

Summary
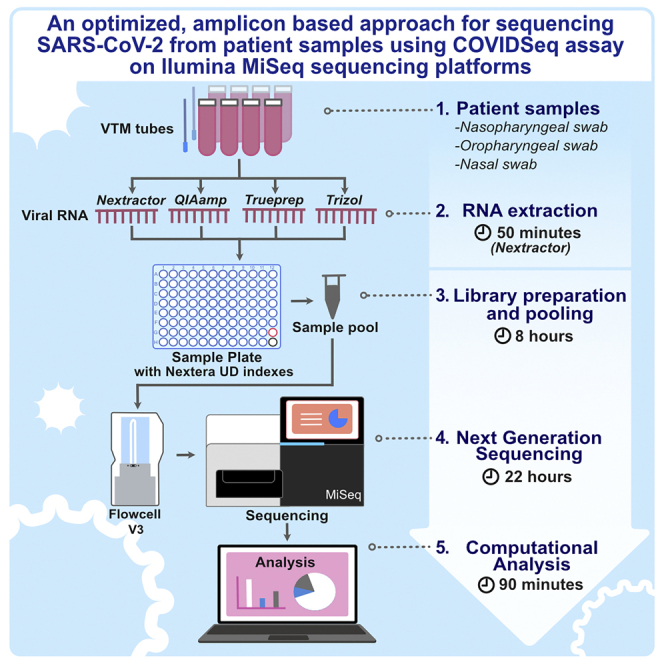
Sequencing of SARS-CoV-2 genomes is crucial for understanding the genetic epidemiology of the COVID-19 pandemic. It is also critical for understanding the evolution of the virus and also for the rapid development of diagnostic tools. The present protocol is a modification of the Illumina COVIDSeq test. We describe an amplicon-based next-generation sequencing approach with short turnaround time, adapted for bench-top sequencers like MiSeq, iSeq, and MiniSeq.
For complete details on the use and execution of this protocol, please refer to Bhoyar et al. (2021).
Subject areas: Bioinformatics, Sequence analysis, Clinical Protocol, Genomics, Sequencing, Immunology, Microbiology
Graphical abstract
Highlights
-
•
An amplicon-based next-generation sequencing approach for detection of SARS-CoV-2 RNA
-
•
Optimized for low-throughput bench-top equipment for smaller batch sizes
-
•
The protocol is compatible with all commonly used viral RNA isolation methods
-
•
An accelerated SARS-CoV-2 sequencing protocol with turnaround time < 30 h
Sequencing of SARS-CoV-2 genomes is crucial for understanding the genetic epidemiology of the COVID-19 pandemic. It is also critical for understanding the evolution of the virus and also for the rapid development of diagnostic tools. The present protocol is a modification of the Illumina COVIDSeq test. We describe an amplicon-based next-generation sequencing approach with short turnaround time, adapted for bench-top sequencers like MiSeq, iSeq, and MiniSeq.
Before you begin
The study was approved by the Institutional Human Ethics Committee (IHEC No. Dated CSIR-IGIB/IHEC/2020-21/01). After obtaining informed consent from patients, samples from nasal, nasopharyngeal, and oropharyngeal swabs were collected according to the standard protocol in 3 mL sterile viral transport medium (VTM) tube. All the samples were transported to the laboratory at a cold temperature (2°C–8°C) within 72 h post collection, and stored at −80°C till further used.
Introduction
The Illumina COVIDSeq assay is an amplicon-based next-generation sequencing (NGS) approach for the detection of SARS-CoV-2 RNA isolated from nasopharyngeal, oropharyngeal, and mid-turbinate nasal swabs from patients. The assay is one of the first NGS based SARS-CoV-2 detection assays approved for use under U.S Food and Drug Administration’s Emergency Use Authorization (EUA). The COVIDSeq test was first standardized and validated for Illumina NovaSeq sequencer and can accommodate 384 to 3072 samples, depending on the configuration of flow cells and the instrument (Bhoyar et al. 2021). RNA required for SARS-CoV-2 sequencing can be extracted from decontaminated nasopharyngeal swabs using the methods mentioned in EUA approval that include; Quick-DNA/RNA Viral MagBead Kit (Zymo Research, # R2141) or QIAamp Viral RNA Mini Kit (Qiagen, part # 52906). However, the protocol is compatible with all commonly used viral RNA isolation methods including, Nextractor® NX-48S (GENOLUTION), Trueprep (Molbio Diagnostics Pvt. Ltd.) and TRIzol (Invitrogen).
While originally adapted and optimized for larger sequencers, this assay could be optimized for low throughput bench-top equipment for smaller batch sizes. This manuscript details the optimized protocol with marginal modifications along with an open-source pipeline for the analysis and interpretation of data. While the protocol has been standardized for the Illumina MiSeq sequencing platform, by considering the final loading concentration of the library and the data output, the proposed protocol can be adopted for any bench-top sequencer from Illumina Inc.
Requirements for library preparation
This protocol consists of the following key steps:
RNA extraction- Prior to RNA extraction, the VTM samples were subjected to heat inactivation at 50°C for 30 min. RNA is extracted from the inactivated specimen (VTM), using any of the following mentioned protocol that includes; QIAmp (QIAGEN), Nextractor® NX-48S (GENOLUTION), TruePrep (Molbio Diagnostics Pvt. Ltd.) and TRIzol (Invitrogen).
cDNA synthesis- Generates complementary DNA to the RNA by reverse transcriptase using random hexamers.
Target amplification- The synthesized cDNA undergoes two separate PCR reactions to amplify the virus genome present in the sample.
Library preparation, Pooling and Quantification- During this process, the pooled amplified products undergo bead-based tagmentation where it gets fragmented and tagged to the adapter sequences. The adapter-tagged fragments undergo another round of PCR amplification, after which indexed tagged libraries will be pooled and cleaned using the purification beads. The pooled library product is quantified using Qubit High Sensitivity dsDNA quantification kit (Invitrogen).
Sequencing- The sequencing-ready libraries are clustered onto a flow cell and sequenced using sequencing by synthesis (SBS) chemistry on the Illumina MiSeq sequencing system.
Analysis- This step includes genome assembly, variant calling and phylogenetic analysis using established protocols (available in the GitHub repository:https://github.com/bnijolly/Genepi).
-
1.Prepare the following reagents
-
a.Isolated SARS-CoV-2 RNA samples.
-
b.A pre-amplification environment with a Biosafety level 2 (BSL-2) facility is required during RNA sample handling and cDNA preparation.
-
c.Personal Protective Equipment (PPE), as recommended by WHO for handling viral samples contagious to humans.
-
d.Two 96-well PCR thermal cyclers.
-
e.Thermo mixer with a 96-well PCR plate holder.
-
f.Qubit HS dsDNA quantification kit along with instrument (Thermofisher).
-
a.
cDNA preparation
Timing: 1 h
During this process, the extracted SARS-CoV-2 RNA is annealed using random hexamers and the RNA fragments primed with random hexamers are reverse-transcribed into first strand cDNA using reverse transcriptase enzyme.
(All steps to be carried out within the Biosafety cabinet of the BSL-2 facility)
-
2.Annealing of RNA
-
a.Label a new 96 well PCR plate as cDNA plate (Figure 1).
-
b.Add 8.5 μL of the isolated SARS-CoV-2 sample into the wells of the cDNA plate.
-
c.Add 8.5 μL of EPH3 HT to each well.
-
d.Mix well with a P10 or P20 multichannel pipette. If plate shaker is available, within the laminar hood, shake the plate at 1600 rpm for 1 min.
-
e.Seal and spin the plate at 1000 × g for 1 min.
-
f.Place the cDNA plate on a thermal cycler and run the following program
-
i.Choose the preheat lid option
-
ii.Set the final volume of the reaction as 17 μL
-
iii.65oC for 3 min
-
iv.Hold at 4oC
-
i.
-
a.
-
3.First strand cDNA synthesis
-
a.Prepare First strand cDNA Mastermix (for 96 samples) by mixing 864 μL of FSM HT and 96 μL of RVT HT in a 1.5mL microcentrifuge tube.
-
b.Add 8 μL of the First strand cDNA mastermix into each well of the cDNA plate and pipette well to mix.
-
c.Seal and spin the plate at 1000 × g for 1 min.
-
d.Place the cDNA plate on a thermal cycler and run the following program
-
i.Choose the preheat lid option
-
ii.Set the final volume of the reaction as 25 μL
-
iii.25oC for 5 min
-
iv.50oC for 10 min
-
v.80oC for 5 min
-
vi.Hold at 4oC
-
i.
-
a.
Pause point: The procedure can be paused here and the cDNA plate can be stored at −20oC for up to 7 days.
Figure 1.

Layout of the sample plate to be used denoting the samples, PC and NC
Up-to 94 samples can be processed at once. A positive control (CPC HT) and Negative Control (ELB HT)is included in every plate that is processed using the COVIDSeq protocol.
Amplification of cDNA
In this step, the SARS-CoV-2 virus genome present in the test samples is amplified using two separate PCR reactions that are then pooled together.
-
4.Amplification of prepared cDNA
-
a.Label two 96-well PCR plates as COV1 and COV2.
-
b.Prepare PCR1 Mastermix and PCR2 Mastermix in two separate 15 mL tubes.
-
i.Add the following to each tube:
S.No Item name Volume for 96 samples Volume for 1 sample 1 Nuclease Free Water 451.2 μL 4.7 μL 2 IPM HT 1440 μL 15μL 3 CPP1 HT (for PCR1)/ CPP2 HT (for PCR2) 412.8μL 4.3μL
-
i.
-
c.Add 5 μL each of first strand cDNA from the cDNA plate to corresponding wells of COV1 and COV2 plates.
-
d.Add 20 μL of PCR1 Mastermix to each well of COV1 plate.
-
e.Add 20 μL of PCR2 Mastermix to each well of COV2 plate.
-
f.Seal and shake the COV1 and COV2 plates at 1600 rpm for 1 min.
-
g.Centrifuge the plates at 1000 × g for 1 min.
-
h.Place the plates on individual thermal cyclers and run the following program
-
i.Choose the preheat lid option
-
ii.Set the final volume of the reaction as 25 μL
-
iii.Set up the PCR with the following conditions
PCR cycling conditions
Steps Temperature Time Cycles Initial Denaturation 98oC 3 min x 1 cycle Denaturation 98oC 15 s x 35 cycles Annealing 65oC 5 min Hold 4oC ∞
-
i.
-
i.The program has a run time of approximately 03 h and 30 min.
-
a.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Ethanol, Molecular Grade | Merck | 100983 |
| Nuclease-Free Water | Ambion | AM9906 |
| Critical commercial assays | ||
| Illumina COVIDSeq Test 3072 samples | Illumina | 20043675 |
| MiSeq Reagent Kit V3 (150 cycles) | Illumina | MS-102-3001 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher | Q32851 |
| Biological samples | ||
| SARS-CoV-2 RNA | NasopharyngealoOropharyngeal swab samples | NA |
| Oligonucleotides | ||
| IDT for Illumina PCR Indexes Set 1–4 | Illumina | 20043137 |
| Software and Algorithms | ||
| bcl2fastq Conversion Software | Illumina | NA |
| Phylogenetic Assignment of Named Global Outbreak Lineages (PANGOLIN) | Center for Genomic Pathogen Surveillance | https://github.com/cov-lineages/pangolin |
| Nextstrain open-source toolkit | Hadfield et al.,2018 | https://github.com/nextstrain |
| Deposited data | ||
| Scripts used for the SARS-CoV-2 sequencing analysis and interpretation | Poojary et al., 2020 | https://github.com/banijolly/Genepi |
| Other | ||
| 96-Well PCR thermal cyclers | Veriti 96-Well Thermal Cycler | 4375786 |
| Thermomixer with a 96-well PCR plate holder | Eppendorf ThermoMixer C | 05–412-503 |
| 96-Well PCR plate | Thermo Fisher | AB0600 |
Materials and equipment
| Reagent | Description | Storage | Total volume required per reaction |
|---|---|---|---|
| SARS-CoV-2 RNA a | Sample | −20oC | 8.5 μL |
| CPC HTb | COVIDSeq Positive Control HT | −20oC | 8.5 μL |
| ELB HTb | Elution Buffer HT | −20oC | 8.5 μL |
| EPH3 HT | Elution Prime Fragment 3HC Mix | −20oC | 8.5 μL |
| FSM HT | First Strand Mix HT | −20oC | 9 μL |
| RVT HT | Reverse Transcriptase HT | −20oC | 1 μL |
| IPM HT | Illumina PCR Mix HT | −20oC | 15 μL |
| CPP1 HT | COVIDSeq Primer Pool HT 1 | −20oC | 4.3 μL |
| CPP2 HT | COVIDSeq Primer Pool HT 2 | −20oC | 4.3 μL |
| TB1 HT | Tagmentation Buffer 1 HT | 4oC | 12 μL |
| EBLTS HT | Enrichment BLT HT | −20oC | 4 μL |
| ST2 HT | Stop TagmentBuffer2 HT | 4oC | 10 μL |
| TWB HT | Tagmentation Wash Buffer HT | 4oC | 200 μL |
| EPM HT | Enhanced PCR Mix HT | −20oC | 24 μL |
| Index adaptera | IDT for Illumina PCR Indexes- Set 1,2,3,4 | −20oC | 10 μL |
| ITB | Illumina Tune Beads | RT | 396 μL (for 96 samples) |
| 80% Ethanola | 80% Ethanol | RT | 2 mL |
| RSB | Resuspension Buffer | 4oC | 55 μL |
| 0.2N NaOH | Denaturation solution | RT | 10 μL |
| HT1 | Neutralizing Buffer | −20oC | 2.4 mL |
| PhiX | Spike in Control | 20oC | 5 μL |
| NFWa | Nuclease Free Water | RT | 25 mL |
Items not included along with the Illumina COVIDSeq test kit
Positive control and Elution Buffer are provided in the COVIDSeq test kit. CPC HT is provided at a stock concentration of 1 million copies/μL. A working solution of 5 copies/μL have to be prepared before adding to the plate. Elution buffer is used directly as a negative control.
Step-by-step method details
Library preparation and library pooling
In this step, the prepared cDNA of the SARS-CoV-2 genome is tagmented (tagged and fragmented), indexed and amplified to become sequencing-ready libraries.
-
1.Tagmentation of PCR amplicons
-
a.Label a new 96-well PCR plate as TAG plate.
-
b.From COV1 and COV2 plates, combine 10 μL of contents from each well to their respective wells in TAG1 plate, and mix well. This results in a total volume of 20 μL in each well of the TAG1 plate.
-
c.To make Tagmentation Mastermix:
-
i.Label a 15 mL tube as Tagmentation Mastermix.
-
ii.Follow the table below to prepare Tagmentation Mastermix.
S.No Item name Volume for 96 samples Volume for 1 sample 1 Nuclease-free water 1920 μL 20 μL 2 TB1 HT 1152 μL 12 μL 3 EBLTS HT 384 μL 4 μL -
iii.Vortex for 30 s to mix well.
-
i.
-
d.Add 30 μL of Tagmentation Mastermix to each well in the TAG1 plate.
-
e.Seal and shake the TAG1 plate at 1600 rpm for 1 min.
-
f.Centrifuge at 1000 × g for 1 min if the contents of the well are found sticking to the walls or the seal of the TAG1 plate.
-
g.Place on a thermal cycler and run the following program.
-
i.Choose the preheat lid option
-
ii.Set the total volume of the reaction as 50 μL.
-
iii.55oC for 5 min
-
iv.Hold at 10oC.
-
i.
-
a.
-
2.Post Tagmentation Clean-up
-
a.Centrifuge the TAG1 plate at 500 × g for 1 min.
-
b.Remove the seal and add 10 μL of ST2 HT to each well of the TAG1 plate.
-
c.Seal and shake at 1600rpm for 1 min.
-
d.Incubate the plate at room temperature (22°C–25°C) for 5 min.
-
e.Centrifuge the TAG1 plate at 500 × g for 1 min.
-
f.Remove the seal carefully and place the plate on a magnetic stand.
-
g.Wait for 3 min/until the liquid is clear.
-
h.Remove and discard all supernatants from the wells.
-
i.Wash the beads as follows
-
i.Remove the plate from the magnetic stand.
-
ii.Add 100 μL of TWB HT to each well.
-
iii.Seal and shake at 1600 rpm for 1 min and centrifuge at 500 × g for 1 min.
-
iv.Remove the seal and place the plate on the magnetic stand. Wait for 3 min/until the liquid is clear.
-
v.Remove and discard the supernatant from each well.
-
i.
-
j.Repeat the wash steps (step i; i-v) one more time.
-
a.
CRITICAL: Do not discard the supernatant after the second wash, to prevent the beads from over drying.
-
3.Amplification of tagmented amplicons
-
a.In a 15 mL tube, prepare Enhanced PCR Mix
-
i.Add 2304 μL of EPM HT (24 μL × 96 samples).
-
ii.Add 2304 μL of Nuclease-free water (24 μL × 96 samples).
-
iii.Vortex the tube to mix.
-
i.
-
b.Remove and discard the supernatant from the TAG1 plate.
-
c.Use a 10 μL or 20 μL pipette to remove any remaining TWB HT from the TAG1 plate.
-
d.Add 40 μL of Enhanced PCR Mix to each well of the TAG1 plate.
-
e.Add 10 μL of index adapters to each well of the TAG1 plate.
-
f.Seal and shake the plate at 1600 rpm for 1 min and centrifuge the plate at 500 × g for 1 min.CRITICAL: Inspect the tubes to make sure the beads are fully resuspended and are not found at the bottom of the tubes.
-
g.Place on a thermal cycler and run the following PCR program
-
i.Choose the preheat lid option and set the temperature to 100oC.
-
ii.Set the total volume of the reaction to be 50 μL.
PCR cycling conditions
Steps Temperature Time Cycles Initialization 72oC 3 min x1 cycle Initial Denaturation 98oC 3 min x1 cycle Denaturation 98oC 20 s x7 cycles Annealing 60oC 30 s Extension 72oC 1 min Final extension 72oC 3 min x1 cycle Hold 10oC ∞
-
i.
-
a.
-
4.Library pooling and Clean up
-
a.Centrifuge the TAG1 plate at 500 × g for 1 min.
-
b.Remove the seal and place the plate on a magnetic stand and wait for 3 min/until the liquid is clear.
-
c.Transfer 5 μL of library from each well into an 8-tubes strip. This results in a pooled volume of 60 μL in each tube of the 8-tubes strip.CRITICAL: Discard and change tips for each column of samples while transferring.
-
d.Transfer 55 μL of pooled library from each tube to a 1.5 mL microcentrifuge tube. This results in a total of 440 μL of pooled libraries (from 96 samples).
-
e.Add 396 μL of ITB to each tube (ITB volume is calculated by multiplying the total volume of the pooled libraries with 0.9 i.e., for 96 samples: 440 μL × 0.9 = 396 μL)
-
f.Vortex the tube to mix well.
-
g.Incubate the tube at room temperature for 5 min.
-
h.Spin briefly and place the tube on a 1.5mL tube magnetic stand and wait for 5 min.
-
i.Remove and discard all supernatants.
-
j.Wash the beads as follows:
-
i.Add 1000 μL of freshly prepared 80% ethanol to the tube and wait for 30 s.
-
ii.Remove and discard the supernatant.
-
iii.Repeat the above-mentioned wash steps one more time for a total of 2 washes with 80% ethanol.
-
i.
-
k.Remove any residual ethanol left on the tube.
-
l.Air dry the beads for 2 min.
-
m.Add 55 μL of Resuspension Buffer to the tube and vortex to mix.
-
n.Incubate at room temperature for 2 min.
-
o.Spin briefly and place the tube in the magnetic stand and wait for 2 min.
-
p.Label a new 1.5 mL tube as Final Pool.
-
q.Transfer 50 μL of the supernatant into the Final Pool tube.
-
a.
-
5.Quantification and Normalization of the library pool
-
a.Using the Qubit High Sensitivity dsDNA quantification kit (Invitrogen), quantify the library pool
-
i.Dilute 1 μL of the pooled library sample in the ratio of 1:3.
-
ii.Use 2 μL of the diluted library for quantification.
-
iii.Calculate the actual concentration of the library by factoring in the dilution.
-
i.
-
b.Load 2 μL of the pooled library (undiluted) onto a 2% agarose gel. The libraries should be observed between the size 300–400 bp.
-
c.Using the formula below, calculate the concentration of the library in Molar units.
-
d.Dilute the library with Resuspension Buffer to a final concentration of 4nM.
-
e.The formula for calculating Molarity:
-
a.
[Library Conc (ng/μL)]/ [660(g/mol) x average library size (bp)]×106= Conc. of Library (nM)
Sequencing the libraries
The prepared COVIDSeq libraries will be sequenced in this step using the MiSeq sequencing platform
-
6.Preparing PhiX
-
a.Label a new 1.5 mL tube as PhiX Control.
-
b.Combine 2 μL of PhiX stock library (10nM) and 3 μL of Nuclease-free water to make 4nM PhiX libraryCRITICAL: Prepare a fresh PhiX dilution and use it within 12 hours of preparation.
-
c.Label a new 1.5 mL tube as PhiX-Final and add 5 μL of diluted PhiX (4nM) and 5 μL of 0.2N freshly prepared NaOH.
-
d.Vortex the tube briefly to mix. Spin and incubate the tube at room temperature for 5 min.
-
e.Add 990 μL of prechilled HT1 to the PhiX-Final tube resulting in 1mL of 20pM PhiX library.
-
a.
-
7.Denaturation of COVIDSeq library
-
a.Label a new 1.5 mL tube as Library-Final and add 5 μL of diluted library (4nM) and 5 μL of 0.2N freshly prepared NaOH.
-
b.Vortex the tube briefly to mix. Spin and incubate the tube at room temperature for 5 min.
-
c.Add 990 μL of pre-chilled HT1 to the tube resulting in 1mL of 20pM Final library.CRITICAL: For instructions regarding final loading concentration and PhiX spike-in for MiSeq system, refer to the MiSeq system Denature and Dilute Libraries Guide.
-
d.To prepare 6pM final loading concentration library
-
i.Label a new 1.5mL tube as 6pM Final Library tube.
-
ii.Add 180 μL of the 20pM library from the Library-Final tube.
-
iii.Add 420 μL of pre-chilled HT1 and mix well.
-
iv.Discard 6 μL of the mixture.
-
v.Add 6 μL of the 20pM PhiX library for 1% spike in as recommended by Illumina Inc.
-
vi.Mix well with a pipette, spin briefly and place the tube on ice. The library is ready to be loaded onto the flow cell.
-
i.
-
a.
Note: To reduce the handling time, the denaturation of the library and PhiX can be performed simultaneously.
-
8.Sequencing and expected Output
-
a.Load the library into the MiSeq Sequencing cartridge and initiate sequencing as per the MiSeq Sequencing guide by Illumina Inc.
-
b.For sequencing using MiSeq v3 2×75 cycles kit, the estimated yield is about 3.3–3.8 Gb as per Illumina Inc.
-
a.
Expected outcomes
Upon successful tagmentation and size selection the final library pool will appear as ~300 bp size band on 2% agarose gel (Figure 2). Any deviation in the fragment size may be the result of erroneous tagmentation or size selection, which can be minimized with proper handling and pipetting practices.
Figure 2.

Quality of the final library pool was analyzed by agarose gel electrophoresis (2%), the expected fragment of ~300 bp size was observed
Quantification and statistical analysis
Genome assembly, variant calling, and phylogenetic analysis
This step describes the basic bioinformatics pipeline for genome assembly and identification of variants in the SARS-CoV-2 genome. The raw sequencing data generated by Illumina sequencing platforms are in the form of Binary Base Call (BCL) files and require conversion to FASTQ format before further processing.
-
1.
The BCL files generated by the sequencer are demultiplexed to FASTQ files using the bcl2fastq conversion software provided by Illumina using the command:
bcl2fastq --runfolder-dir<path to the directory of the sequencing run files> --sample-sheet <path to the sample sheet> --output-dir<path to demultiplexed output folder>
-
2.
The raw FASTQ files need to undergo additional processing steps, including quality control, trimming, alignment against the SARS-CoV-2 reference genome and sorting. The resulting BAM file containing mapped and sorted reads are then used to call variants and generate a consensus sequence of the genomes in FASTA file format. The steps to assemble SARS-CoV-2 genomes from raw FASTQ files are detailed in a recent protocol (Poojary et al 2020). Sample parameters and metrics that may be used to assess the quality of the assembled genomes are given in Table 1.
-
3.
For assignment of lineages to the assembled genome FASTA sequences, we use the Phylogenetic Assignment of Named Global Outbreak Lineages (PANGOLIN) software command line tool using the command:
Table 1.
Data generated from a single sample of the 96 samples sequenced using the MiSeq platform
| Sample ID | Number of raw reads R1 | Number of raw reads R2 | Number of reads after trimming R1 | Number of reads after trimming R2 | % Of reads Aligned with reference | Number of base Pairs Covered | X coverage | Genome coverage | Number of variants called | Pango lineage | Time taken |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CSXXXX | 1431919 | 1431919 | 1221580 | 1221580 | 99.52% | 84310380 | 2819.46 | 99.84% | 11 | B.1.36 | 318 s |
pangolin <FASTA sequence file name>.fasta
Alternatively, the web version of PANGOLIN (available at https://pangolin.cog-uk.io/) can be used to assign lineages to the generated FASTA sequences (Rambaut et al 2020).
-
4.
The FASTA sequence files can also be used to analyze and visualize evolutionary relationships through phylogenetic clustering. For performing the phylogenetic analysis, we use the open-source tool-kit Nextstrain following a recently detailed protocol (Jolly and Scaria 2021). The comparison of data output, coverage and time taken for analysis between MiSeq and NovaSeq 6000 is provided in the Tables S1 and S2.
Limitations
As explained in our previous report (Bhoyar et al 2021), the Illumina COVIDSeq assay can detect RNA from SARS-CoV-2 in nasopharyngeal, oropharyngeal, and mid-turbinate nasal swabs from patients suspected to have COVID-19 with the highest CT 37 to the lowest CT samples. However, we have noticed that in order to capture the whole SARS-CoV-2 genome, RNA samples below CT 22 should be considered.
Troubleshooting
Problem 1
Amplification in the negative control (NC) (Step: cDNA Preparation)
In some cases, users may get amplification in the negative control well in the COVIDSeq assay.
Potential solution
To avoid amplification in the negative control we encourage the users to stick to the following practices.
-
1.
Autoclave all the pipettes before initiating a new experiment.
-
2.
Thoroughly decontaminate the Biosafety cabinet before the experiment.
-
3.
Use fresh pipette tips and fresh consumables between samples and dispensing reagents.
-
4.
Use aerosol resistant pipette tips to minimize the risk of carry over and sample to sample cross contamination.
-
5.
Use fresh PCR plate sealing films every time.
Problem 2
Low genome coverage in samples (Step-Genome Assembly, Variant Calling and Phylogenetic Analysis).
Users may find the genome coverage in certain samples is low and the samples do not return any variant after the analysis.
Potential solution
Users may ensure that the starting material has a high viral load i.e., we recommend samples with a CT value of <25 for 99% genome coverage.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Sridhar Sivasubbu (sridhar@igib.in).
Materials availability
All the materials used in the protocol are available without any restrictions; the details of the kits used in the protocol are given in the “key resources table”.
Data and code availability
All the scripts used for the SARS-CoV-2 sequencing analysis and interpretation are adopted from Poojary et al 2020 (GitHub repository: https://github.com/banijolly/Genepi).
Acknowledgments
Authors acknowledge funding from the Council of Scientific and Industrial Research (CSIR), India, through grants CODEST and MLP2005. The funders had no role in the design of experiment, analysis, or decision to publish.
Author contributions
All of the authors contributed substantially to the conception and design of the study, the acquisition of data, analysis, and interpretation. S.S. and V.S. contributed with funding acquisition.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100755.
Contributor Information
Vinod Scaria, Email: vinods@igib.in.
Sridhar Sivasubbu, Email: sridhar@igib.in.
Supplemental information
References
- Bhoyar R.C., Jain A., Sehgal P., Divakar M.K., Sharma D., Imran M., Jolly B., Ranjan G., Rophina M., Sharma S. High throughput detection and genetic epidemiology of SARS-CoV-2 using COVIDSeq next-generation sequencing. PLoS One. 2021;16:e0247115. doi: 10.1371/journal.pone.0247115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poojary M., Shantaraman A., Jolly B., Scaria V. Computational protocol for assembly and analysis of SARS-nCoV-2 genomes. Res. Rep. 2020;4:e1–e14doi. doi: 10.9777/rr.2020.10001. [DOI] [Google Scholar]
- Rambaut A., Holmes E.C., O’Toole Á., Hill V., McCrone J.T., Ruis C., du Plessis L., Pybus O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020;5:1403–1407. doi: 10.1038/s41564-020-0770-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadfield J., Megill C., Bell S.M., Huddleston J., Potter B., Callender C., Sagulenko P., Bedford T., Neher R.A. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. 2018;1:4121–4123. doi: 10.1093/bioinformatics/bty407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly B., Scaria V. Computational analysis and phylogenetic clustering of SARS-CoV-2 genomes. Bio Protoc. 2021;11:e3999. doi: 10.21769/BioProtoc.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the scripts used for the SARS-CoV-2 sequencing analysis and interpretation are adopted from Poojary et al 2020 (GitHub repository: https://github.com/banijolly/Genepi).