

Abstract
While sustained nuclear factor-κB (NF-κB) activation is critical for proinflammatory molecule expression, regulators of NF-κB activity during chronic inflammation are not known. We investigated the role of focal adhesion kinase (FAK) on sustained NF-κB activation in tumor necrosis factor-α (TNF-α)–stimulated endothelial cells (ECs) both in vitro and in vivo. We found that FAK inhibition abolished TNF-α-mediated sustained NF-κB activity in ECs by disrupting formation of TNF-α receptor complex-I (TNFRC-I). Additionally, FAK inhibition diminished recruitment of receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and the inhibitor of NF-κB (IκB) kinase (IKK) complex to TNFRC-I, resulting in elevated stability of IκBα protein. In mice given TNF-α, pharmacological and genetic FAK inhibition blocked TNF-α-induced IKK-NF-κB activation in aortic ECs. Mechanistically, TNF-α activated and redistributed FAK from the nucleus to the cytoplasm, causing elevated IKK-NF-κB activation. On the other hand, FAK inhibition trapped FAK in the nucleus of ECs even upon TNF-α stimulation, leading to reduced IKK-NF-κB activity. Together, these findings support a potential use for FAK inhibitors in treating chronic inflammatory diseases.
Keywords: FAK, TNF-α, RIPK1, NF-κB, IκB, IKK
INTRODUCTION
Several chronic inflammatory diseases including atherosclerosis and rheumatoid arthritis (RA) have been shown to have increased expression of cytokines such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and IL-6. As such, antibody therapies that target these cytokines or small molecule inhibitors that block their receptors have been investigated as therapeutic options [1, 2]. While these antibody therapies have benefited some patients [3], they carry a major risk of increased susceptibility to fatal infections and sepsis. Therefore, there is a need to better understand the chronic proinflammatory pathways underlying these cytokines to find common therapeutic targets that can alleviate their chronic inflammatory signaling without increasing risk of infection.
Focal adhesion kinase (FAK) is an integrin-associated protein tyrosine kinase that has been implicated in chronic inflammation [4–6]. FAK is activated by stimuli from integrins, growth factors, and cytokines, which increase FAK autophosphorylation at tyrosine 397 (pY397) [7]. In addition, recent studies demonstrated that FAK can shuttle from the cytosol to the nucleus, regulating nuclear protein stability and gene expression [8, 9]. FAK inhibition blocks the expression of a broad range of proinflammatory molecules in response to TNF-α or IL-1β stimulation in endothelial cells (ECs) [5, 10]. Although FAK inhibition reduced proinflammatory molecule expression, in part through decreased mitogen-activated protein kinase (MAPK) activation, FAK-mediated proinflammatory gene expression was not fully elucidated [5]. Several stimuli associated with chronic inflammation require FAK for activation of the key proinflammatory transcription factor nuclear factor-κB (NF-κB) [4, 11–13]. However, it is unknown how FAK regulates NF-κB activation in proinflammatory gene expression in ECs.
NF-κB is typically inactive and sequestered in the cytoplasm by association with inhibitor of NF-κB (IκB) proteins. TNF-α activates NF-κB through trimerization of TNF-α receptor 1 (TNFR1) and formation of TNFR complex-I (TNFRC-I) [14]. TNFRC-I is comprised of receptor-interacting serine/threonine-protein kinase 1 (RIPK1), the IκB kinase (IKK) complex, various kinases, and ubiquitination complexes [14]. The IKK complex, which includes IKKα, IKKβ, and IKKγ (a.k.a. NEMO; NF-κB essential modulator), phosphorylates IκB, which marks IκB for proteasomal degradation and enables NF-κB to enter the nucleus. IKK further promotes NF-κB transcriptional activity through phosphorylation of serine 536 (pS536 NF-κB) [15]. Typically, a negative feedback loop between NF-κB and IκBα, an early NF-κB target gene, occurs. However, under chronic inflammatory conditions such as continued TNF-α stimulation, IκBα is constantly synthesized and degraded, leading to oscillations in NF-κB nuclear localization and transcriptional ability [16]. This oscillatory pattern of NF-κB signaling may be important in ensuring that essential proinflammatory genes are induced only during chronic inflammatory conditions [17, 18]. However, limited knowledge is available about the key signaling factors behind sustained NF-κB activity in chronic inflammation.
In the present study, we investigated the molecular function of FAK in sustained activation of the master proinflammatory transcription factor NF-κB under chronic TNF-α stimulation in ECs using in vitro and in vivo pharmacological and genetic FAK inhibition.
MATERIALS AND METHODS
Antibodies and Reagents
Antibodies used in this study are listed in Table 1. Recombinant human TNF-α (Cat# 210-TA) and recombinant mouse TNF-α (Cat# 410-MT) were purchased from R&D Systems (Minneapolis, MN, USA). Recombinant FLAG-TNF-α and recombinant GST-IκBα (1–55) were purified in-house from bacteria. FAK inhibitor PF-271 (Cat# 206808) was purchased from MedKoo Biosciences (Morrisville, NC, USA). Src inhibitor Dasatinib (Cat# S1021) was purchased from Selleckchem (Houston, TX, USA).
Table 1.
Antibodies Used in This Study
| Name of antibody | Manufacturer, catalog# | Species raised in | Dilution used | RRID |
|---|---|---|---|---|
| pY397 FAK | Thermo Fisher Scientific, 44-624G | Rabbit | 1:3000 (WB) 1:300 (IF) |
AB_2533701 |
| pS32/36 IκBα | Thermo Fisher Scientific, MA5-15224 | Mouse | 1:2000 (WB) | AB_10981266 |
| NF-κB p65 | Thermo Fisher Scientific, 51-0500 | Rabbit | 2 μg (ChIP) | AB_2533893 |
| pS176/177 IKKα/β | Thermo Fisher Scientific, MA5-14857 | Mouse | 1:100 (IF) | AB_10986824 |
| Alexa 488 anti-mouse | Thermo Fisher Scientific, A11001 | Goat | 1:1000 (IF) | AB_2534069 |
| Alexa 594 anti-mouse | Thermo Fisher Scientific, A11005 | Goat | 1:1000 (IF) | AB_2534073 |
| Alexa 488 anti-rabbit | Thermo Fisher Scientific, A11008 | Goat | 1:1000 (IF) | AB_143165 |
| Alexa 594 anti-rabbit | Thermo Fisher Scientific, A11012 | Goat | 1:1000 (IF) | AB_2534079 |
| Anti-mouse HRP | Thermo Fisher Scientific, 31430 | Goat | 1:5000 (WB) | AB_228307 |
| Anti-rabbit HRP | Thermo Fisher Scientific, 31460 | Goat | 1:5000 (WB) | AB_228341 |
| VCAM-1 | Santa Cruz Biotechnology, sc-8304 | Rabbit | 1:2000 (WB) 1:200 (IF) |
AB_2214058 |
| pS311NF-κB | Santa Cruz Biotechnology, sc-33039 | Rabbit | 1:2000 (WB) | AB_2238379 |
| pS529 NF-κB | Santa Cruz Biotechnology, sc-101751 | Rabbit | 1:2000 (WB) | AB_1128538 |
| NF-κB | Santa Cruz Biotechnology, sc-372 | Rabbit | 1:2000 (WB) 1:200 (IF) |
AB_632037 |
| IκBα | Santa Cruz Biotechnology, sc-371 | Rabbit | 1:2000 (WB) | AB_2235952 |
| Src | Santa Cruz Biotechnology, sc-18 | Rabbit | 1:2000 (WB) | AB_631324 |
| vWF | Santa Cruz Biotechnology, sc-271409 | Mouse | 1:200 (IF) | AB_10610681 |
| RIPK1 | Santa Cruz Biotechnology, sc-133102 | Mouse | 1:2000 (WB) | AB_1568814 |
| NEMO | Santa Cruz Biotechnology, sc-8032 | Mouse | 1:2000 (WB) | AB_627786 |
| TNFR1 | Santa Cruz Biotechnology, sc-374186 | Mouse | 1:200 (IF) | AB_10992436 |
| β-actin | Sigma-Aldrich, A5316 | Mouse | 1:5000 (WB) | AB_476743 |
| FLAG | Sigma-Aldrich, F3165 | Mouse | 1:2000 (WB) 2 μg (IP) |
AB_259529 |
| FAK | Millipore, 05–537 | Mouse | 1:2000 (WB) 1:300 (IF) |
AB_2173817 |
| GAPDH | Millipore, MAB374 | Mouse | 1:5000 (WB) | AB_2107445 |
| TNFR1 | Novus Biologicals, NBP1-97453 | Rabbit | 1:2000 (WB) 1:200 (IF) |
AB_11188877 |
| pS176/177 IKKα/β | Cell Signal Technology, 2697 | Rabbit | 1:2000 (WB) 1:200 (IF) |
AB_2079382 |
| pS536 NF-κB | Cell Signal Technology, 3033 | Rabbit | 1:2000 (WB) 1:200 (IF) |
AB_331284 |
| RIPK1 | Cell Signal Technology, 4926 | Rabbit | 1:200 (IF) | AB_2224503 |
| pY418 Src | Cell Signal Technology, 2101 | Rabbit | 1:2000 (WB) | AB_331697 |
| IKKα/β | Bethyl Laboratories, A301-827A | Rabbit | 2 μg (IP) | AB_1264322 |
| vWF | Agilent Technologies, A0082 | Rabbit | 1:500 (IF) | AB_2315602 |
| RIPK1 | BD Biosciences, 610459 | Mouse | 1:200 (IF) | AB_397832 |
| GST | In-house against full-length recombinant GST (Thermo Fisher Pierce Antibodies) | Rabbit | 1:2000 (WB) | N/A |
| FAK | In-house against amino acids 896–912 of human FAK (DSYNEGVKLQPQEISPP; GenScript) | Rabbit | 1:500 (IF) | N/A |
RRID research resource identifier, WB western blot, IF immunofluorescence, ChIP chromatin immunoprecipitation, IP immunoprecipitation
Cell Culture
Human aortic endothelial cells (HAoECs) and human umbilical vein endothelial cells (HUVECs) were purchased and grown in EC medium (VascuLife VEGF Endothelial Medium Kit; Lifeline Cell Technology, Carlsbad, CA, USA). HAoECs and HUVECs were used for up to 11 passages for experiments.
Mouse pulmonary ECs were isolated as previously described, and validated by staining for vascular endothelial cadherin and CD31, and their response to VEGF [19]. Mouse ECs were maintained in Dulbecco Modified Eagle Medium (DMEM; Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS; Omega Scientific, Tarzana, CA, USA). Mouse ECs were used up to passage 5.
Luciferase Assay
HUVECs were co-transfected with 0.2 μg Renilla luciferase and 0.5 μg pGL3 control (Promega, Madison, WI, USA) or NF-κB promoter luciferase construct (pNF-κB-Luc, Cat# 219078; Agilent Technologies, Santa Clara, CA, USA). After 24 h, cells were treated with or without PF-271 for 1 h followed by TNF-α for various times. Luciferase activity was measured using a dual luciferase assay kit (Promega, Madison, WI, USA).
Chromatin Immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed as previously described [20]. HAoECs were fixed with 1% formaldehyde for 10 min prior to quenching with 250 mmol/L glycine for 10 min. Cells were collected in lysis buffer (5 mmol/L PIPES pH 8.0, 85 mmol/L KCl, 0.5% NP-40, 1 × Complete Protease Inhibitor Cocktail (Roche, Basel, Switzerland)), and crude nuclear fraction was collected by centrifugation for 5 min at 3000g. Nuclear pellets were then lysed in RIPA buffer (150 mmol/L NaCl, 50 mmol/L Tris-HCl pH 8.0, 1 mmol/L EDTA, 1% sodium deoxycholate, 0.1% SDS, 1% Triton X-100, 1 × Complete Protease Inhibitor Cocktail). Chromatin was sheared to lengths ranging between 200 and 500 base pairs. Sonicated DNA from each sample were incubated at 4 °C overnight with 2 μg of NF-κB antibody with magnetic Protein A Dynabeads (Thermo Fisher Scientific, Waltham, MA, USA). Beads were washed 5 times in wash buffer (100 mmol/L Tris-HCl pH 7.5, 500 mmol/L LiCl, 1% NP-40, 1% sodium deoxycholate) and once with TE (10 mmol/L Tris-HCl pH 7.5, 0.1 mmol/L EDTA). After washing, beads were incubated with Proteinase K (Roche, Basel, Switzerland) to free DNA, and DNA was recovered using PCR Clean Up kit (Qiagen, Hilden, Germany). DNA was eluted in sterile water and stored at − 20 °C until needed for quantitative PCR.
Generation of Recombinant GST-IκBα (1–55) and FLAG-TNF-α Protein
GST-IκBα (1–55) fusion plasmid was created by polymerase chain reaction (PCR) amplification of the first 55 amino acids of human IκBα from pBabe-GFP-IKBalpha-wt (RRID:Addgene_15263, Addgene, Watertown, MA, USA). 6× His-FLAG-TNF-α fusion plasmid was created by PCR amplification of amino acids 85–233 of human TNF-α and cloning it into pET-28a(+)-6xHis (Cat# 69864; Millipore, Burlington, MA). GST-IκBα (1–55) or GST-FLAG-TNF-α protein expression was induced by treating Escherichia coli transformed with GST-IκBα (1–55) or GST-FLAG-TNF-α plasmids with 0.1 mmol/L isopropyl-β-D-thiogalactoside (IPTG) at 37 °C overnight and purified.
IκB Kinase Assay
In vitro IKK assay was performed as previously described [4]. IKK complex was immunoprecipitated using IKKβ antibody from stimulated HAoECs for 3 h at 4 °C. IKKβ immunoprecipitation beads were incubated with 200 μmol/L adenosine triphosphate (ATP) and 1 μg recombinant GST-IκBα (1–55) protein. IKK kinase activity was determined via immunoblot of phospho-serine32/36 of IκBα.
Tumor Necrosis Factor-α Receptor Complex-I Immunoprecipitation
Mouse pulmonary ECs (3 × 15 cm per group) were treated with FLAG-TNF-α (1 μg/mL), and TNFRC-I was immunoprecipitated with anti-FLAG agarose beads as previously described [21]. The cells were treated with the proteasome inhibitor MG-132 (10 μmol/L) immediately prior to FLAG-TNF-α stimulation to prevent degradation of proteins associated with TNFRC-I.
Immunoblot
Cells or hearts were lysed in modified RIPA buffer (pH 7.4) that contained 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES; 50 mmol/L), sodium chloride (150 mmol/L), Triton X-100 (1%), sodium deoxycholate (1%), SDS (0.1%), glycerol (10%), and protease inhibitors (Complete Protease Inhibitor Cocktail, Roche, Basel, Switzerland). Lysates were cleared by centrifugation, and supernatants were boiled in SDS loading buffer. Samples were separated by SDS polyacrylamide gel electrophoresis (PAGE), transferred to a polyvinylidene difluoride (PVDF; Millipore, Burlington, MA, USA) membrane, and immunoblot performed with various antibodies. Enhanced chemiluminescence was performed and blots were visualized (Chemidoc; BioRad, Hercules, CA, USA). All immunoblots were repeated at least 3 times.
Immunostaining
HAoECs on fibronectin-coated coverslips were fixed with paraformaldehyde (PFA) and permeabilized with 0.3% Triton X-100. Frozen tissue sections were fixed with cold acetone for 15 min. Samples were blocked (3% bovine serum albumin and 1% goat serum) for 1 h at room temperature and incubated with primary antibody overnight at 4 °C. Samples were incubated with secondary antibody (1:1000) for 1 h at room temperature. Species-specific immunoglobulin G or secondary antibodies were used as negative control. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich, St. Louis, MO, USA). Slides were mounted (Fluoromount-G; SouthernBiotech, Birmingham, AL, USA), and images were acquired with a confocal microscope (Nikon A1R; Nikon, Minato City, Tokyo, Japan). Images were processed (Photoshop CS5, RRID:SCR_014199; Adobe, San Jose, CA, USA). Quantification of fluorescence intensity was analyzed with (ImageJ, RRID:SCR_003070; National Institutes of Health, Bethesda, MD, USA). At least 3 section images were acquired from each sample.
Animal Experiments
Animal experiments were approved by the University of South Alabama Institutional Animal Care and Use Committee and performed in accordance with the committee guidelines. For EC-specific FAK kinase-dead (KD) and FAK wild-type (WT) experiments, FAK-WT/KD mice were crossed with FAK flox/flox SCL-CreERT mice to generate FAK flox/WT SCL-CreERT and FAK flox/KD SCL-CreERT mice [22, 23]. EC-specific FAK flox/WT and flox/KD mice (n = 8) were treated with tamoxifen (2 mg in corn oil) every other day for 2 weeks to generate EC-specific FAK-WT and FAK-KD mice. For FAK inhibitor experiments, mice were treated twice daily with vehicle (30% [2-hydroxypropyl]-β-cyclodextrin/3%dextrose; Sigma-Aldrich, St. Louis, MO, USA) or PF-271 (35 mg/kg) by oral gavage for 2 days prior to TNF-α injection.
C57BL/6 and EC-specific FAK-WT and FAK-KD mice were used for TNF-α tail vein injections. Vehicle-treated, PF-271-treated, EC-specific FAK-WT and FAK-KD mice were injected with 100 μL PBS or mouse TNF-α (0.02 mg/kg) via tail vein. After 0.5 h, aortas were isolated for immunostaining and hearts for immunoblot to monitor PF-271 or tamoxifen efficacy.
Statistical Analysis
Data sets underwent Shapiro-Wilk test for normality, and statistical significance between experimental groups was determined with student t test or 1-way analysis of variance (ANOVA) with Sidak multiple comparisons test (GraphPad Prism, RRID:SCR_002798, GraphPad, San Diego, CA, USA). Power analyses were performed to determine sample size for 1-way ANOVA.
RESULTS
Pharmacological FAK Inhibition Decreases Sustained NF-κB Activation and Proinflammatory Gene Expression in Endothelial Cells
FAK activity promotes proinflammatory gene expression through MAPKs, but we found that inhibition of MAPKs alone was not sufficient to completely block proinflammatory cytokine signaling [5]. As long-term NF-κB activation is critical for proinflammatory gene expression [16–18, 24, 25], and there were implications that FAK may regulate NF-κB activity in different types of cells [4, 12], we investigated the temporal correlation of FAK activity with sustained NF-κB activation in ECs. In human aortic ECs (HAoECs), a pharmacological FAK inhibitor (PF-271) decreased active phospho-serine (pS)536 NF-κB at 6 h, but not 0.5 h, after TNF-α stimulation (Fig. 1a). Although elevated pS536 NF-κB was observed after 12-h TNF-α stimulation, PF-271 began to abolish pS536 NF-κB after 3 h (Supplementary Fig. 1a). PF-271 also decreased pS311 and pS529 NF-κB (Fig. 1a), which are important for NF-κB transcriptional activation [15]. PF-271-treated cells showed significantly less NF-κB promoter luciferase activity compared with control (Fig. 1b), which was associated with decreased vascular cell adhesion molecule-1 (VCAM-1) expression (Fig. 1a). These results are consistent with the observation that sustained NF-κB nuclear localization and pS536 NF-κB staining were decreased in PF-271-treated HAoECs compared with TNF-α alone (Fig. 1c). NF-κB chromatin immunoprecipitation (ChIP) revealed that TNF-α increased NF-κB binding to the IκBα promoter (Supplementary Fig. 1b). Interestingly, PF-271 reduced the amount of NF-κB associated to the IκBα promoter (Supplementary Fig. 1b), suggesting that FAK activity is important for promoting NF-κB nuclear translocation and transcriptional activity in ECs. As Src family kinases often form a complex with FAK to promote activation of downstream target proteins [26], we treated HAoECs with a Src inhibitor (Dasatinib) to evaluate the role of Src on sustained NF-κB activation. Even though Dasatinib completely blocked active Src phosphorylation at tyrosine 418 (pY418 Src), it did not reduce sustained phosphorylation of pS536 NF-κB (Supplementary Fig. 1c). These data further support that FAK, but not Src, is important for TNF-α-mediated sustained NF-κB activation and proinflammatory gene expression in HAoECs.
Fig. 1.
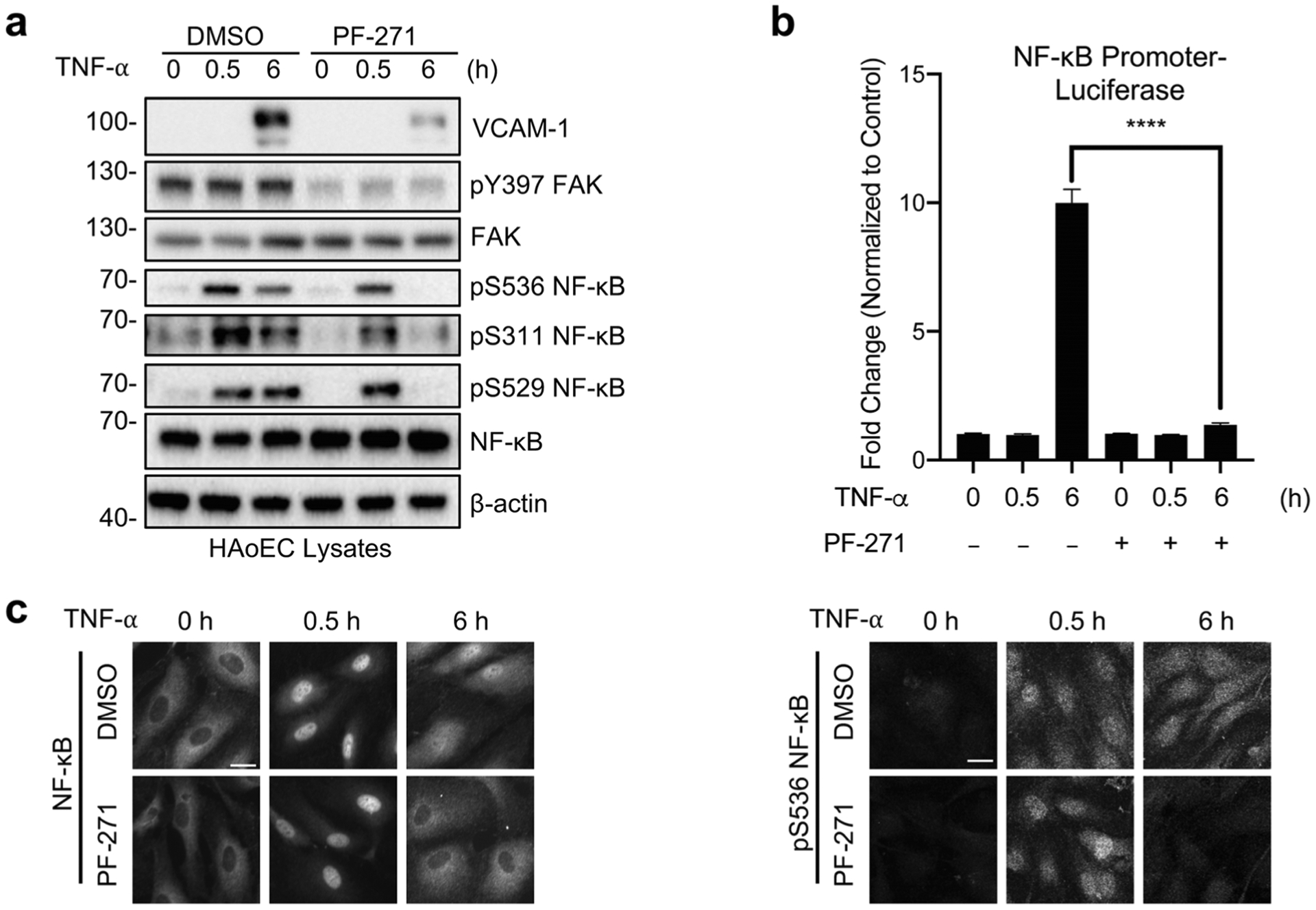
FAK inhibition decreases TNF-α-induced sustained NF-κB activation in endothelial cells. a–c HAoECs were treated for 1 h with DMSO or PF-271 (2.5 μmol/L) prior to TNF-α (10 ng/mL) stimulation for the indicated times. a Western blot analysis of VCAM-1, pY397 FAK, FAK, pS536 NF-κB, pS311 NF-κB, pS529 NF-κB, NF-κB, and β-actin (n = 3). b HAoECs were transfected with NF-κB promoter luciferase construct. Results are expressed as fold change over unstimulated control (n = 3). c Localization of NF-κB (rabbit) and expression of pS536 NF-κB (rabbit) were evaluated by immunostaining (n = 3). Scale bar, 20 μm. ****P < 0.0001.
FAK Inhibition Decreases Sustained IKK Activation
As subcellular localization of NF-κB is tightly regulated by IκB proteins, we evaluated whether PF-271 altered IκBα stability. TNF-α rapidly led to loss of IκBα protein, which remained low even after 12 h (Fig. 2a, Supplementary Fig. 1a). On the other hand, PF-271-treated HAoECs showed increased IκBα starting at 3 h, which persisted even after 12 h TNF-α stimulation (Fig. 2a, Supplementary Fig. 1a). However, PF-271 did not block TNF-α-induced loss of IκBα within 1 h (Fig. 2a, Supplementary Fig. 1a), suggesting that FAK inhibition increases IκBα protein stability at later times, which increases NF-κB cytoplasmic localization and blocks sustained activation of NF-κB.
Fig. 2.
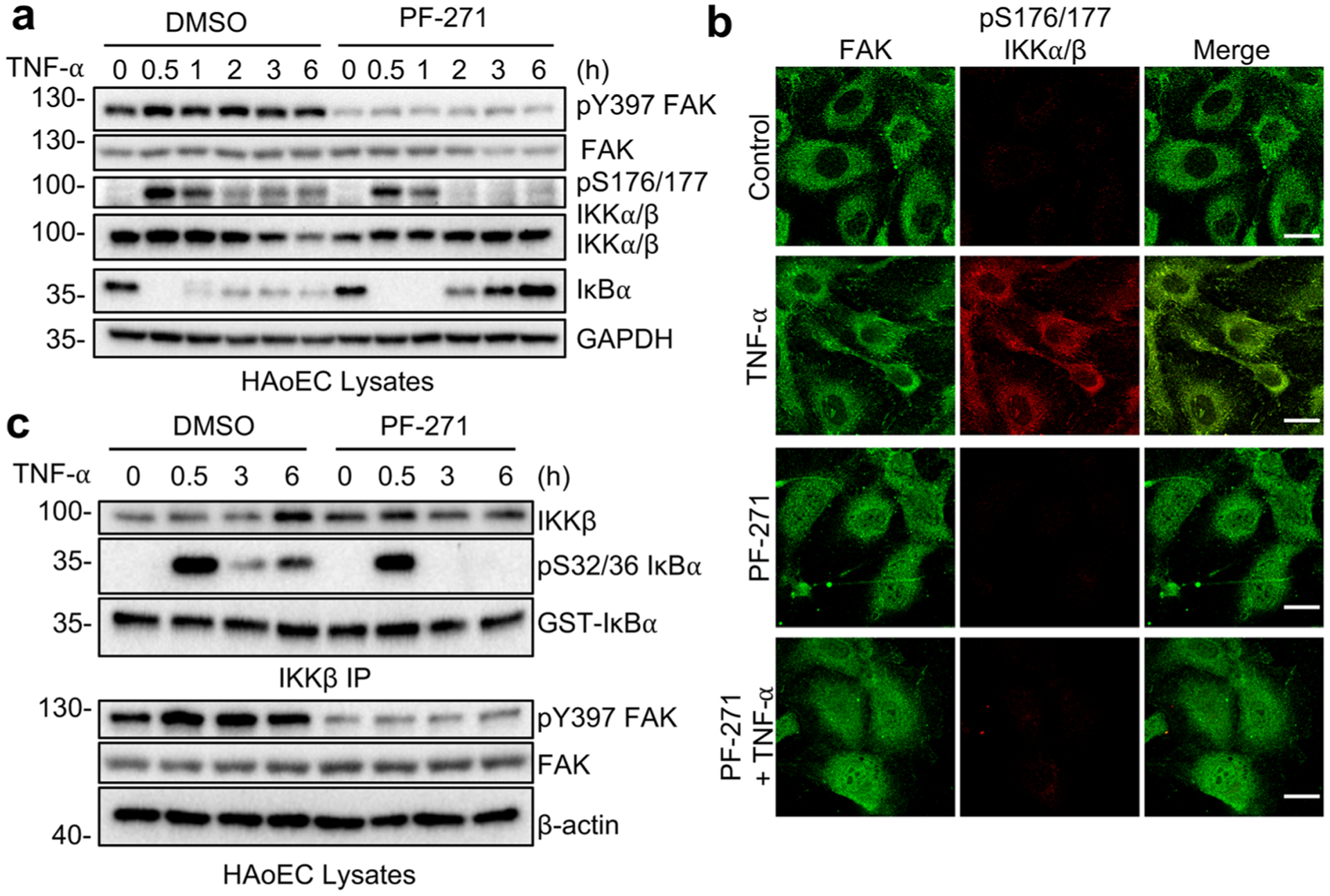
FAK inhibition decreases IKK activation and activity in endothelial cells. a–c HAoECs were treated for 1 h with DMSO or PF-271 (2.5 μmol/L) prior to TNF-α (10 ng/mL) stimulation for the indicated times. a Western blot analysis of pY397 FAK, FAK, pS176/177 IKKα/β, IKKα/β, IκBα, and GAPDH (n = 3). b FAK localization (green; mouse) and active pS176/177 IKKα/β (red; rabbit) were evaluated by immunostaining (n = 3). Scale bar, 20 μm. c IKKβ was immunoprecipitated (IP) from stimulated HAoECs and in vitro IKK assay was performed. Western blot analysis of IKKβ, pS32/36 IκBα, and GST-IκBα in IKK kinase assay and pY397 FAK, FAK, and β-actin of inputs (n = 3).
As the IKK complex is important in phosphorylating IκBα protein and regulating its stability [27], we tested whether FAK inhibition altered the activation status of the IKK complex. TNF-α rapidly increased active pS176/177 IKKα/β levels, and the increased levels persisted after 6 h and were associated with decreased IκBα protein (Fig. 2a). Although PF-271 did not completely block early activation of the IKK complex, it completely decreased pS176/177 IKKα/β and increased IκBα 2 h after TNF-α stimulation (Fig. 2a). Interestingly, TNF-α stimulation significantly increased staining of active pS176/177 IKKα/β levels in HAoECs (Fig. 2b).
To determine whether intrinsic IKK activity was decreased by PF-271, we performed an in vitro IKK assay using recombinant GST-IκBα containing the IKKα/β phosphorylation sites S32 and S36 [28]. No difference in phosphorylation of GST-IκBα (pS32/36 IκBα) was observed 0.5 h after TNF-α stimulation in cells treated with vehicle or PF-271 (Fig. 2c). Although IKK remained active out to 6 h in vehicle-treated cells, PF-271 blocked IKK-mediated phosphorylation of GST-IκBα at 3 h (Fig. 2c). The phosphorylation levels of endogenous IκBα were consistent with the results seen from in vitro IKK assays (Supplementary Fig. 1d). These data demonstrated that FAK activity controls sustained activation of the IKK complex leading to prolonged NF-κB activation.
FAK Inhibition Disrupts RIPK1 and IKK Recruitment to the TNF-α Receptor
To identify the molecular link between FAK activity and IKK/NF-κB activation upon TNF-α stimulation, we investigated whether FAK inhibition altered formation of TNFRC-I, which would lead to failure of IKK activation in ECs. In order to observe only active TNFRC-I, we used recombinant FLAG-TNF-α immunoprecipitation (IP) instead of using TNFR1 antibody IP, which could pull down both active and inactive TNFR1. We first attempted to use FLAG-TNF-α IP using HAoECs, but we were unable to obtain IP complexes (data not shown). Since mouse cells are more commonly used when investigating active TNFRC-I using FLAG-TNF-α, we tested this possibility in mouse pulmonary ECs (mECs). First, we checked if FAK inhibition blocked sustained activation IKK/NF-κB in response to TNF-α stimulation in mECs. PF-271 blocked active pS176/177 IKKα/β and pS536 NF-κB at 6 h and had increased IκBα compared to TNF-α alone (Fig. 3a).
Fig. 3.
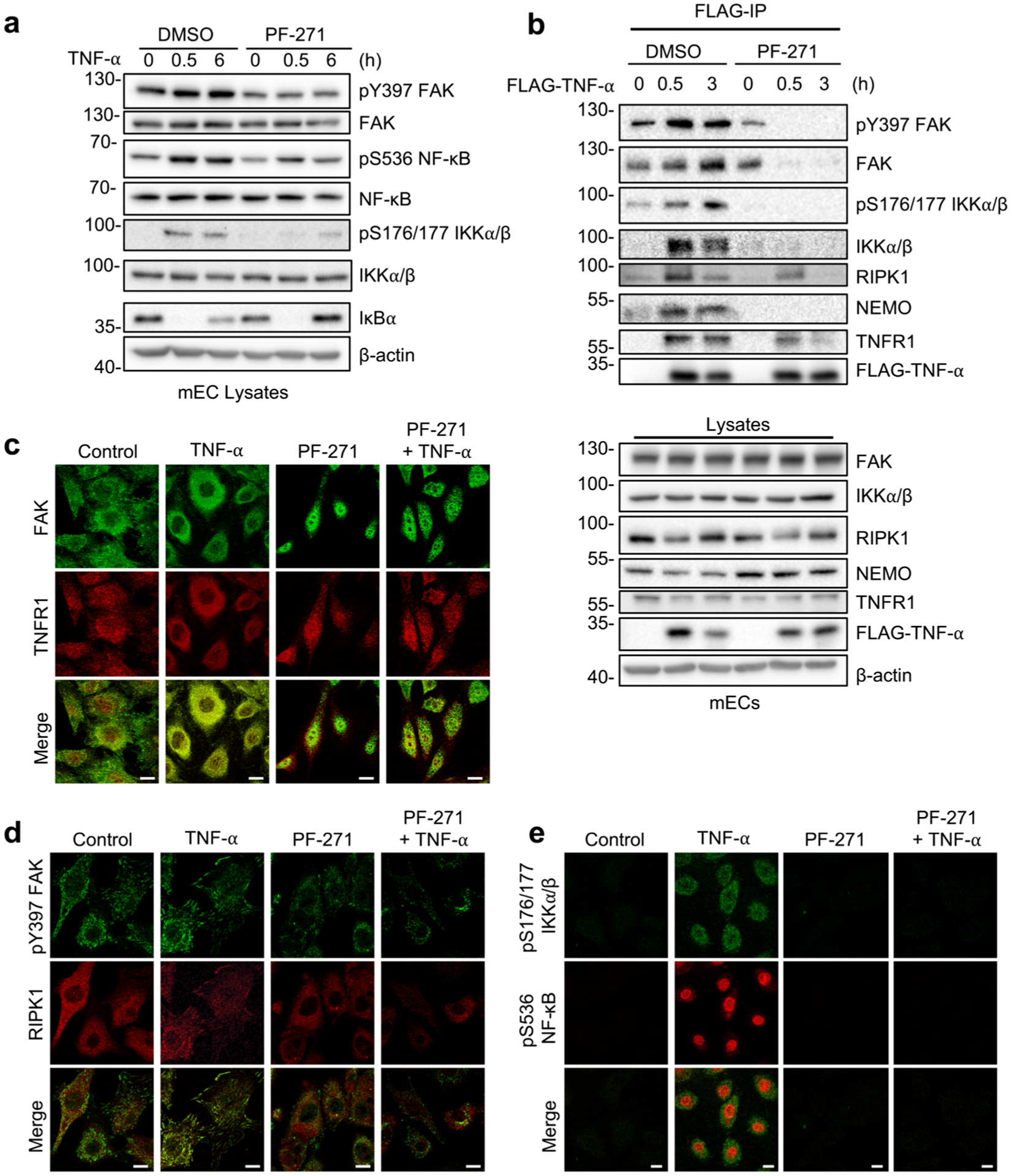
FAK inhibition disrupts RIPK1 and IKK recruitment to the TNF-α receptor. a, b mECs were treated for 1 h with DMSO or PF-271 (2.5 μmol/L) prior to TNF-α (10 ng/mL) stimulation for indicated times. a Western blot analysis of pY397 FAK, FAK, pS536 NF-κB, NF-κB, pS176/177 IKKα/β, IKKα/β, IκBα, and β-actin (n = 3). b Flag immunoprecipitation (IP) of FLAG-TNF-α stimulated mECs was performed. Western blot analysis for pY397 FAK, FAK, pS176/177 IKKα/β, IKKα/β, RIPK1, NEMO, TNFR1, FLAG-TNF-α, and β-actin in either FLAG-IP or lysates (n = 3). c–e mECs were treated for 1 h with DMSO or PF-271 (2.5 μmol/L) prior to TNF-α (10 ng/mL) stimulation for 3 h. c Immunostaining for FAK (green; rabbit) and TNFR1 (red; mouse) (n = 3). d Immunostaining for pY397 FAK (green; rabbit) and RIPK1 (red; mouse) (n = 3). e Immunostaining for pS176/177 IKKα/β (green; mouse) and pS536 NF-κB (red; rabbit) (n = 3). Scale bar, 20 μm.
Active TNFRC-I immunoprecipitated using FLAG-TNF-α contains active pY397 FAK, RIPK1, active pS176/177 IKKα/β, and NEMO (Fig. 3b). Interestingly, PF-271 reduced RIPK1 recruitment to TNFR1 at 0.5 h, and completely blocked RIPK1 association after 3 h FLAG-TNF-α (Fig. 3b). Several TNFRC-I components, such as RIPK1, undergo M1- and/or K63-linked ubiquitination to facilitate recruitment of downstream signaling complexes including IKK [29, 30]. FAK inhibition blocked recruitment of the IKK complex (IKKα/β, NEMO) to TNFR1 (Fig. 3b), which is consistent with previous reports that RIPK1 is important for recruitment of the IKK complex to TNFRC-I [31]. Interestingly, PF-271 decreased the amount of TNFR1 immunoprecipitated with FLAG-TNF-α despite the equal expression in lysates (Fig. 3b), which could explain the reduction in both RIPK1 and IKK complex with FLAG-TNF-α. To better understand the role of FAK in TNFRC-I formation, we performed immunofluorescence of TNFR1 and RIPK1 on mEC. While FAK and TNFR1 showed very little co-localization in unstimulated mECs, TNF-α increased both FAK and TNFR1 co-localization at the cell periphery (Fig. 3c). However, PF-271 increased FAK nuclear localization, and this reduced TNFR1 redistribution to the cell membrane (Fig. 3c). Immunostaining also revealed increased active pY397 FAK near the cell periphery and focal adhesions following TNF-α stimulation (Fig. 3d). TNF-α induced redistribution of RIPK1 from the cytoplasm to the cell periphery or focal adhesions, which colocalized with active pY397 FAK (Fig. 3d). PF-271 reduced both active pY397 FAK staining and blocked RIPK1 localization to the cell periphery (Fig. 3d). Reduced co-localization of FAK with both RIPK1 and TNFR1 in PF-271-treated mECs was associated with decreased staining of both active pS176/177 IKKα/β and pS536 NF-κB (Fig. 3e). A similar pattern of increased co-localization of FAK with either TNFR1 or RIPK1 following TNF-α stimulation of HAoECs was also observed via immunostaining (Supplementary Fig. 2). PF-271 increased FAK nuclear localization, and reduced redistribution of TNFR1 and RIPK1 (Supplementary Fig. 2). These results suggest that FAK cytoplasmic localization and activity is required for TNFR1 redistribution and formation of TNFRC-I in ECs.
TNF-α Activates and Redistributes FAK from the Nucleus to Cytoplasm In vivo
To evaluate FAK function in TNF-α-mediated IKK-NF-κB activation within ECs in vivo, mice were treated with either vehicle or PF-271 and challenged with TNF-α via tail vein injection (Fig. 4a). Active pY397 FAK was increased 0.5 h after TNF-α stimulation in heart lysates and PF-271 efficacy was verified by pY397 FAK immunoblot (Fig. 4b). Increased FAK activation was associated with increased pS536 NF-κB and pS176/177 IKKα/β, but decreased IκBα (Fig. 4b). In contrast to observations in cultured HAoECs or mECs (Figs. 1a and 3a), FAK inhibition in vivo completely blocked TNF-α-induced activation NF-κB and IKKα/β at 0.5 h (Fig. 4b). This difference may be associated with lower levels of active pY397 FAK at 0 h in vivo than in cultured ECs.
Fig. 4.
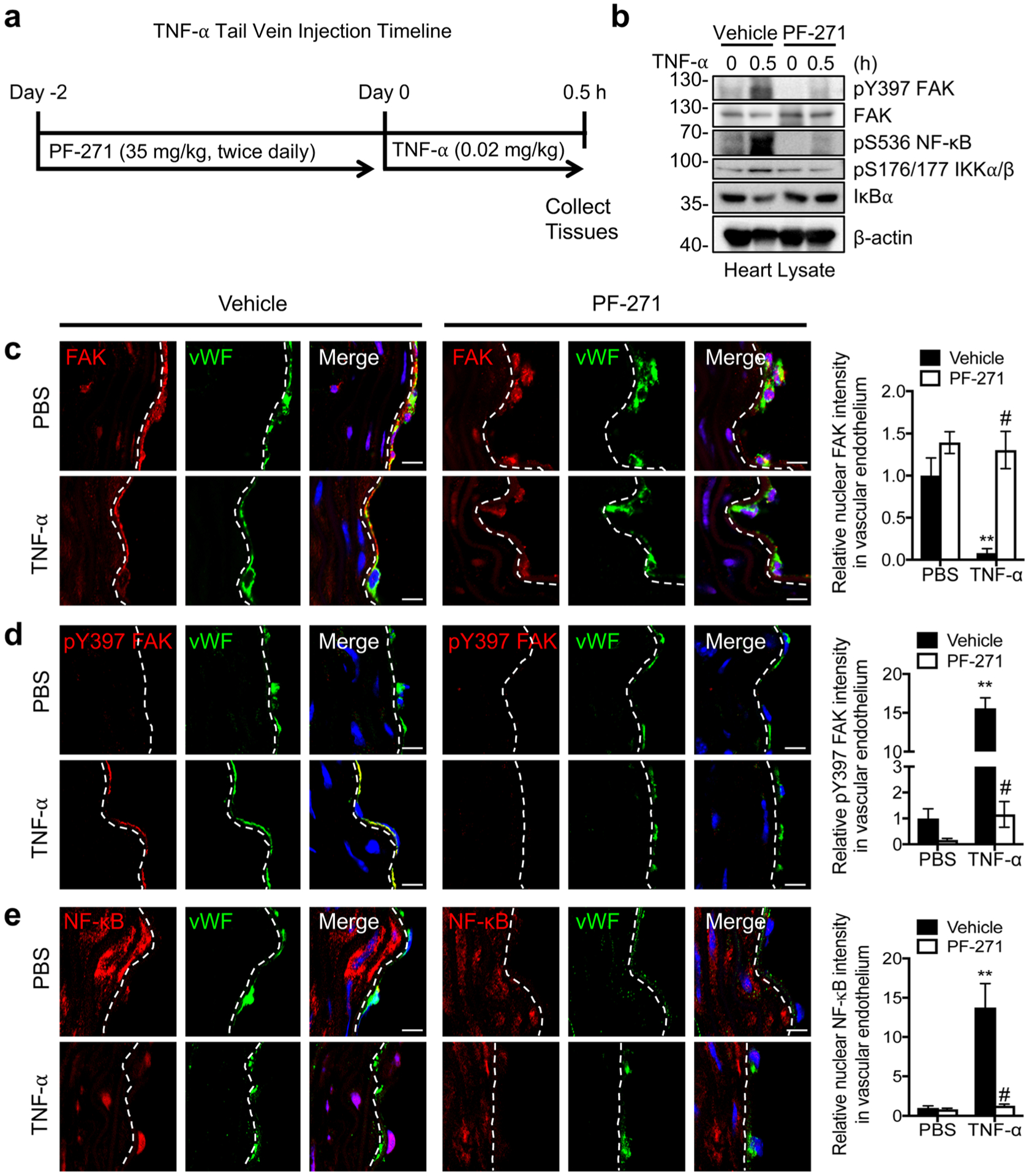
Immunostaining of mouse aortas showing that nuclear-localized FAK decreases TNF-α-induced NF-κB activation in mice. a Timeline for when C57BL/6 mice were treated with vehicle or PF-271 (35 mg/kg) before injection with PBS or mouse TNF-α (0.02 mg/kg) for 0.5 h. b Western blot analysis of heart lysates for pY397 FAK, FAK, pS536 NF-κB, pS176/177 IKKα/β, IκBα, and β-actin as loading control (n=4). Aorta sections were stained for FAK (red; mouse (c), pY397 FAK (red; rabbit) (d), or NF-κB (red; rabbit) (e). ECs were stained with vWF (green; rabbit or mouse) and nuclei with DAPI (blue). Relative fluorescence intensity in vWF-positive ECs was reported as mean ± SD (n = 4). Dashed lines, boundary between media and EC based on vWF staining. Scale bars, 10 μm. **P < 0.01 vs vehicle PBS; #P < 0.01 vs TNF-α.
Analysis of FAK activation in ECs in situ showed that FAK was largely inactive and primarily localized to the nucleus of ECs in unstimulated controls (Fig. 4c, d). Stimulation with TNF-α rapidly activated FAK and redistributed FAK into the cytoplasm (Fig. 4c, d). TNF-α also induced nuclear NF-κB localization and increased active pS536 NF-κB levels (Fig. 4d, Supplementary Fig. 3a). In addition, TNF-α increased pS176/17 IKKα/β (Supplementary Fig. 3b), suggesting that TNFRC-I is formed and activated following TNF-α stimulation in vivo. Conversely, mice pretreated with PF-271 failed to show any differences in FAK or NF-κB signaling following TNF-α injection. TNF-α failed to induce FAK activation or cytoplasmic localization in mice treated with PF-271 (Fig. 4c, d). Forced FAK nuclear localization was associated with cytoplasmic retained NF-κB and reduction of both active pS536 NF-κB and pS176/177 IKKα/β (Fig. 4e, Supplementary Fig. 3). The results from in vivo TNF-α challenge experiment are consistent with those of in vitro culture ECs, at least in that FAK cytoplasmic localization and activity is required for TNFRC-I formation and activation of IKKα/β and NF-κB.
EC-Specific FAK Activity is Required for TNF-α-Induced Activation of NF-κB In vivo
As FAK inhibitors would affect all cell types in the vasculature, we evaluated the effect of EC-specific FAK activity on TNF-α-induced IKK/NF-κB activation. To do this, we used mice that express a catalytically inactive form of FAK known as FAK kinase-dead (KD). As homozygous FAK-KD is embryonic lethal [23], we generated EC-specific FAK-WT and FAK-KD by crossing EC-specific SCL-Cre FAK flox/flox mice with FAK-WT/KD mice (Fig. 5a) [22, 23]. This allows us to generate EC-specific catalytically inactive FAK-KD in adult mice by deleting endogenous flox allele following tamoxifen injections (Fig. 5a). We then injected EC-specific FAK-WT and FAK-KD mice with TNF-α via tail vein. FAK-KD mice showed decreased levels of pY397 FAK, pS536 NF-κB, and pS176/177 IKKα/β in heart lysates after 0.5 h TNF-α compared with FAK-WT (Fig. 5b). In addition, FAK-KD mice showed higher levels of IκBα compared to FAKWT, suggesting that FAK activity in ECs is important for TNF-α-induced IKK/NF-κB activation (Fig. 5b). Immunostaining showed that FAK was localized to the nuclei of ECs with very low levels of active pY397 FAK levels in both FAK-WT and FAK-KD mice in unstimulated controls (Fig. 5c). TNF-α treatment redistributed and activated FAK in cytoplasm, resulting in NF-κB activation seen by NF-κB nuclear localization and active pS536 NF-κB immunostaining in FAK-WT mice (Fig. 5c–e, Supplementary Fig. 4). However, FAK remained inactive and in the nucleus following TNF-α stimulation in FAK-KD mice (Fig. 5c, d). Nuclear FAK localization in FAK-KD ECs was associated with decreased NF-κB nuclear localization and pS536 NF-κB levels (Fig. 5e, Supplementary Fig. 4). These results align with the observation from pharmacological FAK inhibitor experiments in mice. Therefore, FAK localization and FAK activity in ECs are important in promoting TNF-α-induced activation of NF-κB in vivo.
Fig. 5.
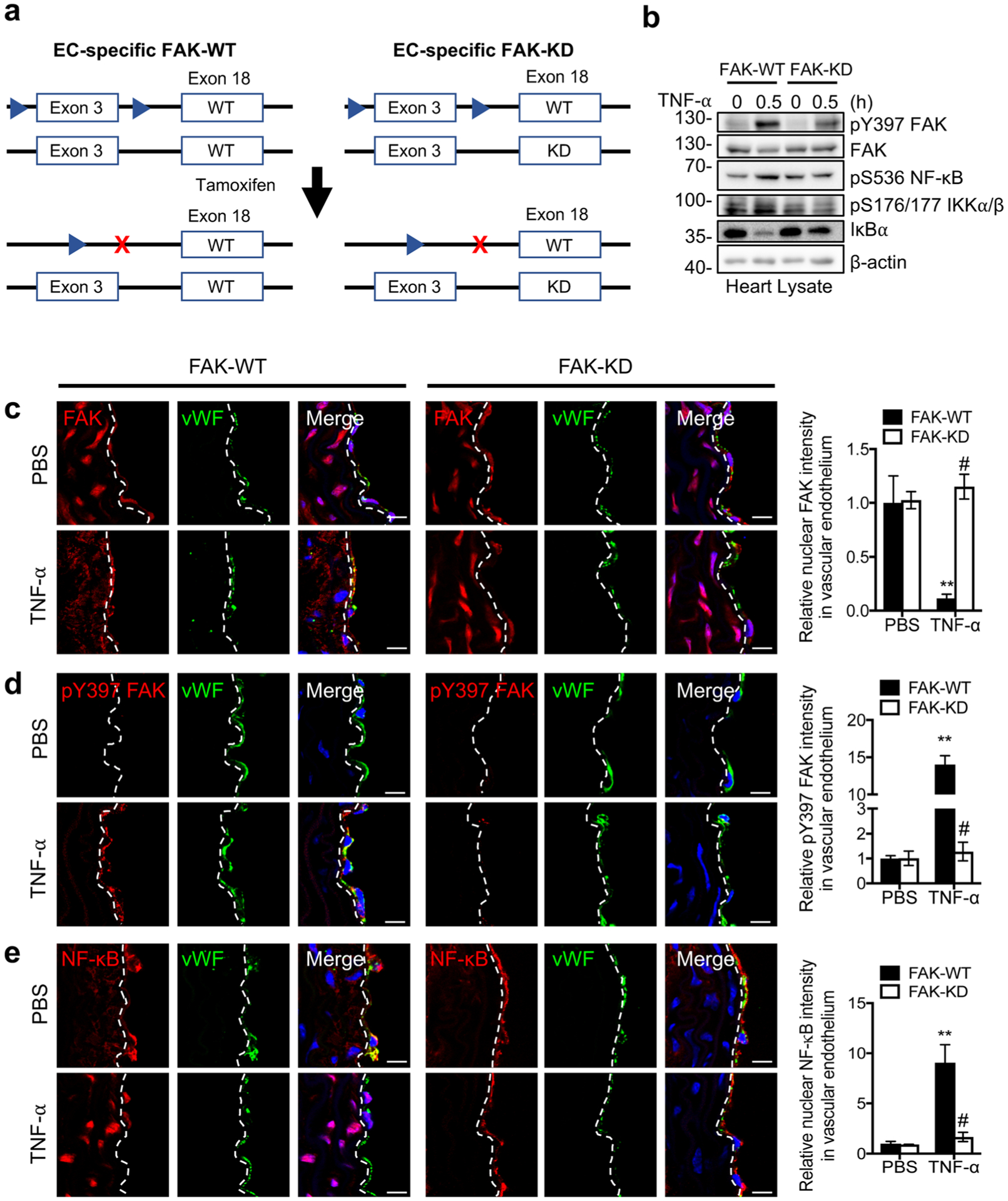
Immunostaining of mouse aortas showing that EC-specific FAK inhibition decreases TNF-α-induced NF-κB activation in mice. a Schematic of the deletion of the FAK flox allele and generation of EC-specific FAK-WT and FAK-KD mice. EC-specific FAK-WT and FAK-KD mice were injected with PBS or mouse TNF-α (0.02 mg/kg) for 0.5 h. b Western blot analysis of heart lysates for pY397 FAK, FAK, pS536 NF-κB, pS176/177 IKKα/β, IκBα, and β-actin as loading control (n=4). Aortas sections were stained for FAK (red; mouse) (c), pY397 FAK (red; rabbit) (d), or NF-κB (red; rabbit) (e). ECs were stained with vWF (green; rabbit or mouse) and nuclei with DAPI (blue). Relative fluorescence intensity in vWF-positive ECs was reported as mean ± SD (n = 4). Dashed lines, boundary between media and EC on the basis of vWF staining. Scale bars, 10 μm. **P < 0.01 vs FAK-WT PBS; #P < 0.01 vs FAK-WT TNF-α.
DISCUSSION
Sustained activation of the proinflammatory transcription factor NF-κB is thought to play a major role in several chronic inflammatory diseases including RA, atherosclerosis, and cancer. In the present study, we have demonstrated that FAK activity is important for sustained activation of NF-κB in ECs following TNF-α stimulation both in vivo and in vitro. While a majority of studies have used FLAG-TNF-α to investigate TNFRC-I formation in mouse embryonic fibroblasts (MEFs) [29, 32, 33], to our knowledge, we are the first to do so in ECs (Fig. 3b). In addition to using mouse pulmonary ECs to investigate TNFRC-I component recruitment, we investigated TNFRC-I formation in human ECs and observed that human ECs responded more rapidly to TNF-α stimulation, possibly due to increased amplitude or higher expression levels of TNFR signaling components compared to mECs. Mechanistically, TNF-α stimulation increased active pY397 FAK recruitment to TNFR1. However, pharmacological FAK inhibition blocked FAK recruitment to TNFR1, resulting in loss of NF-κB activation.
Intriguingly, we observed that FAK was primarily inactive and localized to the nuclei of unstimulated ECs in vivo. Additionally, our previous study demonstrated that FAK also primarily localized to the nuclei of healthy smooth muscle cells in vivo [34]. While these suggest that dominant nuclear FAK localization may be specific to cells of the vessel wall, FAK localization in other cell types in vivo still needs to be investigated. TNF-α stimulation promotes FAK activation and cytoplasmic localization where it associates with TNFR1, to recruit RIPK1 and the IKK complex. However, catalytic FAK inhibition enhances FAK retention in the nucleus, thus preventing FAK recruitment of RIPK1 to TNFR1, which blocks IKK/NF-κB activation and inflammatory gene expression (Fig. 6). Therefore, this model suggests that FAK activity may be important for maintaining sustained activation of TNFRC-I, and FAK inhibitors may have potential in treating chronic inflammatory diseases.
Fig. 6.
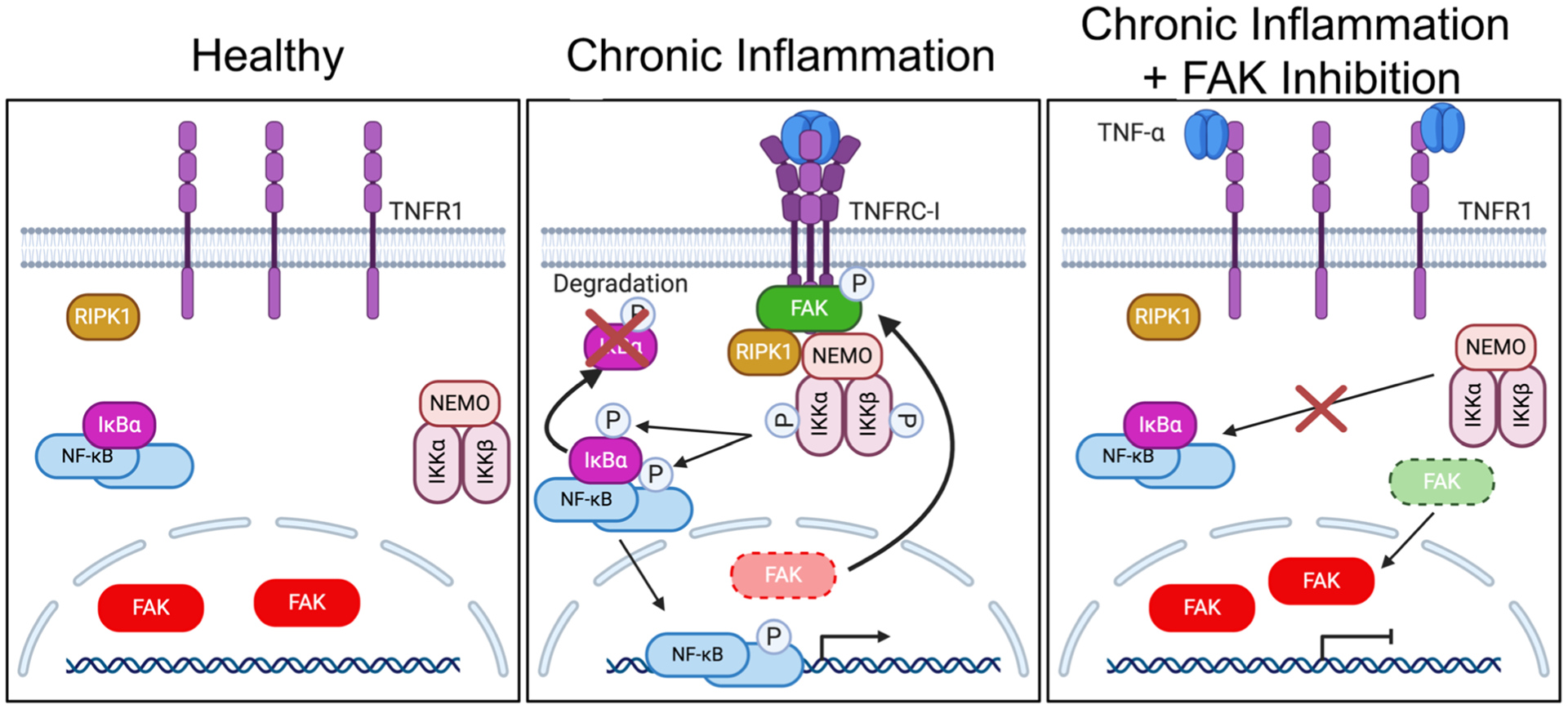
FAK cytoplasmic localization and activity are important for TNFRC-I formation and IKK-NF-κB activation. Left: Under healthy conditions, FAK is mainly localized to the nucleus of ECs. This increased nuclear FAK is associated with inactive NF-κB sequestered by IκBα proteins in the cytoplasm. Middle: Under chronic inflammation, such as continued TNF-α stimulation, trimerization of TNFR1 leads to FAK activation and redistribution of FAK from the nucleus to the cytoplasm. Active FAK increases recruitment of RIPK1 and the IKK complex to TNFRC-I. After activation, the IKK complex promotes IκBα degradation and activates NF-κB, which enters the nucleus and promotes inflammatory gene transcription. Right: FAK inhibition decreases recruitment of RIPK1 and the IKK complex to TNFR1, thus blocking formation of TNFRC-1 and decreasing sustained activation of the IKK complex and NF-κB.
Our present study also demonstrated that there appears to be some discrepancies in FAK localization within ECs in vivo and in vitro. In vitro, FAK showed a mix of both nuclear and cytoplasmic localization in unstimulated ECs, which is in contrast to the primarily nuclear localization of FAK observed in vivo. These discrepancies may account for our observations that TNF-α was still effective at promoting early activation of the IKK/NF-κB pathway in vitro but not in vivo. However, pharmacological FAK inhibition eventually promoted enhanced FAK nuclear localization within ECs in vitro which then resulted in decreased TNFRC-I signaling. It is possible that several environmental cues may contribute to the elevated cytoplasmic FAK observed in vitro. ECs are grown in EC growth medium that contains numerous growth factors and are cultured on plastic dishes which is a high stiffness environment. Both growth factors and high stiffness have been shown to promote FAK activation and cell proliferation in numerous cell types. However, ECs in vivo are not typically stimulated with growth factors and are usually exposed to low laminar flow, which is known to inhibit FAK activation. As such, FAK is more predisposed to be located within the nuclei of ECs in vivo compared to in vitro. Further studies to better mimic physiological EC environment are needed to fully understand the role of FAK in inflammatory signaling.
ECs play a fundamental role in regulating the inflammatory environment of the surrounding tissue and reducing EC inflammatory signaling in chronic inflammatory diseases could prove beneficial. While it is known that sustained NF-κB activation plays an important role in chronic inflammatory diseases, most of the focus on therapies have been designed to target proinflammatory cytokines such as TNF-α and IL-1β that promote NF-κB activation. As such, TNF-α and IL-1β antibody therapies have been approved for treating RA; however, their use in other chronic inflammatory diseases such as atherosclerosis and cancer has not shown enough benefit to outweigh the risks [3]. The major drawbacks to TNF-α and IL-1β antibody therapies include increased risk of fatal infections and sepsis. Thus, better understanding of proinflammatory signaling pathways is needed to find more promising therapeutic targets.
Recently, it was found that high levels of active cytoplasmic FAK within tumor ECs of prechemotherapy breast cancer patients were associated with resistance to neoadjuvant chemotherapy and decreased 5-year survival [39]. FAK expression within tumor ECs was also shown to FAK and NF-κB Activation be important for tumor resistance to DNA-damaging therapies through increased activation of EC NF-κB [40]. However, the role of FAK inhibitors on the inflammatory environment of tumors is not well understood. While FAK inhibitors are currently being tested as anti-cancer agents in clinical settings [35–38], there are currently no clinical studies that have investigated the effects of FAK inhibitors on other chronic inflammatory diseases such as vascular inflammation and atherosclerosis. Here, we have demonstrated that FAK inhibition is effective at reducing EC inflammation by blocking formation of TNFRC-I, thus preventing activation of the IKK/NF-κB pathway. Therefore, the potential of FAK inhibitors to treat chronic inflammatory diseases should be further investigated.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Elly Trepman for critical reading of the manuscript, and Drs. Mary I. Townsley and John V. Marymont for editorial support.
FUNDING
This work was supported by American Heart Association grants 12SDG10970000 and 16GRNT30960007 to SL, National Institutes of Health grants R01CA190688 to EA and R01HL136432 to SL, and 2019 College of Medicine Intramural grant from University of South Alabama to SL. The confocal microscope was supported by National Institutes of Health grant S10RR027535.
Abbreviations
- ECs
Endothelial cells
- FAK
Focal adhesion kinase
- HAoEC
Human aortic endothelial cell
- HUVEC
Human umbilical vein endothelial cell
- IκBα
Inhibitor of NF-κBα
- IKK
IκB kinase
- KD
Kinase-dead
- NEMO
Nuclear factor-κB essential modulator (IKKγ)
- NF-κB
Nuclear factor-κB
- pS536
Phospho-serine 536 NF-κB
- pY397
Autophosphorylation at tyrosine 397 of FAK
- RIPK1
Receptor-interacting serine/threonine-protein kinase 1
- TNF-α
Tumor necrosis factor-α
- TNFR1
Tumor necrosis factor-α receptor 1
- TNFRC-I
TNF-α receptor complex-I
- VCAM-1
Vascular cell adhesion molecule-1
- WT
Wild-type
Footnotes
Conflict of Interest. The authors declare that they have no conflicts of interest.
SUPPLEMENTARY INFORMATION
The online version contains supplementary material available at https://doi.org/10.1007/s10753-020-01408-5.
REFERENCES
- 1.Ursini F, Leporini C, Bene F, D’Angelo S, Mauro D, Russo E, De Sarro G, Olivieri I, Pitzalis C, Lewis M, and Grembiale RD. 2017. Anti-TNF-alpha agents and endothelial function in rheumatoid arthritis: a systematic review and meta-analysis. Scientific Reports 7 (1): 5346. 10.1038/s41598-017-05759-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Angel K, Provan SA, Fagerhol MK, Mowinckel P, Kvien TK, and Atar D. 2012. Effect of 1-year anti-TNF-alpha therapy on aortic stiffness, carotid atherosclerosis, and calprotectin in inflammatory arthropathies: a controlled study. American Journal of Hypertension 25 (6): 644–650. 10.1038/ajh.2012.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baylis RA, Gomez D, Mallat Z, Pasterkamp G, and Owens GK. 2017. The CANTOS Trial: one important step for clinical cardiology but a giant leap for vascular biology. Arteriosclerosis, Thrombosis, and Vascular Biology 37 (11): e174–e177. 10.1161/ATVBAHA.117.310097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yurdagul A Jr., Sulzmaier FJ, Chen XL, Pattillo CB, Schlaepfer DD, and Orr AW. 2016. Oxidized LDL induces FAK-dependent RSK signaling to drive NF-kappaB activation and VCAM-1 expression. Journal of Cell Science 129 (8): 1580–1591. 10.1242/jcs.182097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murphy JM, Jeong K, Rodriguez YAR, Kim JH, Ahn EE, and Lim SS. 2019. FAK and Pyk2 activity promote TNF-alpha and IL-1beta-mediated pro-inflammatory gene expression and vascular inflammation. Scientific Reports 9 (1): 7617. 10.1038/s41598-019-44098-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamaura T, Kasaoka T, Iijima N, Kimura M, and Hatakeyama S. 2019. Evaluation of therapeutic effects of FAK inhibition in murine models of atherosclerosis. BMC Research Notes 12 (1): 200. 10.1186/s13104-019-4220-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schlaepfer DD, and Mitra SK. 2004. Multiple connections link FAK to cell motility and invasion. Current Opinion in Genetics & Development 14 (1): 92–101. 10.1016/j.gde.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 8.Murphy JM, Rodriguez YAR, Jeong K, Ahn EE, and Lim SS. 2020. Targeting focal adhesion kinase in cancer cells and the tumor microenvironment. Experimental & Molecular Medicine 52 (6): 877–886. 10.1038/s12276-020-0447-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murphy JM, Jeong K, and Lim SS. 2020. FAK family kinases in vascular diseases. International Journal of Molecular Sciences 21 (10). 10.3390/ijms21103630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lim ST, Miller NL, Chen XL, Tancioni I, Walsh CT, Lawson C, Uryu S, Weis SM, Cheresh DA, and Schlaepfer DD. 2012. Nuclear-localized focal adhesion kinase regulates inflammatory VCAM-1 expression. The Journal of Cell Biology 197 (7): 907–919. 10.1083/jcb.201109067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petzold T, Orr AW, Hahn C, Jhaveri KA, Parsons JT, and Schwartz MA. 2009. Focal adhesion kinase modulates activation of NF-kappaB by flow in endothelial cells. American Journal of Physiology. Cell Physiology 297 (4): C814–C822. 10.1152/ajpcell.00226.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Funakoshi-Tago M, Sonoda Y, Tanaka S, Hashimoto K, Tago K, Tominaga S, and Kasahara T. 2003. Tumor necrosis factor-induced nuclear factor kappaB activation is impaired in focal adhesion kinase-deficient fibroblasts. The Journal of Biological Chemistry 278 (31): 29359–29365. 10.1074/jbc.M213115200. [DOI] [PubMed] [Google Scholar]
- 13.Schlaepfer DD, Hou S, Lim ST, Tomar A, Yu H, Lim Y, Hanson DA, Uryu SA, Molina J, and Mitra SK. 2007. Tumor necrosis factor-alpha stimulates focal adhesion kinase activity required for mitogen-activated kinase-associated interleukin 6 expression. The Journal of Biological Chemistry 282 (24): 17450–17459. [DOI] [PubMed] [Google Scholar]
- 14.Wajant H, and Scheurich P. 2011. TNFR1-induced activation of the classical NF-kappaB pathway. The FEBS Journal 278 (6): 862–876. 10.1111/j.1742-4658.2011.08015.x. [DOI] [PubMed] [Google Scholar]
- 15.Viatour P, Merville MP, Bours V, and Chariot A. 2005. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends in Biochemical Sciences 30 (1): 43–52. 10.1016/j.tibs.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 16.Hoffmann A, Levchenko A, Scott ML, and Baltimore D. 2002. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science 298 (5596): 1241–1245. 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- 17.Zambrano S, Bianchi ME, and Agresti A. 2014. High-throughput analysis of NF-kappaB dynamics in single cells reveals basal nuclear localization of NF-kappaB and spontaneous activation of oscillations. PLoS One 9 (3): e90104. 10.1371/journal.pone.0090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zambrano S, De Toma I, Piffer A, Bianchi ME, and Agresti A. 2016. NF-kappaB oscillations translate into functionally related patterns of gene expression. Elife 5: e09100. 10.7554/eLife.09100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stevens T, Creighton J, and Thompson WJ. 1999. Control of cAMP in lung endothelial cell phenotypes. Implications for control of barrier function. The American Journal of Physiology 277 (1): L119–L126. 10.1152/ajplung.1999.277.1.L119. [DOI] [PubMed] [Google Scholar]
- 20.Kim JH, Baddoo MC, Park EY, Stone JK, Park H, Butler TW, Huang G, Yan X, Pauli-Behn F, Myers RM, Tan M, Flemington EK, Lim ST, and Ahn EY. 2016. SON and its alternatively spliced isoforms control mll complex-mediated H3K4me3 and transcription of leukemia-associated genes. Molecular Cell 61 (6): 859–873. 10.1016/j.molcel.2016.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pescatore A, Esposito E, Draber P, Walczak H, and Ursini MV. 2016. NEMO regulates a cell death switch in TNF signaling by inhibiting recruitment of RIPK3 to the cell death-inducing complex II. Cell Death & Disease 7 (8): e2346. 10.1038/cddis.2016.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gothert JR, Gustin SE, van Eekelen JA, Schmidt U, Hall MA, Jane SM, Green AR, Gottgens B, Izon DJ, and Begley CG. 2004. Genetically tagging endothelial cells in vivo: bone marrow-derived cells do not contribute to tumor endothelium. Blood 104 (6): 1769–1777. 10.1182/blood-2003-11-3952. [DOI] [PubMed] [Google Scholar]
- 23.Lim ST, Chen XL, Tomar A, Miller NL, Yoo J, and Schlaepfer DD. 2010. Knock-in mutation reveals an essential role for focal adhesion kinase activity in blood vessel morphogenesis and cell motility-polarity but not cell proliferation. The Journal of Biological Chemistry 285 (28): 21526–21536. 10.1074/jbc.M110.129999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nelson DE, Ihekwaba AE, Elliott M, Johnson JR, Gibney CA, Foreman BE, Nelson G, See V, Horton CA, Spiller DG, Edwards SW, McDowell HP, Unitt JF, Sullivan E, Grimley R, Benson N, Broomhead D, Kell DB, and White MR. 2004. Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science 306 (5696): 704–708. 10.1126/science.1099962. [DOI] [PubMed] [Google Scholar]
- 25.Sung MH, Salvatore L, De Lorenzi R, Indrawan A, Pasparakis M, Hager GL, Bianchi ME, and Agresti A. 2009. Sustained oscillations of NF-kappaB produce distinct genome scanning and gene expression profiles. PLoS One 4 (9): e7163. 10.1371/journal.pone.0007163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Westhoff MA, Serrels B, Fincham VJ, Frame MC, and Carragher NO. 2004. SRC-mediated phosphorylation of focal adhesion kinase couples actin and adhesion dynamics to survival signaling. Molecular and Cellular Biology 24 (18): 8113–8133. 10.1128/MCB.24.18.8113-8133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Israel A 2010. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harbor Perspectives in Biology 2 (3): a000158. 10.1101/cshperspect.a000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, and Karin M. 1997. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature 388 (6642): 548–554. 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 29.Dziedzic SA, Su Z, Jean Barrett V, Najafov A, Mookhtiar AK, Amin P, Pan H, Sun L, Zhu H, Ma A, Abbott DW, and Yuan J. 2018. ABIN-1 regulates RIPK1 activation by linking Met1 ubiquitylation with Lys63 deubiquitylation in TNF-RSC. Nature Cell Biology 20 (1): 58–68. 10.1038/s41556-017-0003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peltzer N, Darding M, and Walczak H. 2016. Holding RIPK1 on the ubiquitin leash in TNFR1 signaling. Trends in Cell Biology 26 (6): 445–461. 10.1016/j.tcb.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 31.Blackwell K, Zhang L, Workman LM, Ting AT, Iwai K, and Habelhah H. 2013. Two coordinated mechanisms underlie tumor necrosis factor alpha-induced immediate and delayed IkappaB kinase activation. Molecular and Cellular Biology 33 (10): 1901–1915. 10.1128/MCB.01416-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Annibaldi A, Wicky John S, Vanden Berghe T, Swatek KN, Ruan J, Liccardi G, Bianchi K, Elliott PR, Choi SM, Van Coillie S, Bertin J, Wu H, Komander D, Vandenabeele P, Silke J, and Meier P. 2018. Ubiquitin-mediated regulation of RIPK1 kinase activity independent of IKK and MK2. Molecular Cell 69 (4): 566–580 e565. 10.1016/j.molcel.2018.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang H, Meng H, Li X, Zhu K, Dong K, Mookhtiar AK, Wei H, Li Y, Sun SC, and Yuan J. 2017. PELI1 functions as a dual modulator of necroptosis and apoptosis by regulating ubiquitination of RIPK1 and mRNA levels of c-FLIP. Proceedings of the National Academy of Sciences of the United States of America 114 (45): 11944–11949. 10.1073/pnas.1715742114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jeong K, Kim JH, Murphy JM, Park H, Kim SJ, Rodriguez YAR, Kong H, Choi C, Guan JL, Taylor JM, Lincoln TM, Gerthoffer WT, Kim JS, Ahn EE, Schlaepfer DD, and Lim SS. 2019. Nuclear focal adhesion kinase controls vascular smooth muscle cell proliferation and neointimal hyperplasia through GATA 4-mediated cyclin D1 transcription. Circulation Research 125 (2): 152–166. 10.1161/CIRCRESAHA.118.314344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hirt UA, Waizenegger IC, Schweifer N, Haslinger C, Gerlach D, Braunger J, Weyer-Czernilofsky U, Stadtmuller H, Sapountzis I, Bader G, Zoephel A, Bister B, Baum A, Quant J, Kraut N, Garin-Chesa P, and Adolf GR. 2018. Efficacy of the highly selective focal adhesion kinase inhibitor BI 853520 in adenocarcinoma xenograft models is linked to a mesenchymal tumor phenotype. Oncogenesis 7 (2): 21. 10.1038/s41389-018-0032-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soria JC, Gan HK, Blagden SP, Plummer R, Arkenau HT, Ranson M, Evans TR, Zalcman G, Bahleda R, Hollebecque A, Lemech C, Dean E, Brown J, Gibson D, Peddareddigari V, Murray S, Nebot N, Mazumdar J, Swartz L, Auger KR, Fleming RA, Singh R, and Millward M. 2016. A phase I, pharmacokinetic and pharmacodynamic study of GSK2256098, a focal adhesion kinase inhibitor, in patients with advanced solid tumors. Annals of Oncology 27 (12): 2268–2274. 10.1093/annonc/mdw427. [DOI] [PubMed] [Google Scholar]
- 37.Jones SF, Siu LL, Bendell JC, Cleary JM, Razak AR, Infante JR, Pandya SS, Bedard PL, Pierce KJ, Houk B, Roberts WG, Shreeve SM, and Shapiro GI. 2015. A phase I study of VS-6063, a second-generation focal adhesion kinase inhibitor, in patients with advanced solid tumors. Investigational New Drugs 33 (5): 1100–1107. 10.1007/s10637-015-0282-y. [DOI] [PubMed] [Google Scholar]
- 38.de Jonge MJA, Steeghs N, Lolkema MP, Hotte SJ, Hirte HW, van der Biessen DAJ, Abdul Razak AR, De Vos F, Verheijen RB, Schnell D, Pronk LC, Jansen M, and Siu LL. 2019. Phase I Study of BI 853520, an inhibitor of focal adhesion kinase, in patients with advanced or metastatic nonhematologic malignancies. Targeted Oncology 14 (1): 43–55. 10.1007/s11523-018-00617-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roy-Luzarraga M, Abdel-Fatah T, Reynolds LE, Clear A, Taylor JG, Gribben JG, Chan S, Jones L, and Hodivala-Dilke K. 2020. Association of low tumor endothelial cell pY397-focal adhesion kinase expression with survival in patients with neoadjuvant-treated locally advanced breast cancer. JAMA Network Open 3 (10): e2019304. 10.1001/jamanetworkopen.2020.19304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tavora B, Reynolds LE, Batista S, Demircioglu F, Fernandez I, Lechertier T, Lees DM, Wong PP, Alexopoulou A, Elia G, Clear A, Ledoux A, Hunter J, Perkins N, Gribben JG, and Hodivala-Dilke KM. 2014. Endothelial-cell FAK targeting sensitizes tumours to DNA-damaging therapy. Nature 514 (7520): 112–116. 10.1038/nature13541. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.