

Abstract
Tangier disease is a genetic disorder characterized by an absence or extremely low level of high-density lipoprotein (HDL)-cholesterol (HDL-C). It is caused by a dysfunctional mutation of the ATP-binding cassette transporter A1 ( ABCA1 ) gene, the mandatory gene for generation of HDL particles from cellular cholesterol and phospholipids, and it appears in an autosomal recessive hereditary profile. To date, 35 cases have been reported in Japan and 109 cases outside Japan. With dysfunctional mutations in both alleles (homozygotes or compound heterozygotes), the HDL-C level is mostly less than 5 mg/dL and there is 10 mg/dL or less of apolipoprotein A-I (apoA-I), the major protein component of HDL. In patients with Tangier disease, major physical findings are orange-colored pharyngeal tonsils, hepatosplenomegaly, corneal opacity, lymphadenopathy, and peripheral neuropathy. Although patients tend to have decreased low-density lipoprotein (LDL)-cholesterol (LDL-C) levels, premature coronary artery disease is frequently observed. No specific curative treatment is currently available, so early identification of patients and preventing atherosclerosis development are crucial. Management of risk factors other than low HDL-C is also important, such as LDL-C levels, hypertension and smoking. Additionally, treatment for glucose intolerance might be required because impaired insulin secretion from pancreatic beta cells has occasionally been reported.
Keywords: Tangier disease, HDL, Reverse cholesterol transport, ABCA1, Cholesterol efflux, Orange tonsil, Atherosclerosis
Introduction
Tangier disease is an autosomal recessive disease characterized by extremely low levels or absence of high-density lipoprotein (HDL)-cholesterol (HDL-C) and apolipoprotein A-I (apoA-I) 1) . The disease was named after Tangier Island in Chesapeake Bay, Virginia, USA, where the first case was discovered in 1960 and reported in 1961 2) . In 1991, generation of HDL particles through the direct action of helical HDL apoproteins on cells was first reported 3) , and this was found to be deficient in cells derived from a patient with Tangier disease in 1995 4) . Eventually, ATP binding cassette transporter A1 ( ABCA1 ) was identified as the gene responsible for this action and for Tangier disease in 1999 5 - 7) . Sequential progress in the investigation of HDL biosynthesis showed that HDL particles generated through ABCA1-dependent interaction of apolipoproteins with cells are the main source of plasma HDL. Patients with homozygous or compound heterozygous mutations in the ABCA1 gene display the phenotype of Tangier disease and heterozygotes have decreases in HDL-cholesterol to various extents.
1. Disease Frequency
The number of cases of Tangier disease reported by 2020 was 35 in Japan and 109 in other countries (possibly including duplicates), indicating that it is a rather rare disease 8 , 9) . However, the frequency of dysfunctional mutations in the ABCA1 gene in the general population is not clear. A recent article using the Exome Aggregation Consortium database reported that 1 in 400 individuals in the general population is a heterozygote for a loss-of-function variant in the ABCA1 gene on the basis of allele frequencies (frameshift, nonsense and splicing only; not missense), indicating a global prevalence of Tangier disease of at least 1 in 640,000 10) . It therefore seems that a substantial number of cases might go undiagnosed.
2. Genetic Backgrounds and Pathophysiology
ABCA1 is a member of the ATP-binding cassette transporter membrane protein family. It is an essential factor for generation of nascent HDL particles with extracellular helical apolipoproteins, through transport of cellular phospholipids and cholesterol ( Fig.1 ) . This process is the major source of plasma HDL and one of the major mechanisms of cholesterol export from cells. It may be essential for final processing of cholesterol in mammalian somatic cells that are unable to catabolize cholesterol molecules. Peripheral cells sense intra-cellular cholesterol levels and increase ABCA1 expression for its excretion 11) , while it undergoes bidirectional control in hepatocytes to prevent cholesterol recovered from peripheral cells flowing back into blood plasma 12 , 13) . With functional deficiency in ABCA1, spherical HDL particles are not produced resulting in extremely low plasma HDL-C levels, which, in Tangier disease patients, are about one third of the normal level. The reason for this is unclear but it has been postulated that a substantial portion of cholesterol molecules in LDL in human plasma are those which have been acyl-esterified in HDL and transferred to VLDL/LDL and that a severe decrease in HDL cholesterol may lead to a decrease in LDL-C level.
Fig.1.Roles of ABCA1 in formation of HDL particles, reverse cholesterol transport and pathogenesis of Tangier disease.
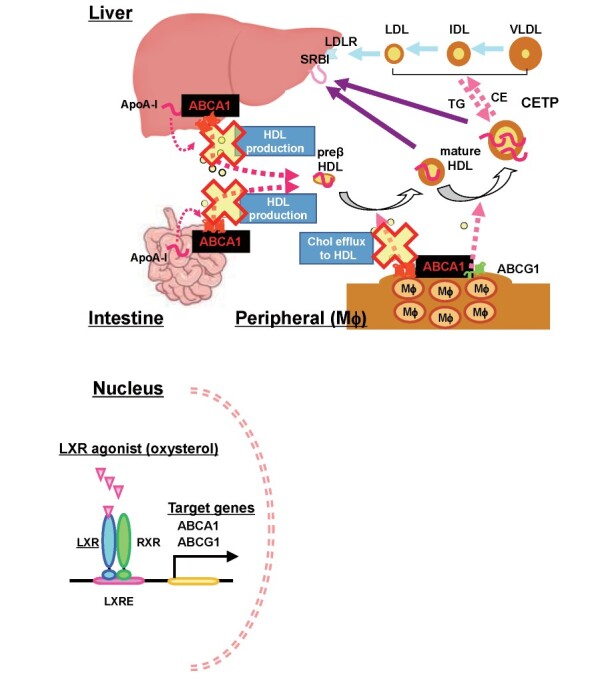
In Tangier disease patients, cellular cholesterol export is impaired due to ABCA1 deficiency in peripheral cells, including macrophages and Schwann cells. Cholesterol therefore accumulates in these cells, causing orange-colored pharyngeal tonsillar swelling, corneal opacity, hepatosplenomegaly, lymphadenopathy and peripheral neuropathy. However, impairment of the initial stage of reverse cholesterol transport should be considered to be a risk for developing atherosclerotic diseases even though plasma LDL-C concentrations could be reduced.
ABCA1 appears to destabilize the raft structure, a cholesterol-rich domain of the plasma membrane 14 , 15) , its deficiency leads to an increase in “lipid” rafts and it has been suggested that this increases secretion of inflammatory cytokines 16) . It has also been reported that the insulinogenic index decreases due to cholesterol accumulation in pancreatic β-cells, which often accompanies glucose intolerance 17 , 18) . These metabolic disorders are collectively involved in the development of premature coronary artery disease 8) .
In early studies of Tangier disease, Schaefer et al. revealed the kinetics of plasma lipoprotein metabolism using externally labeled injected HDL and found that that apoA-I was catabolized at a much greater fractional rate in patients 19) . Their data, however, should be reinterpreted in terms of external HDL catabolism on the basis of ABCA1 deficiency. A recent study using human pluripotent stem cell-derived hepatocytes has demonstrated that ABCA1 deficiency increases angiopoietin-like protein 3 secretion, which is consistent with increased triglyceride in the plasma of Tangier patients 20) .
3. Clinical Manifestations
3.1. Abnormal Plasma Lipoproteins
Plasma HDL-C is mostly low, at 5 mg/dL or less (mean of identified cases 3±3 mg/dL), and the apoA-I concentration is 10 mg/dL or less 21) . Plasma LDL-C is also reduced, to around 37% of the average normal level. The appearance of remnant lipoprotein particles (intermediate products between VLDL and LDL) rich in triglycerides has been reported and this was found to result in abnormal small, dense LDL particles 21) . In subjects with heterozygous mutations of the ABCA1 gene, plasma HDL-C and apoA-I levels are often reduced to about 50% of that in normal subjects, though the extent of HDL-C decline is not consistent.
3.2. Physical Findings
Impairment of HDL biogenesis results in reduced cholesterol export, which leads to lipid accumulation in cells. A typical finding of this disorder is orange-colored tonsils ( Fig.2 ) 8) . The tonsils of patients are lobulated and swollen and present a bright orange or yellow-grey surface. A history of recurrent tonsillitis or tonsillectomy is often noted. In addition, splenomegaly and associated thrombocytopenia and/or reticulocyte hyperplasia may be present ( Fig.3 ) . Hepatomegaly is also observed in about one-third of cases, but liver dysfunction is not usually present 22) . There is also cholesterol accumulation in other organs, such as lymph nodes, thymus, intestinal mucosa, and skin. Its accumulation in the cornea causes corneal opacity.
Fig.2. Orange-colored tonsils observed in male patient with Tangier disease in his 50s.
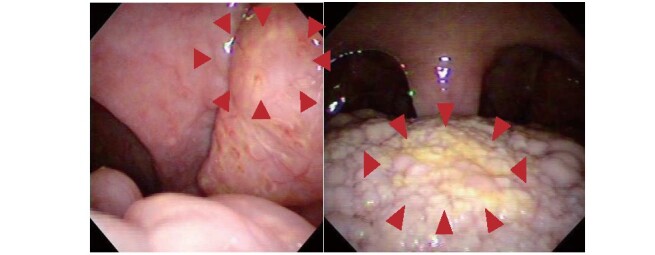
Arrow heads indicate palatine tonsil (left panel) and lingual tonsil (right panel). Reproduced from reference [8].
Fig.3. Abdominal CT scan of splenomegaly in female patient with Tangier disease in her 40s.
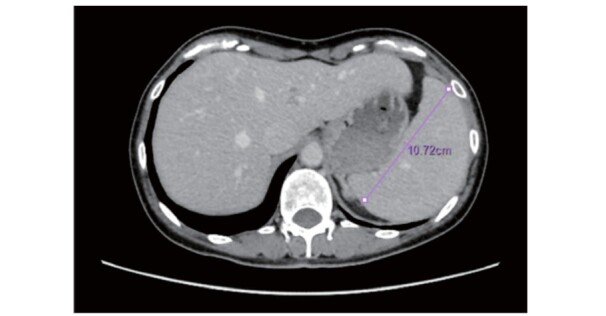
The photo was kindly provided by one of the co-authors, Prof. Yasushi Ishigaki (Iwate Medical University).
3.3. Peripheral Neuropathy
Various peripheral neuropathies, ranging from mild to severe, have been reported. Sensory, motor or mixed disorders appear transiently or persistently. Reduced deep perception and tendon reflexes are rare. Peripheral neuropathy appears as a recurrent asymmetric disorder of peripheral nerves including cranial nerves, as neuropathy with symmetry in the lower limbs, or syringomyelia-like peripheral neuropathy 23 , 24) .
3.4. Cardiovascular Diseases
It has been reported that 12 out of 35 patients (34.3%) in Japan and 34 out of 109 patients (31.2%) in other countries had some type of cardiovascular disease, suggesting accelerated atherogenicity in Tangier disease ( Fig.4 ) 8) . A previous case study using intravascular ultrasound (IVUS) revealed diffuse calcified coronary artery lesions 25) , which might have been affected by HDL deficiency and glucose intolerance 17) .
Fig.4. Advanced systemic atherosclerotic lesions in male patient with Tangier disease in his 50s.

Arrows indicate stenosis or occlusion of the artery.
A: left coronary artery, B: right coronary artery, C: brachiocephalic artery, D: left iliac artery, E: right external iliac artery [8].
4. Diagnostic Criteria and Differential Diagnosis
Diagnostic criteria for Tangier disease are given in Table 1 and the flow chart for differential diagnosis of hypo-HDL-cholesterolemia is shown in Fig.5 , based on discussions by the Committee on Primary Dyslipidemia under the Research Program on Rare and Intractable Diseases of Japan’s Ministry of Health, Labour and Welfare.
Table 1. Diagnostic criteria.
|
A.Required laboratory test results 1.Plasma (serum) HDL-cholesterol less than 25 mg/dL 2. Plasma (serum) apoA-I concentration less than 20 mg/dL B.Clinical symptoms 1.Orange-colored tonsillar swelling 2.Hepatomegaly and/or splenomegaly 3.Corneal opacity 4.Peripheral neuropathy 5.Cardiovascular disease C.Differential diagnosis The following diseases should be excluded; LCAT deficiency, apoA-I deficiency and secondary hypo-HDL-cholesterolemia * D.Genetic testing ** Identification of pathogenic mutations in the ABCA1 gene < Diagnostic category > Definite: Patients should satisfy both of required laboratory test results (A) AND at least one clinical symptom of (B) AND should be excluded for the diseases of differential diagnosis (C) AND should be positive for genetic testing (D). Probable: Patients should satisfy both of required laboratory test results (A) AND at least two clinical symptoms of (B) AND should be excluded for the diseases of differential diagnosis (C). Tangier disease can be diagnosed if patients are categorized “Definite” or “Probable”. |
* Secondary hypo-HDL-cholesterolemia: After surgery, liver disorders (especially liver cirrhosis and severe hepatitis, including convalescent stage), acute phase of systemic inflammatory disease, debilitating diseases such as cancer, history of oral probucol within the past 6 months, and combined probucol and fibrate (including fibrate administration after discontinuation of probucol).
** When differential diagnosis is difficult, genetic testing for ABCA1 mutations should be performed. The diagnosis can be definite if pathogenic mutations in the ABCA1 gene are identified.
Fig.5. Differential diagnosis flow chart for hypo-HDL-cholesterolemia.
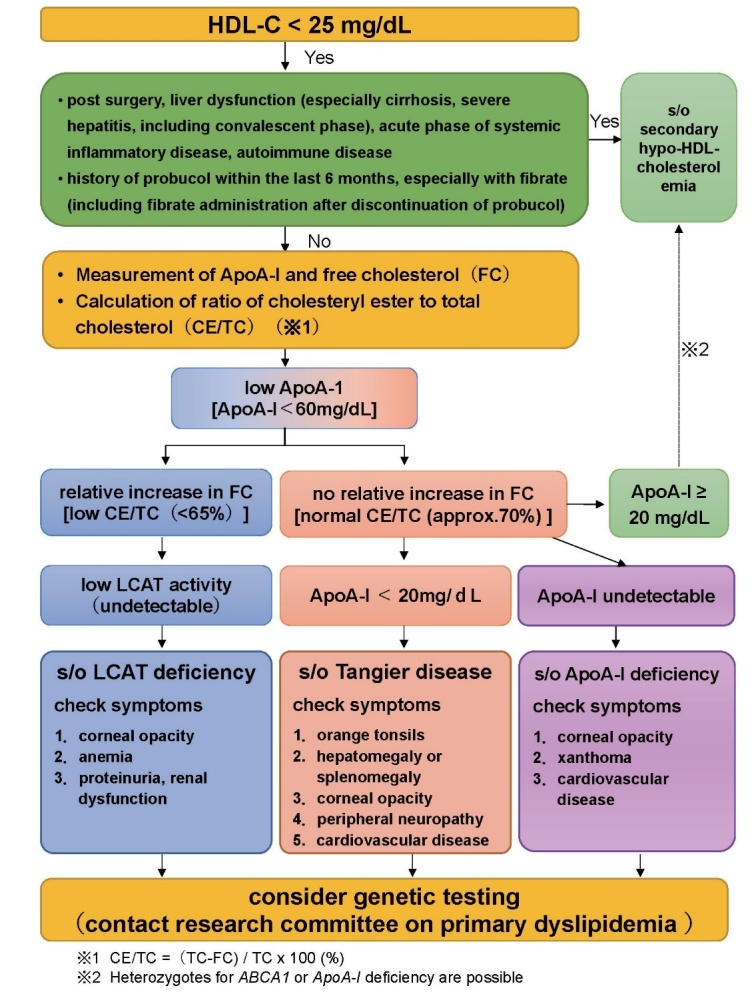
Inherited diseases that lead to hypo-HDL-cholesterolemia (Familial hypoalphalipoproteinemia) include classical lecithin: cholesterol acyltransferase (LCAT) deficiency, fish-eye disease, and familial apoA-I deficiency. Corneal opacity is commonly observed in these diseases, but tonsillar swelling and peripheral neuropathy are specific to Tangier disease, and xanthomas are found only in familial apoA-I deficiency 24 , 26) .
Care should be taken to exclude secondary hypo-HDL-cholesterolemia, such as in severe liver diseases, especially liver cirrhosis, and drug-induced hypo-HDL-cholesterolemia, most frequently due to combination of probucol and fibrates. It should be noted that probucol may continue to influence lipid profiles for several months after discontinuation, so HDL-C may be significantly reduced by switching from probucol to fibrates or permafibrate, a selective peroxisome proliferator-activated receptor alpha modulator (SPPARMα), in some cases.
5. Current Management of Tangier Disease
Based on the patients identified in Japan to date, there seem to be no distinct differences in clinical or genetic profiles with patients in other countries 8 , 9) . No curative treatment, such as gene therapy for the ABCA1 gene, has yet been established. Since extremely enhanced risk of atherosclerotic diseases is the major clinical problem, patients should be carefully monitored for presence of atherosclerotic lesions through regular testing including exercise electrocardiography, echocardiography and computed tomography coronary angiography 8) . The management of atherosclerotic risk factors, such as hypertension, smoking and diabetes mellitus, is crucial, 27) . Plasma LDL-C levels are generally low in patients with Tangier disease but if this is not the case, they should be reduced through administration of statins or other means. Impairment of the insulinogenic index should be estimated using a 75 g oral glucose tolerance test 17) .
Conclusions and Future Perspectives
Gene therapy for ABCA1 gene may have the greatest potential for Tangier disease. There is the possibility that restoration of ABCA1 expression in the liver would raise serum HDL-C levels but this might not be enough to recover cellular cholesterol efflux and suppress extra lipid accumulation in cells in atherogenic lesions such as macrophages, smooth muscle cells and endothelial cells. It may not be easy to develop a fundamental therapy for Tangier disease.
Acknowledgments and Notice of Grant Support
This work has been supported by Health, Labour and Welfare Sciences Research Grant for Research on Rare and Intractable Diseases (H30-nanji-ippan-003).
Conflicts of Interest
Atsushi Nohara has nothing to disclose. Hayato Tada has nothing to disclose. Masatsune Ogura has received honoraria from Amgen Inc., Astellas Pharma Inc. Sachiko Okazaki has received scholarship grants from Minophagen Pharmaceutical Co., Ltd., Kowa Company, Ltd. Koh Ono has nothing to disclose. Hitoshi Shimano has nothing to disclose. Hiroyuki Daida has received honoraria from Amgen Inc., Daiichi-Sankyo Co., Ltd., Kowa Co., Ltd., and MSD K.K., Novartis Pharma K.K., Bayer Yakuhin, Ltd. and received clinical research funding from Canon Medical Systems Corporation, Philips Japan, Ltd., Toho Holdings Co., Ltd., Asahi Kasei Corporation, and Inter Reha Co., Ltd. HD has also received scholarship grants from Nippon Boehringer Ingelheim Co., Ltd., Otsuka Pharmaceutical Co., Ltd., Sanofi K.K., MSD K.K., Daiichi-Sankyo Co., Ltd., Pfizer Co., Ltd., Mitsubishi Tanabe Pharma Corp., Astellas Pharma Inc., Takeda Pharmaceutical Co., Ltd., Teijin Pharma, Ltd., Shionogi & Co., Ltd., Actelion Pharmaceuticals, Ltd., Actelion Ltd., Kowa Co., Ltd., Bayer Yakuhin, Ltd. HD has also courses endowed by companies, including Philips Japan, Ltd., ResMed, Fukuda Denshi Co., Ltd., and Paramount Bed Co., Ltd. Kazushige Dobashi has nothing to disclose. Toshio Hayashi has nothing to disclose. Mika Hori has nothing to disclose. Kota Matsuki has nothing to disclose. Tetsuo Minamino has nothing to disclose. Shinji Yokoyama has nothing to disclose. Mariko Harada-Shiba has received stock holdings or options from Liid Pharma, honoraria from Amgen Inc., Astellas Pharma Inc., Sanofi, and scholarship grants from Aegerion Pharmaceuticals, Inc., Recordati Rare Diseases Japan, and Kaneka Corporation. Katsunori Ikewaki has nothing to disclose. Yasushi Ishigaki has nothing to disclose. Shun Ishibashi has received honoraria from Kowa Co., Ltd., and a scholarship grant from Ono Pharmaceutical Co., Ltd. Kyoko Inagaki has nothing to disclose. Hirotoshi Ohmura has nothing to disclose. Hiroaki Okazaki has received scholarship grants from Minophagen Pharmaceutical Co., Ltd., Kowa Company, Ltd. Masa-aki Kawashiri has nothing to disclose. Masayuki Kuroda has nothing to disclose. Masahiro Koseki has received clinical research funding from Kowa Company, Ltd., Rohto Pharmaceutical Co., Ltd. Takanari Gotoda has nothing to disclose. Shingo Koyama has nothing to disclose. Yoshiki Sekijima has nothing to disclose. Manabu Takahashi has nothing to disclose. Yasuo Takeuchi has nothing to disclose. Misa Takegami has nothing to disclose. Kazuhisa Tsukamoto has received honoraria from Bayer Yakuhin, Ltd., MSD Ltd., Takeda Pharmaceutical Company Ltd., and scholarship grants from Mitsubishi Tanabe Pharma Corporation., Bayer Yakuhin, Ltd., Sanofi K.K. Atsuko Nakatsuka has nothing to disclose. Kimitoshi Nakamura has nothing to disclose. Satoshi Hirayama has nothing to disclose. Hideaki Bujo has nothing to disclose. Daisaku Masuda has received clinical research funding from MSD K.K., Ono Pharmaceutical Co., Ltd., Takeda Pharmaceutical Co., Ltd., Kowa Co., Ltd. Takashi Miida has nothing to disclose. Yoshihiro Miyamoto has nothing to disclose. Takeyoshi Murano has nothing to disclose. Takashi Yamaguchi has nothing to disclose. Shizuya Yamashita has received honoraria from Kowa Company, Ltd., MSD K.K. Masashi Yamamoto has nothing to disclose. Koutaro Yokote has received honoraria from Kowa Company, Ltd., MSD K.K., Astellas Pharma Inc., Mitsubishi Tanabe Pharma Corp., Amgen K.K., Takeda Pharmaceutical Company Limited, Sanofi K.K., Ono Pharmaceutical Co., Ltd., AstraZeneca K.K., Daiichi-Sankyo Co., Ltd., Novartis Pharma K.K., Sumitomo Dainippon Pharma Co., Ltd., Kyowa Kirin Co., Ltd., Pfizer Japan Inc., Novo Nordisk Pharma Ltd., Nippon Boehringer Ingelheim Co., Ltd., Eli Lilly Japan K.K., Taisho Pharmaceutical Co., Ltd., Janssen Pharmaceutical K.K., and received clinical research funding from Taisho Pharmaceutical Co., Ltd. KY has also received scholarship grants from Mitsubishi Tanabe Pharma Corp., Takeda Pharmaceutical Co., Ltd., MSD K.K., Pfizer Japan Inc., Novo Nordisk Pharma Ltd., Taisho Pharmaceutical Co., Ltd., Kao Corporation, Ono Pharmaceutical Co., Ltd., Eli Lilly Japan K.K., Sumitomo Dainippon Pharma Co., Ltd., Nippon Boehringer Ingelheim Co., Ltd., Daiichi-Sankyo Co., Ltd., Teijin Pharma, Ltd., Shionogi Co., Ltd., Bayer Yakuhin, Ltd. Jun Wada has nothing to disclose.
References
- 1).Assmann G, Eckardstein A, Brewer HB. Familial analphalipoproteinemia: Tangier disease. The Metabolic and Molecular Bases of Inherited Diseases (McGraw Hill), 2001; 8th ed. vol 2: 2937-2960 [Google Scholar]
- 2). Fredrickson DS, Altrocchi PH, Avioli LV, Goodman DS, Goodman HC. Tangier disease.Combined clinical staff conference at the National Institutes of Health. Ann Intern Med, 1961; 55: 1016-1031 [Google Scholar]
- 3).Hara H, Yokoyama S. Interaction of free apolipoproteins with macrophages. Formation of high-density lipoprotein-like lipoproteins and reduction of cellular cholesterol. J Biol Chem, 1991; 266: 3080-3086 [PubMed] [Google Scholar]
- 4).Francis GA, Knopp RH, Oram JF. Defective removal of cellular cholesterol and phospholipids by apolipoprotein A-I in Tangier disease. J Clin Invest, 1995; 96: 78-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).Brooks-Wilson A, Marcil M, Clee SM, Zhang LH, Roomp K, van Dam M, Yu L, Brewer C, Collins JA, Molhuizen HO, Loubser O, Ouelette BF, Fichter K, Ashbourne-Excoffon KJ, Sensen CW, Scherer S, Mott S, Denis M, Martindale D, Frohlich J, Morgan K, Koop B, Pimstone S, Kastelein JJ, Genest J Jr, Hayden MR . Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat Genet, 1999; 22: 336-345 [DOI] [PubMed] [Google Scholar]
- 6).Bodzioch M, Orsó E, Klucken J, Langmann T, Böttcher A, Diederich W, Drobnik W, Barlage S, Büchler C, Porsch-Ozcürümez M, Kaminski WE, Hahmann HW, Oette K, Rothe G, Aslanidis C, Lackner KJ, Schmitz G. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat Genet, 1999; 22: 347-351 [DOI] [PubMed] [Google Scholar]
- 7).Rust S, Rosier M, Funke H, Real J, Amoura Z, Piette JC, Deleuze JF, Brewer HB, Duverger N, Denèfle P, Assmann G. Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat Genet, 1999; 22: 352-355 [DOI] [PubMed] [Google Scholar]
- 8).Muratsu J, Koseki M, Masuda D, Yasuga Y, Tomoyama S, Ataka K, Yagi Y, Nakagawa A, Hamada H, Fujita S, Hattori H, Ohama T, Nishida M, Hiraoka H, Matsuzawa Y, Yamashita S. Accelerated atherogenicity in Tangier disease. J Atheroscler Thromb, 2018; 25: 1076-1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9).Maruyama T, Yamashita S, Matsuzawa Y, Bujo H, Takahashi K, Saito Y, Ishibashi S, Ohashi K, Shionoiri F, Gotoda T, Yamada N, Kita T; Research Committee on Primary Hyperlipidemia of the Ministry of Health and Welfare of Japan. Mutations in Japanese subjects with primary hyperlipidemia; Results from the Research Committee of the Ministry of Health and Welfare of Japan since 1996. J Atheroscler Thromb, 2004; 11: 131-145 [DOI] [PubMed] [Google Scholar]
- 10).Hooper AJ, McCormick S, Hegele RA, Burnett JR. Clinical utility gene card for: Tangier disease. Eur J Hum Genet, 2017; 25: e1-e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).Costet P, Luo Y, Wang N, Tall AR. Sterol-dependent transactivation of the ABC1 promoter by the liver X receptor/retinoid X receptor. J Biol Chem, 2000; 275: 28240-28245 [DOI] [PubMed] [Google Scholar]
- 12).Tamehiro N, Shigemoto-Mogami Y, Kakeya T, Okuhira K, Suzuki K, Sato R, Nagao T, Nishimaki-Mogami T. Sterol regulatory element-binding protein-2- and liver X receptor-driven dual promoter regulation of hepatic ABC transporter A1 gene expression: mechanism underlying the unique response to cellular cholesterol status. J Biol Chem, 2007; 282: 21090-21099 [DOI] [PubMed] [Google Scholar]
- 13).Ohoka N, Okuhira K, Cui H, Wu W, Sato R, Naito M, Nishimaki-Mogami T. HNF4α increases liver-specific human ATP-binding cassette transporter A1 expression and cholesterol efflux to apolipoprotein A-I in response to cholesterol depletion. Arterioscler Thromb Vasc Biol, 2012; 32: 1005-1014 [DOI] [PubMed] [Google Scholar]
- 14).Ito J, Nagayasu Y, Yokoyama S. Cholesterol-sphingomyelin interaction in membrane and apolipoprotein-mediated cellular cholesterol efflux. J Lipid Res, 2000; 41: 894-904 [PubMed] [Google Scholar]
- 15).Yamauchi Y, Iwamoto N, Rogers MA, Abe-Dohmae S, Fujimoto T, Chang CC, Ishigami M, Kishimoto T, Kobayashi T, Ueda K, Furukawa K, Chang TY, Yokoyama S. Deficiency in the lipid exporter ABCA1 impairs retrograde sterol movement and disrupts sterol sensing at the endoplasmic reticulum. J Biol Chem, 2015; 290: 23464-23477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).Koseki M, Hirano K, Masuda D, Ikegami C, Tanaka M, Ota A, Sandoval JC, Nakagawa-Toyama Y, Sato SB, Kobayashi T, Shimada Y, Ohno-Iwashita Y, Matsuura F, Shimomura I, Yamashita S. Increased lipid rafts and accelerated lipopolysaccharide-induced tumor necrosis factor-alpha secretion in Abca1-deficient macrophages. J Lipid Res, 2007; 48: 299-306 [DOI] [PubMed] [Google Scholar]
- 17).Koseki M, Matsuyama A, Nakatani K, Inagaki M, Nakaoka H, Kawase R, Yuasa-Kawase M, Tsubakio-Yamamoto K, Masuda D, Sandoval JC, Ohama T, Nakagawa-Toyama Y, Matsuura F, Nishida M, Ishigami M, Hirano K, Sakane N, Kumon Y, Suehiro T, Nakamura T, Shimomura I, Yamashita S. Impaired insulin secretion in four Tangier disease patients with ABCA1 mutations. J Atheroscler Thromb, 2009; 16: 292-296 [DOI] [PubMed] [Google Scholar]
- 18).Brunham LR, Kruit JK, Pape TD, Timmins JM, Reuwer AQ, Vasanji Z, Marsh BJ, Rodrigues B, Johnson JD, Parks JS, Verchere CB, Hayden MR. Beta-cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment. Nat Med, 2007; 13: 340-347 [DOI] [PubMed] [Google Scholar]
- 19).Schaefer EJ, Blum CB, Levy RI, Jenkins LL, Alaupovic P, Foster DM, Brewer HB Jr. Metabolism of high-density lipoprotein apolipoproteins in Tangier disease. N Engl J Med, 1978; 299: 905-910 [DOI] [PubMed] [Google Scholar]
- 20).Bi X, Pashos EE, Cuchel M, Lyssenko NN, Hernandez M, Picataggi A, McParland J, Yang W, Liu Y, Yan R, Yu C, DerOhannessian SL, Phillips MC, Morrisey EE, Duncan SA, Rader DJ. ATP-binding cassette transporter A1 deficiency in human induced pluripotent stem cell-derived hepatocytes abrogates HDL biogenesis and enhances triglyceride secretion. EBioMedicine, 2017; 18: 139-145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21).Serfaty-Lacrosniere C, Civeira F, Lanzberg A, Isaia P, Berg J, Janus ED, Smith MP Jr, Pritchard PH, Frohlich J, Lees RS, Barnard FF, Ordovas JM, Schaefer EJ. Homozygous Tangier disease and cardiovascular disease. Atherosclerosis, 1994; 107: 85-98 [DOI] [PubMed] [Google Scholar]
- 22).Puntoni M, Sbrana F, Bigazzi F, Sampietro T. Tangier disease: epidemiology, pathophysiology, and management. Am J Cardiovasc Drugs, 2012; 12: 303-311 [DOI] [PubMed] [Google Scholar]
- 23).Zyss J, Béhin A, Couvert P, Bouhour F, Sassolas A, Kolev I, Denys V, Vial C, Lacour A, Carrié A, Stojkovic T. Clinical and electrophysiological characteristics of neuropathy associated with Tangier disease. J Neurol, 2012; 259, 1222-1226 [DOI] [PubMed] [Google Scholar]
- 24).Amanda AJ, Hegele RA, Burnett JR. Tangier disease: update for 2020. Curr Opin Lipidol, 2020; 31: 80-84 [DOI] [PubMed] [Google Scholar]
- 25).Komuro R, Yamashita S, Sumitsuji S, Hirano K, Maruyama T, Nishida M, Matsuura F, Matsuyama A, Sugimoto T, Ouchi N, Sakai N, Nakamura T, Funahashi T, Matsuzawa Y. Tangier disease with continuous massive and longitudinal diffuse calcification in the coronary arteries: demonstration by the sagittal images of intravascular ultrasonography. Circulation, 2000; 101: 2446-2448 [DOI] [PubMed] [Google Scholar]
- 26).Zanoni P, von Eckardstein A. Inborn errors of apolipoprotein A-I metabolism: implications for disease, research and development. Curr Opin Lipidol, 2020; 31: 62-70 [DOI] [PubMed] [Google Scholar]
- 27).Kinoshita M, Yokote K, Arai H, Iida M, Ishigaki Y, Ishibashi S, Umemoto S, Egusa G, Ohmura H, Okamura T, Kihara S, Koba S, Saito I, Shoji T, Daida H, Tsukamoto K, Deguchi J, Dohi S, Dobashi K, Hamaguchi H, Hara M, Hiro T, Biro S, Fujioka Y, Maruyama C, Miyamoto Y, Murakami Y, Yokode M, Yoshida H, Rakugi H, Wakatsuki A, Yamashita S; Committee for Epidemiology and Clinical Management of Atherosclerosis. Japan Atherosclerosis Society (JAS) Guidelines for Prevention of Atherosclerotic Cardiovascular Diseases 2017. J Atheroscler Thromb, 2018; 25: 846-984 [DOI] [PMC free article] [PubMed] [Google Scholar]