson S, Yang L, Powell J, Wilson WH, Jaffe ES, Simon R, Klausner RD, Montserrat E, Bosch F, Greiner TC, Weisenburger DD, Sanger WG, Dave BJ, Lynch JC, Vose J, Armitage JO, Fisher RI, Miller TP, LeBlanc M, Ott G, Kvaloy S, Holte H, Delabie J, Staudt LM. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell 3: 185–197, 2003. doi: 10.1016/s1535-6108(03)00028-x. [DOI] [PubMed] [Google Scholar]
- 4.Liu S, Edgerton SM, Moore DH 2nd, Thor AD. Measures of cell turnover (proliferation and apoptosis) and their association with survival in breast cancer. Clin Cancer Res 7: 1716–1723, 2001. [PubMed] [Google Scholar]
- 5.Tayek JA, Blackburn GL, Bistrian BR. Alterations in whole body, muscle, liver, and tumor tissue protein synthesis and degradation in Novikoff hepatoma and Yoshida sarcoma tumor growth studied in vivo. Cancer Res 48: 1554–1558, 1988. [PubMed] [Google Scholar]
- 6.Argilés JM, Busquets S, López-Soriano FJ, Costelli P, Penna F. Are there any benefits of exercise training in cancer cachexia? J Cachexia Sarcopenia Muscle 3: 73–76, 2012. doi: 10.1007/s13539-012-0067-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Argilés JM, Busquets S, Stemmler B, López-Soriano FJ. Cancer cachexia: understanding the molecular basis. Nat Rev Cancer 14: 754–762, 2014. doi: 10.1038/nrc3829. [DOI] [PubMed] [Google Scholar]
- 8.Schutz Y. Protein turnover, ureagenesis and gluconeogenesis. Int J Vitam Nutr Res 81: 101–107, 2011. doi: 10.1024/0300-9831/a000064. [DOI] [PubMed] [Google Scholar]
- 9.Waterlow JC. Protein turnover with special reference to man. Q J Exp Physiol 69: 409–438, 1984. doi: 10.1113/expphysiol.1984.sp002829. [DOI] [PubMed] [Google Scholar]
- 10.Rolfe DF, Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev 77: 731–758, 1997. doi: 10.1152/physrev.1997.77.3.731. [DOI] [PubMed] [Google Scholar]
- 11.Bolster DR, Crozier SJ, Kimball SR, Jefferson LS. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J Biol Chem 277: 23977–23980, 2002. doi: 10.1074/jbc.C200171200. [DOI] [PubMed] [Google Scholar]
- 12.Viollet B, Foretz M, Guigas B, Horman S, Dentin R, Bertrand L, Hue L, Andreelli F. Activation of AMP-activated protein kinase in the liver: a new strategy for the management of metabolic hepatic disorders. J Physiol 574: 41–53, 2006. doi: 10.1113/jphysiol.2006.108506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ronnett GV, Ramamurthy S, Kleman AM, Landree LE, Aja S. AMPK in the brain: its roles in energy balance and neuroprotection. J Neurochem 109, Suppl 1: 17–23, 2009. doi: 10.1111/j.1471-4159.2009.05916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dyck JRB, Lopaschuk GD. AMPK alterations in cardiac physiology and pathology: enemy or ally? J Physiol 574: 95–112, 2006. doi: 10.1113/jphysiol.2006.109389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Labbadia J, Morimoto RI. The biology of proteostasis in aging and disease. Annu Rev Biochem 84: 435–464, 2015. doi: 10.1146/annurev-biochem-060614-033955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baiceanu A, Mesdom P, Lagouge M, Foufelle F. Endoplasmic reticulum proteostasis in hepatic steatosis. Nat Rev Endocrinol 12: 710–722, 2016. doi: 10.1038/nrendo.2016.124. [DOI] [PubMed] [Google Scholar]
- 17.Jaisson S, Gillery P. Impaired proteostasis: role in the pathogenesis of diabetes mellitus. Diabetologia 57: 1517–1527, 2014. doi: 10.1007/s00125-014-3257-1. [DOI] [PubMed] [Google Scholar]
- 18.Kaushik S, Cuervo AM. Proteostasis and aging. Nat Med 21: 1406–1415, 2015. doi: 10.1038/nm.4001. [DOI] [PubMed] [Google Scholar]
- 19.Morimoto RI, Cuervo AM. Proteostasis and the aging proteome in health and disease. J Gerontol A Biol Sci Med Sci 69, Suppl 1: S33–S38, 2014. doi: 10.1093/gerona/glu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hipp MS, Kasturi P, Hartl FU. The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol 20: 421–435, 2019. doi: 10.1038/s41580-019-0101-y. [DOI] [PubMed] [Google Scholar]
- 21.Klaips CL, Jayaraj GG, Hartl FU. Pathways of cellular proteostasis in aging and disease. J Cell Biol 217: 51–63, 2018. doi: 10.1083/jcb.201709072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siddiq T, Richardson PJ, Sherwood RA, Preedy VR. Protein synthesis in pulmonary, cardiac, and skeletal muscle in acute hypertension induced by aortic constriction in the rat. Cardiovasc Res 26: 72–81, 1992. doi: 10.1093/cvr/26.1.72. [DOI] [PubMed] [Google Scholar]
- 23.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science 319: 916–919, 2008. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 24.Hamilton KL, Miller BF. Mitochondrial proteostasis as a shared characteristic of slowed aging: the importance of considering cell proliferation. J Physiol 595: 6401–6407, 2017. doi: 10.1113/JP274335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller BF, Drake JC, Naylor B, Price JC, Hamilton KL. The measurement of protein synthesis for assessing proteostasis in studies of slowed aging. Ageing Res Rev 18: 106–111, 2014. doi: 10.1016/j.arr.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown JL, Lee DE, Rosa-Caldwell ME, Brown LA, Perry RA, Haynie WS, Huseman K, Sataranatarajan K, Van Remmen H, Washington TA, Wiggs MP, Greene NP. Protein imbalance in the development of skeletal muscle wasting in tumour-bearing mice. J Cachexia Sarcopenia Muscle 9: 987–1002, 2018. [Erratum in J Cachexia Sarcopenia Muscle 10: 712, 2019]. doi: 10.1002/jcsm.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hardee JP, Counts BR, Gao S, VanderVeen BN, Fix DK, Koh HJ, Carson JA. Inflammatory signalling regulates eccentric contraction-induced protein synthesis in cachectic skeletal muscle. J Cachexia Sarcopenia Muscle 9: 369–383, 2018. doi: 10.1002/jcsm.12271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lima M, Sato S, Enos RT, Baynes JW, Carson JA. Development of an UPLC mass spectrometry method for measurement of myofibrillar protein synthesis: application to analysis of murine muscles during cancer cachexia. J Appl Physiol (1985) 114: 824–828, 2013. doi: 10.1152/japplphysiol.01141.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.White JP, Baynes JW, Welle SL, Kostek MC, Matesic LE, Sato S, Carson JA. The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the ApcMin/+ mouse. PLoS One 6: e24650, 2011. doi: 10.1371/journal.pone.0024650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith HJ, Greenberg NA, Tisdale MJ. Effect of eicosapentaenoic acid, protein and amino acids on protein synthesis and degradation in skeletal muscle of cachectic mice. Br J Cancer 91: 408–412, 2004. doi: 10.1038/sj.bjc.6601981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Puppa MJ, Gao S, Narsale AA, Carson JA. Skeletal muscle glycoprotein 130’s role in Lewis lung carcinoma-induced cachexia. FASEB J 28: 998–1009, 2014. doi: 10.1096/fj.13-240580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown JL, Lawrence MM, Ahn B, Kneis P, Piekarz KM, Qaisar R, Ranjit R, Bian J, Pharaoh G, Brown C, Peelor Ff 3rd, Kinter MT, Miller BF, Richardson A, Van Remmen H. Cancer cachexia in a mouse model of oxidative stress. J Cachexia Sarcopenia Muscle 11: 1688–1704, 2020. doi: 10.1002/jcsm.12615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Narsale AA, Puppa MJ, Hardee JP, VanderVeen BN, Enos RT, Murphy EA, Carson JA. Short-term pyrrolidine dithiocarbamate administration attenuates cachexia-induced alterations to muscle and liver in ApcMin/+ mice. Oncotarget 7: 59482–59502, 2016. doi: 10.18632/oncotarget.10699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller BF, Wolff CA, Peelor FF 3rd, Shipman PD, Hamilton KL. Modeling the contribution of individual proteins to mixed skeletal muscle protein synthetic rates over increasing periods of label incorporation. J Appl Physiol (1985) 118: 655–661, 2015. doi: 10.1152/japplphysiol.00987.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee DE, Brown JL, Rosa-Caldwell ME, Perry RA, Brown LA, Haynie WS, Washington TA, Wiggs MP, Rajaram N, Greene NP. Cancer-induced cardiac atrophy adversely affects myocardial redox state and mitochondrial oxidative characteristics. JCSM Rapid Commun 4: 3–15, 2021. doi: 10.1002/rco2.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown JL, Rosa-Caldwell ME, Lee DE, Blackwell TA, Brown LA, Perry RA, Haynie WS, Hardee JP, Carson JA, Wiggs MP, Washington TA, Greene NP. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour-bearing mice. J Cachexia Sarcopenia Muscle 8: 926–938, 2017. doi: 10.1002/jcsm.12232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Drake JC, Bruns DR, Peelor FF 3rd, Biela LM, Miller RA, Hamilton KL, Miller BF. Long-lived crowded-litter mice have an age-dependent increase in protein synthesis to DNA synthesis ratio and mTORC1 substrate phosphorylation. Am J Physiol Endocrinol Metab 307: E813–E821, 2014. doi: 10.1152/ajpendo.00256.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Drake JC, Peelor FF 3rd, Biela LM, Watkins MK, Miller RA, Hamilton KL, Miller BF. Assessment of mitochondrial biogenesis and mTORC1 signaling during chronic rapamycin feeding in male and female mice. J Gerontol A Biol Sci Med Sci 68: 1493–1501, 2013. doi: 10.1093/gerona/glt047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Drake JC, Bruns DR, Peelor FF 3rd, Biela LM, Miller RA, Miller BF, Hamilton KL. Long-lived Snell dwarf mice display increased proteostatic mechanisms that are not dependent on decreased mTORC1 activity. Aging Cell 14: 474–482, 2015. doi: 10.1111/acel.12329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller BF, Reid JJ, Price JC, Lin H-JL, Atherton PJ, Smith K. CORP: The use of deuterated water for the measurement of protein synthesis. J Appl Physiol (1985) 128: 1163–1176, 2020. doi: 10.1152/japplphysiol.00855.2019. [DOI] [PubMed] [Google Scholar]
- 41.Miller BF, Baehr LM, Musci RV, Reid JJ, Peelor FF 3rd, Hamilton KL, Bodine SC. Muscle-specific changes in protein synthesis with aging and reloading after disuse atrophy. J Cachexia Sarcopenia Muscle 10: 1195–1209, 2019. doi: 10.1002/jcsm.12470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee DE, Brown JL, Rosa-Caldwell ME, Blackwell TA, Perry RA Jr, Brown LA, Khatri B, Seo D, Bottje WG, Washington TA, Wiggs MP, Kong BW, Greene NP. Cancer cachexia-induced muscle atrophy: evidence for alterations in microRNAs important for muscle size. Physiol Genomics 49: 253–260, 2017. doi: 10.1152/physiolgenomics.00006.2017. [DOI] [PubMed] [Google Scholar]
- 43.Reid JJ, Linden MA, Peelor FF, Miller RA, Hamilton KL, Miller BF. Brain protein synthesis rates in the UM-HET3 mouse following treatment with rapamycin or rapamycin with metformin. J Gerontol A Biol Sci Med Sci 75: 40–49, 2020. doi: 10.1093/gerona/glz069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Busch R, Neese RA, Awada M, Hayes GM, Hellerstein MK. Measurement of cell proliferation by heavy water labeling. Nat Protoc 2: 3045–3057, 2007. doi: 10.1038/nprot.2007.420. [DOI] [PubMed] [Google Scholar]
- 45.Robinson MM, Turner SM, Hellerstein MK, Hamilton KL, Miller BF. Long-term synthesis rates of skeletal muscle DNA and protein are higher during aerobic training in older humans than in sedentary young subjects but are not altered by protein supplementation. FASEB J 25: 3240–3249, 2011. doi: 10.1096/fj.11-186437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller BF, Robinson MM, Bruss MD, Hellerstein M, Hamilton KL. A comprehensive assessment of mitochondrial protein synthesis and cellular proliferation with age and caloric restriction. Aging Cell 11: 150–161, 2012. doi: 10.1111/j.1474-9726.2011.00769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Groennebaek T, Sieljacks P, Nielsen R, Pryds K, Jespersen NR, Wang J, Carlsen CR, Schmidt MR, de Paoli FV, Miller BF, Vissing K, Bøtker HE. Effect of blood flow restricted resistance exercise and remote ischemic conditioning on functional capacity and myocellular adaptations in patients with heart failure. Circ Heart Fail 12: e006427, 2019. doi: 10.1161/CIRCHEARTFAILURE.119.006427. [DOI] [PubMed] [Google Scholar]
- 48.Smith LR, Hammers DW, Sweeney HL, Barton ER. Increased collagen cross-linking is a signature of dystrophin-deficient muscle. Muscle Nerve 54: 71–78, 2016. doi: 10.1002/mus.24998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim YO, Popov Y, Schuppan D. Optimized mouse models for liver fibrosis. Methods Mol Biol 1559: 279–296, 2017. doi: 10.1007/978-1-4939-6786-5_19. [DOI] [PubMed] [Google Scholar]
- 50.Brown JL, Rosa-Caldwell ME, Lee DE, Brown LA, Perry RA, Shimkus KL, Blackwell TA, Fluckey JD, Carson JA, Dridi S, Washington TA, Greene NP. PGC-1α4 gene expression is suppressed by the IL-6-MEK-ERK 1/2 MAPK signalling axis and altered by resistance exercise, obesity and muscle injury. Acta Physiol (Oxf) 220: 275–288, 2017. doi: 10.1111/apha.12826. [DOI] [PubMed] [Google Scholar]
- 51.Polymenis M, Aramayo R. Translate to divide: сontrol of the cell cycle by protein synthesis. Microb Cell 2: 94–104, 2015. doi: 10.15698/mic2015.04.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wolff CA, Reid JJ, Musci RV, Bruns DR, Linden MA, Konopka AR, Peelor FF, Miller BF, Hamilton KL, Bruns DR. Differential effects of rapamycin and metformin in combination with rapamycin on mechanisms of proteostasis in cultured skeletal myotubes. J Gerontol A Biol Sci Med Sci 75: 32–39, 2020. doi: 10.1093/gerona/glz058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Deshaies RJ. Drug discovery: fresh target for cancer therapy. Nature 458: 709–710, 2009. doi: 10.1038/458709a. [DOI] [PubMed] [Google Scholar]
- 54.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 136: 823–837, 2009. [Erratum in Cell 138: 807, 2009]. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen HL, Tung YT, Tsai CL, Lai CW, Lai ZL, Tsai HC, Lin YL, Wang CH, Chen CM. Kefir improves fatty liver syndrome by inhibiting the lipogenesis pathway in leptin-deficient ob/ob knockout mice. Int J Obes 38: 1172–1179, 2014. doi: 10.1038/ijo.2013.236. [DOI] [PubMed] [Google Scholar]
- 56.Park EJ, Lee JH, Yu G-Y, He G, Ali SR, Holzer RG, Österreicher CH, Takahashi H, Karin M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 140: 197–208, 2010. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Burt AD, Lackner C, Tiniakos DG. Diagnosis and assessment of NAFLD: definitions and histopathological classification. Semin Liver Dis 35: 207–220, 2015. doi: 10.1055/s-0035-1562942. [DOI] [PubMed] [Google Scholar]
- 58.Rosa-Caldwell ME, Brown JL, Lee DE, Wiggs MP, Perry RA, Jr, Haynie WS, Caldwell AR, Washington TA, Lo WJ, Greene NP. Hepatic alterations during the development and progression of cancer cachexia. Appl Physiol Nutr Metab 45: 500–512, 2020. doi: 10.1139/apnm-2019-0407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lieffers JR, Mourtzakis M, Hall KD, McCargar LJ, Prado CM, Baracos VE. A viscerally driven cachexia syndrome in patients with advanced colorectal cancer: contributions of organ and tumor mass to whole-body energy demands. Am J Clin Nutr 89: 1173–1179, 2009. doi: 10.3945/ajcn.2008.27273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tisdale MJ. Cachexia in cancer patients. Nat Rev Cancer 2: 862–871, 2002. doi: 10.1038/nrc927. [DOI] [PubMed] [Google Scholar]
- 61.Thompson KJ, McKillop IH, Schrum LW. Targeting collagen expression in alcoholic liver disease. World J Gastroenterol 17: 2473–2481, 2011. doi: 10.3748/wjg.v17.i20.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Luo Y, Oseini A, Gagnon R, Charles ED, Sidik K, Vincent R, Collen R, Idowu M, Contos MJ, Mirshahi F, Daita K, Asgharpour A, Siddiqui MS, Jarai G, Rosen G, Christian R, Sanyal AJ. An evaluation of the collagen fragments related to fibrogenesis and fibrolysis in nonalcoholic steatohepatitis. Sci Rep 8: 12414–12414, 2018. doi: 10.1038/s41598-018-30457-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, Mills PR, Keach JC, Lafferty HD, Stahler A, Haflidadottir S, Bendtsen F. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 149: 389–397.e10, 2015. doi: 10.1053/j.gastro.2015.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Anstee QM, Seth D, Day CP. Genetic factors that affect risk of alcoholic and nonalcoholic fatty liver disease. Gastroenterology 150: 1728–1744.e7, 2016. doi: 10.1053/j.gastro.2016.01.037. [DOI] [PubMed] [Google Scholar]
- 65.Kakisaka K, Kataoka K, Onodera M, Suzuki A, Endo K, Tatemichi Y, Kuroda H, Ishida K, Takikawa Y. Alpha-fetoprotein: a biomarker for the recruitment of progenitor cells in the liver in patients with acute liver injury or failure. Hepatol Res 45: E12–E20, 2015. doi: 10.1111/hepr.12448. [DOI] [PubMed] [Google Scholar]
- 66.Alison MR, Islam S, Lim S. Stem cells in liver regeneration, fibrosis and cancer: the good, the bad and the ugly. J Pathol 217: 282–298, 2009. doi: 10.1002/path.2453. [DOI] [PubMed] [Google Scholar]
- 67.Karaca G, Swiderska-Syn M, Xie G, Syn WK, Krüger L, Machado MV, Garman K, Choi SS, Michelotti GA, Burkly LC, Ochoa B, Diehl AM. TWEAK/Fn14 signaling is required for liver regeneration after partial hepatectomy in mice. PLoS One 9: e83987, 2014. doi: 10.1371/journal.pone.0083987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Anker SD, Negassa A, Coats AJ, Afzal R, Poole-Wilson PA, Cohn JN, Yusuf S. Prognostic importance of weight loss in chronic heart failure and the effect of treatment with angiotensin-converting-enzyme inhibitors: an observational study. Lancet 361: 1077–1083, 2003. doi: 10.1016/s0140-6736(03)12892-9. [DOI] [PubMed] [Google Scholar]
- 69.Christensen HM, Kistorp C, Schou M, Keller N, Zerahn B, Frystyk J, Schwarz P, Faber J. Prevalence of cachexia in chronic heart failure and characteristics of body composition and metabolic status. Endocrine 43: 626–634, 2013. doi: 10.1007/s12020-012-9836-3. [DOI] [PubMed] [Google Scholar]
- 70.Tian M, Asp ML, Nishijima Y, Belury MA. Evidence for cardiac atrophic remodeling in cancer-induced cachexia in mice. Int J Oncol 39: 1321–1326, 2011. doi: 10.3892/ijo.2011.1150. [DOI] [PubMed] [Google Scholar]
- 71.Springer J, Tschirner A, Haghikia A, von Haehling S, Lal H, Grzesiak A, Kaschina E, Palus S, Pötsch M, von Websky K, Hocher B, Latouche C, Jaisser F, Morawietz L, Coats AJ, Beadle J, Argiles JM, Thum T, Földes G, Doehner W, Hilfiker-Kleiner D, Force T, Anker SD. Prevention of liver cancer cachexia-induced cardiac wasting and heart failure. Eur Heart J 35: 932–941, 2014. doi: 10.1093/eurheartj/eht302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fearon Kenneth CH, Glass David J, Guttridge Denis C. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 16: 153–166, 2012. doi: 10.1016/j.cmet.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 73.Onesti JK, Guttridge DC. Inflammation based regulation of cancer cachexia. Biomed Res Int 2014: 168407, 2014. doi: 10.1155/2014/168407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, Jatoi A, Loprinzi C, MacDonald N, Mantovani G, Davis M, Muscaritoli M, Ottery F, Radbruch L, Ravasco P, Walsh D, Wilcock A, Kaasa S, Baracos VE. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 12: 489–495, 2011. doi: 10.1016/s1470-2045(10)70218-7. [DOI] [PubMed] [Google Scholar]
- 75.Ciciliot S, Rossi AC, Dyar KA, Blaauw B, Schiaffino S. Muscle type and fiber type specificity in muscle wasting. Int J Biochem Cell Biol 45: 2191–2199, 2013. doi: 10.1016/j.biocel.2013.05.016. [DOI] [PubMed] [Google Scholar]
- 76.Augusto V, Padovani C, Eduardo G, Campos R. Skeletal muscule fiber types in C57BL6J mice J Morphol Sci 21: 0, 2004. http://www.jms.periodikos.com.br/journal/jms/article/587cb4537f8c9d0d058b45ea. [Google Scholar]
- 77.Rubenstein AB, Smith GR, Raue U, Begue G, Minchev K, Ruf-Zamojski F, Nair VD, Wang X, Zhou L, Zaslavsky E, Trappe TA, Trappe S, Sealfon SC. Single-cell transcriptional profiles in human skeletal muscle. Sci Rep 10: 229, 2020. doi: 10.1038/s41598-019-57110-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bentzinger CF, Wang YX, Dumont NA, Rudnicki MA. Cellular dynamics in the muscle satellite cell niche. EMBO Rep 14: 1062–1072, 2013. doi: 10.1038/embor.2013.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tirziu D, Giordano FJ, Simons M. Cell communications in the heart. Circulation 122: 928–937, 2010. doi: 10.1161/CIRCULATIONAHA.108.847731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bu L, Jiang X, Martin-Puig S, Caron L, Zhu S, Shao Y, Roberts DJ, Huang PL, Domian IJ, Chien KR. Human ISL1 heart progenitors generate diverse multipotent cardiovascular cell lineages. Nature 460: 113–117, 2009. doi: 10.1038/nature08191. [DOI] [PubMed] [Google Scholar]
- 81.He WA, Berardi E, Cardillo VM, Acharyya S, Aulino P, Thomas-Ahner J, Wang J, Bloomston M, Muscarella P, Nau P, Shah N, Butchbach ME, Ladner K, Adamo S, Rudnicki MA, Keller C, Coletti D, Montanaro F, Guttridge DC. NF-κB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J Clin Invest 123: 4821–4835, 2013. doi: 10.1172/JCI68523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zebrack BJ, Gurney JG, Oeffinger K, Whitton J, Packer RJ, Mertens A, Turk N, Castleberry R, Dreyer Z, Robison LL, Zeltzer LK. Psychological outcomes in long-term survivors of childhood brain cancer: a report from the childhood cancer survivor study. J Clin Oncol 22: 999–1006, 2004. doi: 10.1200/JCO.2004.06.148. [DOI] [PubMed] [Google Scholar]
- 83.Hudson MM, Mertens AC, Yasui Y, Hobbie W, Chen H, Gurney JG, Yeazel M, Recklitis CJ, Marina N, Robison LR, Oeffinger KC. Health status of adult long-term survivors of childhood cancer: a report from the Childhood Cancer Survivor Study. JAMA 290: 1583–1592, 2003. doi: 10.1001/jama.290.12.1583. [DOI] [PubMed] [Google Scholar]
- 84.Hudson MM, Oeffinger KC, Jones K, Brinkman TM, Krull KR, Mulrooney DA, Mertens A, Castellino SM, Casillas J, Gurney JG, Nathan PC, Leisenring W, Robison LL, Ness KK. Age-dependent changes in health status in the Childhood Cancer Survivor cohort. J Clin Oncol 33: 479–491, 2015. doi: 10.1200/JCO.2014.57.4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Du F, Zhu X-H, Zhang Y, Friedman M, Zhang N, Ugurbil K, Chen W. Tightly coupled brain activity and cerebral ATP metabolic rate. Proc Natl Acad Sci USA 105: 6409–6414, 2008. doi: 10.1073/pnas.0710766105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Scheen AJ. Central nervous system: a conductor orchestrating metabolic regulations harmed by both hyperglycaemia and hypoglycaemia. Diabetes Metab 36, Suppl 3: S31–S38, 2010. doi: 10.1016/s1262-3636(10)70464-x. [DOI] [PubMed] [Google Scholar]
- 87.Mergenthaler P, Lindauer U, Dienel GA, Meisel A. Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci 36: 587–597, 2013. doi: 10.1016/j.tins.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fogarty S, Hardie DG. Development of protein kinase activators: AMPK as a target in metabolic disorders and cancer. Biochimica Biophysica Acta 1804: 581–591, 2010. doi: 10.1016/j.bbapap.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 89.Vara-Ciruelos D, Russell FM, Hardie DG. The strange case of AMPK and cancer: Dr Jekyll or Mr Hyde?†. Open Biol 9: 190099, 2019. doi: 10.1098/rsob.190099. [DOI] [PMC free article] [PubMed] [Google Scholar]