

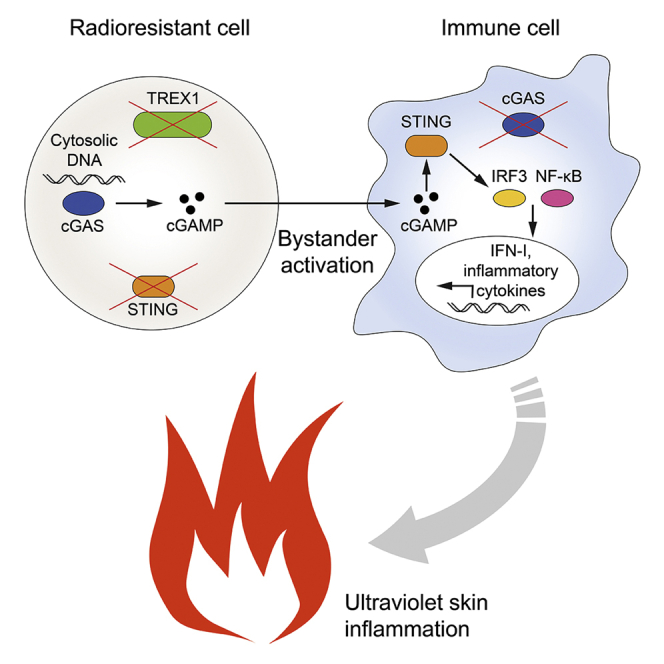
Summary
Intercellular transmission of the second messenger 2′,3′-cGAMP, synthesized by the viral DNA sensor cGAMP synthase (cGAS), is a potent mode of bystander activation during host defense. However, whether this mechanism also contributes to cGAS-dependent autoimmunity remains unknown. Here, using a murine bone marrow transplantation strategy, we demonstrate that, in Trex1−/−-associated autoimmunity, cGAMP shuttling from radioresistant to immune cells induces NF-κB activation, interferon regulatory factor 3 (IRF3) phosphorylation, and subsequent interferon signaling. cGAMP travel prevented myeloid cell and lymphocyte death, promoting their accumulation in secondary lymphoid tissue. Nonetheless, it did not stimulate B cell differentiation into autoantibody-producing plasmablasts or aberrant T cell priming. Although cGAMP-mediated bystander activation did not induce spontaneous organ disease, it did trigger interface dermatitis after UV light exposure, similar to cutaneous lupus erythematosus. These findings reveal that, in Trex1-deficiency, intercellular cGAMP transfer propagates cGAS signaling and, under conducive conditions, causes tissue inflammation.
Subject areas: Immunology, Immunity, Immune response, Cell biology
Graphical abstract
Highlights
-
•
In Trex1−/−-associated autoimmunity radioresistant cells transfer cGAMP to immune cells
-
•
cGAMP shuttling induces NF-κB activation, IRF3 and IFN signaling in vivo
-
•
Intercellular cGAMP transmission is sufficient to cause UV skin inflammation
Immunology; Immunity; Immune response; Cell biology
Introduction
In the context of an antimicrobial response, bystander activation describes a form of intercellular communication in which infected cells trigger immune signaling in previously uninvolved neighboring cells. This mechanism can rapidly amplify an immune response and counters the ability of pathogens to avoid detection by disabling immune signaling in directly infected cells (Holmgren et al., 2017). Although beneficial during an infection, bystander activation is also widely considered to participate in the pathogenesis of autoimmune diseases with deleterious consequences for the affected person (Pacheco et al., 2019).
Erroneous recognition of self-nucleic acids is a central mechanism by which autoimmunity can be instigated. Several germline encoded pattern recognition receptors (PRRs) initiate immune responses upon recognition of nucleic acids. These nucleic acid sensors include Toll-like receptors (TLRs), retinoic acid-inducible gene I-like helicases (RLHs), absent in melanoma 2 (AIM2), and cyclic GMP-AMP (cGAMP) synthase (cGAS). Nucleic acid detecting PRRs are limited in their ability to distinguish between microbial and self-nucleic acids (Schlee and Hartmann, 2016). Therefore, numerous control systems exist to prevent detrimental activation of PRRs by self-nucleic acids. A key mechanism is the degradation of nucleic acids by a set of nucleases that surveil distinct cellular compartments. In the cytosol, three prime repair exonuclease 1 (Trex1) is critically involved in the clearance of DNA species that arise during DNA repair or DNA replication or from endogenous retroelements. In humans, loss-of-function mutations in TREX1 are associated with Aicardi-Goutières syndrome, familial chilblain lupus, and systemic lupus erythematosus (Crow and Manel, 2015). In mice, Trex1 deficiency leads to autoantibody formation and multiorgan inflammation (Stetson et al., 2008).
Immune activation in mice lacking Trex1 results from the recognition of unmetabolized cytosolic DNA by cGAS. cGAS catalyzes the formation of the cyclic dinucleotide 2′,3′-cGAMP (called cGAMP hereafter) (Sun et al., 2013), which acts as a second messenger. cGAMP binds the endoplasmic reticulum-localized protein stimulator of interferon (IFN) genes (STING, encoded by the gene Sting1), which then forms a complex with TBK1 and translocates to the perinuclear compartment. The STING-TBK1 complex subsequently activates transcription factors IFN regulatory factor 3 (IRF3) and nuclear factor kappa B (NF-κB) to induce type I IFN and inflammatory cytokine production. STING has also been shown to initiate lysosomal cell death and NLRP3 activation in certain human myeloid cells (Gaidt et al., 2017). A series of in vitro studies have established that cGAMP can be transferred to neighboring cells, thereby bypassing the need for cell-intrinsic PRR engagement in recipient cells. Initially, it was demonstrated that cGAMP can be shuttled between cells through gap junctions (Ablasser et al., 2013). Later studies provided evidence that delivery may also occur via virions, HIV-1 Env-induced membrane fusion, the folate transporter SLC19A1, or LRRC8 Volume-Regulated Anion Channels (Bridgeman et al., 2015; Gentili et al., 2015; Luteijn et al., 2019; Xu et al., 2016; Zhou et al., 2020).
Altogether, these studies have revealed a role for cGAMP-mediated bystander activation in inducing a robust type I IFN response in viral and bacterial infections. Nonetheless, there is a paucity of information regarding its importance in autoimmunity. Support for the idea that intercellular cGAMP transmission is indeed conducive to the development of autoimmunity comes from the observation that Trex1-deficient mouse embryonic fibroblasts induce cGAS-dependent bystander activation in co-culture experiments (Ablasser et al., 2014). However, the in vivo relevance of this phenomenon has yet to be elucidated. Of note, very few studies to date have investigated the role of cGAMP transfer in vivo (Chen et al., 2016; Marcus et al., 2018; Schadt et al., 2019). Even if cell-cell transmission of cGAMP occurs in autoimmune disease, it is unclear between which cell types this would transpire. The concept that cells can be activated by delivery of the messenger cGAMP has largely been established through the use of immortalized cell lines. Only recently has evidence emerged that primary immune cells are also activated through this process (Marcus et al., 2018; Schadt et al., 2019). Moreover, assuming that immune cells are stimulated by cGAMP travel in autoimmunity, it is unclear whether this would enable professional antigen-presenting cells to prime naive or expand pre-activated self-reactive T cells. Similarly, whether cGAMP transmission to self-reactive B cells would facilitate auto-antibody formation is not known.
In the present study, to clarify these unresolved issues, we generated mice in which cells were either double deficient for Trex1 and STING (dKO) or lacked cGAS. In the chimera, dKO cells produce cGAMP in response to the accumulation of cytosolic DNA but are not activated due to absence of STING. However, cGAMP from dKO cells can be delivered to neighboring cGAS-deficient cells that express intact STING. In this system downstream signaling thus critically depends on the transfer of cGAMP to bystander cells. Employing this strategy allowed us to unequivocally establish the relevance of intercellular cGAMP transfer in Trex1−/−-mediated autoimmunity, with implications for cGAS-mediated autoimmunity in general.
Results
Cell-extrinsic cGAMP activates the IRF3 pathway in DCs
To investigate the contribution of cGAMP-mediated bystander activation to the development of Trex1−/−-associated autoimmunity, we utilized a bone marrow transplantation approach (Figure 1A). Specifically, mice double deficient for Trex1 and STING (Trex1−/−;Sting1gt/gt, hereafter called dKO) were lethally irradiated and reconstituted with bone marrow (BM) from cGAS-deficient mice (Cgas−/− BM), Trex1 and STING double-deficient mice (dKO BM), or a 50:50 mixture of cells from both strains (Cgas−/− + dKO BM). In dKO cells, unmetabolized cytosolic DNA is sensed by cGAS. Owing to the lack of STING, the produced cGAMP does not lead to cell-intrinsic immune activation. However, it can still be shuttled to neighboring Cgas−/− cells that have functional STING and induce downstream signaling. In Cgas−/− BM mice, cGAMP is only produced by non-hematopoietic radioresistant cells (such as epithelial cells and fibroblasts) and a small number of hematopoietic radioresistant cells, such as a subset of Kupffer cells and alveolar macrophages. In contrast, cGAMP in Cgas−/− + dKO BM mice can additionally be generated by half of the transplanted hematopoietic cells.
Figure 1.

Cell-extrinsic cGAMP activates the IRF3 pathway in DCs
(A) Schematic of bone marrow transplantation experiments. Lethally irradiated Trex1−/−;Sting1gt/gt (dKO) mice received bone marrow from Cgas−/- or dKO donor mice for groups 1 and 3, respectively. For group 2, recipients were transplanted with a mixture of bone marrow from both donor types.
(B) Spontaneous IRF3 signaling in splenic cDCs. Histograms display representative results for p-IRF3 (Ser396) of gated cDCs in instantly fixed spleens. Each histogram shows an overlay of λ protein phosphatase-treated (gray shaded) and untreated (colored) cells.
(C and D) Fold change of p-IRF3 mean fluorescence intensity (MFI) of cDCs (C) and pDCs (D) in instantly fixed spleens from Cgas−/− BM (n = 5) and Cgas−/− + dKO BM (n = 4) mice relative to those from dKO BM mice (n = 4). Individual fold change values were calculated as ΔMFIi = (MFIi, untreated – MFIi, λPP) divided by the average of ΔMFIi values obtained from dKO BM mice.
Data are represented as mean + SEM. Experiments in (B–D) were performed twice, and data are representative of a single experiment. Statistically significant differences were determined by two-tailed unpaired Mann-Whitney U test (∗p < 0.05).
See also Figure S1.
First, we assessed whether there was selective pressure favoring Cgas−/− or dKO cells in Cgas−/− + dKO BM mice. Across all examined immune cell subsets sorted from spleens, we found that 12 weeks after transplantation, in Cgas−/− + dKO BM mice deletion of Cgas was 45%–54% (Table S1) as determined by qRT-PCR. Thus, the genetic composition was virtually the same as in the BM mixture used for transplantation, revealing that neither Cgas−/− nor dKO cells had a competitive advantage over each other.
Autoimmunity in Trex1 deficiency depends on the induction of type I IFN by the transcription factor IRF3 together with its transcriptional coactivator CBP/p300 (Stetson et al., 2008). Therefore, we investigated whether cGAMP shuttling between cells stimulates IRF3 phosphorylation. We used phospho-flow to determine the native phosphorylation state of IRF3 directly ex vivo. Type 1 and 2 conventional dendritic cells (cDC1, cDC2) in Cgas−/− BM and Cgas−/− + dKO BM had substantially higher p-IRF3 (Ser396) levels compared with their counterparts in dKO BM mice (Figures 1B and 1C). The increase was more prominent in cDCs from Cgas−/− BM than in those from Cgas−/− + dKO BM mice, likely reflecting the doubled frequency of cGAMP-responsive Cgas−/− cells. As in cDCs, the p-IRF3 signal was also stronger in pDCs from Cgas−/− BM than in those from dKO mice (Figure 1D). For reference, we additionally compared p-IRF3 levels in untreated wild-type C57BL/6J (WT) mice, WT mice transplanted with WT bone marrow, untreated dKO mice, and dKO BM mice. These were similar in all groups (Figure S1A) demonstrating that STING deficiency does not per se alter basal IRF3 activity. Together, these results were compatible with the idea that cGAMP is delivered from dKO cells to Cgas−/− DCs where it engages STING and initiates downstream IRF3 signaling. However, it remained possible that STING in Cgas−/− BM mice is stimulated in a cGAMP-independent fashion. Conceivably, the transplantation procedure provokes the release of bacterial cyclic di-nucleotides (CDNs) that bind STING or induce STING activation in a non-canonical fashion. Non-canonical STING activation by IFI16/ATM has been described in human cells upon DNA damage with etoposide (Dunphy et al., 2018). To investigate this, we compared IRF3 phosphorylation in Cgas−/− mice transplanted with Cgas−/− bone marrow and untreated Cgas−/− mice. We did not find differences in IRF3 phosphorylation between those two groups (Figure S1B) arguing against the notion of cGAMP-independent STING activation in transplanted mice.
We conclude that, in the context of autoimmunity, cGAMP mediates activation of bystander cDCs and pDCs in vivo. Of interest, cGAMP derived from radioresistant cells is sufficient to elicit this response.
Delivery of cGAMP from radioresistant cells to immune cells induces tonic IFN and NF-κB signaling in Trex1 deficiency
To test whether IRF3 activation resulting from cGAMP transmission provoked functional reprogramming of immune cells, we chose to conduct global transcriptional analysis. Given that the greatest difference in p-IRF3 was observed between Cgas−/− BM and dKO BM mice, we performed 3′-mRNA sequencing of splenocytes from these animals. Unsupervised hierarchical clustering of the most variable genes (Figure 2A) and principal component analysis (Figure 2B) demonstrated that intercellular cGAMP travel led to changes in the expression of 660 genes (non-adjusted p value <0.05). Of note, we found Trex1 to be significantly upregulated in Cgas−/− BM mice and Cgas to be significantly upregulated in dKO BM mice confirming the validity of the analysis (Figure 2C). To better define how cGAMP travel shapes gene transcription, we investigated which Hallmark gene sets were enriched in Cgas−/− BM compared with dKO BM mice (Figure 2D). Genes upregulated in Cgas−/− BM mice were associated with IFN-α and IFN-γ, which induce largely overlapping gene sets, and NF-κB signaling, consistent with STING activation in these mice. A 3′-mRNA sequencing analysis of the bone marrow used for transplantation showed that the observed gene expression differences between Cgas−/− BM and dKO BM mice 12 weeks after transplantation were acquired and not intrinsic to the transplanted bone marrow. Only 66 of the 1,276 differentially expressed genes pre-transplantation (non-adjusted p value <0.05) were also differentially expressed post-transplantation (Figure S2A). Of the 66 genes, 18 were contra-regulated (Table S2). Of importance, no upregulation of IFN- or NF-κB-induced genes was detected in Cgas−/− bone marrow pre-transplantation (Figure S2B). On the contrary, IFN-stimulated genes were transcribed at a lower level than in dKO bone marrow pre-transplantation.
Figure 2.

Delivery of cGAMP from radioresistant cells to immune cells induces tonic IFN and NF-κB signaling in Trex1 deficiency
(A) Global gene expression analysis by 3′-mRNA seq. Gene and sample-wise hierarchical clustering based on differentially expressed genes (non-adjusted p value <0.05, n = 660) within the dataset of spleen tissue from Cgas−/− BM and dKO BM mice. Gene expression values are Z score standardized. For visualization purposes Z scores were limited to numbers between −2 and 2.
(B) PCA plot of spleen tissue data from Cgas−/− BM and dKO BM mice. PCA is based on differentially expressed genes (non-adjusted p value <0.05, n = 684).
(C) Volcano plot indicating transcriptomic changes between Cgas−/− BM and dKO BM mice. Genes with a non-adjusted p value <0.05 and a signed fold change >2 are colored in red (upregulated in Cgas−/− KO BM mice) and blue (upregulated in dKO BM mice). Differentially expressed genes with an adjusted p value < 0.05 are labeled.
(D) Normalized Enrichment Scores (NES) of Hallmark gene sets significantly upregulated (FDR q value <0.01) in spleen tissue from Cgas−/− BM mice compared with that from dKO BM mice in a pre-ranked Gene Set Enrichment Analysis (GSEA) based on a metric score calculated by log2(fold change) x (-log10(p value)). n = 5 mice per group.
(E) Percentage of splenic classical monocytes and red pulp macrophages with high expression of IκBα (n = 7 for Cgas−/− BM mice, n = 6 for dKO BM mice).
(F) Fold change of p-RelA mean fluorescence intensity (MFI) of classical monocytes and red pulp macrophages in instantly fixed spleens from Cgas−/− BM (n = 7) and dKO BM (n = 6) mice. Individual fold change values were calculated as ΔMFIi = (MFIi, untreated – MFIi, λPP) divided by the average of ΔMFIi values obtained from dKO BM mice.
(G) Histograms display representative results for IκBα staining of gated classical monocytes and red pulp macrophages.
Data in bar graphs are represented as mean + SEM. Data in (E-G) are pooled from two independent experiments. Statistically significant differences were determined by two-tailed unpaired Mann-Whitney U test (∗p < 0.05, ∗∗p < 0.01).
See also Figure S2.
To verify the activation of NF-κB in Cgas−/− BM mice and to determine the cell subgroups in which it occurred, we evaluated NF-κB signaling events such as IκBα degradation and RelA phosphorylation by flow cytometry. The frequency of cells with high IκBα expression among classical monocytes and red pulp macrophages was substantially lower in Cgas−/− BM than in dKO BM mice (Figures 2E and 2G) indicating that cGAMP transmission initiated NF-κB signaling. In line with this, we also found increased basal RelA (p536) phosphorylation in classical monocytes and red pulp macrophages in Cgas−/− BM compared with dKO BM mice (Figure 2F). Of note, we have not observed these effects in cDCs. cGAMP-independent STING activation induced by the transplantation procedure was ruled out as a cause for NF-κB activation (Figure S2C). We also measured relative expression of cytokine proteins in the serum of Cgas−/− BM and dKO BM mice using a cytokine array, which yielded no obvious differences (Figure S2D).
Altogether, these data support the notion that cGAMP-mediated bystander activation leads to constitutive IFN and NF-κB signaling in Trex1 deficiency.
cGAMP transfer promotes T cell accrual in lymphoid tissue but not spontaneous T cell priming
We next sought to elucidate the contributions of intercellular cGAMP travel to the spontaneous self-directed T cell response in Trex1 deficiency. CD4+ and CD8+ T cell numbers in spleens were considerably higher in Cgas−/− BM and Cgas−/− + dKO BM mice than in dKO mice (Figure 3A), indicating that cGAMP transmission facilitates T cell accumulation. Expression of stem cell antigen 1 (Sca-1) on lymphocytes is associated with persistent IFN signaling (Lee et al., 2008). Consistent with our finding that cGAMP transmission induces an IFN gene signature (Figure 2), we found Sca-1 protein expression to be increased in T cells from Cgas−/− BM and Cgas−/− + dKO BM compared with those from dKO BM controls (Figures 3B and 3C). This increase was significant, although Sca-1 levels were higher on lymphocytes from dKO BM mice than on those from WT mice transplanted with WT bone marrow (Figure S3A). cGAMP delivery to bystander cells did not promote T cell activation. There was no appreciable difference in the frequency of phenotypically naive CD4+ and CD8+ T cells as well as memory subsets in all three groups of mice (Figure 3D). In Trex1-deficient mice, T cells preferentially differentiate into Th1/Tc1 cells, whereas Th2/Tc2 and Th17/Tc17 cells are rare (Figure S3B). To determine whether cGAMP spread promotes polarization of CD4+ and CD8+ T cells into IFN-γ and TNF-α producing effectors, we intracellularly stained splenocytes for these cytokines after culture with PMA and ionomycin. cGAMP-mediated bystander activation had no detectable effect on the percentage of IFN-γ/TNF-α-producing CD4+ and CD8+ T cells (Figures 3E and S3C). We also did not find differences in the frequency of regulatory Foxp3+CD25+ cells among CD4+ T cells (Figure 3F).
Figure 3.

cGAMP transfer promotes T cell accrual in lymphoid tissue but not spontaneous T cell priming
(A) Number of CD4+ and CD8+ T cells in spleens from Cgas−/− BM (n = 14), Cgas−/− + dKO BM (n = 14), and dKO BM mice (n = 16).
(B) Mean fluorescence intensity (MFI) for Sca-1 of splenic CD4+ and CD8a+ T cells in Cgas−/− BM (n = 7), Cgas−/− + dKO BM (n = 7), and dKO BM mice (n = 6).
(C) Representative Sca-1 staining histograms of splenic CD4+ and CD8a+ T cells.
(D) Distribution of CD4+ and CD8a+ T cell subsets in spleens from Cgas−/− BM (n = 14), Cgas−/− + dKO BM (n = 14), and dKO BM mice (n = 16).
(E) IFN-γ and TNF-α staining profiles of gated CD4+ T cells of PMA plus ionomycin stimulated splenocytes from Cgas−/− BM (n = 14), Cgas−/− + dKO BM (n = 14) and dKO BM mice (n = 16). Values indicate percentage of CD4+ T cells (mean).
(F) Frequency of regulatory Foxp3+CD25+ cells among CD4+ T cells in spleens from Cgas−/− BM (n = 14), Cgas−/− + dKO BM (n = 14), and dKO BM mice (n = 16).
Bar graphs show mean + SEM. Data are pooled from four (A, D, E) or two (B, C) independent experiments. Statistically significant differences were determined by two-tailed unpaired Mann-Whitney U test (∗p < 0.05, ∗∗p < 0.01).
See also Figure S3.
These observations demonstrate that, although cGAMP transfer increased overall T cell abundance, it did not provoke spontaneous activation or differentiation of T cells.
Transmission of cGAMP is unable to induce B cell differentiation into plasmablasts and autoantibody production
The activation of self-reactive B cells and induction of autoantibody formation is a defining characteristic of autoimmunity and readily detectable in Trex1 deficiency. Thus, we investigated whether cGAMP shuttling facilitates B cell autoimmunity. First, we determined splenic B cell numbers and found that B cells were elevated in Cgas−/− BM mice compared with dKO mice (Figure 4A). Sca-1 protein expression levels were higher in B cells from Cgas−/− BM and Cgas−/− + dKO BM mice than in their counterparts from dKO mice (Figures 4B and 4C). We then enumerated splenic antibody-forming cells (AFCs), identifying them as CD138+CD44+intracellular-kappahiBlimp1+ cells by flow cytometry (Figure 4D). Of note, AFCs in spleens of Trex1−/− mice showed robust proliferation and had a high turnover rate (Figure 4E). They therefore corresponded to short-lived plasmablasts rather than long-lived plasma cells. There was a trend toward more plasmablasts in Cgas−/− BM and Cgas−/− + dKO BM mice than in dKO controls (Figure 4F), but this effect did not reach statistical significance despite the large number of mice analyzed (n = 14–16). Trex1−/− mice produce autoantibodies that bind cardiac proteins (Gall et al., 2012) and contents of the cell nucleus. To quantitate serum IgG autoantibodies against cardiac myosin and nucleosomes we performed ELISAs (Figures 4G and 4H). Both autoantibody specificities were invariably detectable in sera from MRL.Faslpr mice, a polygenic model of systemic lupus, and Trex1−/− mice. Serum concentrations in the BM transplant models, however, were not different from those in non-autoimmune WT mice. Furthermore, the indirect immune fluorescence test on HEp-2 cells did not show IgG antinuclear antibodies in BM transplanted mice but did so in MRL.Faslpr and Trex1−/− mice (Table S3). cGAMP shuttling thus failed to recapitulate the self-directed humoral response in Trex1−/− mice.
Figure 4.

Transmission of cGAMP is unable to induce B cell differentiation into plasmablasts and autoantibody production
(A) Number of B cells in spleens from Cgas−/− BM (n = 14), Cgas−/− + dKO BM (n = 14), and dKO BM mice (n = 16).
(B) Mean fluorescence intensity (MFI) for Sca-1 of splenic B cells in Cgas−/− BM (n = 7), Cgas−/− + dKO BM (n = 7), and dKO BM mice (n = 6).
(C) Representative Sca-1 staining histograms of splenic B cells.
(D) Contour plots show the gating strategy for B cells (orange gate) and AFCs (green gate). The histogram illustrates expression of Blimp-1, which drives the terminal differentiation of B cells to plasma cells, in B cells and AFCs.
(E) Percentage of splenic AFCs that are proliferating (Ki67+) or undergoing cell death (EMA+) in Trex1−/− mice (n = 5).
(F) Total numbers of AFCs in spleens from Cgas−/− BM (n = 14), Cgas−/− + dKO BM (n = 14), and dKO BM mice (n = 16).
(G and H) Serum concentrations of anti-nucleosome IgG (G) and anti-myosin IgG (H) in Cgas−/− BM, Cgas−/− + dKO BM, dKO BM, Trex1−/−, lupus-prone MRL.Faslpr, and WT mice. Each dot represents an individual mouse; horizontal lines represent median values.
Data in bar graphs are represented as mean + SEM. Data are pooled from four (A, F) or two (B) independent experiments. Statistically significant differences were determined by two-tailed unpaired Mann-Whitney U test (∗p < 0.05).
Overall, our results argue against the notion that cGAMP-mediated bystander activation triggers autoantibody production.
cGAMP travel inhibits lymphocyte and myeloid cell death
cGAMP augments antiviral immunity after vaccination, and this adjuvanticity has been ascribed to its ability to mature DCs (Gutjahr et al., 2019; Li et al., 2016; Wang et al., 2017). Based on our finding that intercellular cGAMP transmission induced IRF3 signaling in cDCs (Figures 1B and 1C), it could be assumed that this would also license them to effectively present antigen. However, we did not find evidence that cGAMP travel promoted priming of self-reactive T cells (Figure 3). Hence, the effect of cDC stimulation by cGAMP transfer on cDC maturation was uncertain. Unexpectedly, increased IRF3 signaling in Cgas−/− BM and Cgas−/− + dKO BM did not lead to the upregulation of MHC class II and the costimulatory ligand CD86 (Figure 5A). Thus, cell-extrinsic cGAMP did not convert immature cDCs into immunologically competent antigen-presenting cells in Trex1 deficiency.
Figure 5.

cGAMP travel inhibits lymphocyte and myeloid cell death
(A) Representative staining histograms for MHC-II and CD86 of gated cDCs from Cgas−/− BM (n = 9), Cgas−/− + dKO BM (n = 10), and dKO BM mice (n = 11). Values indicate the mean of the mean fluorescence intensity (MFI).
(B) Number of myeloid cell subsets in spleens from Cgas−/− BM (n = 14), Cgas−/− + dKO BM (n = 14), and dKO BM mice (n = 16).
(C) Percentage of dying cells (FSCloFITC-VAD-FMK+) among lymphocyte and myeloid cell subsets in spleens from Cgas−/− BM (n = 6) and dKO BM mice (n = 6).
(D) Flow cytometry plots show gating of dying T cells in spleens of Cgas−/− BM and dKO BM mice. Numbers indicate percentage of T cells (mean). Results are summarized in (C, left).
(E) Splenocytes from WT and Sting1gt/gt mice were treated with various concentrations of cGAMP (0, 0.2, 1.0, 10, and 50 μg/mL). After 12h, dying cells (FSCloFITC-VAD-FMK+) were quantified by flow cytometry.
Data in (A–D) are pooled from three (A), four (B) or two (C and D) independent experiments. Bar graphs show mean + SEM. Data in (E) display mean ± SEM of technical replicates (n = 4) of one representative experiment out of two. Statistically significant differences were determined by two-tailed unpaired Mann-Whitney U test (∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001).
See also Figure S4.
Because cGAMP shuttling increased T and B cell numbers in the spleen, we asked whether it also led to quantitative changes in the myeloid compartment. Numbers of cDCs1 and cDCs2 in spleen were markedly elevated in Cgas−/− BM compared with dKO BM mice, whereas pDC numbers were similar in all groups (Figure 5B). Neutrophil and classical monocyte numbers were also higher in Cgas−/− BM than in dKO BM mice (Figure 5B). As expected from the immune cell subset data, the total number of spleen cells in Cgas−/− BM mice was larger than in dKO BM mice, but spleen weight remained the same (Figures S4A and S4B), To understand how cGAMP transfer controls the abundance of different immune cell populations, we investigated its effect on cell survival. We used the FITC-labeled pan-caspase inhibitor VAD-FMK and scatter characteristics to detect dying cells by flow cytometry. Unexpectedly, in Cgas−/− BM mice splenic T cells, B cells, cDCs, and neutrophils underwent cell death at a considerably lower rate than in dKO BM mice (Figures 5C and 5D). Direct administration of cGAMP to WT splenocyte cultures moderately promoted B and T cell survival at low concentrations (Figure 5E). This effect was lost or reversed at high concentrations. In neutrophils, cGAMP stimulated survival over the entire dose range tested. As expected, cGAMP had no effect on cell death in Sting1gt/gt splenocyte cultures. We also assessed proliferative activity by staining for Ki67 but did not find differences between the BM transplant models (Figures S4C and S4D).
These data demonstrate that cGAMP transmission stimulates survival of several lymphoid and myeloid cell subsets in the spleen, thereby promoting their accumulation.
Intercellular cGAMP passage induces interface dermatitis after UV light exposure
Trex1−/− mice spontaneously develop multiorgan inflammation, which is evident on a C57BL/6 background already at the age of 6 weeks. Cardiac and tongue muscle are reproducibly affected. To establish the impact of cGAMP-mediated bystander activation on organ disease, we scored severity of myocarditis and glossitis by histology. At 12 weeks after transplantation all groups showed only minimal signs of inflammation compared with Trex1−/− mice (Figures 6A–6D and S5A–S5D). There was no discernible difference between the bone marrow transplant models (Figures 6C and 6D). In order not to miss subtle differences in immune cell infiltration or composition, we additionally quantified immune cell infiltration in the heart by flow cytometry (Figures S5E and S5F), which was similar in all analyzed groups.
Figure 6.

Intercellular cGAMP passage induces interface dermatitis after UV light exposure
(A and B) Representative images of H&E-stained sections of hearts (A) and tongues (B) from Cgas−/− BM, Cgas−/− + dKO BM, and dKO BM mice. Scale bars equal 50 μm for hearts and 100 μm for tongues.
(C and D) Inflammation in hearts (C) and tongues (D) was scored from 1 to 3. Shown are scores of Cgas−/− BM, Cgas−/− + dKO BM, and dKO BM mice.
(E) Representative images of H&E-stained skin sections from Cgas−/− BM, Cgas−/− + dKO BM, and dKO BM mice after UV stimulation. Yellow arrows highlight areas of cell-poor interface dermatitis with vacuolization at the dermoepidermal junction as it occurs in cutaneous lupus erythematosus. Scale bar equals 50 μm.
(F) Interface dermatitis was scored from 0 to 3. Shown are scores of Cgas−/− BM, Cgas−/− + dKO BM, and dKO BM mice.
(G) Representative images of Ly6G/Ly6C (Gr-1) staining (pink) of skin sections from Cgas−/− BM, Cgas−/− + dKO BM, and dKO BM mice after UV stimulation. Scale bar equals 20 μm.
In scatter dot plots each dot depicts an individual mouse and horizontal lines represent median values. Statistically significant differences were determined by two-tailed unpaired Mann-Whitney U test (∗p < 0.05, ∗∗∗p < 0.001).
See also Figures S5 and S6.
Oxidized DNA is less sensitive to Trex1 degradation, and sunlight is a major source of oxidation damage to DNA in the skin (Gehrke et al., 2013). UV light causes lupus erythematosus-like skin lesions with interface dermatitis in Trex1-deficient mice (Scholtissek et al., 2017). We therefore investigated whether the passage of cGAMP between cells could trigger UV-dependent autoimmune dermatitis. It was striking that, after UV exposure, only Cgas−/− BM and Cgas−/− + dKO BM but not dKO BM mice displayed clear histological signs of cell-poor interface dermatitis with basal hydropic degeneration of the epidermis (Figures 6E and 6F). Skin areas of Cgas−/− BM and Cgas−/− + dKO BM with immune cell infiltrates stained positive for Ly6G/Ly6C (Gr-1) antigen, which labels neutrophils and monocytes (Figure 6G).
Because skin pathology in Cgas−/− BM mice required UV light we revisited DC maturation and T cell priming after UV stimulation. The frequency of migratory and resident cDC subsets in skin draining lymph nodes did not vary between UV-treated Cgas−/− BM and dKO BM mice (Figure S6A). Furthermore, in both groups of mice MHC-II and CD86 expression levels of cDCs in skin draining lymph nodes and spleens were similar (Figures S6B and S6C). We also did not detect differences in T cell activation (Figure S6D) or differentiation into IFN-γ- and TNF-α-producing effector cells (Figure S6E) between Cgas−/− BM and dKO BM mice after UV exposure, suggesting that interface dermatitis developed independently of T cell priming.
Taken together, UV light stimulation led to lupus erythematosus-like skin pathology that was dependent on cell-to-cell cGAMP transmission.
Discussion
A great number of studies have implicated bystander activation in the development of autoimmunity (Pacheco et al., 2019; Smatti et al., 2019). However, the precise contributions of specific modes of bystander activation are not very well understood. Bystander activation occurs through a variety of mechanisms. These mechanisms include secretion of cytokines (e.g., IL-1β), release of nucleic acids from damaged tissues, ejection of polymeric particles formed by the adaptor protein apoptosis-associated speck-like protein (ASC), exosomal transfer of viral RNA and Ca2+ influx through gap junctions (Holmgren et al., 2017). Here, we evaluated whether dissemination of innate immunity by intercellular travel of the second messenger molecule cGAMP causes autoimmune disease. We provide evidence that, in Trex1 deficiency, transmission of cGAMP leads to innate immune signaling, IFN induction, and, in the presence of environmental triggers, tissue inflammation.
In our study, we tested whether cGAMP transmission has the ability to activate primary immune cells in vivo in autoimmunity. It was intriguing that we found that cell-extrinsic cGAMP induces IRF3 signaling in cDCs and pDCs in Trex1−/−-associated autoimmunity. We surveyed other cell types that are potential recipients of cGAMP, such as red pulp macrophages, neutrophils, and B and T cells, but did not detect increased IRF3 phosphorylation in these populations. However, we cannot exclude subtle increases in IRF3 signaling activity below the detection threshold of our assay.
We also addressed the question as to which cell types deliver cGAMP to immune cells, such as cDCs in Trex1 deficiency. The finding that innate immune induction was clearly detectable in Cgas−/− BM animals, in which Cgas−/− bone marrow was transplanted into dKO mice, establishes that immune cells receive cGAMP from radioresistant cells. These largely consist of non-hematopoietic cells and also include some residual radioresistant hematopoietic cells, for example, certain tissue-resident macrophages and mast cells. Of interest, Cgas−/− + dKO BM mice, which were reconstituted with a mixture of dKO and Cgas−/− bone marrow, and thus had a substantially larger number of hematopoietic cells that might provide cGAMP to neighboring cells compared with Cgas−/− BM mice, did not show more robust immune activation than Cgas−/− BM animals. In fact, Cgas−/− + dKO BM mice displayed an ameliorated phenotype probably due to the 50% lower frequency of cGAMP-responsive Cgas−/− cells in the hematopoietic compartment. If hematopoietic cells were the primary source of cGAMP for bystander activation of immune cells, one would have expected a more pronounced immune response in Cgas−/− + dKO BM compared with Cgas−/− BM mice. However, this was not the case, suggesting that immune cells are activated by cGAMP derived from non-hematopoietic cells. This notion is consistent with data demonstrating that Trex1−/− non-hematopoietic cells such as keratinocytes and fibroblasts spontaneously produce IFN-β (Gall et al., 2012; Peschke et al., 2016).
The ability of DCs to regulate T cell immunity depends on their maturation state. cGAMP induces DC maturation, and it is an effective adjuvant for potentiating an antiviral CD8+ T cell response after vaccination (Gutjahr et al., 2019; Li et al., 2016; Wang et al., 2017). Surprisingly, in our study we found that, although DCs exhibited spontaneous IRF3 signaling due to cGAMP-mediated bystander activation, they did not evolve into mature, immunologically competent antigen-presenting cells. Consistent with this, intercellular cGAMP transmission did not promote CD4+ or CD8+ T cell activation or differentiation. Our data thus indicate that cGAMP levels might have to cross a high threshold level to render DCs immunogenic, which would constitute an important safety mechanism, considering that it is not obvious how DCs matured via cGAMP transfer would select antigen for presentation.
In this work, cGAMP-dependent bystander activation improved survival of several immune cell subsets. In contrast, others have previously reported that STING signaling promotes cell death in a cell type-specific manner (Brault et al., 2018; Gulen et al., 2017; Larkin et al., 2017), which initially appears to contradict our findings. However, most of these studies used pharmacological agents, DNA transfection, or pathogens to activate STING with high intensity. Such robust activation is unlikely to be achieved by cGAMP transmission in our model. In Gülen et al., it was demonstrated that the outcome of STING stimulation depends on signaling strength (Gulen et al., 2017). Although T cell apoptosis was observed for the synthetic STING agonist carboxymethyl-9-acridanone (CMA) this was not the case for cGAMP without the use of a potent cell delivery system. Our in vitro data support the idea that, although cGAMP may well promote cell death at high concentrations, it nonetheless impedes it at low concentrations. Furthermore, the pro-survival effect of STING observed in Cgas−/− BM mice and cGAMP stimulated WT splenocyte cultures might also be in part indirect. It is notable that IFN-I drives myelopoiesis (Buechler et al., 2013) and delays apoptosis in human neutrophils (Aga et al., 2018). In the case of T cells, a possible scenario would be that antigen-presenting cells are initially activated by cGAMP transfer and subsequently promote T cell survival. T helper cells might then relay signals to B cells that support their persistence. Overall, it is important to emphasize that cGAMP boosts the immune response in vaccination studies rather than impairing it (Gutjahr et al., 2019; Li et al., 2016; Wang et al., 2017), in line with the important role of the cGAS-STING pathway in the antiviral response.
Although cGAMP shuttling did not cause organ inflammation under steady-state conditions, it did mediate interface dermatitis after UV exposure. The pathogenesis of interface dermatitis is not well understood. There is evidence for the involvement of cytotoxic T cells mediating keratinocytic cell death (Wenzel, 2019). However, even after UV exposure there were no differences in DC maturation and T cell priming between Cgas−/− BM and dKO BM mice. Of note, Page et al. have shown that interface dermatitis can develop in mice lacking any lymphocytes (Page et al., 2010). In this study, interface dermatitis was characterized by abundant macrophage infiltration. Similarly, dermoepidermal immune cell infiltrates in UV-treated Cgas−/− BM and Cgas−/− + dKO BM mice stained positive for the neutrophil and monocyte marker Ly6G/Ly6C. We interpret this to mean that recruitment of T cells to the skin is a late event in interface dermatitis and is not required in the early stages of disease.
In our study, cGAMP travel facilitated neither B cell differentiation into AFCs nor autoantibody formation. Several explanations may account for this observation. First, B cells might not serve as recipient cells for cGAMP. Second, B cell-intrinsic STING signaling might not lead to the activation of self-reactive B cells. There are limited data available regarding the role of cGAS-STING in B cells. An early report indicated that B cell-expressed cGAS was critically involved in T-independent B cell responses, but this article was later retracted (Zeng et al., 2014). Later work revealed that B cells can be directly activated by cyclic-di-GMP (CDG) in a STING-dependent manner (Walker et al., 2018). Mice deficient for STING selectively in B cells had reduced antigen-specific IgG and IgA serum concentrations after intranasal immunization with ovalbumin and CDG, although globally STING-deficient mice had a much more pronounced decrement. Unlike for TLR-driven models of autoimmunity in which it has been shown that autoantibody production entirely relies on B cell-intrinsic signaling of the TLR adaptor MyD88 (Teichmann et al., 2013), the contributions of B cell-intrinsic cGAS-STING signaling to the humoral response in STING-mediated autoimmunity remain to be specified.
In Cgas−/− BM mice cGAMP is produced by radioresistant host dKO cells. A caveat of our study is that inactivating STING in Trex1-deficient cells may increase cGAMP levels above those that are present in Trex1 deficiency alone, possibly because STING facilitates the clearance of cGAMP (Gao et al., 2015). It is conceivable that these levels exceed those normally occurring in autoimmunity, which may have contributed to the phenotype observed in Cgas−/− BM mice. However, to what extent STING deletion raises cGAMP concentrations likely depends on the native STING expression level of the respective cell type. More importantly, there is also reason to assume that the phenotype in Cgas−/− BM mice underestimates the consequences of cGAMP transfer. In our setting, all radioresistant cells are double deficient for Trex1 and STING. Thus, cGAMP transfer in between radioresistant cells was excluded as a mechanism of immune activation.
In summary, we have established that, in cGAS-dependent autoimmune disease, cGAMP-mediated bystander activation serves as a mechanism to broaden innate immune activation, which can lead to organ inflammation when provocation factors, such as UV light, are encountered. However, we also found that intercellular cGAMP transfer was insufficient to induce adaptive immunity against self. This highlights the stringent requirements that have to be met for the activation of autoreactive T and B cells. Altogether, our findings define the pathological consequences of cGAMP transmission in the development of systemic autoimmunity and provide a rationale to target it therapeutically.
Limitations of the study
In the study presented here, we provide comprehensive evidence for immune activation by intercellular cGAMP transfer in Trex1-related autoimmunity. A limitation of the work is that it was not possible to directly measure cGAMP in Cgas−/− bystander cells. Detection of cGAMP in complex biological samples remains a major challenge, especially in chronic diseases with only low-level activation of cGAS. We and others have successfully detected cGAMP in hearts from Trex1−/− mice (Gao et al., 2015). Because the heart is the most affected organ in Trex1−/− mice, it is reasonable to assume that cGAMP production in this organ must be particularly high, facilitating detection. Here, we sought to measure cGAMP in bone marrow-transplant derived hematopoietic cells from dKO BM and Cgas−/− BM mice. Using two methods, ELISA and LC/MS-analysis, cGAMP could not be detected in splenocytes or peripheral blood mononuclear cells in either group of mice. This shows that, with the methods used, cGAMP levels are already below the limit of detection in hematopoietic cGAMP-producing dKO cells and accordingly, of course, in Cgas−/− bystander cells. For future research and clinical use, it will be important to develop assays for cGAMP measurement that combine high sensitivity and low susceptibility to interference.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse Kappa, IC (BV421) | Biolegend | Cat#409511 RRID:AB_2563586 |
| Anti-mouse Kappa, SF (unconj.) | Biolegend | Cat#409502 RRID:AB_2563297 |
| Blimp1 (AF647) | Biolegend | Cat#150003 RRID:AB_2565617 |
| Bst-2 (eFlour 450) | eBioscience | Cat#48-3172-80 RRID:AB_2043880 |
| Cardiac myosin heavy chain antibody (ELISA standard) | Thermo Fisher | Cat#MA1-26180 RRID:AB_2147173 |
| CD4 (PE/Cy7) | Biolegend | Cat#100421 RRID:AB_312706 |
| CD4 (BV421) | Biolegend | Cat#100437 RRID:AB_10900241 |
| CD8a (PE) | Life Technologies | Cat#MCD0804 RRID:AB_10373415 |
| CD8a (APC/Cy7) | Biolegend | Cat#100713 RRID:AB_312752 |
| CD8a (BV421) | Biolegend | Cat#100753 RRID:AB_2562558 |
| CD11b (BV510) | Biolegend | Cat#101263 RRID:AB_2629529 |
| CD11c (AF488) | Biolegend | Cat#117313 RRID:AB_492849 |
| CD11c (PE/Cy7) | Biolegend | Cat#117317 RRID:AB_493569 |
| CD19 (PerCP-Cy5.5) | Biolegend | Cat#115534 RRID:AB_2072925 |
| CD19 (PE/Cy7) | Biolegend | Cat#115519 RRID:AB_313654 |
| CD19 (AF647) | Biolegend | Cat#115525 RRID:AB_493340 |
| CD26 (PE) | Biolegend | Cat#137804 RRID:AB_2293047 |
| CD44 (AF488) | Biolegend | Cat#103016 RRID:AB_493679 |
| CD44 (APC/Cy7) | Biolegend | Cat#103027 RRID:AB_830784 |
| CD44 (BV510) | Biolegend | Cat#103043 RRID:AB_2561391 |
| CD45 (APC/Cy7) | Biolegend | Cat#103116 RRID:AB_312981 |
| CD62L (APC/Cy7) | Biolegend | Cat#104428 RRID:AB_830799 |
| CD86 (FITC) | Biolegend | Cat#105005 RRID:AB_313148 |
| CD115 (PE) | Biolegend | Cat#135505 RRID:AB_1937254 |
| CD138 (PE) | Biolegend | Cat#142503 RRID:AB_10915989 |
| CX3CR1 (BV421) | Biolegend | Cat#149023 RRID:AB_2565706 |
| F4/80 (Pacific Blue) | Biolegend | Cat#123123 RRID:AB_893487 |
| FoxP3 (PE) | eBioscience | Cat#12-5773-80 RRID:AB_465935 |
| Goat Anti-Mouse IgG (Alkaline Phosphatase) | SouthernBiotech | Cat#1030-04; RRID: AB_2794293 |
| I-A/I-E (AF488) | Biolegend | Cat#107616 RRID:AB_493523 |
| I-A/I-E (APC/Cy7) | Biolegend | Cat#107628 RRID:AB_2069377 |
| IFNγ (BV421) | Biolegend | Cat#505830 RRID:AB_2563105 |
| IκBα (PE) | Cell Signaling | Cat#7523 RRID:AB_10950821 |
| Ki67 (AF488) | Biolegend | Cat#652417 RRID:AB_2564236 |
| Ki67 (AF647) | Biolegend | Cat#652408 RRID:AB_2562139 |
| Ly6B.2 (AF647) | AbD Serotec | Cat#MCA771A647T RRID:AB_1102791 |
| Ly6G/Ly6C | BD Biosciences | Cat#550291; RRID AB_393586 |
| Ly6G (PE/Cy7) | Biolegend | Cat#127618 RRID:AB_1877261 |
| NK1.1 (PerCP-Cy5.5) | Biolegend | Cat#108728 RRID:AB_2132705 |
| PDC-Trem (PE) | Biolegend | Cat#139203 RRID:AB_10613646 |
| Phospho-IRF-3 (Ser396) (PE) | Cell Signaling Technology | Cat#83611 RRID:AB_2800022 |
| Phospho-NK-κB p65 (Ser536) | Cell Signaling Technology | Cat#3033T RRID:AB_331284 |
| PL2-3 (ELISA standard) | Gift from Mark Shlomchik, University of Pittsburgh | N/A |
| Sca-1 (Ly6A/E) (PE/Cy7) | Biolegend | Cat#108113 RRID:AB_493597 |
| SiglecH (eFlour 660) | eBioscience | Cat#50-0333-80 RRID:AB_11151702 |
| TCRß (BV421) | Biolegend | Cat#109229 RRID:AB_10933263 |
| TCRß (PerCP/Cy5.5) | Biolegend | Cat#109228 RRID:AB_1575173 |
| TNFalpha (PE) | Biolegend | Cat#506305 RRID:AB_315426 |
| Chemicals, peptides, and recombinant proteins | ||
| λ Protein Phosphatase | New England Biolabs, Ipswich, MA, USA | Cat#P0753 |
| Brefeldin A Solution (1,000X) | Biolegend | Cat#420601 |
| Cardiac myosin protein (bovine) | Cytoskeleton Inc. | Cat#MY03-A |
| 2′,3′-cGAMP | Jena Bioscience | Cat#NU-249L |
| Collagenase D | Roche, Basel, Switzerland | Cat#11088866001 |
| DNAse I | Roche | Cat#11284932001 |
| Ethidium Monoazide Bromide | Biotium, Fremont, USA | Cat#40015 |
| FITC-VAD-FMK | R&D Systems | Cat#FMK012 |
| Fixation Buffer | Biolegend, San Diego, USA | Cat#420801 |
| FoxP3 Staining Kit | Invitrogen, Carlsbad, USA | Cat#50-112-8857 |
| Histone from calf thymus | Sigma-Aldrich | Cat#H9250 |
| Ionomycin | Sigma-Aldrich | Cat#I0634 |
| NEBuffer for Protein MetalloPhosphatases | New England Biolabs, Ipswich, MA, USA | Cat#B0761 |
| iTaq Universal SYBR Green Supermix | Bio-Rad, Hercules, USA | Cat#1725120 |
| Intracellular Staining Permeabilization Wash Buffer | Biolegend | Cat#421002 |
| Phorbol-12-myristat-13-acetat (PMA) | Sigma-Aldrich | Cat#P8139 |
| Red Blood Cell Lysis Buffer | Biolegend | Cat#420301 |
| S1 Nuclease | Promega | Cat#E576B |
| SPLIT RNA extraction kit | Lexogen, Vienna, Austria | Cat#008.48 |
| Critical commercial assays | ||
| Mouse Cytokine Array C1 | RayBiotech Inc., Peachtree Corners, USA | Cat#AAM-CYT-1-2 |
| QuantSeq 3’-mRNA-Seq Library Prep Kit (FWD) | Lexogen, Vienna, Austria | Cat#015.96 |
| Deposited data | ||
| Raw and analyzed 3’-mRNA seq data | This paper | GEO: GSE176225 |
| Experimental models: organisms/strains | ||
| C57BL/6J | The Jackson Laboratory | Stock No. 000664 |
| B6;129P2-Trex1tm1Tld (Trex1-/-) | Tomas Lindahl (Cancer Research UK, London) | N/A |
| B6(C)-Cgastm1d(EUCOMM)Hmgu/J | The Jackson Laboratory | Stock No. 026554 |
| C57BL/6J-Sting1gt/J | The Jackson Laboratory | Stock No: 017537 |
| Oligonucleotides | ||
| ATA TTT CCC CCT GTG TTG GA Cgas_common_fwd |
IDT, Leuven, Belgium | N/A |
| CGG ATG GAT GAA CAA ACA GA Cgas_mutant_rev |
IDT, Leuven, Belgium | N/A |
| TTA CCT CAG TTC ATC CAC GG CCR2_fwd |
IDT, Leuven, Belgium | N/A |
| ACA TGT TGC CCA CAA AAC CA CCR2_rev |
IDT, Leuven, Belgium | N/A |
| Software and algorithms | ||
| FlowJo v9 | BD, Becton Dickinson GmbH, Heidelberg | https://www.flowjo.com/ |
| GraphPad Prism 6 | Graphpad Software, LaJolla, USA | https://www.graphpad.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Lino L. Teichmann (lino.teichmann@uni-bonn.de).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
3’-mRNA seq data have been deposited at GEO and are publicly available as of the date of publication. The accession number is GSE176225.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
Mice
B6;129P2-Trex1tm1Tld mice (Morita et al., 2004) were kindly provided by Tomas Lindahl and backcrossed at least 10 generations to C57BL/6J. C57BL/6J-Sting1gt/J and B6(C)-Cgastm1d(EUCOMM)Hmgu/J mice were purchased from Jackson Laboratories. Mice double-deficient for Trex1 and STING (Trex1-/-;Sting1gt/gt) were generated by interbreeding. Mice were maintained under special pathogen free (SPF) conditions and randomly assigned to treatment groups. Experimental procedures were performed in accordance with the German Animal Welfare Act and approved by the State Agency for Nature, Environment and Consumer Protection, NRW.
Methods details
Bone marrow transplantation
To generate bone marrow transplanted mice 10-12 wk old recipients were irradiated with 5.5 Gy x 2 with a 4h interval and injected intravenously with 1 x 107 bone marrow cells harvested from 6-8 wk old donors. Mice of both sexes were used as recipients. In all groups, care was taken to balance the ratio of female to male recipients. Male mice were used as donors to exclude that female donor cells recognize the H-Y antigens of the male recipients. Analysis was performed 12 wks after transplantation.
Preparation of single-cell suspensions
Spleens were removed, minced, pressed through a metal strainer and treated with Red Blood Cell Lysis buffer (Biolegend). After perfusion of mice with PBS, hearts were also removed, minced and digested in HBSS with 100 Mandl U/ml collagenase D (Roche) and 50 Kunitz U/ml DNase I (Sigmal-Aldrich) for 45 min at 37 °C. Tissue pieces were then pressed through a metal strainer. All cell suspensions were filtered through 70 μm Nitex nylon mesh before further use.
Flow cytometry
Surface staining was performed on ice in PBS containing 0.5% BSA and 0.05% sodium azide. Intracellular staining was carried out using the Biolegend Fixation Buffer and Permeabilization Buffer or, for Ki67 and FoxP3 staining, the Invitrogen FoxP3 Staining Kit. For intracellular cytokine staining, 1x107 splenocytes were stimulated for 5h at 37 °C with 750 ng/ml ionomycin and 20 ng/ml PMA in RPMI containing 10% FCS. After 1h of stimulation 5 mg/ml brefeldin was added to the cultures. For exclusion of dead cells ethidium monoazide bromide (EMA) was used. To determine the endogenous phosphorylation state of IRF-3 and NK-kB p65 Phospho-Flow analysis was performed as previously described (Teichmann et al., 2015). Spleens were directly disrupted in RPMI containing 1% paraformaldehyde. Fixed cells were then permeabilized with 90% methanol and washed. Aliquots of all samples were incubated with λ protein phosphatase (New England Biolabs). Cells were stained with antibodies against phospho-IRF-3 (Ser396) or phospho-NK-κB p65 (Ser536) (Cell Signaling). Cells were analyzed on a FACS Canto II (BD) or sorted on a FACS Aria II (BD). Antibody clones are detailed in the key resources table.
ELISAs and cytokine array
Serum anti-nucleosome IgG concentrations were determined by ELISA as previously described (Nickerson et al., 2010). Polystyrene plates were coated with 10 μg/mL poly-L-lysine. Plates were then incubated with 15 μg/mL phenol-extracted and S1 nuclease-treated (Promega) calf thymus dsDNA, which was followed by 10 μg/mL calf thymus histones type II-A (Sigma Aldrich). For the anti-cardiac myosin IgG ELISA, polystyrene plates were coated with 10 μg/mL bovine cardiac myosin protein (Cytoskeleton). After blocking with 1% BSA in PBS, serum was applied in serial dilutions and specific antibodies were detected by alkaline phosphatase conjugated goat anti-mouse IgG. P-nitrophenyl phosphate (Sigma Aldrich) was used as substrate and absorbance was read at 405/630 nm. The nucleosome-specific antibody clone PL2-3 and anti-cardiac myosin heavy chain antibody clone 3-48 (Thermo Fisher) were used as standards. The Mouse Cytokine Array C1 (RayBiotech) was performed according to the manufacturer’s protocol.
3’-mRNA seq and gene expression analysis
For 3’-mRNA seq RNA was extracted from spleen or bone marrow single cell suspensions using the SPLIT RNA Extraction kit (Lexogen). Sequencing was performed with 200 ng of RNA per library on a HiSeq 2500 v4 machine (Illumina) with 1 x 50 bp, single read, employing the Lexogen QuantSeq 3’-mRNA-Seq Library Prep Kit (FWD). Pre-processing of 3’-mRNA seq data was performed using the Partek Genomics Suite (PGS, build 8.0.19.0819). Briefly, adapter-trimmed raw reads were uploaded to PGS, trimmed for a Phred quality score > 20 and aligned to the mouse reference genome mm10 by STAR (version 2.6.1) with slightly modified standard settings (max mismatches = 14) as recommended by Lexogen (Dobin et al., 2013). For mRNA quantification mm10 Ensembl transcripts (release 98) were used as annotation files. Afterwards, raw gene counts were normalized and differentially expressed genes (defined by p value < 0.05) identified by the DESeq2 (package version 1.25.16) algorithm in R (Love et al., 2014). Heatmaps and several plots were generated using the ggplot2 library. Principal component analysis was performed using the ggbiplot library. For gene set enrichment analysis (GSEA) a pre-ranked list was generated based on a metric score calculated by: and applied to the GSEA java desktop application (version 4.0.3) provided by the Broad Institute (Subramanian et al., 2005).
qPCR on genomic DNA
To analyze the genetic composition of bone marrow transplanted mice based on relative Cgas content, genomic DNA was extracted from FACS purified cells and used as template. qPCR was performed using the iTaq Universal SYBR Green Supermix (Bio-Rad) on a realplex2 Cycler (Eppendorf). The amount of Cgas in each sample was normalized to the unaffected gene Ccr2. Primer sequences are listed in the key resources table.
UV stimulation
To induce dermatitis, mice 12 wks after bone marrow transplantation were shaved on the back and subjected to UV irradiation on 3 consecutive days with 450 mJ/cm2 of UVB per day using the UV 801 KL system (Waldmann) equipped with UV21 UVB lamps (Philips). Skin biopsy samples were taken after sacrificing the animals on day 12 after start of UV exposure.
Histology and immunohistochemistry
Tissues were formalin-fixed, embedded in paraffin and sectioned. Organ inflammation was scored in a blinded fashion on hematoxylin and eosin stained sections. For hearts and tongues, inflammation was scored from 0 to 3, where 0 is no inflammation, 1 depicts slight inflammation with few scattered infiltrates, 2 is medium inflammation characterized by several scattered small infiltrates and less than three large infiltrates, and 3 represents extensive inflammation with many scattered small infiltrates and three or more large infiltrates or diffuse inflammation. For skin, interface dermatitis including basal hydropic degeneration of the epidermis and colloid bodies was scored from 0 to 3 (0 none, 3 severe). Immunohistochemistry was performed using an antibody specific for Ly-6G/Ly-6C (Gr-1).
Quantification and statistical analysis
Statistical analysis was performed by GraphPad (Prism) software to determine p values by two-tailed unpaired Mann-Whitney U test. p values are indicated throughout as ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Independent experiments were pooled and analyzed together whenever possible as detailed in figure legends.
Acknowledgments
We thank HET (Haus für Experimentelle Therapie) for outstanding animal husbandry. This study was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy–EXC2151–390873048 of which E.B. and L.L.T are members. It was also supported by other grants of the DFG, including Te 785/3-1 (B.B.J. and L.L.T.), TRR237-369799452 (C.K., E.B., and L.L.T.), TRR57 (P. Boor), TRR219 (P. Boor), Bo 3755/3-1 (P. Boor), Bo 3755/6-1 (P. Boor), as well as the German Federal Ministry of Education and Research (BMBF: STOP-FSGS-01GM1901A to P. Boor) and the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant 741912 to T.C.). We thank Gunther Hartmann and Mark Shlomchik for helpful discussion and critical reading of the manuscript.
Author contributions
B.B.J. and L.L.T. designed the experiments, analyzed the data, and wrote the manuscript. B.B.J., K.C., C.K., R.-M.K., and M.W. performed experiments. P. Boor, P.A., J.L., and J.W. performed the histological analysis. C.M.G. performed the bioinformatic analysis on 3′-mRNA seq data. P. Brossart and T.C. provided resources. E.B. provided feedback on and edited the manuscript. L.L.T. conceived and supervised the study. All authors had the opportunity to discuss the results and comment on the manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We worked to ensure sex balance in the selection of non-human subjects.
Published: August 20, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102833.
Supplemental information
References
- Ablasser A., Schmid-Burgk J.L., Hemmerling I., Horvath G.L., Schmidt T., Latz E., Hornung V. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature. 2013;503:530–534. doi: 10.1038/nature12640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ablasser A., Hemmerling I., Schmid-Burgk J.L., Behrendt R., Roers A., Hornung V. TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J. Immunol. 2014;192:5993–5997. doi: 10.4049/jimmunol.1400737. [DOI] [PubMed] [Google Scholar]
- Aga E., Mukherjee A., Rane D., More V., Patil T., van Zandbergen G., Solbach W., Dandapat J., Tackenberg H., Ohms M. Type-1 interferons prolong the lifespan of neutrophils by interfering with members of the apoptotic cascade. Cytokine. 2018;112:21–26. doi: 10.1016/j.cyto.2018.06.027. [DOI] [PubMed] [Google Scholar]
- Brault M., Olsen T.M., Martinez J., Stetson D.B., Oberst A. Intracellular nucleic acid sensing triggers necroptosis through synergistic type I IFN and TNF signaling. J. Immunol. 2018;200:2748–2756. doi: 10.4049/jimmunol.1701492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridgeman A., Maelfait J., Davenne T., Partridge T., Peng Y., Mayer A., Dong T., Kaever V., Borrow P., Rehwinkel J. Viruses transfer the antiviral second messenger cGAMP between cells. Science. 2015;349:1228–1232. doi: 10.1126/science.aab3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buechler M.B., Teal T.H., Elkon K.B., Hamerman J.A. Cutting edge: type I IFN drives emergency myelopoiesis and peripheral myeloid expansion during chronic TLR7 signaling. J. Immunother. 2013;190:886–891. doi: 10.4049/jimmunol.1202739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q., Boire A., Jin X., Valiente M., Er E.E., Lopez-Soto A., Jacob L.S., Patwa R., Shah H., Xu K. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. 2016;533:493–498. doi: 10.1038/nature18268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow Y.J., Manel N. Aicardi-Goutières syndrome and the type I interferonopathies. Nat. Rev. Immunol. 2015;15:429–440. doi: 10.1038/nri3850. [DOI] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunphy G., Flannery S.M., Almine J.F., Connolly D.J., Paulus C., Jønsson K.L., Jakobsen M.R., Nevels M.M., Bowie A.G., Unterholzner L. Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF-κB signaling after nuclear DNA damage. Mol. Cell. 2018;71:745–760.e5. doi: 10.1016/j.molcel.2018.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidt M.M., Ebert T.S., Chauhan D., Ramshorn K., Pinci F., Zuber S., O’Duill F., Schmid-Burgk J.L., Hoss F., Buhmann R. The DNA inflammasome in human myeloid cells is initiated by a STING-cell death program upstream of NLRP3. Cell. 2017;171:1110–1124.e18. doi: 10.1016/j.cell.2017.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall A., Treuting P., Elkon K.B., Loo Y.M., Gale M., Barber G.N., Stetson D.B. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity. 2012;36:120–131. doi: 10.1016/j.immuni.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D., Li T., Li X.-D.D., Chen X., Li Q.-Z.Z., Wight-Carter M., Chen Z.J. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc. Natl. Acad. Sci. U S A. 2015;112:E5699–E5705. doi: 10.1073/pnas.1516465112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrke N., Mertens C., Zillinger T., Wenzel J., Bald T., Zahn S., Tüting T., Hartmann G., Barchet W. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity. 2013;39:482–495. doi: 10.1016/j.immuni.2013.08.004. [DOI] [PubMed] [Google Scholar]
- Gentili M., Kowal J., Tkach M., Satoh T., Lahaye X., Conrad C., Boyron M., Lombard B., Durand S., Kroemer G. Transmission of innate immune signaling by packaging of cGAMP in viral particles. Science. 2015;349:1232–1236. doi: 10.1126/science.aab3628. [DOI] [PubMed] [Google Scholar]
- Gulen M.F., Koch U., Haag S.M., Schuler F., Apetoh L., Villunger A., Radtke F., Ablasser A. Signalling strength determines proapoptotic functions of STING. Nat. Commun. 2017;8:427. doi: 10.1038/s41467-017-00573-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutjahr A., Papagno L., Nicoli F., Kanuma T., Kuse N., Cabral-Piccin M.P., Rochereau N., Gostick E., Lioux T., Perouzel E. The STING ligand cGAMP potentiates the efficacy of vaccine-induced CD8+ T cells. JCI Insight. 2019;4:e125107. doi: 10.1172/jci.insight.125107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren A.M., McConkey C.A., Shin S. Outrunning the Red Queen: bystander activation as a means of outpacing innate immune subversion by intracellular pathogens. Cell Mol. Immunol. 2017;14:14–21. doi: 10.1038/cmi.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin B., Ilyukha V., Sorokin M., Buzdin A., Vannier E., Poltorak A. Cutting edge: activation of STING in T cells induces type I IFN responses and cell death. J. Immunother. 2017;199:397–402. doi: 10.4049/jimmunol.1601999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P.Y., Kumagai Y., Li Y., Takeuchi O., Yoshida H., Weinstein J., Kellner E.S., Nacionales D., Barker T., Kelly-Scumpia K. TLR7-dependent and FcγR-independent production of type I interferon in experimental mouse lupus. J. Exp. Med. 2008;205:2995–3006. doi: 10.1084/jem.20080462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T., Cheng H., Yuan H., Xu Q., Shu C., Zhang Y., Xu P., Tan J., Rui Y., Li P. Antitumor activity of cGAMP via stimulation of cGAS-cGAMP-STING-IRF3 mediated innate immune response. Sci. Rep. 2016;6:19049. doi: 10.1038/srep19049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15 doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luteijn R.D., Zaver S.A., Gowen B.G., Wyman S.K., Garelis N.E., Onia L., McWhirter S.M., Katibah G.E., Corn J.E., Woodward J.J. SLC19A1 transports immunoreactive cyclic dinucleotides. Nature. 2019;573:434–438. doi: 10.1038/s41586-019-1553-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus A., Mao A.J., Lensink-Vasan M., Wang L., Vance R.E., Raulet D.H. Tumor-derived cGAMP triggers a STING-mediated interferon response in non-tumor cells to activate the NK cell response. Immunity. 2018;49:754–763.e4. doi: 10.1016/j.immuni.2018.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M., Stamp G., Robins P., Dulic A., Rosewell I., Hrivnak G., Daly G., Lindahl T., Barnes D.E. Gene-targeted mice lacking the Trex1 (DNase III) 3’-->5’ DNA exonuclease develop inflammatory myocarditis. Mol. Cell. Biol. 2004;24:6719–6727. doi: 10.1128/MCB.24.15.6719-6727.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson K.M., Christensen S.R., Shupe J., Kashgarian M., Kim D., Elkon K., Shlomchik M.J. TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J. Immunol. 2010;184:1840–1848. doi: 10.4049/jimmunol.0902592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco Y., Acosta-Ampudia Y., Monsalve D.M., Chang C., Gershwin M.E., Anaya J.M. Bystander activation and autoimmunity. J. Autoimmun. 2019;103:102301. doi: 10.1016/j.jaut.2019.06.012. [DOI] [PubMed] [Google Scholar]
- Page A., Navarro M., Garín M., Pérez P., Casanova M.L., Moreno R., Jorcano J.L., Cascallana J.L., Bravo A., Ramírez A. IKKbeta leads to an inflammatory skin disease resembling interface dermatitis. J. Invest. Dermatol. 2010;130:1598–1610. doi: 10.1038/jid.2010.28. [DOI] [PubMed] [Google Scholar]
- Peschke K., Achleitner M., Frenzel K., Gerbaulet A., Ada S.R., Zeller N., Lienenklaus S., Lesche M., Poulet C., Naumann R. Loss of Trex1 in dendritic cells is sufficient to trigger systemic autoimmunity. J. Immunother. 2016;197:2157–2166. doi: 10.4049/jimmunol.1600722. [DOI] [PubMed] [Google Scholar]
- Schadt L., Sparano C., Schweiger N.A., Silina K., Cecconi V., Lucchiari G., Yagita H., Guggisberg E., Saba S., Nascakova Z. Cancer-cell-intrinsic cGAS expression mediates tumor immunogenicity. Cell Rep. 2019;29:1236–1248.e7. doi: 10.1016/j.celrep.2019.09.065. [DOI] [PubMed] [Google Scholar]
- Schlee M., Hartmann G. Discriminating self from non-self in nucleic acid sensing. Nat. Rev. Immunol. 2016;16:566–580. doi: 10.1038/nri.2016.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholtissek B., Zahn S., Maier J., Klaeschen S., Braegelmann C., Hoelzel M., Bieber T., Barchet W., Wenzel J. Immunostimulatory endogenous nucleic acids drive the lesional inflammation in cutaneous lupus erythematosus. J. Invest. Dermatol. 2017;137:1484–1492. doi: 10.1016/j.jid.2017.03.018. [DOI] [PubMed] [Google Scholar]
- Smatti M.K., Cyprian F.S., Nasrallah G.K., Al Thani A.A., Almishal R.O., Yassine H.M. Viruses and autoimmunity: a Review on the potential Interaction and molecular mechanisms. Viruses. 2019;11:762. doi: 10.3390/v11080762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson D.B., Ko J.S., Heidmann T., Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134:587–598. doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L., Wu J., Du F., Chen X., Chen Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichmann L.L., Schenten D., Medzhitov R., Kashgarian M., Shlomchik M.J. Signals via the adaptor MyD88 in B cells and DCs make distinct and synergistic contributions to immune activation and tissue damage in lupus. Immunity. 2013;38:528–540. doi: 10.1016/j.immuni.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichmann L.L., Cullen J.L., Kashgarian M., Dong C., Craft J., Shlomchik M.J. Local triggering of the ICOS coreceptor by CD11c(+) myeloid cells drives organ inflammation in lupus. Immunity. 2015;42:552–565. doi: 10.1016/j.immuni.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker M.M., Crute B.W., Cambier J.C., Getahun A. B cell–intrinsic STING signaling triggers cell activation, synergizes with B cell receptor signals, and promotes antibody responses. J. Immunother. 2018;201:2641–2653. doi: 10.4049/jimmunol.1701405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Hu S., Chen X., Shi H., Chen C., Sun L., Chen Z.J. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc. Natl. Acad. Sci. U S A. 2017;114:1637–1642. doi: 10.1073/pnas.1621363114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel J. Cutaneous lupus erythematosus: new insights into pathogenesis and therapeutic strategies. Nat. Rev. Rheumatol. 2019;15:519–532. doi: 10.1038/s41584-019-0272-0. [DOI] [PubMed] [Google Scholar]
- Xu S., Ducroux A., Ponnurangam A., Vieyres G., Franz S., Müsken M., Zillinger T., Malassa A., Ewald E., Hornung V. cGAS-mediated innate immunity spreads intercellularly through HIV-1 env-induced membrane fusion sites. Cell Host Microbe. 2016;20:443–457. doi: 10.1016/j.chom.2016.09.003. [DOI] [PubMed] [Google Scholar]
- Zeng M., Hu Z., Shi X., Li X., Zhan X., Li X.D., Wang J., Choi J.H., Wang K.W., Purrington T. MAVS, cGAS, and endogenous retroviruses in T-independent B cell responses. Science. 2014;346:1486–1492. doi: 10.1126/science.346.6216.1486. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhou C., Chen X., Planells-Cases R., Chu J., Wang L., Cao L., Li Z., López-Cayuqueo K.I., Xie Y., Ye S. Transfer of cGAMP into bystander cells via LRRC8 volume-regulated anion Channels augments STING-mediated interferon responses and anti-viral immunity. Immunity. 2020;52:767–781.e6. doi: 10.1016/j.immuni.2020.03.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
3’-mRNA seq data have been deposited at GEO and are publicly available as of the date of publication. The accession number is GSE176225.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.