

Summary
New strategies are urgently needed to characterize the functions of the lipid droplet (LD). Here, adiposome, an artificial LD mimetic platform, was validated by comparative in vitro bioassays. Scatchard analysis found that the binding of perilipin 2 (PLIN2) to the adiposome surface was saturable. Phosphatidylinositol (PtdIns) was found to inhibit PLIN2 binding while it did not impede perilipin 3 (PLIN3). Structural analysis combined with mutagenesis revealed that the 73rd glutamic acid of PLIN2 is significant for the effect of PtdIns on the PLIN2 binding. Furthermore, adiposome was also found to be an ideal platform for in situ enzymatic activity measurement of adipose triglyceride lipase (ATGL). The significant serine mutants of ATGL were found to cause the loss of lipase activity. Our study demonstrates the adiposome as a powerful, manipulatable model system that mimics the function of LD for binding and enzymatic activity studies of LD proteins in vitro.
Subject areas: Cell biology, Functional aspects of cell biology, Biophysics
Graphical abstract
Highlights
-
•
An artificial organelle was validated for the targeting and regulation of LD proteins
-
•
Binding affinity analysis of PLIN2 targeting on artificial LD was performed
-
•
PtdIns and the 73rd glutamic acid in PLIN2 affect the targeting of PLIN2 to LD
-
•
In situ enzymatic activity of ATGL was demonstrated and measured via artificial LD
Cell biology; Functional aspects of cell biology; Biophysics
Introduction
Constructing biomimetic models of organelles is essential for focused studies of specific biophysical and biochemical processes and is thus used in areas of biological science, material science, chemistry, and medicine (Einfalt et al., 2018; Tanner et al., 2013; Thingholm et al., 2016). Using natural or synthetic materials to construct artificial nano-assemblies that mimic the organelles has two main aims: To explore effective therapies and diagnostic strategies and to improve current understanding of molecular life processes (Hammer and Kamat, 2012; Palivan et al., 2012, 2016). Liposomes and polymersomes have been widely used to study molecular cell functions and processes in vitro, using an artificial membranous structure similar to intracellular bilayer compartments (Rideau et al., 2018). However, other than the bilayer structures, there are also monolayer phospholipid structures that maintain separation of oil and aqua phases.
Lipid droplet (LD) is a unique organelle conserved in most organisms from bacteria to humans (Martin and Parton, 2006; Zhang and Liu, 2017). The LD consists of a neutral lipid core of mostly triacylglycerol (TAG), sterol esters, retinyl ester, and/or polyhydroxyalkanoate, surrounded by a monolayer phospholipid membrane and associated proteins (Farese and Walther, 2009). It is certain that it plays a significant role in energy homeostasis, in particular lipid metabolism, storage, and transportation, but in-depth understanding of LD as an organelle is still developing (Bartz et al., 2007a; Liu et al., 2004; Walther et al., 2017; Yao et al., 2019). The specific targeting of LD-associated proteins is an unresolved issue, as well as the details of in situ enzymatic activity of LD-associated lipases.
Most LD-associated proteins are found at multiple locations in cells. However, a smaller number are resident proteins that specifically and directly localize on the organelle (Na et al., 2015; Ohsaki et al., 2014; Wolins et al., 2006; Zhang and Liu, 2019). With few exceptions, these proteins are only found on the LD surface, and therefore it is believed that they have binding mechanism(s) specific for monolayer phospholipid membranes, the nature of which is under open debate. Specific domains have been identified for targeting the LD; these are integral monotopic structures including hydrophobic domains or/and amphipathic α-helixes (Bersuker and Olzmann, 2017; Kory et al., 2016; Zehmer et al., 2008). The targeting formats include hydrophobic hairpins, terminal hydrophobic domains, and non-terminal hydrophobic domains (Boeszoermenyi et al., 2015; Huang and Huang, 2017; Na et al., 2015). They involve secondary bonds such as van der Waals force, hydrogen bond, hydrophobic interaction, charge-charge interaction, and they are proven to work on driving LD-associated protein targeting (Olarte et al., 2020; Prévost et al., 2018). These targeting domains have been identified using protein truncation mutants (Nakamura and Fujimoto, 2003). However, there is no definitive explanation to the mechanism(s) that permits selective LD binding of these proteins.
The LD is a reservoir of neutral lipids, especially TAG, and also is a site of lipid synthesis and lipolysis (Walther and Farese, 2012). Therefore, a change in lipase activity on LDs will affect cellular lipid metabolism and homeostasis. Adipose triglyceride lipase (ATGL) is a TAG hydrolytic enzyme with multiple sites of active serine (Ahmadian et al., 2011; Bartz et al., 2007b; Pagnon et al., 2012; Xie et al., 2014). Active ATGL targeting to LDs can reduce their volume, even to the point where they become undetectable, making the study of ATGL targeting and function challenging. Also, the intracellular environment makes the characterization of ATGL lipase activity difficult.
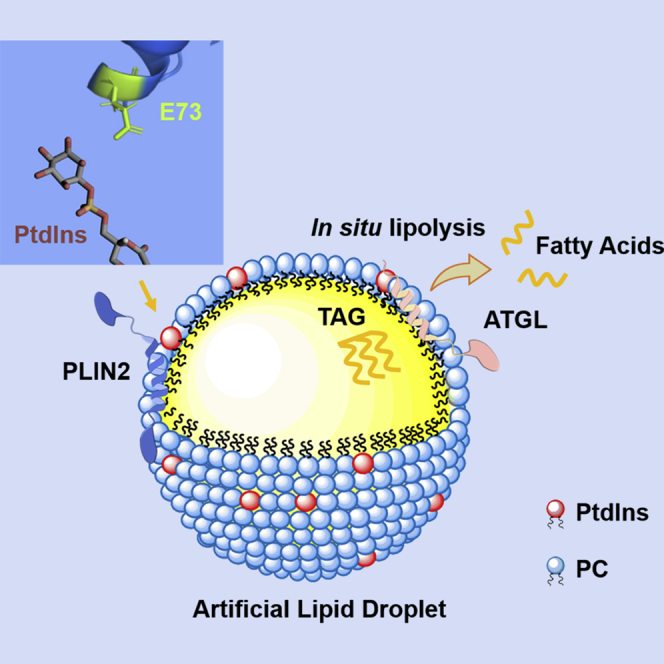
Adiposomes are artificial nanostructures containing a neutral lipid core coated with a monolayer phospholipid membrane, which can be used to mimic the structure and function of LDs to allow in vitro LD assays (Wang et al., 2016; Zhang et al., 2017). Compared to previous methods used to prepare LD-like emulsions, our technique separates adiposomes from impurities and keeps the diameter of adiposomes homogeneous, allowing the structure to more effectively model LDs (Chen et al., 2015; Fei et al., 2011; Krahmer et al., 2011; Tzen and Huang, 1992; Wang et al., 2016). Importantly, the neutral and polar lipid constituents of adiposomes can be controlled as required for a particular experiment. For example, the role of protein-phospholipid binding in membrane targeting can be explored through manipulation of the adiposome phospholipid composition (Lemmon, 2008; Yan et al., 2018). The role of phosphatidic acid in LD protein targeting has been reported (Barneda et al., 2015; Yan et al., 2018), but the role of PtdIns, which is abundant on the LD, has attracted little attention (Bartz et al., 2007a; Tauchi-Sato et al., 2002). However, PtdIns is known to be involved in protein binding in other contexts (Phan et al., 2016), so further investigation is well warranted.
The study was performed in two parts: The LD protein targeting mechanism of LD-specific proteins and TAG hydrolase activity on the adiposome platform, proving adiposome as a useful LD mimetic artificial organelle. This is the first report of the characterization of LD-specific protein binding affinity on in vitro LD model. Compared to previous studies on LD-specific protein binding mechanism, it provides new insights for quantitative dynamic analysis, which is different from the qualitative analyses such as microscopy imaging of fluoresce labeled proteins or truncations, and immunoblotting (Nakamura and Fujimoto, 2003; Rowe et al., 2016; Sletten et al., 2014). The LD resident protein PLIN2 was selected as a model for the binding affinity study, where the role of anionic PtdIns was assessed using Scatchard plotting. For the in vitro determination of TAG hydrolase activity, ATGL was used as a model protein. Our results demonstrate that the adiposomes closely mimic the actual LDs for in vitro studies of protein targeting and activity measurement.
Results
Construction of artificial lipid droplet organelle for in vitro protein binding
The preparation of adiposome (the precursor of artificial LD) followed the method reported by our group previously (Wang et al., 2016). Adiposome provided an ideal spherical model of phospholipid monolayer enveloping neutral lipids, which was close to the structure of LD. To verify the availability of adiposome on mimicking LD-associated protein targeting to LD, the targeting of GFP-tagged PLIN2 to natural LDs and to adiposomes was compared morphologically and quantitatively. A PLIN2-GFP knock-in C2C12 cell line was constructed to study the distribution of PLIN2 in vivo (Xu et al., 2019). The PLIN2-GFP knock-in cell line reduced the artifacts from the overexpression of proteins so that the proteins could be quantified. GFP-tagged PLIN2 fusion proteins appeared as ring-like structures surrounding intracellular LDs (Figure 1A), LDs isolated from cells (Figure 1B), and adiposomes (Figure 1C), confirming that both endogenous and recombinant PLIN2 were able to target to LDs and adiposomes, respectively. The size analysis of LDs and adiposomes also supported that adiposomes were able to mimic the size and shape of intracellular LD (Figure 1D). Lipid emulsions were manufactured using vortex and they were further purified to produce adiposomes (Wang et al., 2016). Figure S1A shows the morphological difference of adiposomes and lipid emulsions using TEM with ultrathin section. Clearly, lipid emulsions contained multiplayer structures and LD-like droplets, while adiposomes were almost spherical LD-like droplets. To study the interaction between LD-associated protein and LD, an artificial LD platform suitable for quantification analysis was developed. First, the average diameter of purified LDs was determined to be approximately 540.2 nm using dynamic light scattering and the number density of LD sample was determined to be approximately 2.26 × 108 per ml using field-flow fractionation multi-angle light scattering (FFF-MALS) (Figures S1B and S1D). The average diameter of DOPC adiposomes was determined to be 161.1 nm using dynamic light scattering and the number density of adiposomes was estimated as 8.53 × 109 per ml by FFF-MALS (Figures S1C and S1E). The recombinant SMT3-PLIN2-GFP was expressed, purified, and utilized to study protein binding on adiposomes. SMT3 tag was added into the sequence to increase the solubility of PLIN2-GFP in aqueous buffer. The purified proteins were analyzed by Colloidal Blue staining and Western blot (Figure S2A). The purities of SMT3-PLIN2-GFP were determined by scanning Colloidal Brilliant Blue-stained gel and measuring the intensity of individual lane using ImageJ (Schneider et al., 2012). In Figure S2A b, based on ImageJ scanning of gel bands in Figure S2A a, the purity of PLIN2-GFP (band I and II) was calculated as 59 ± 4% (mean ± SD, n = 3). In terms of Western blot and molecular weight (MW), band I was the protein SMT3-PLIN2-GFP and band II was PLIN2-GFP with no SMT3. SMT tag was not included into the LD binding domains and the fluorescence tag of protein; hence, the recombinants would not affect this targeting experiment.
Figure 1.

PLIN2 targets to lipid droplets and adiposomes
Lipid droplets or adiposomes were stained with LipidTOX Red (1:1,000, v/v) and endogenous PLIN2 or recombinant PLIN2 were labeled by GFP (green).
(A) PLIN2-GFP targets on surface of LDs. PLIN2–GFP was knocked in C2C12 cells, and the cells were treated with 100 μM oleate for 12 h and then imaged by Olympus FV1200 confocal microscope.
(B) PLIN2-GFP targets on lipid droplets isolated from C2C12 cells. The lipid droplets were isolated from C2C12 cells with the same treatment and imaged by Zeiss Image M2 microscope.
(C) Recombinant SMT3-PLIN2-GFP targets on adiposomes. The adiposomes (30 μl) prepared from DOPC and TAG were incubated with 5 μg SMT3-PLIN2-GFP at 37°C for 5 min. After reisolation, fluorescence images of adiopsomes were captured using a DeltaVision OMX (SIM) microscope. The images were grouped as (a) endogenous PLIN2-GFP or SMT3-PLIN2-GFP, (b) LDs or adiposomes both stained with LipidTOX Red, (c) merged signals. Scale bar = 2 μm.
(D) The diameter distribution analysis of intracellular LDs, isolated LDs (intracellular) and adiposomes in fluorescence images. The diameter of LD or adiposome was measured and analyzed using ImageJ to scan three images for each group.
See also Figures S1–S3 and Table S1.
We next determined the density of expressed PLIN2 on LDs and recombinant PLIN2 on adiposomes. First, the concentration of purified recombinant PLIN2 was approximated by comparing the density of protein bands from recombinant PLIN2 to a standard curve of BSA in Coomassie stained gels using ImageJ (Figure S2C) (Poppelreuther et al., 2018). The binding density of PLIN2 on the adiposomes was then calculated using the following the Equation 1 (Figure S2E):
| (Equation 1) |
where PDensity is the binding density of protein, d is the average diameter of the LDs/adiposomes, PBound is the maximum amount of the protein on the surface of LDs/adiposomes, NA is Avogadro's constant, and N is the absolute number of LDs/adiposomes. The concentration of PLIN2-GFP on LDs isolated from PLIN2-GFP knock-in (KI) cells was determined using a similar approach. The quantity of PLIN2-GFP on LDs was estimated by comparing the density of protein band from Western blot of LD bound PLIN2 to a standard curve of recombinant PLIN2 (Figure S2D) (Poppelreuther et al., 2018). Multiplying the average surface area of an LD by the LD concentration gave a total surface area of 2.07 × 108 μm2 per ml. Therefore, the estimated binding amount of PLIN2 on LDs was 1.93 × 1016 per ml, derived from the quantification of the Western blot. Hence, the density of endogenous GFP-tagged PLIN2 on LDs was 9.33 × 107 per μm2. Similarly, the estimated density of SMT3-PLIN2-GFP bound on the adiposomes was calculated as 1.02 × 106 per μm2. In both cases, even distribution was observed in the fluorescence images that led to a ring-like appearance.
To construct the quantitative relation between fluorescence intensity and the protein-bound adiposomes, the range of PLIN2-GFP protein concentration was determined and a linear fluorescence response was observed from 0.005 μM to 0.25 μM (Figure S3A). Triton X-100 was mixed with the pure protein to prevent protein aggregation, which resulted in slightly increased fluorescence intensity compared to the aqueous solution. The relationship between adiposome concentration and optical density at 600 nm (OD600) was also studied (Figure S3B). OD600 decreased linearly when the adiposomes were diluted. Therefore, the OD600 value could be used to unify the amount of adiposomes for each assay. Furthermore, the adiposomes were diluted to test the regression between RFU and adiposome concentration when they were bound with proteins (Figure S3C). A linear regression showed that the fluorescence intensity of PLIN2 bound on adiposomes was close to that of PLIN2 striped by Triton X-100 from adiposomes. It indicated that the direct scanning of PLIN2 bound on adiposomes was feasible for the binding assay. These experiments were conducted to determine the binding saturation of PLIN2 on adiposomes. According to the results in Figures S1, S2C, and S2D, the binding density of PLIN2 on adiposomes was comparable to that on LDs. Therefore, from both lipid structural aspect and peripheral protein binding aspect, adiposome is a suitably LD-mimic platform for in vitro studies.
PtdIns inhibits the binding of PLIN2 to adiposomes
By using adiposomes, the mixture of phospholipids in the monolayer can be adjusted freely, unlike in cellular assay. PtdIns is the dominant negatively charged phospholipid on the LD monolayer membrane (Bartz et al., 2007a). We investigated whether PtdIns plays a role in the targeting of PLIN2, which is one of the major resident proteins on LDs (Miura et al., 2002; Nakamura and Fujimoto, 2003). The average diameter of adiposomes prepared with and without PtdIns was close, guaranteeing a consistent adiposome quantity and similar size of binding surface for each assay (Figure 2A). The adiposome concentration of different preparations could be normalized using the aforementioned method (measuring OD600). The OD600 values of the adiposome samples were measured before they were incubated with proteins to ensure equivalence. Adiposomes composed of DOPC and DOPE were used as a control for the binding study, the diameter of which was also proved to be close to the diameter of adiposomes with DOPC only (Figure 2B). The physiological ratios of the primary membrane lipids are known: The molar ratios of PC, PE, and PtdIns to total phospholipids on LD are roughly 46%, 17%, and 8%, respectively (Bartz et al., 2007a). Since it was hard to regulate the change of different phospholipids on intracellular LDs in vivo, the adiposome was designed to solve this issue in vitro. Thus, a dose of increasing PtdIns ratio in total phospholipid of adiposomes, from 0 to 9.6%, was applied to evaluate the feasibility of adiposomes on PLIN2 binding, approaching its physiological ratio (Figure 2C). The same procedure was conducted for DOPE, from 0 to 19% (Figure 2D). Western blot showed that the binding of PLIN2 to adiposomes was increased proportionally with DOPE content, whereas it was gradually reduced with increasing PtdIns content. Fluorescence imaging confirmed this trend: Figure 3 shows that the green rings formed by the recombinant PLIN2-GFP were gradually reduced with an increasing PtdIns ratio (from 0 to 7.6%) (Figures 3A a–3C a), while the number density of adiposomes (red dots) were kept roughly constant, OD600 = 20 (Figures 3A b-3C b). The colocalization analysis of adiposomes in each group also confirmed the same trend (Figures 3A c–3C c). Figure 3A d, 3B d, and 3C d show the Pearson coefficient of correlation values for PLIN2-GFP targeting on adiposomes with pure DOPC (0.68), 99.6% DOPC +0.4% PtdIns prepared adiposomes (0.52), and 92.4% DOPC +7.6% PtdIns prepared adiposomes (0.02), respectively. Thus, the binding of PLIN2 on adiposomes is inhibited by PtdIns.
Figure 2.

Phospholipids affect PLIN2 binding to adiposomes
Adiposomes were prepared using different amount of PtdIns or DOPE with DOPC in molar ratio. And then, the size of these adiposomes were measured using dynamic light scattering (DLS) (Delsa Nano C Particle Analyzer, Beckman Coulter). After preparation, adiposome preparation (OD600 = 20) was aliquoted equally to 30 μl each and incubated with 1.2 μg PLIN2 per aliquot, supplementing corresponded volume of buffer B to prepare an equal 30 μl specimen. After incubation, the adiposomes were reisolated and washed to remove nonspecific binding proteins. The adiopsome-bound PLIN2 was determined using Western blot with anti-PLIN2 antibody and the density of protein band was quantified by ImageJ.
(A) The average diameters of adiposomes with dose of PtdIns (from 0 to 9.6%, mol/mol).
(B) The average diameters of adiposomes with dose of DOPE (from 0 to 19%, mol/mol). Inputs of 7.6% of PtdIns and 19% of DOPE were corresponded to the physiological ratio of the two phospholipids on LDs (Bartz et al., 2007a).
(C) The binding of PLIN2 on adiposomes with increased doses of PtdIns. (a) The Western blot. (b) The statistical analysis of Western blots using ImageJ.
(D) The binding of PLIN2 on adiposomes with increased doses of DOPE. (a) The Western blot of PLIN2. (b) The statistical analysis of Western blots using ImageJ. Data represent mean ± s.e.m., n = 3. ∗p< 0.05, ∗∗p< 0.01, ∗∗∗p< 0.001, two-tailed t-test.
Figure 3.

Reduction of PLIN2 binding to adiposomes by PtdIns
Adiposome preparation with dose of PtdIns (OD600 = 20) was aliquoted equally to 30 μl each and incubated with 0.5 μg SMT3-PLIN2-GFP per aliquot at 37°C for 5 min. After reisolation, washing and stained with LipidTOX Red (red), the PLIN2 (green)-coated adiposomes were visualized using confocal microscopy, and the colocalization PLIN2 (green) and adiposomes (red) were quantified by Pearson correlation analysis using ImageJ. The images were grouped as (a) SMT3-PLIN2-GFP, (b) LipidTOX Red, (c) merged signals, and (d) Pearson coefficient of correlation plot.
(A) Adiposomes were made with DOPC only (DOPC).
(B) Adiposomes were made with 99.6% DOPC and 0.4% PtdIns, in molar ratio (0.4% PtdIns).
(C) Adiposomes were made with 92.4% DOPC and 7.6% PtdIns, in molar ratio (7.6% PtdIns). Scale bar = 2 μm.
To further prove that PtdIns was affecting the binding of PLIN2, another major LD-associated protein in the perilipin family, known as PLIN3, was compared to PLIN2. PLIN3-APPLE was expressed and purified as a control of SMT3-PLIN2-GFP for the binding assay. The purities of PLIN3-APPLE were also determined using the method for SMT3-PLIN2-GFP (Figure S2B). In Figure S2B b, based on ImageJ scanning, the purity of PLIN3-APPLE (band III) was 88 ± 4%, mean ± SD, n = 3. For PLIN3, band III was the protein PLIN3-APPLE, the band IV was degraded PLIN3-APPLE fraction. Recombinant PLIN3 was incubated with adiposomes with varying PtdIns content using the same method as for PLIN2. Figure 4A summarizes the procedure of the binding experiments. The binding kinetic charts and Scatchard plots showed that both PLIN2 and PLIN3 were able to bind to adiposomes in a saturable pattern. Based on the Scatchard analysis, the Bmax value for SMT3-PLIN2-GFP binding to adiposomes in the absence of PtdIns (roughly 1.29 μM) was higher than that binding on the adiposomes in the presence of PtdIns (roughly 0.62 μM) (Figures 4B and 4C). This was consistent with the fluorescence microscopy results showing that PtdIns reduced the binding of SMT3-PLIN2-GFP on adiposomes. In contrast, the Bmax value of PLIN3-APPLE on adiposomes with DOPC was approximately 2.56 μM while on adiposomes with DOPC (92.4%) + PtdIns (7.6%) was 2.97 μM, overall higher values than those of SMT3-PLIN2-GFP (Figures 4D and 4E). The KD values for SMT3-PLIN2-GFP either in the presence or absence of PtdIns was lower than the values of PLIN3-APPLE, suggesting a higher affinity of SMT3-PLIN2-GFP for adiposomes than PLIN3-APPLE (Figures 4B–4E).
Figure 4.

Reduction of PLIN2 saturated binding on adiposomes by PtdIns
Adiposomes were prepared using a dose of PtdIns and DOPC in a molar ratio. Then, adiposome preparation was aliquoted equally to 30 μl each and incubated with increase doses of PLIN2, supplementing corresponded volume of Tris-NaCl buffer (50 mM Tris-HCl, 150 mM NaCl, pH 7.4) to prepare an equal 60 μl specimen. After incubation, the adiposomes were reisolated and washed to remove nonspecific binding proteins. The fluorescence intensity of adiopsome-bound PLIN2 was determined using an EnSpire Multimode Plate Reader (PerkinElmer) with excitation and emission wavelength at 488 nm and 530 nm for GFP tag, respectively; or 550 nm and 580 nm for APPLE tag, respectively.
(A) Diagram of the experimental design. The Scatchard plot was analyzed using GraphPad 7.0. The xaxis represents the concentration of PLIN2 or PLIN3 in both figures. The concentration of purified proteins was determined by BCA Protein Assay Kit and the fluorescence intensity represented their concentrations on adiposomes. A series of doses of SMT3-PLIN2-GFP and PLIN3-APPLE were incubated with adiposomes at 4°C for 12 h and the fluorescence intensity was detected using EnSpire Multimode Plate Reader.
(B) The saturation curves with Scatchard plot of bound SMT3-PLIN2-GFP on adiposomes with DOPC only.
(C) The saturation curves with Scatchard plots of bound SMT3-PLIN2-GFP on adiposomes with 92.4% DOPC and 7.6% PtdIns, molar ratio.
(D) The saturation curves with Scatchard plots of bound PLIN3-APPLE on adiposomes with DOPC only.
(E) The saturation curves with Scatchard plots of bound PLIN3-APPLE on adiposomes with 92.4% DOPC and 7.6% PtdIns, molar ratio. The equilibrium dissociation constant KD and the maximum saturation concentration of the binding sites on the adiposomes, Bmax, were determined using Scatchard plots. Data represent mean ± s.e.m., n = 3.
See also Figures S2 and S3 and Table S1.
The 73rd glutamic acid in PLIN2 significantly affects the binding of PLIN2
To investigate the mechanism of their differences in binding, the structures of PLIN2 and PLIN3 were compared. The full native structures of PLIN2 and PLIN3 (Homo sapiens) have not yet been solved. Therefore, the known crystal structure of PLIN3 (C-terminal) (Mus musculus)aa 191-437 (PDB: 1SZI) was used as a template to predict the structures of PLIN2 and PLIN3 (Homo sapiens) using I-TASSER (Yang and Zhang, 2015). The structures with the highest confidence scores for the proteins were selected for structural comparison using PyMOL (Hickenbottom et al., 2004). Figure 5A shows the optimized structures of PLIN2 (blue) and PLIN3 (red). The binding difference of these two proteins was speculated to be caused by the negatively charged amino acid in the α-helix of proteins. Therefore, the distinctions of α-helix in PLIN2 and PLIN3 were highlighted and the locations of significant glutamic acid residues in PLIN2 and PLIN3 were found (Figure 5B). The amphiphilic helical wheels that included E73 of PLIN2 and E86 of PLIN3 were predicted by Heliquest, which indicated that E73 in PLIN2 probably affected its binding on adiposomes (Figure 5C) (Gautier et al., 2008; Liu et al., 2015). A close-up comparison is shown in Figure 5D where the amino acid sequences of PLIN2 and PLIN3 were compared by Clustal X and shown by ESPript 3.0 (Larkin et al., 2007; Robert and Gouet, 2014). The sequences marked by a-f were structurally different regions between PLIN2 and PLIN3. The red triangle pointed to the location of E73 in the PLIN2 sequence. E73 of PLIN2 was located in the potential membrane binding α-helix, whereas E83 of PLIN3 was in a coil in a spatial configuration away from the phospholipid surface (Rowe et al., 2016). Therefore, E73 may contribute to the targeting of PLIN2 but E83 cannot serve the same purpose in PLIN3.
Figure 5.

Structural analysis reveals the differences between PLIN2 and PLIN3
The structures of PLIN2 and PLIN3 were compared using PyMOL.
(A) The overall structural comparison of PLIN2 (blue) and PLIN3 (red). The pink regions were the structurally different sequences between PLIN2 and PLIN3, while the yellow regions were negatively charged amino acids in amphiphilic α-helixes of PLIN2. The difference of two proteins were compared using Chimera 1.13.
(B) The location of residue E73 in PLIN2 sequence and residue E86 in PLIN3 sequence.
(C) Helical wheel projections of PLIN2 sequence including E73 and PLIN3 sequence including E86 were predicted by Heliquest.
(D) The amino acid sequence alignment of PLIN2 and PLIN3. The amino acid sequences of PLIN2 and PLIN3 were compared by Clustal X and shown by ESPript 3.0. The structurally different regions in PLIN2 and PLIN3 were marked from a tof. Those light blue triangles pointed the negatively charged amino acids in the amphiphilic helixes of PLIN2 sequence and the red triangle highlighted the E73.
See also Figures S4 and S5.
To test this hypothesis, various mutants of PLIN2 were constructed where negatively charged amino acids were replaced by positively charged or neutral amino acids, i.e., asparagine, glutamine, or lysine. These negatively charged amino acids were labeled in the α-helixes of PLIN2 sequence alignment (Figure 5D). The bacteria lysate expressing PLIN2 mutants was incubated with adiposomes. In Figure 6A, Western blot results of each PLIN2 mutant were compared to the wild type PLIN2. In Figure 6B, the chart summarizes the change ratio of each mutant compared to the control, quantified by density of the bands in the Western blot, using the following equation:
| (Equation 2) |
where DensityMutant-PtdIns is the intensity of mutant sample in the presence of PtdIns, and Densitymutant-NO PtdIns is the intensity of mutant sample in the absence of PtdIns. The binding was significantly enhanced when the E73 was replaced by lysine (Figure 6B). This result suggests that there is a potential role of charge interactions in PLIN2 binding to PtdIns containing monolayer membrane. The binding was slightly enhanced when E73 was mutated to glutamine, which also suggests that the binding at this site was affected by charge. The results confirm that E73 has an inhibitory role in PLIN2 binding on PtdIns containing adiposome membranes. The binding characters of PLIN2 mutants, i.e., E48K, E48Q, E73K, and E73Q, on adiposomes with the absence or presence of PtdIns were also verified by fluorescence imaging and the PLIN2 recombinant protein was the control (Figure S4). The LD binding ability of the PLIN2 mutants was further tested in cells as a comparison. Mutants at residues E48, E73, and D341 were overexpressed in Huh7 cells and all of them were targeted to LDs (Figure S5).
Figure 6.

Glutamic acid 73 of PLIN2 negatively affects PLIN2 binding to adiposomes
Adiposomes were prepared according to Transparent Methods. PLIN2 and its mutants were expressed in Transetta (DE3). The bacteria were cultured in the same condition. Each 800 ml of bacteria suspension was collected and removed the medium. The bacteria were mixed with 10 ml Tris-NaCl buffer (50 mM Tris-HCl, 150 mM NaCl, pH = 7.4) to resuspended and sonicated on ice for 15 min (6 s on and 6 s off) using a probe with a power of 210 W, to prepare the bacterial homogenate. The bacterial lysate was obtained by collecting 1 ml homogenate to perform centrifugation at 21,130g for 10 min, and the supernatant was the lysate. Thirty μl of adiposomes (OD600 = 20) was incubated with 10 μl of bacterial lysate containing PLIN2 or mutants, the adiposomes were reisolated and bound proteins were determined using Western blot with anti-PLIN2 antibody. The density of protein band was quantified by ImageJ from three independent experiments.
(A) Western blots of adiposome-bound PLIN2 and its mutants in the (1) absence or (2) presence of PtdIns using anti-PLIN2 antibody.
(B) Comparative quantification using ratio of protein band in lane 2 versus lane 1 within the same mutant. Data represent mean ± s.e.m., n = 3. ∗p< 0.05, two-tailed t-test.
See also Figures S4 and S5, Tables S1 and S2.
TAG hydrolase activity of ATGL is reproduced on adiposomes
As adiposome has been proved to be feasible for the validation of LD-associated protein binding, we next study whether it supports the function of LD-associated proteins in vitro. Lipase enzymatic activity is one of the key LD functions and hence it is used in this model study. Due to the perfect LD mimicking structure of adiposomes, they offer a more accurate model for evaluating in situ TAG hydrolysis than traditional emulsion assays, avoiding the interference of bilayer contaminants in emulsions. ATGL is a well-known TAG lipase that can translocate to LDs and the main function of which on LDs is to catalyze the first step of TAG hydrolysis (Bartz et al., 2007b; Zimmermann et al., 2004). Firstly, the binding of ATGL on adiposomes was studied. ATGL is abundant and it can be activated in the cytosol of brown adipose tissue (BAT) (Liu et al., 2015), therefore, BAT cytosol was widely used as a source of ATGL in the following binding and enzymatic assays. Figures 7A and 7B show that endogenous ATGL from BAT cytosol bound to adiposomes which were prepared using DOPC with the presence of PtdIns or DOPE. However, there was no significant difference in the ATGL bound to adiposomes when an increasing dose of DOPE or PtdIns was applied to produce adiposomes. The recombinant SMT3-ATGL was also expressed to study the binding behavior of ATGL on adiposomes. Figure S6 showed that ATGL and PLIN2 could bind to adiposomes independently, indicating that ATGL recognized the adiposome monolayer membrane as PLIN2. Furthermore, ATGL and PLIN2 were able to colocalize on adiposomes alternately. In another experiment, ATGL was knocked out in C2C12 cells using the CRISPR-Cas9 system to prove the function of ATGL (Figure S7). Monoclone line KO-2-15 was the cells transfected with a target sequence but did not show frameshift mutation (control), whereas the monoclone line KO-2-16 was transfected with the same target sequence and found missing one nucleotide and causing the frameshift mutation. The cells were fractionated and analyzed by silver stained SDS-PAGE with same volume loading and Western blot using same protein loading (Figures S7A, S7B, and S7C). The plots showed ATGL was successfully knocked out in KO-2-16 cell lines and the LDs were larger in the KO-2-16, also demonstrating the knockout of ATGL inhibited the lipolysis (Figure S7D). The expression of PLIN2 in KO-2-15 and KO-2-16 was also shown in Figure S7C. Next, the pre-measurement of ATGL enzymatic activity was performed using lipid emulsion. A diagram of ATGL enzymatic activity assay is shown in Figure S8A. An increased dose of PtdIns promoted the enzymatic activity of ATGL from BAT cytosol. The activity was significantly increased when the ratio of PtdIns reached 25%, and the ratio of triolein and phospholipid followed the literatures (Lass et al., 2006; Schreiber et al., 2017; Schweiger et al., 2014). This result indicates that PtdIns plays a role in stimulating the activity of ATGL.
Figure 7.

ATGL binding on adiposomes that mimic lipid droplets for measuring activity of ATGL
Adiposomes prepared using different amount of PtdIns or DOPE were mixed with DOPC in molar ratio. The BAT cytosol containing endogenous ATGL was used as the source of ATGL. The adiposome preparation (OD600 = 20) was aliquoted equally to 30 μl each and incubated with 10 μl BAT cytosol per aliquot, to prepare an equal 40 μl specimen. After incubation, the adiposomes were reisolated and washed to remove nonspecific binding proteins. The adiopsome-bound ATGL was determined using Western blot with anti-ATGL antibody and the density of protein band was quantified by ImageJ. The ATGL binding on adiposomes with DOPC only (0) was set as the control.
(A) The binding of ATGL on adiposomes with increasing doses of PtdIns. (a) The Western blot of ATGL with increasing doses of PtdIns. (b) The statistical analysis of Western blots.
(B) The binding of ATGL on adiposomes with increasing doses of DOPE. (a) The Western blot of ATGL with increasing doses of DOPE. (b) The statistical analysis of Western blots. Triolein [9, 10-3H(N)] was used as a radio labeled substrate to measure enzymatic activity of ATGL. The phospholipid composition for adiposomes was DOPC:liver PtdIns:DOPE = 11:3:5, molar ratio. 100 μl solution A (0.25 M sucrose, 1 mM EDTA, 1 mM DTT) were added to resuspend adiposomes in the last preparation step and the adiposomes were further adjusted to OD600 = 20. The bacteria lysate containing recombinant ATGL or its mutants was used as the source of ATGL. 25 μl adiposomes were mixed with 25 μl bacterial lysate as the specimen. The radioactivity was analyzed by gamma counter with 1 ml scintillation cocktails (PerkinElmer).
(C) Schematic of the experimental design.
(D) Radio labeledadiposomes were prepared and incubated with ATGL mutants. Data represent mean ± s.e.m., n = 3. ∗p< 0.05, ∗∗p< 0.01, two-tailed t-test.
See also Figures S6–S9, Tables S1 and S2.
However, high PtdIns content significantly decreased the yield of the adiposomes (data not show) so the ratio of phospholipid preparing adiposomes for determining the in situ lipolysis activity of ATGL were fixed as DOPC:liver PtdIns:DOPE = 11:3:5, molar ratio. The diagram of experiment is shown in Figure 7C. It is known that the enzymatic activity of ATGL is regulated by phosphorylation with many phosphorylation sites (Ahmadian et al., 2011; Bartz et al., 2007b; Pagnon et al., 2012; Xie et al., 2014). Hence, ATGL mutants were constructed by site-directed mutation to mimic the phosphorylated (aspartic acid) or non-phosphorylated (alanine) protein. Compared with the wild type ATGL, the activities of mutants S47A, S47D, S87A, S87D, S430A, and S430D were measured using both lipid emulsion and adiposomes (Figure 7D). Notably, S47 is not a phosphorylation site of ATGL, but a catalytic dyad in the GXSXG motif (Duncan et al., 2010; Xie et al., 2014). Therefore, it is also important for the lipase activity of ATGL and included in this study. The activity of mutants decreased in all cases and decreased most significantly for mutants S47A, S47D, and S87A. The S47A, S47D, S87A, and S87D mutants were overexpressed in C2C12 cells followed by immunofluorescence detection using anti-Flag antibody. Fluorescent micrographs revealed that the S47A and the S47D mutants had caused the loss of lipolytic activity compared to wild type ATGL, based on the size of LDs. Lipolytic activity of the S47D mutant was particularly suppressed (Figure S9).
Discussion
The characterization of adiposome structure, the phospholipid monolayer covering neutral lipid core, was reported in our former work (Wang et al., 2016). Previous studies of in vitro LD-specific protein targeting usually resorted to lipid emulsions and bilayer structures such as large unilamellar vesicles as the LD-mimicking model (Krahmer et al., 2011; Prévost et al., 2018; Sletten et al., 2014). In their structure and physicochemical properties, such structures are closer to emulsion rather than the LD organelle, making them dubious models for precise quantitative determination of LD protein functions in vitro. The high purity, structural LD similarity, and diameter homogeneity of adiposomes offer an incomparably better LD mimicking artificial organelle. They, therefore, underlie the new methodology to characterize the interaction between LD and proteins.
As a test problem, to validate adiposome as a functionally LD mimetic platform, it was assessed whether the differences in targeting of PtdIns containing adiposomes by PLIN2 and PLIN3 originated from the negatively charged amino acids in the membrane binding α-helixes. The binding dynamics of protein on adiposome was analyzed using the Scatchard plot, by applying the ligand-receptor binding model on the binding of protein on adiposome. The Scatchard plots clearly show that the two LD-associated proteins are able to saturate the surface of adiposomes and thus the binding behavior can be quantitively analyzed using adiposome. The difference of KD for the two wild type recombinant proteins is in accordance with previous publications that indicate the binding hierarchy between PLIN2 and PLIN3 (Bell et al., 2008; Listenberger et al., 2007). By analyzing the binding dynamics, PtdIns was found to be a significant factor affecting the binding of PLIN2 on adiposomes. The behavior of PLIN2 mutants showed the importance of E73 in regulating binding to adiposomes in the presence of PtdIns, which may be related to charge repulsion. Under physiological pH, the PC surface of adiposomes is a zwitterion that carries a negative charge on the phosphate moiety and positive charge on the choline group. PE is also a zwitterion on the surface of LD like PC. Compared to PC and PE, PtdIns is more negatively charged at physiological pH (≈7.4) and can carry multiple negative charges once phosphorylated (Marsh, 2013). Cations in the cellular milieu can penetrate membranes past choline head groups to interact with phosphate and ester groups. However, PtdIns is more accessible due to its bulk, which may result in the negative charge of PtdIns dominating interactions with large charged species such as proteins. PtdIns contributes a great density of negative charge and a bulky sugar ring containing five hydroxyl groups to the membrane (Mak, 2013). Since the targeting of proteins to LDs is likely influenced by the ionized phospholipid head group on the LD monolayer membrane, the excess charge of PtdIns could potentially repel E73 in the α-helix of PLIN2.
According to previous research, the α-helix bundles play an important role in the binding of PLIN2 to LDs (Najt et al., 2014). Although the predicted structure suggests that E73 is in a position to interact with the LD membrane, it is not in a domain that was implicated in membrane binding. The structure reveals several sites of negatively charged amino acids in the amphiphilic α-helixes that may influence binding in the presence of the anionic PtdIns. Protein interaction with PtdIns is a complex phenomenon, involving multiple factors such as electrostatic interaction, repulsive desolvation, and the hydrophobic-hydrophobic interaction between proteins and the phospholipid layer. It is believed that electrostatic interaction is the most dominant in this scheme (Mulgrew-Nesbitt et al., 2006). Truncation studies showed that the amino-terminal sequence (aa 1-181) and the carboxyl-terminal sequence (aa 277-426) are pivotal for PLIN2 to target LDs (Nakamura and Fujimoto, 2003). The conserved PAT domain, a region more than 100 amino acids long close to the N-termini of PAT family proteins, is unnecessary for PLIN2 targeting (Garcia et al., 2003; McManaman et al., 2003; Najt et al., 2014). E73 localizes in the PAT domain of PLIN2, and not the domains identified as directly responsible for LD binding. Thus, it is an unexpected outcome of the study that this amino acid influences PLIN2 targeting to adiposomes. This result may derive from the balance of nonspecific electrostatic, desolvation, and non-polar interactions between the protein and phospholipid layer (Mulgrew-Nesbitt et al., 2006). The latter two effects act on a short range while the electrostatic interaction works on long range. Hence, glutamic acid residues localized in the binding domains of PLIN2 are more affected by desolvation and hydrophobic interaction since these hydrophobic helixes are likely embedded in the phospholipid acyl chain region. This could potentially affect the protonation (charged) state as the buffering effect of the physiological solution is lost (Kory et al., 2016; Mulgrew-Nesbitt et al., 2006). In contrast, the E73 of the PAT domain of PLIN2 is more exposed and thus retains charge. Therefore, it would be more affected by Coulombic force, such that the mutants show significant changes in targeting when the charge-charge repulsion is altered.
As a second validation approach, lipase activity of ATGL was studied. Previously cytosolic extracts containing the enzyme were incubated with either the purified LD, LD mimics (i.e. a neutral lipid and buffer mixture), or neutral lipid and phospholipid microdroplets (Duncan et al., 2008; Schweiger et al., 2008; Zimmermann et al., 2004). However, these methods have limitations in that the composition of isolated LDs cannot be manipulated easily and the surfaces of the synthetic substrates are dissimilar of natural LDs. In contrast, the adiposome offers a surface which is close to the structure of LDs with a homogeneous size distribution such that it may more closely reflect the native lipase activity (Figure S1). In this study, the results obtained with ATGL mutants suggest that the active serine of ATGL affects the in vitro lipolysis capacity. The 47th serine has been reported as an important part of S47-D166 catalytic dyad that is critical for TAG hydrolysis (Duncan et al., 2010; Lake et al., 2005). Our results support this finding since the lipase activity of the S47 mutants lost significantly (Figure 7D); the fluorescence images (Figure S9) show no decrease in the size of LDs, which proves the availability of this method in comparison to literature (Lass et al., 2006; Smirnova et al., 2006). The 430th serine was found to be neither critical for LD targeting nor necessary for TAG hydrolysis (Duncan et al., 2010). However, our data indicates that S430 mutation can result in the loss of in vitro lipase activity of ATGL. The S87A mutant also showed a decline in lipase activity.
In our previous work, the design of the adiposome platform was described in detail, and the structure and LD protein binding ability of adiposomes was verified (Wang et al., 2016). Our present work delivers the biochemical validation of the adiposome platform as a suitable model for in vitro study of LD proteins. Compared to the emulsion prepared by homogenizing phospholipids and neutral lipids, purified adiposomes show a substantial advantage by mimicking the actual structure of LDs.
In summary, adiposomes were used as an in vitro model of LDs for the study of LD-associated proteins. The proteins studied, PLIN2, PLIN3, and ATGL, bound to adiposomes. The addition of PtdIns to the phospholipid composition decreased PLIN2 binding, but not PLIN3 or ATGL. This suggests that different mechanisms are responsible for the binding of these proteins. PLIN2 and PLIN3 binding was saturable and the binding properties were analyzed with Scatchard plots. A comparison of the structures of PLIN2 and PLIN3 suggests that the E73 residue in the PAT domain of PLIN2 influences the binding of PtdIns containing membranes. In contrast, the analogous E86 residue in PLIN3 is located in a coil, which may explain the different response to PtdIns content. ATGL displayed increasing lipolytic activity against PtdIns containing lipid emulsions. Important active serine mutants in ATGL were examined and S47 was found to be a significant enzyme active site and phosphorylation of S87 was required to maintain activity. This work showcases the utility of using adiposomes as an in vitro LD model to study the targeting of LD-associated proteins and the determination of lipase activity.
Limitations of the study
In this present study, we validate adiposome as the LD in vitro model to develop a method determining the LD specific protein binding affinity. We also prove the availability of adiposome for determining the in situ lipase activity of ATGL. However, it is a proof of concept study and therefore a vast amount of proteins may need to be studied using this platform to further support this proposition. Likewise, we do not study a broader range of lipid components of adiposome than PtdIns and PE in the present study.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Recombinant anti-ADFP antibody | Abcam | Cat#ab108323, RRID:AB_10863476 |
| Anti-Perilipin 3/TIP47 antibody | Abclonal | Cat#A6822, RRID:AB_2767397 |
| Monoclonal ANTI-FLAG® M2 antibody | Sigma-Aldrich | Cat#F1804, RRID: AB_262044 |
| FITC labelled goat anti-mouse IgG antibody | ZSGB-Bio | Cat#ZF-0312, RRID:AB_2716306 |
| ATGL antibody | Cell Signaling Technology | Cat#2138, RRID:AB_2167955 |
| Bacterial and virus strains | ||
| Transetta (DE3) Chemically Competent Cell | TransGen Biotech | Cat#CD801 |
| TOP10 Competent Cell | CoWin Biosciences | Cat#CW0807 |
| Biological samples | ||
| Rat fat pad tissue | This paper | N/A |
| Mouse brown adipose tissue | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) | Avanti Polar Lipids | Cat#850375 |
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) | Avanti Polar Lipids | Cat#850725 |
| L-α-phosphatidylinositol (Liver, Bovine) (sodium salt) (Liver PtdIns) | Avanti Polar Lipids | Cat#840042 |
| Triacylglycerol | Isolated from rat fat pad (Wang et al., 2016) | N/A |
| LipidTOX Red Neutral Lipid Stain | Thermo Fisher Scientific | Cat#H34476 |
| LipidTOX Green Neutral Lipid Stain | Thermo Fisher Scientific | Cat#H34475 |
| MitoTracker Red CMXRos | Thermo Fisher Scientific | Cat#M7512 |
| Hoechst 33258 | Thermo Fisher Scientific | Cat#H21491 |
| Triolein [9, 10-3H(N)] | PerkinElmer | Cat#NET431001MC |
| Phenylmethylsulfonyl fluoride (PMSF) | Sigma-Aldrich | Cat#P7626 |
| Puromycin dihydrochloride | Invitrogen | Cat#A1113803 |
| isopropyl β-D-1-thiogalatopyranoside (IPTG) | Amresco | Cat#1328-0487 |
| Glutaraldehyde (25% Aqueous Solution, EM grade) | Electron Microscopy Sciences | Cat#16220 |
| Uranyl acetate | Electron Microscopy Sciences | Cat#22400 |
| Lead citrate | Electron Microscopy Sciences | Cat#17800 |
| Osmium tetroxide | Nacalai Tesque Inc. | Cat#29532 |
| Sodium oleate | Sigma-Aldrich | Cat#143-19-1 |
| Triton X-100 | Sigma-Aldrich | Cat#9002-93-1 |
| Bovine serum albumin | Sigma-Aldrich | Cat#9048-46-8 |
| Phusion® High-Fidelity DNA Polymerase | New England Biolabs | Cat#M0530S |
| Recombinant SMT3-hPLIN2-GFP | This paper | N/A |
| Recombinant SMT3-hPLIN2-GFP mutants (PLIN2: E48K, E48Q, E51K, E51Q, E73K, E73Q, E177K, E177Q, D341K, D341Q, E383K, E383Q, E386K, E386N, E387K, E387N, E390K, E390N) | This paper | N/A |
| Recombinant hPLIN3-APPLE | This paper | N/A |
| Recombinant SMT3-mATGL | This paper | N/A |
| Recombinant SMT3-mATGL (ATGL: S47A, S47D, S87A, S87D, S430A, S430D) | This paper | N/A |
| Critical commercial assays | ||
| EMbed 812 Kit | Electron Microscopy Sciences | Cat#14120 |
| BCA protein assay kit | Thermo Fisher Scientific | Cat#PI23227 |
| Colloidal blue staining kit | Invitrogen | Cat#LC6025 |
| Deposited data | ||
| Mendeley Data: Original Western bot data for Figures 2, 6, 7, and S6 | This paper | https://doi.org/10.17632/yyvxtx3bmc.1 |
| Experimental models: Cell lines | ||
| Mouse: C2C12 myoblasts | ATCC | CRL-1772, RRID:CVCL_0188 |
| Human: Huh7 hepatocarcinoma cells | Shanghai Institutes for Biological Sciences | SCSP-526 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6 | Vital River Laboratories | Strain code: 219 |
| Rat: Sprague-Dawley (SD) | Vital River Laboratories | Strain code: 400 |
| Oligonucleotides | ||
| See Table S1 for primers | This paper | N/A |
| Recombinant DNA | ||
| pET-28a | Gift from Dr. Yang Wang (Institute of Biophysics, CAS, Beijing) | N/A |
| pET-28a-SMT3-N | Gift from Dr. Sarah Perret (Institute of Biophysics, CAS, Beijing) | N/A |
| pEGFP-N1 | Gift from Dr. Yufeng Ding (Institute of Biophysics, CAS, Beijing) | Cat#6085-1 |
| pFlag-CMV4 | Gift from Dr. Shimeng Xu (Institute of Biophysics, CAS, Beijing) | N/A |
| pX260a | Gift from Dr. Feng Zhang (Massachusetts Institute of Technology, Boston) | N/A |
| See Table S2 for recombinant plasmids | This paper | N/A |
| Software and algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Origin 2019 | OriginLab | https://www.originlab.com/ |
| GraphPad Prism 7.0 | GraphPad Software | https://www.graphpad.com/ |
| I-TASSER | Yang and Zhang, 2015 | https://zhanglab.dcmb.med.umich.edu/I-TASSER/ |
| Heliquest | Gautier et al., 2008 | https://heliquest.ipmc.cnrs.fr/ |
| PyMOL | Schrödinger, LLC. | https://pymol.org/2/ |
| Chimera 1.13 | Resource for Biocomputing, Visualization, and Informatics | https://www.cgl.ucsf.edu/chimera/ |
| Clustal X | Larkin et al., 2007 | http://www.clustal.org/ |
| QuteMol | Tarini et al., 2006 | http://qutemol.sourceforge.net |
| ChemBioDraw Ultra 12.0 | PerkinElmer | https://scistore.cambridgesoft.com/chembiodraw/ |
| ESPript 3.0 | Robert and Gouet, 2014 | https://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi |
| Other | ||
| SuperdexTM 200 Increase 10/300 GL column | GE Healthcare | Cat#28-9909-44 |
| Optiphase supermix (high flash-point scintillation cocktail) | PerkinElmer | Cat#1200-439 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Pingsheng Liu, (pliu@ibp.ac.cn).
Materials availability
Plasmids, cell lines, and triacylglycerol used in this study can be obtained in Dr. Pingsheng Liu's laboratory, National Laboratory of Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences.
Data and code availability
-
•
Original Western blot images reported in this paper have been deposited at Mendeley and are publicly available as the date of publication. The DOI is listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
Cell culture
Mouse C2C12 myoblasts and human Huh7 hepatocarcinoma cells were purchased from American Type Culture Collections and Shanghai Institutes for Biological Sciences, respectively. Both cell lines were maintained in high glucose Dulbecco’s-Modified Eagle Medium (DMEM, C11965500BT, Invitrogen) containing 2 mM L-glutamine, 100 U ml-1 penicillin, 100 μg ml-1 streptomycin, and 10% heat-inactivated fetal bovine serum (FBS, Gibco). Cultures were grown at 37°C under 5% CO2.
Animals
Eight-week-old male C57BK/6 mice (Mus musculus) and ten-week-old male Sprague-Dawley (SD) rats (Rattus norvegicus) were purchased from Vital River Laboratories, Beijing. Six C57BK/6 mice were sacrificed to extract the brown adipose tissue and three SD rats were sacrificed to extract the fat pad. All animal protocols were approved by the Animal Care and Use Committee of the Institute of Biophysics and University of Chinese Academy of Sciences under the permission number SYXK (Jing) 2016-0026.
Plasmids and primers
The primers used in this study can be found in Table S1. The plasmids can be found in Table S2.
Method details
Experimental strains and culture details
Transetta (DE3) strain was used for protein expression. The strain was purchased from TransGen Biotech and the competent cells were prepared according to the method slightly modified in Molecular Cloning. The strains were screened from newly activated Transetta (DE3) single colonies, and cultured for 12 h at 37°C with shaking until OD600 = 0.6-0.8. The bacteria suspension was incubated in 100 ml LB medium (10 g NaCl, 10 g tryptone, 5 g yeast extract in 1000 ml double-distilled water) at a ratio of 1:100-1:50 (v/v). The bacteria were incubated at 37°C for 2-3 h until OD600 = 0.35. Then the bacteria suspension was cooled on ice for 10 min and centrifuged at 3,220g for 10 min at 4°C. The supernatant was carefully removed and resuspended in ice-cold 0.1 M CaCl2-MgCl2solution (0.08 M MgCl2 and 0.02 M CaCl2, each 50 ml bacterial medium corresponded to 30 ml CaCl2-MgCl2 solution). The mixture was put on ice bath for 15 min and then centrifuged at 3,220g for 10 min at 4°C. After removing the supernatant, ice-cold 0.1 M CaCl2 solution with 15% glycerol (v/v) was added to resuspend the bacteria (each 50 ml bacteria medium corresponded to 2 ml CaCl2 solution) to produce the Transetta (DE3) competent cells. After a few minutes on ice, the competent cells were stored at −80°C. TOP10 strain was purchased from CWbiotech Co., Ltd and used to construct recombinant DNA. The TOP10 strains were maintained at −80°C in 25% glycerol.
Preparation of adiposomes and lipid emulsions
The preparation of adiposomes followed the method reported previously (Wang et al., 2016). Two mg of total phospholipids in chloroform were added to a 1.5 ml microcentrifuge tube, and the solvent was dried under a stream of nitrogen. Then 100 μl of Buffer B (20 mM HEPES, 100 mM KCl, 2 mM MgCl2, pH 7.4) was added and followed by 5 μl of TAG (extracted from rat fat pad in the laboratory). The tube containing lipids and buffer was vortexed for 24 cycles of 10 seconds on and 10 seconds off, to prepare the milky emulsion. Adiposomes in the milky emulsion were then isolated using centrifugation as described previously (Wang et al., 2016). In brief, the milky lipid emulsion was firstly centrifuged at 20,000g for 5 min at 4°C, and then the underneath transparent solution as well as the precipitated pellet were removed, while the floating white lipid layer was preserved. Corresponding volume of Buffer B was mixed with the lipid layer and suspended to obtain a 100 μl of emulsion. The same procedure was conducted once to acquire emulsion without precipitated pellet. The emulsion was then centrifuged at 1,000g for 5 min at 4°C. The milky emulsion underneath a floating white lipid layer was collected as adiposomes. The diameter distribution of adiposomes was analyzed by dynamic light scattering (DLS; Delsa Nano C Particle Analyzer, Beckman Coulter). The concentration of adiposomes was measured by optical density at 600 nm (OD600) using an Eppendorf Biophotometer (Eppendorf). The number density of adiposomes was determined using a field-flow fractionation multi-angle light scattering system (Eclipse, Wyatt Technology). The lipid emulsion for TEM analysis was prepared using the same materials while the mixture was sonicated using water bath sonicator at 20°C for 6 min (1 min on and 10 seconds off), modified from the published method (Wang et al., 2016).
Isolation of lipid droplets from C2C12 cells
The isolation of lipid droplets (LDs) from C2C12 cells was conducted using the modified method described in literature (Ding et al., 2013). The cells were treated with 50 μM oleate for 24 h if needed. C2C12 cells were rinsed by ice-cold PBS buffer (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) for three times and the cells in one 10-cm dish were scraped into 1 ml PBS buffer. The cell suspension from 20 dishes, except for KO-2-16, the ATGL knockout cell, the suspension of which was collected from 30 dishes, was centrifuged at 1,000g for 10 min at 4°C. The precipitated cells were harvested and resuspended in 20 ml ice-cold Buffer A (250 mM sucrose, 25 mM tricine, pH = 7.8) with 0.5 mM PMSF for 20 min in ice bath, respectively. Then they were transferred into a nitrogen bomb at a pressure of 700 psi for 15 min in ice bath and followed by slow release. The cell lysate was centrifuged at 1,000g for 10 min at 4°C. Several hundred microliters of supernatant were collected as post nuclear supernatant (PNS) sample for gel electrophoresis analysis. 10 ml of supernatant was transferred into SW 40 Ti tubes and 2 ml Buffer B was carefully loaded onto the top of the supernatant in each tube. Then the gradient was centrifuged at 182,348g (average RCF) for 1 h at 4°C and the top LD fraction was carefully collected. A few volumes of liquid underneath LD fraction were collected as cytosol (Cyto) sample for gel electrophoresis. The precipitates were washed three times by Buffer B and resuspended using Buffer B as the total membrane (TM) sample for gel electrophoresis. The LD fraction was centrifuged at 21,130g for 5 min at 4°C and the aqueous phase underneath was removed. The LDs were resuspended using 100 μl Buffer B in each 1.5 ml Eppendorf tube, and washed three times by performing centrifugation at 20,000g for 5 min at 4°C, to remove other membrane contaminations. For gel electrophoresis analysis, LD samples were mixed with 1 ml acetone and thoroughly vortexed to precipitate the proteins. The protein precipitations were collected as the LD sample.
Ultrastructural analysis of adiposomes and emulsions by transmission electron microscopy
Briefly, adiposomes or emulsions were fixed with an equal volume of 2% glutaraldehyde in 0.1 M sodium phosphate buffer (0.02 M NaH2PO4, 0.08 M Na2HPO4, pH 7.2) for 30 min at room temperature. Next an equal volume of 2% osmium tetroxide was added to further fix the sample for 30 min at room temperature. The fixed adiposomes or emulsions were collected by centrifugation and processed for dehydration in an ascending concentration series of ethanol and infiltration subsequently in Embed 812. Afterwards, 70 nm sections were prepared with a Leica EM UC6 Ultramicrotome. The sections were then stained with uranyl acetate and lead citrate. The samples were observed with Tecnai Spirit electron microscope (EM) (FEI, Netherlands).
Expression and purification of proteins
A 6×-His tag was inserted at the N-terminus of a SMT3-PLIN2-GFP fusion protein expression vector which was constructed as previously reported (Wang et al., 2016). Briefly, standard molecular cloning techniques were applied to fuse the gene of PLIN2 and GFP, and then the PLIN2-GFP was cloned into pET28a-SMT3 expression vector, for being expressed with N-terminal 6×-His tag and SMT3 domain. Standard molecular cloning techniques were also used to fuse genes of PLIN3 and APPLE. PLIN3-APPLE was cloned into the pET28a expression vector and was expressed with an N-terminal 6×-His tag. Mutagenesis was conducted on the SMT3-PLIN2-GFP and SMT3-ATGL fusion protein expression vectors. All proteins were expressed using Transetta (DE3). The expression and purification of proteins followed the published methods (Wang et al., 2016). In brief, the bacteria were cultured in 2 × yeast extract-Tryptone media until OD600 = 0.6 and 0.4 mM isopropyl β-D-1-thiogalatopyranoside was added into the medium to induce the expression of proteins at 16°C for 24 h. The bacterial cells were collected to remove the medium and resuspended in Tris-NaCl buffer (50 mM Tris-HCl, 150 mM NaCl, pH = 7.4). Then it was lysed and centrifuged to collect the supernatants (bacterial lysate). The proteins were purified using a nickel affinity chromatography column (Chelating Sepharose Fast Flow, Amersham Biosciences). Then, they were further purified using a SuperdexTM 200 Increase 10/300 GL column (GE, Sweden) and the purified proteins were collected.
Protein binding to adiposomes for fluorescence imaging and Western blot analysis
The methods to characterize protein binding to adiposomes were slightly modified from our previous publication, depending on the requirements of the different assays (Wang et al., 2016). Briefly, defined quantities of purified proteins were added to 30 μladiposome preparations (OD600 = 20) and corresponding volume of Tris-NaCl buffer (50 mM Tris-HCl, 150 mM NaCl, pH 7.4) was supplemented for a final volume of 50 μl except in Scatchard analysis assays, which used a 60 μl volume system. The mixture was gently vortexed three times, centrifuged at 1,000g for 10 seconds, and then incubated at 37°C in a water bath for 5 min or at room temperature for 1 h. The adiposome suspension was centrifuged at 21,130g for 5 min and the solution underneath was removed, while the top adiposome layer was reserved. The reisolated adiposomes were then resuspended in 30 μl of Buffer B and centrifuged again. The wash procedure was repeated three times to remove any nonspecifically binding proteins. The reisolated adiposomes were used for microscopy imaging or protein analysis. To determine whether ATGL affected PLIN2 binding to adiposomes, 2.5 μg SMT3-ATGL was firstly incubated with 30 μladiposomes at room temperature for 1 h. The adiposomes were reisolated using centrifuge at 21,130g for 5 min at 4°C and washed three times to remove the nonspecifically bound proteins. Then, 2.5 μg SMT3-PLIN2 was incubated with ATGL-coated adiposomes at room temperature for 1 h, and the adiposomes were reisolated and washed three times. Likewise, to determine whether PLIN2 affects the binding of ATGL to adiposomes, 2.5 μg SMT3-PLIN2 was first incubated with 25 μladiposomes at room temperature for 1 h. The adiposomes were reisolated and washed to remove the nonspecifically bound proteins three times. Then, 2.5 μg SMT3-ATGL was incubated with PLIN2-coated adiposomes at room temperature for 1 h, and the same isolation and washing procedures were conducted.
Fluorescence microscopy
Adiposomes were incubated with LipidTOX Red (H34476, Invitrogen, 1:1,000 dilution, this ratio was fixed unless specifically mentioned) for 30 min at room temperature in dark and then mounted on a slide. For protein bound adiposome samples, 0.5 μg, 1 μg or 5 μg SMT3-PLIN2-GFP was incubated with 30 μladiposomes at 37°C for 5 min followed by three washes with 30 μl Buffer B. The adiposomes were reisolated with centrifugation at 21,130g for 5 min at 4°C. Fluorescence images were obtained using an Olympus FV1000 confocal microscope, an Olympus FV1200 confocal microscope or a DeltaVision OMX V3 super resolution microscope.
C2C12 cells or Huh7 cells were incubated in the medium containing LipidTOX Red (1:1,000 dilution, v/v) at 37°C for 30 min. Fluorescence signals were captured using an Olympus FV1000 confocal microscope to visualize LDs. The ATGL knockout C2C12 cells were treated with 50 μM oleate for 24 h and incubated in the medium including 50 nM MitoTracker Red, LipidTOX Green (1:1,000 dilution, v/v), and Hoechst 33258 (1:1,000 dilution, v/v) at 37°C for 30 min (Cui et al., 2019). The cells were then imaged by Olympus FV1000 confocal microscope.
For the immunofluorescence microscopy, C2C12 ATGL-Flag cells were seeded and grown overnight on a glass-bottomed plate. Then cells were placed on ice and washed three times with ice-cold PBS for 5 min each. Each additional step described below was followed by three washes. The cells were fixed in 4% paraformaldehyde at room temperature for 30 min and then permeabilized with 0.01% Triton X-100 in PBS at room temperature for 30 min. After blocking with 1% BSA in PBS at room temperature for 1 h, the cells were incubated with anti-Flag monoclonal antibody (1:100 diluted in 0.25% BSA/PBS) at room temperature for 1 h. Afterwards, the cells were washed using PBS and incubated with FITC labelled goat anti-mouse IgG antibody (1:100 diluted in 0.25% BSA/PBS) for 1 h at room temperature. LDs were stained using LipidTOX Red for 30 min. The coverslips were applied to a slide, mixed with 2 μl of mounting media, and sealed with nail polish. Cells were examined using an Olympus FV1000 confocal fluorescence microscope. The colocalization analysis of protein targeting on adiposomes was conducted using ImageJ to provide Pearson coefficient of correlation.
Protein binding assay for binding dynamics analysis
Adiposomes prepared in individual tubes were mixed by gentle vortex and were adjusted to OD600 = 20 using an Eppendorf Biophotometer. Adiposomes were aliquoted equally to 30 μl each and incubated with proteins (SMT3-PLIN2-GFP or PLIN3-APPLE) in varying concentrations, supplementing corresponded volume of Tris-NaCl buffer (50 mM Tris-HCl, 150 mM NaCl, pH 7.4) to an equal 60 μl specimen. The mixture was vortexed gently and centrifuged at 1,000g for 10 s. Then, the samples were incubated at 4°C in the dark for 12 h. After incubation, the adiposomes were centrifuged at 21,130g for 5 min to be reisolated. The adiposomes were then resuspended in 30 μl of Buffer B and centrifuged again. The wash procedure was repeated three times to remove the nonspecifically binding proteins. Finally, the reisolated adiposomes were resuspended in 800 μl of protein dissolving buffer and vortexed for at least 15 s. Then, the samples were distributed equally into wells of a 96-microwell plate (200 μl for each well) in three technical replicates. At the same time, proteins were diluted into different concentrations with dissolving buffer containing Triton X-100 as standards. The final volume of each tube was also 800 μl and the final concentration of Triton X-100 was 1%. The standards were also distributed into the same 96-microwell plate using the same procedure. The plate was centrifuged at 3,220g for 1 min to remove bubbles and analyzed using an EnSpire Multimode Plate Reader to read the absorption at 600 nm. Then, fluorescence intensity (FI) was measured with excitation and emission at 488 nm and 530 nm, respectively, for GFP or 550 nm and 580 nm, respectively, for APPLE. One standard curve and one saturation curve were recorded in each experiment. Three independent experiments were conducted for the binding of PLIN2.
Saturation analysis
Protein purity was estimated using SDS-PAGE gels stained overnight using a Colloidal Blue Staining Kit (Invitrogen) and the density of protein band was quantified by ImageJ. The percent purity was used in protein concentration normalization. A standard curve of fluorescence intensity versus protein concentration was constructed and later used to determine the amount of protein bound to adiposomes. Background fluorescence, as measured in buffer, was subtracted from standards and samples. The experiments were conducted in adiposome preparations with a starting OD600 of 20. Prior to the fluorescence measurement of adiposomes preparations, they were diluted 13.3-fold to get into the linear range of the assay. The OD600 values of the diluted samples were measured and the ratio of OD600Measured to OD600Theoretical was used to correct for losses during washing when calculating the concentration of bound protein. Finally, the data was analyzed by nonlinear regression of bound protein concentration versus total protein concentration.
Structural analysis of PLIN2 and PLIN3
The structures of PLIN2 (Homo sapiens) and PLIN3 (Homo sapiens) were modeled by I-TASSER (Yang and Zhang, 2015). PLIN3 (Mus musculus) was used as the template for homology modeling since the C-terminal structure (191-437) of PLIN3 (Mus musculus) has been characterized (PDB: 1SZI, http://www.rcsb.org/) (Hickenbottom et al., 2004). The sequences of PLIN2 and PLIN3 that exhibited the highest confidence values were predicted using Heliquest and the structures were compared using PyMOL (Chong et al., 2011).
Single site-directed mutation of PLIN2 and ATGL
The cloning and mutagenesis primers designed by Vector NTI were listed in Table S1 and the constructed plasmids were listed in Table S2. C2C12/Huh7 cells were used as the templates to perform the reverse transcription of RNA. The reaction system and procedure are shown briefly. 1 μl of enzyme Dpn1 was added to the vector mixture and incubated at 37°C water bath for 1 h. The TOP10 competent cells were mixed with the vector after enzymatic reaction in the ice bath for 30 min. They underwent the heat stimulus (42°C) for 90 seconds and then were placed on ice for 2 min. Five-hundred μl of LB medium was mixed with them and the cells were revived at 37°C for 45 min with 200 rpm shaking. Fifty to one hundred μl of bacterial cells were coated on the resistant plates and cultured. pET-28a-SMT3-N plasmid was obtained from Dr. Sarah Perret’s lab. All plasmids were sequenced to confirm successful mutagenesis.
Gene overexpression and knockout
Huh7 cells were cultured in confocal dishes. The vector was dispersed in 500 μl of Opti-MEM medium and 5 μl of Lipofectamine 2000 was also dispersed in 500 μl of Opti-MEM medium. The vector suspension and Lipofectamine 2000 suspension were then mixed gently and incubated for 15-20 min. Cells were detected using fluorescence microscopy after well-cultured C2C12 cells were digested by trypsin. The digested cells were collected and centrifuged at 500g for 5 min. The supernatant was removed and the cells were resuspended using NT buffer. Cells were mixed with vector and electroporated. Those cells were resuspended and cultured for fluorescence microscope observation.
CRISPR/Cas9 technique was used to knock out ATGL. The mRNA sequence and genomic sequence were introduced into Vector NTI database to design the target. The target genome was identified using CRISPR tools and the primers with the highest score were selected. The test primer was designed by introducing genome sequence into Vector NTI and screening the sequences (500-550 bp) that covered target sequence. The pX260a vector (a gift from Prof. Feng Zhang), T4 ligase, T4 ligase buffer and annealing product were used to construct the vector. The product was introduced into TOP10 competent cells and the sequence was verified after culturing. Afterwards, the correct vectors were collected and transfected into C2C12 cells. The gene knockout cells were cultured and screened using 1 μg ml-1 Puromycin for 2 weeks. The cells were further diluted to gradients and cultured for 2-3 weeks. Next, monoclone was selected and cultured to screen the positive cells by immunoblotting and PCR. The positive cells were cultured and fed with oleic acid 50 μM for 24 h, and then they were observed using an Olympus FV1000 confocal microscope.
Triacylglycerol hydrolase assay
The reagents were prepared as previously published method with the following modifications (Schweiger et al., 2014). Solution A (0.25 M sucrose, 1 mM EDTA, 1 mM DTT) with 0.5 mM PMSF was prepared and the n-heptane in extraction solution I (methanol:chloroform:n-heptane = 10:9:7, v:v:v) was substituted with n-hexane. The lipid emulsion per microliter was prepared using 0.28 μg triolein mixed with 0.01 μCi triolein [9, 10-3H(N)] and 45 μM phospholipid of DOPC and PtdIns (DOPC:PtdIns = 3:1, molar ratio) by ice bath sonication. For PtdIns dose experiment, an increasing dose of PtdIns was applied, from 0 to 25%, in molar ratio. 150 μl lipid emulsions were mixed with 50 μl 20% BSA to prepare the substrate. Brown adipose tissue (BAT) cytosol from a C57BL/6 mouse (Vital River Laboratories) was suspended in Solution A, flash frozen in liquid nitrogen, and stored at −80°C, as the source of ATGL. 25 μl BAT cytosol was incubated with 25 μl substrate in 37°C for 1 h. The ATGL mutants were expressed in Transetta (DE3) following the method described above. Each 800 ml of bacteria suspension was collected and removed the medium. The bacteria precipitates were mixed with 1.6 ml Solution A to resuspend and sonicated on ice (6 s on and 6 s off for 15 min total sonication time) using a probe with a power of 210 W, to prepare the bacterial homogenate. The homogenate was centrifuged at 21,130g for 10 min, and the supernatant was the bacterial lysate. 25 μl BAT cytosol or bacterial lysate was incubated with 25 μl substrate in 37°C for 1 h. For enzymatic activity determination using adiposomes, 100 μl of radioactive labelled adiposomes were prepared using 1,100 μg DOPC, 340 μg liver PtdIns, 520 μg DOPE (DOPC:liver PtdIns:DOPE = 11:3:5, molar ratio) and 5 μl of TAG, mixed with 5 μl Triolein [9, 10-3H(N)] (0.5 μCi μl-1, PerkinElmer). The adiposomes were produced using the method cited previously, but were resuspended in Solution A in the last step (Wang et al., 2016). Twenty-five μl of the bacterial supernatant was mixed with 25 μl of adiposomes and the mixture was incubated for 1 h in at 37°C. The reaction for both determinations was terminated by the addition of 650 μl extraction solution I and followed by adding 200 μl of extraction solution II (0.1 M K2CO3, adjusting pH to 10.5 using boric acid) with vigorous vortex. Samples were centrifuged at 1,000g for 10 min to drive the proteins into the aqueous-organic boundary. 200 μl of the upper aqueous phase was transferred to a scintillation vial containing 1 ml of scintillation cocktails and its radioactivity was analyzed by gamma meter (PerkinElmer). Twenty-five μl of the substrate was measured to determine specific substrate radioactivity. 25 μl of Solution A was used as a blank. The lipase activity was calculated using the equation (Zimmermann et al., 2004):
where V1 is the total volume of upper water phase; V2 is the volume measured in the scintillation counter; and t is the incubation time (h). CpmSample, cpmBlank, and cpmSubstrate are the values of counts per minute for sample, blank and substrate. nFA is the mole value of fatty acid (nmol), mProtein is the mass of protein (mg) and estimated by analyzing the stained gels using ImageJ.
Quantification and statistical analys
Scatchard analysis
Scatchard analysis was used to calculate the binding affinity and the maximum saturation concentration of binding sites on adiposomes. A linear regression of the plot of bound protein/free protein (ordinate) against bound protein (abscissa) yielded the slope = -KD-1, where KD is the equilibrium dissociation constant and the abscissa intercept = Bmax, the maximum saturation concentration of ligand binding sites. The regressions were plotted using Graphpad 7.0.
The significance was evaluated by two-tailed t-tests. A P value inferior to 0.05 was considered significant.
Acknowledgments
The authors thank Dr. John Zehmer for his critical reading and useful suggestions, Dr. Yang Wang for advices on adiposome preparation, and Dr. Liujuan Cui for administrative and technique support. We would like to thank Ms. Shuoguo Li (Center for Biological Imaging, IBP, CAS) for her help of taking and analyzing SIM images, Ms. Yan Teng (Center for Biological Imaging, IBP, CAS) for her help of taking and analyzing Confocal images, Ms. Xiaoxia Yu for her suggestions on protein purification and particle counting, Ms. Zhenwei Yang for her suggestions on fluorescence detection by Enspire, Mr. Hongjie Zhang for his help in radioisotope experiments. Z.Z.gratefully acknowledges his Fong Tam Yuen Leung fellowship. This work was supported by the National Key R&D Program of China (Grant No. 2018YFA0800700 and 2018YFA0800900), National Natural Science Foundation of China (Grant No. 91857201, 91954108, 31671402, 31671233, 31701018 and U1702288). This work was also supported by the "Personalized Medicines—Molecular Signature-based Drug Discovery and Development", Strategic Priority Research Program of the Chinese Academy of Sciences, Grant No. XDA12040218.
Author contributions
A.M. and P.L. conceived the study and designed the experiments. X.M., Z.Z., S.Z., and C.Z. performed the experiments and analyzed the data. P.L. analyzed data and provided guidance and support. X.M., Z.Z., A.M., and P.L. wrote the paper.
Declaration of interests
The authors declare that there is no conflict of interests regarding the publication of this article.
Published: August 20, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102834.
Contributor Information
Adam Mechler, Email: a.mechler@latrobe.edu.au.
Pingsheng Liu, Email: pliu@ibp.ac.cn.
Supplementalinformation
References
- Ahmadian M., Abbott M.J., Tang T., Hudak C.S.S., Kim Y., Bruss M., Hellerstein M.K., Lee H.-Y., Samuel V.T., Shulman G.I. Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab. 2011;13:739–748. doi: 10.1016/j.cmet.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barneda D., Planas-Iglesias J., Gaspar M.L., Mohammadyani D., Prasannan S., Dormann D., Han G.-S., Jesch S.A., Carman G.M., Kagan V. The brown adipocyte protein CIDEA promotes lipid droplet fusion via a phosphatidic acid-binding amphipathic helix. ELife. 2015;4:e07485. doi: 10.7554/eLife.07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartz R., Li W.-H., Venables B., Zehmer J.K., Roth M.R., Welti R., Anderson R.G.W., Liu P., Chapman K.D. Lipidomics reveals that adiposomes store ether lipids and mediate phospholipid traffic. J. Lipid Res. 2007;48:837–847. doi: 10.1194/jlr.M600413-JLR200. [DOI] [PubMed] [Google Scholar]
- Bartz R., Zehmer J.K., Zhu M., Chen Y., Serrero G., Zhao Y., Liu P. Dynamic activity of lipid droplets: protein phosphorylation and GTP-mediated protein translocation. J. Proteome Res. 2007;6:3256–3265. doi: 10.1021/pr070158j. [DOI] [PubMed] [Google Scholar]
- Bell M., Wang H., Chen H., McLenithan J.C., Gong D.-W., Yang R.-Z., Yu D., Fried S.K., Quon M.J., Londos C., Sztalryd C. Consequences of lipid droplet coat protein downregulation in liver cells. Diabetes. 2008;57:2037. doi: 10.2337/db07-1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersuker K., Olzmann J.A. Establishing the lipid droplet proteome: mechanisms of lipid droplet protein targeting and degradation. Biochim.Biophys. Acta Mol.Cell Biol. Lipids. 2017;1862:1166–1177. doi: 10.1016/j.bbalip.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeszoermenyi A., Nagy H.M., Arthanari H., Pillip C.J., Lindermuth H., Luna R.E., Wagner G., Zechner R., Zangger K., Oberer M. Structure of a CGI-58 motif provides the molecular basis of lipid droplet anchoring. J. Biol. Chem. 2015;290:26361–26372. doi: 10.1074/jbc.M115.682203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Jena K.C., Lütgebaucks C., Okur H.I., Roke S. Three dimensional nano “Langmuir trough” for lipid studies. Nano Lett. 2015;15:5558–5563. doi: 10.1021/acs.nanolett.5b02143. [DOI] [PubMed] [Google Scholar]
- Chong B.M., Russell T.D., Schaack J., Orlicky D.J., Reigan P., Ladinsky M., McManaman J.L. The adipophilin C terminus is a self-folding membrane-binding domain that is important for milk lipid secretion. J. Biol. Chem. 2011;286:23254–23265. doi: 10.1074/jbc.M110.217091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L., Mirza A.H., Zhang S., Liang B., Liu P. Lipid droplets and mitochondria are anchored during brown adipocyte differentiation. Protein Cell. 2019;10:921–926. doi: 10.1007/s13238-019-00661-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y., Zhang S., Yang L., Na H., Zhang P., Zhang H., Wang Y., Chen Y., Yu J., Huo C. Isolating lipid droplets from multiple species. Nat. Protoc. 2013;8:43–51. doi: 10.1038/nprot.2012.142. [DOI] [PubMed] [Google Scholar]
- Duncan R.E., Sarkadi-Nagy E., Jaworski K., Ahmadian M., Sul H.S. Identification and functional characterization of adipose-specific phospholipase A2 (AdPLA) J. Biol. Chem. 2008;283:25428–25436. doi: 10.1074/jbc.M804146200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan R.E., Wang Y., Ahmadian M., Lu J., Sarkadi-Nagy E., Sul H.S. Characterization of desnutrin functional domains: critical residues for triacylglycerol hydrolysis in cultured cells. J. Lipid Res. 2010;51:309–317. doi: 10.1194/jlr.M000729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einfalt T., Witzigmann D., Edlinger C., Sieber S., Goers R., Najer A., Spulber M., Onaca-Fischer O., Huwyler J., Palivan C.G. Biomimetic artificial organelles with in vitro and in vivo activity triggered by reduction in microenvironment. Nat. Commun. 2018;9:1127. doi: 10.1038/s41467-018-03560-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farese R.V., Walther T.C. Lipid droplets finally get a little R-E-S-P-E-C-T. Cell. 2009;139:855–860. doi: 10.1016/j.cell.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei W., Shui G., Zhang Y., Krahmer N., Ferguson C., Kapterian T.S., Lin R.C., Dawes I.W., Brown A.J., Li P. A role for phosphatidic acid in the formation of “supersized” lipid droplets. PLoS Genet. 2011;7:e1002201. doi: 10.1371/journal.pgen.1002201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia A., Sekowski A., Subramanian V., Brasaemle D.L. The central domain is required to target and anchor perilipin A to lipid droplets. J. Biol. Chem. 2003;278:625–635. doi: 10.1074/jbc.M206602200. [DOI] [PubMed] [Google Scholar]
- Gautier R., Douguet D., Antonny B., Drin G. HELIQUEST: a web server to screen sequences with specific α-helical properties. Bioinformatics. 2008;24:2101–2102. doi: 10.1093/bioinformatics/btn392. [DOI] [PubMed] [Google Scholar]
- Hammer D.A., Kamat N.P. Towards an artificial cell. FEBS Lett. 2012;586:2882–2890. doi: 10.1016/j.febslet.2012.07.044. [DOI] [PubMed] [Google Scholar]
- Hickenbottom S.J., Kimmel A.R., Londos C., Hurley J.H. Structure of a lipid droplet protein: the PAT family member TIP47. Structure. 2004;12:1199–1207. doi: 10.1016/j.str.2004.04.021. [DOI] [PubMed] [Google Scholar]
- Huang C.Y., Huang A.H.C. Unique motifs and length of hairpin in oleosin target the cytosolic side of endoplasmic reticulum and budding lipid droplet. Plant Physiol. 2017;174:2248–2260. doi: 10.1104/pp.17.00366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kory N., Farese R.V., Jr., Walther T.C. Targeting fat: mechanisms of protein localization to lipid droplets. Trends Cell Biology. 2016;26:535–546. doi: 10.1016/j.tcb.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krahmer N., Guo Y., Wilfling F., Hilger M., Lingrell S., Heger K., Newman H.W., Schmidt-Supprian M., Vance D.E., Mann M. Phosphatidylcholine synthesis for lipid droplet expansion is mediated by localized activation of CTP:phosphocholinecytidylyltransferase. Cell Metab. 2011;14:504–515. doi: 10.1016/j.cmet.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake A.C., Sun Y., Li J.-L., Kim J.E., Johnson J.W., Li D., Revett T., Shih H.H., Liu W., Paulsen J.E., Gimeno R.E. Expression, regulation, and triglyceride hydrolase activity of Adiponutrin family members. J. Lipid Res. 2005;46:2477–2487. doi: 10.1194/jlr.M500290-JLR200. [DOI] [PubMed] [Google Scholar]
- Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R. Clustal W and clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Lass A., Zimmermann R., Haemmerle G., Riederer M., Schoiswohl G., Schweiger M., Kienesberger P., Strauss J.G., Gorkiewicz G., Zechner R. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metab. 2006;3:309–319. doi: 10.1016/j.cmet.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Lemmon M.A. Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 2008;9:99–111. doi: 10.1038/nrm2328. [DOI] [PubMed] [Google Scholar]
- Listenberger L.L., Ostermeyer-Fay A.G., Goldberg E.B., Brown W.J., Brown D.A. Adipocyte differentiation-related protein reduces the lipid droplet association of adipose triglyceride lipase and slows triacylglycerol turnover. J. Lipid Res. 2007;48:2751–2761. doi: 10.1194/jlr.M700359-JLR200. [DOI] [PubMed] [Google Scholar]
- Liu P., Ying Y., Zhao Y., Mundy D.I., Zhu M., Anderson R.G.W. Chinese hamster ovary K2 cell lipid droplets appear to be metabolic organelles involved in membrane traffic. J. Biol. Chem. 2004;279:3787–3792. doi: 10.1074/jbc.M311945200. [DOI] [PubMed] [Google Scholar]
- Liu W., Xie Y., Ma J., Luo X., Nie P., Zuo Z., Lahrmann U., Zhao Q., Zheng Y., Zhao Y. IBS: an illustrator for the presentation and visualization of biological sequences. Bioinformatics. 2015;31:3359–3361. doi: 10.1093/bioinformatics/btv362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak L.H. Lipid signaling and phosphatidylinositols. In: Roberts G.C.K., editor. Encyclopedia of Biophysics. Springer Berlin Heidelberg; 2013. pp. 1286–1289. [DOI] [Google Scholar]
- Marsh D. CRC press; 2013. Handbook of Lipid Bilayers. [DOI] [Google Scholar]
- Martin S., Parton R.G. Lipid droplets: a unified view of a dynamic organelle. Nat. Rev. Mol. Cell Biol. 2006;7:373–378. doi: 10.1038/nrm1912. [DOI] [PubMed] [Google Scholar]
- McManaman J.L., Zabaronick W., Schaack J., Orlicky D.J. Lipid droplet targeting domains of adipophilin. J. Lipid Res. 2003;44:668–673. doi: 10.1194/jlr.C200021-JLR200. [DOI] [PubMed] [Google Scholar]
- Miura S., Gan J.-W., Brzostowski J., Parisi M.J., Schultz C.J., Londos C., Oliver B., Kimmel A.R. Functional conservation for lipid storage droplet association among perilipin, ADRP, and TIP47 (PAT)-related proteins in mammals, drosophila and dictyostelium. J. Biol. Chem. 2002;277:32253–32257. doi: 10.1074/jbc.M204410200. [DOI] [PubMed] [Google Scholar]
- Mulgrew-Nesbitt A., Diraviyam K., Wang J., Singh S., Murray P., Li Z., Rogers L., Mirkovic N., Murray D. The role of electrostatics in protein–membrane interactions. Biochim.Biophys. Acta Mol.Cell Biol. Lipids. 2006;1761:812–826. doi: 10.1016/j.bbalip.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Na H., Zhang P., Chen Y., Zhu X., Liu Y., Liu Y., Xie K., Xu N., Yang F., Yu Y. Identification of lipid droplet structure-like/resident proteins in Caenorhabditis elegans. Biochim.Biophys.Acta. 2015;1853:2481–2491. doi: 10.1016/j.bbamcr.2015.05.020. [DOI] [PubMed] [Google Scholar]
- Najt C.P., Lwande J.S., McIntosh A.L., Senthivinayagam S., Gupta S., Kuhn L.A., Atshaves B.P. Structural and functional assessment of perilipin 2 lipid binding domain(s) Biochemistry. 2014;53:7051–7066. doi: 10.1021/bi500918m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N., Fujimoto T. Adipose differentiation-related protein has two independent domains for targeting to lipid droplets. Biochem. Bioph Res. Commun. 2003;306:333–338. doi: 10.1016/S0006-291X(03)00979-3. [DOI] [PubMed] [Google Scholar]
- Ohsaki Y., Suzuki M., Fujimoto T. Open questions in lipid droplet biology. Chem.Biol. 2014;21:86–96. doi: 10.1016/j.chembiol.2013.08.009. [DOI] [PubMed] [Google Scholar]
- Olarte M.-J., Kim S., Sharp M.E., Swanson J.M.J., Farese R.V., Jr., Walther T.C. Determinants of endoplasmic reticulum-to-lipid droplet protein targeting. Dev.Cell. 2020;54:471–487.e477. doi: 10.1016/j.devcel.2020.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagnon J., Matzaris M., Stark R., Meex R.C.R., Macaulay S.L., Brown W., O'Brien P.E., Tiganis T., Watt M.J. Identification and functional characterization of protein kinase A phosphorylation sites in the major lipolytic protein, adipose triglyceride lipase. Endocrinology. 2012;153:4278–4289. doi: 10.1210/en.2012-1127. [DOI] [PubMed] [Google Scholar]
- Palivan C.G., Fischer-Onaca O., Delcea M., Itel F., Meier W. Protein–polymer nanoreactors for medical applications. Chem. Soc. Rev. 2012;41:2800–2823. doi: 10.1039/C1CS15240H. [DOI] [PubMed] [Google Scholar]
- Palivan C.G., Goers R., Najer A., Zhang X., Car A., Meier W. Bioinspired polymer vesicles and membranes for biological and medical applications. Chem. Soc. Rev. 2016;45:377–411. doi: 10.1039/C5CS00569H. [DOI] [PubMed] [Google Scholar]
- Phan T.K., Lay F.T., Poon I.K.H., Hinds M.G., Kvansakul M., Hulett M.D. Human β-defensin 3 contains an oncolytic motif that binds PI(4,5)P2 to mediate tumour cell permeabilisation. Oncotarget. 2016;7:2054–2069. doi: 10.18632/oncotarget.6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poppelreuther M., Sander S., Minden F., Dietz M.S., Exner T., Du C., Zhang I., Ehehalt F., Knüppel L., Domschke S. The metabolic capacity of lipid droplet localized acyl-CoA synthetase 3 is not sufficient to support local triglyceride synthesis independent of the endoplasmic reticulum in A431 cells. Biochim.Biophys. Acta Mol.Cell Biol. Lipids. 2018;1863:614–624. doi: 10.1016/j.bbalip.2018.03.003. [DOI] [PubMed] [Google Scholar]
- Prévost C., Sharp M.E., Kory N., Lin Q., Voth G.A., Farese R.V., Jr., Walther T.C. Mechanism and determinants of amphipathic helix-containing protein targeting to lipid droplets. Dev.Cell. 2018;44:73–86.e74. doi: 10.1016/j.devcel.2017.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rideau E., Dimova R., Schwille P., Wurm F.R., Landfester K. Liposomes and polymersomes: a comparative review towards cell mimicking. Chem. Soc. Rev. 2018;47:8572–8610. doi: 10.1039/C8CS00162F. [DOI] [PubMed] [Google Scholar]
- Robert X., Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe E.R., Mimmack M.L., Barbosa A.D., Haider A., Isaac I., Ouberai M.M., Thiam A.R., Patel S., Saudek V., Siniossoglou S., Savage D.B. Conserved amphipathic helices mediate lipid droplet targeting of perilipins 1-3. J. Biol. Chem. 2016;291:6664–6678. doi: 10.1074/jbc.M115.691048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber R., Diwoky C., Schoiswohl G., Feiler U., Wongsiriroj N., Abdellatif M., Kolb D., Hoeks J., Kershaw E.E., Sedej S. Cold-induced thermogenesis depends on ATGL-mediated lipolysis in cardiac muscle, but not Brown adipose tissue. Cell Metab. 2017;26:753–763.e757. doi: 10.1016/j.cmet.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweiger M., Eichmann T.O., Taschler U., Zimmermann R., Zechner R., Lass A. Chapter ten - measurement of lipolysis. In: MacDougald O.A., editor. Methods Enzymol. Academic Press; 2014. pp. 171–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweiger M., Schoiswohl G., Lass A., Radner F.P.W., Haemmerle G., Malli R., Graier W., Cornaciu I., Oberer M., Salvayre R. The C-terminal region of human adipose triglyceride lipase affects enzyme activity and lipid droplet binding. J. Biol. Chem. 2008;283:17211–17220. doi: 10.1074/jbc.M710566200. [DOI] [PubMed] [Google Scholar]
- Sletten A., Seline A., Rudd A., Logsdon M., Listenberger L.L. Surface features of the lipid droplet mediate perilipin 2 localization. Biochem. Bioph Res. Commun. 2014;452:422–427. doi: 10.1016/j.bbrc.2014.08.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova E., Goldberg E.B., Makarova K.S., Lin L., Brown W.J., Jackson C.L. ATGL has a key role in lipid droplet/adiposome degradation in mammalian cells. EMBO Rep. 2006;7:106–113. doi: 10.1038/sj.embor.7400559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner P., Balasubramanian V., Palivan C.G. Aiding nature’s organelles: artificial peroxisomes play their role. Nano Lett. 2013;13:2875–2883. doi: 10.1021/nl401215n. [DOI] [PubMed] [Google Scholar]
- Tarini M., Cignoni P., Montani C. Ambient occlusion and edge cueing for enhancing real time molecular visualization. IEEE Trans. Vis. Comput. Graphics. 2006;12:1237–1244. doi: 10.1109/TVCG.2006.115. [DOI] [PubMed] [Google Scholar]
- Tauchi-Sato K., Ozeki S., Houjou T., Taguchi R., Fujimoto T. The surface of lipid droplets is a phospholipid monolayer with a unique fatty acid composition. J. Biol. Chem. 2002;277:44507–44512. doi: 10.1074/jbc.M207712200. [DOI] [PubMed] [Google Scholar]
- Thingholm B., Schattling P., Zhang Y., Städler B. Subcompartmentalized nanoreactors as artificial organelle with intracellular activity. Small. 2016;12:1806–1814. doi: 10.1002/smll.201502109. [DOI] [PubMed] [Google Scholar]
- Tzen J.T., Huang A.H. Surface structure and properties of plant seed oil bodies. J. Cell Biol. 1992;117:327. doi: 10.1083/jcb.117.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther T.C., Chung J., R.V.F. Lipid droplet biogenesis. Curr.Opin.Cell Biol. 2017;33:491–510. doi: 10.1146/annurev-cellbio-100616-060608. [DOI] [Google Scholar]
- Walther T.C., Farese R.V., Jr. Lipid droplets and cellular lipid metabolism. Annu.Rev.Biochem. 2012;81:687–714. doi: 10.1146/annurev-biochem-061009-102430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Zhou X.-M., Ma X., Du Y., Zheng L., Liu P. Construction of nanodroplet/adiposome and artificial lipid droplets. ACS Nano. 2016;10:3312–3322. doi: 10.1021/acsnano.5b06852. [DOI] [PubMed] [Google Scholar]
- Wolins N.E., Brasaemle D.L., Bickel P.E. A proposed model of fat packaging by exchangeable lipid droplet proteins. FEBS Lett. 2006;580:5484–5491. doi: 10.1016/j.febslet.2006.08.040. [DOI] [PubMed] [Google Scholar]
- Xie X., Langlais P., Zhang X., Heckmann B.L., Saarinen A.M., Mandarino L.J., Liu J. Identification of a novel phosphorylation site in adipose triglyceride lipase as a regulator of lipid droplet localization. Am. J. Physiol. Endocrinol. Metab. 2014;306:E1449–E1459. doi: 10.1152/ajpendo.00663.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S., Zou F., Diao Z., Zhang S., Deng Y., Zhu X., Cui L., Yu J., Zhang Z., Bamigbade A.T. Perilipin 2 and lipid droplets provide reciprocal stabilization. Biophys. Rep. 2019;5:145–160. doi: 10.1007/s41048-019-0091-5. [DOI] [Google Scholar]
- Yan R., Qian H., Lukmantara I., Gao M., Du X., Yan N., Yang H. Human SEIPIN binds anionic phospholipids. Dev.Cell. 2018;47:248–256.e244. doi: 10.1016/j.devcel.2018.09.010. [DOI] [PubMed] [Google Scholar]
- Yang J., Zhang Y. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 2015;43:W174–W181. doi: 10.1093/nar/gkv342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y., Ding L., Huang X. Diverse functions of lipids and lipid metabolism in development. Small Methods. 2019:1900564. doi: 10.1002/smtd.201900564. [DOI] [Google Scholar]
- Zehmer J.K., Bartz R., Liu P., Anderson R.G.W. Identification of a novel N-terminal hydrophobic sequence that targets proteins to lipid droplets. J. Cell Sci. 2008;121:1852–1860. doi: 10.1242/jcs.012013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Liu P. The lipid droplet: a conserved cellular organelle. Protein Cell. 2017;8:796–800. doi: 10.1007/s13238-017-0467-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Liu P. The new face of the lipid droplet: lipid droplet proteins. Proteomics. 2019;19:e1700223. doi: 10.1002/pmic.201700223. [DOI] [PubMed] [Google Scholar]
- Zhang C., Yang L., Ding Y., Wang Y., Lan L., Ma Q., Chi X., Wei P., Zhao Y., Steinbüchel A. Bacterial lipid droplets bind to DNA via an intermediary protein that enhances survival under stress. Nat. Commun. 2017;8:15979. doi: 10.1038/ncomms15979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann R., Strauss J.G., Haemmerle G., Schoiswohl G., Birner-Gruenberger R., Riederer M., Lass A., Neuberger G., Eisenhaber F., Hermetter A., Zechner R. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306:1383–1386. doi: 10.1126/science.1100747. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Original Western blot images reported in this paper have been deposited at Mendeley and are publicly available as the date of publication. The DOI is listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.