

Abstract
The traditional view is that the occurrence and development of hallux valgus (HV) are mainly due to environmental factors. Recent studies have suggested the large contribution of genetic heritability to HV, but it remains elusive about the genetic variants underlying the development of HV. To gain knowledge about the molecular mechanisms of HV pathogenesis by genetic approach, whole exome sequencing studies were performed in 10 individuals (7 affected by HV and 3 unaffected) from three independent families. Specific mutations were found to be related to the pathogenesis of HV and conform to the laws of inheritance. A total of 36 genes with functional candidate single nucleotide variants were identified. Genetic predisposition plays an important role in the development of HV. Interestingly, some of these genes are related to chronic arthritis, such as the complement encoding gene C7, or are related to long toe or long fingers, such as TTN, COL6A3, LARS, FIG4, and CBS. This study identified rare potentially pathogenic mutations represented by genes related to digital anomalies and chronic arthritis underlying the familial types of HV, which acquired new insights into the genetic and physiological foundations of HV, thereby might improve accurate prevention and drug development for HV.
Keywords: Hallux valgus, whole exome sequencing, pedigree, susceptibility gene, single nucleotide mutation, digital anomalies
Impact statement
So far, orthomorphia was still the main treatment for hallux valgus. If non-invasive therapeutic approaches can be used to intervene hallux valgus at an early stage, some patients may have the opportunity to be exempt from surgery. This study identified potentially pathogenic mutations underlying the familial types of hallux valgus, and acquired insights into the genetic basis and molecular mechanism of hallux valgus. It helps to provide targets to facilitate the development of novel therapeutic approaches and identify susceptible populations early to improve primary and secondary prevention of hallux valgus.
Introduction
Hallux valgus (HV), also known as a bunion, which refers to the lateral deviation of first toe at the metatarsophalangeal joint, is the most common forefoot deformity.1–3 HV usually develops with age progressively,3,4 and can cause pain and decreased mobility. 5 The prevalence of HV is estimated to be 23% (95% CI:16.3%–29.6%) in adults aged 18–65 years, and 35.7% (95% CI: 29.5–42.0) in adults aged over 65 years. 2 The prevalence of HV is higher in women [30% (95% CI: 22–%38%)] than that in men [13% (95% CI: 9%–17%)]. 2 The pain of HV mainly focuses on the bunion, the medial surface of the foot, the load-carrying subface of the foot, and the little toes. 1 Progressive subluxation of the first metatarsophalangeal joint may occur at the later stage of HV development.3,6 Several types of non-operative treatment may alleviate symptoms of HV. However, none of them can fully reverse the HV deformity, and surgery is usually recommended if the pain persists.1,3
The pathogenesis of HV is complex, which is attributable to both extrinsic and intrinsic factors. HV is subjected to genetic predisposition and is related to ligamentous laxity, other foot deformities, age, and neuromuscular disorders.3,4 Nonlinear osseous alignment or a laxity of the static stabilizers due to genetic predisposition may contribute to HV development, while restrictive footwear can accelerate the process of HV. 4 As genetic predisposition plays an important role in the development of HV as evidenced by the results from family studies, important knowledge could be gained about the molecular mechanisms of HV pathogenesis. A research including three-generation pedigrees of 350 patients showed that 90% of probands had at least one family member affected, which suggested autosomal dominant inheritance with incomplete penetrance. 7 The heritability of HV in men and women of European descent ranges from 0.29 to 0.89 depending on age and sex, 8 and that in Korean monozygotic twins is estimated to be 0.51 (CI: 0.42–0.59). 9 The genome-wide genotyped single nucleotide polymorphisms (SNP) explained 50% of HV variance in men and 48% in women. 10 Common genetic variants showed sex specific association with HV, peaking at the SNP rs9675316 near the axin 2 gene (AXIN2) in males (p = 5.46E–07) and rs7996797 (p = 7.21E–07) near the esterase D gene (ESD) in females. Genome-wide significant SNP-by-sex interaction was identified for the SNP rs1563374 near the MAS related GPR family member X3 gene (MRGPRX3) (interaction p = 4.07E–09). 10 Important knowledge was acquired by the discoveries of this study, highlighting molecular pathways related to skeletal development and inflammation in HV. However, the knowledge about the genetic basis of HV is far from complete, e.g. rare mutations underlying the familial types of HV cannot be identified by a GWAS study. 11
Although the exome accounts for only ∼1% of the whole human genome, 85% of the pathogenic mutations related to Mendelian diseases were harbored in this region.12,13 The whole exome sequencing (WES) technology thus presents an opportunity to discover rare causative variants related to familial types of HV. Families with significant HV history are an essential resource for rare causative genetic variant study. Here, we present the first WES study on three HV families to search for rare variants related to HV.
Materials and methods
Participants
The study was approved by the institutional review board and the informed consents were obtained from all the participants. Ten Chinese individuals were enrolled in this study. Three unrelated participants had been diagnosed with HV from October 2014 to December 2018, and all of them (i.e. the three probands in this study) had a positive family history of HV (i.e. first-degree relative(s) with HV). A total of seven direct family members (spouses and children/biological parents) of the probands were also enrolled into the study (Figure 1). The diagnoses were made by orthopedic surgeons of foot and ankle expert based on the expert consensus from the Foot and Ankle Surgery Group of Orthopedics Branch of Chinese Medical Association. The diagnosis of HV was based on a comprehensive evaluation of clinical manifestations, medical history, physical examination, and imaging findings. The severity of HV was graded into three levels in terms of hallux valgus angle (HVA, also known as hallux abductus angle, normally <16°) and intermetatarsal angle (IMA, normally < 10°): mild (HVA ≤ 20°, IMA ≤ 13°), moderate (20° < HVA ≤ 40°, 13° < IMA ≤ 16°), and severe (HVA > 40°, IMA > 16°).
Figure 1.

The pedigree charts of the three families. The relationships among subjects from three families included in this study are shown in standard pedigree charts. Elements filled with horizontal hachures indicate patients with mild hallux valgus. Elements filled with diagonal lines indicate patients with moderate hallux valgus. Blank elements indicate family members without hallux valgus.
Whole-exome capture and sequencing
Genomic DNA was extracted from whole blood. Exome capture was performed using human exome capture kit TargetSeq™ Enrichment Kit (iGeneTech™) following the manufacturer’s instructions. Paired-end next generation sequencing was performed on the Illumina NovaSeq™ 6000 Sequencing System.
WES data analyses
The analysis pipeline is shown in Supplementary Figure 1. The quality of raw exome sequencing reads was assessed using FastQC (version 0.11.7, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Automated quality and adapter trimming were performed using Trim-Galore (version 0.6.4_dev, https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) with Cutadapt (version 1.18), 14 and the quality was reassessed using FastQC. Analysis-ready sequencing reads were aligned to the human reference genome (GRCh37/hg19) using BWA (Burrows-Wheeler Aligner)-MEM (version 0.7.17). Next, the alignment outputs were sorted by genomic coordinates, and polymerase chain reaction (PCR) duplicates were marked using GATK15–17 (version 4.1.6.0). Quality control of intermediary binary alignment map file was performed using Samtools 18 (version 1.58). Base quality score recalibrations (BQSRs) were performed using GATK. After BQSRs, we confirmed no contamination of cross-samples prior to variants calling using verifyBamID 19 (version 1.1.3). The kinship coefficient between samples was calculated using the software VCFtools 20 (version 0.1.17) based on VCF files to verify the self-reported relationship among participants of the study. Then, variants calling, joint genotyping and basic hard-filtration of SNPs and indels were performed using GATK coherently. The basic hard-filter parameters were as follow: (a) for SNPs: quality by depth (QD) < 2.0, fisher strand (FS) > 60.0, mapping quality (MQ) < 40.0, mapping quality rank sum (MQRankSum) < –12.5, read position rank sum (ReadPosRankSum) < –8.0, strand odds ratio (SOR) > 3.0; b) for indels: QD < 2.0, FS > 200.0, ReadPosRankSum < –20.0, SOR > 10.0, InbreedingCoeff <–0.8. Quality control of variant call format file 20 was performed using SNPEff 21 (version 4.3t). MultiQC 22 (version 1.9) was used to integrate the quality control parameters of the whole process.
Variant annotation
Functional annotation of variants and in silico functional prediction of SNVs were performed using ANNOVAR 23 (version 20180416), based on different databases. The variant allele frequency was annotated by the 1000 Genomes Project 24 (1000G, release August 2015) data, the Exome Aggregation Consortium 25 (ExAC, version 0.3) databases, the exome sequencing data of the Genome Aggregation Database (gnomAD, version 2.1.1). 26 The predicted mutation effect scores were annotated by SIFT 27 and Polyphen2. 28 The human gene mutation database (HGMD) 29 was searched for any known mutations or genes in the results of our study. The variant allele frequency was also obtained from the ChinaMAP 30 and the KOREAN population from KRGDB. 31
Identification of potential pathogenic variants
The following filters were adopted to identify potentially pathogenic genetic variations: (a) Based on the pedigree information of each family in our analysis, we screened for the variants according to the likely inheritance pattern. If HV is inherited in an autosomal dominant pattern, then the affected child(ren) should carry heterozygous mutations, the affected parent should carry the same mutations, and unaffected parent should be wild-type. Conversely, if HV is inherited in an autosomal recessive pattern, the affected child(ren) and the affected parent should carry the same homozygous mutations, and the unaffected parent should carry the heterozygous mutations; (b) variants are located at exonic or splicing sites; (c) functional variants, including nonsynonymous SNVs, stop loss, stop gain, frameshift indels, variants at splicing donor/recipient sites; (d) SNVs predicted to be deleterious by both SIFT and PolyPhen-2 HDIV; (e) variants with mAF > 0.001 in East Asian Population (EAS) of the 1000G, ExAC or gnomAD, and variants with mAF > 0.01 in ChinaMAP databases or the KOREAN population from KRGDB were excluded from further analysis; (f) allelic depths (ADs) of alternative alleles >4.
Sanger sequencing validation
Sanger sequencing was conducted to verify the mutations we found. The genomic DNA of patients and their unaffected relatives in each pedigree was extracted (QIAamp DNA Blood Mini Kit, Germany). The primers of each mutation site were designed with a length of approximately 200bp from upstream and downstream (Supplementary Table 1). All sequences information was extracted from NCBI database. Phanta max super-fidelity DNA polymerase (Vazyme, China) was used to perform PCR (95°C 3 min; 35 cycles: 95°C 15 s, 56°C 15 s, 72°C 30 s; 72°C 5 min). All PCR products were verified by DNA agarose gel electrophoresis and purified by gel DNA extraction. The product after purification and matching primers were used for sanger sequencing.
Results
To identify causative variants for HV, we performed WES (Supplementary Figure 1) on the three probands and their family members (Figure 1). The primary clinical characteristics of all participants are shown in Table 1. The proband in each family had first-degree relatives affected, and most probands self-reported that they developed HV around teenage. The appearance of the patients’ and direct relatives’ feet and the X-rays of the probands’ affected feet are shown in Supplementary Figures 2 to 7.
Table 1.
The primary clinical characteristics of all participants.
| Family | ID | Kinship | Sex | Current age | Hallux valgus | Degree | Age of onset | Surgery | Other conditions | BMI | Wearing unfit shoes |
|---|---|---|---|---|---|---|---|---|---|---|---|
| F1 | I-1 | Father | Male | 64 | × | N/A | N/A | N/A | Coronary heart disease and diabetes | 27.10 | × |
| I-2 | Mother | Female | 64 | √ | Mild | 45 | × | High blood pressure | 26.67 | √ | |
| II-1 | Proband | Female | 29 | √ | Moderate | 5 | √ | None | 25.15 | √ | |
| II-2 | Sister | Female | 37 | √ | Moderate | 10 | × | None | 30.11 | √ | |
| F2 | I-1 | Wife | Female | 48 | × | N/A | N/A | N/A | None | 25.39 | × |
| I-2 | Proband | Male | 45 | √ | Moderate | Congenital | √ | None | 27.68 | × | |
| II-1 | Daughter | Female | 23 | √ | Moderate | Congenital | × | None | 24.22 | × | |
| F3 | I-1 | Mother | Female | 50 | × | N/A | N/A | N/A | None | 27.14 | × |
| I-2 | Father | Male | 54 | √ | Moderate | 10 | × | None | 23.53 | × | |
| II-1 | Proband | Female | 26 | √ | Moderate | 10 | √ | Fourth metatarsal short deformity | 20.70 | × |
The table shows a series of clinical information of the subjects enrolled in this study.Family: the serial number of the family which the subject belongs to. ID: the subject's anonymized number. Kinship: the relationship between the family member and the proband. Current age: the age at which the subject participated in the study, received orthopedic evaluation, and blood samples were collected. Hallux valgus: whether the subject was diagnosed with hallux valgus when participating in this study. Degree: the degree of hallux valgus of the patient. Age of onset: approximate onset age of hallux valgus reported by the patient and his/her family members. Surgery: whether the patient has undergone an orthomorphia. Other conditions: other diseases that the patient has been diagnosed with in the past. BMI: the subject's body mass index. Wearing unfit shoes: whether the subject wears unfit shoes. N/A: not applicable.
The average value of median coverages in the capture area of all samples was ∼139.4X. Coverage more than 30X was obtained in ∼95.76% capture area averagely. More than 99.9% reads were aligned to reference genome. Transitions/transversions ratios varied from 2.365 to 2.386. The detailed quality parameters of each sample are shown in Supplementary Table 2.
The numbers of remaining SNVs and indels after each screening step are shown in Table 2. A total of 36 SNVs in the exon regions meeting the aforementioned screening criteria were identified from the families (Table 3). All SNVs that meet the filter criteria are in line with the classic Mendelian autosomal or X chromosome homologous regions dominant inheritance mode, and no recessive mode SNVs (i.e. homozygous) that meet the filter conditions have been obtained, which is basically consistent with the suggested dominant inheritance of HV. 7 In addition, all indels and variants in splicing sites were finally eliminated due to not meeting certain filter criteria.
Table 2.
The number of remaining SNVs and indels after each filtering step.
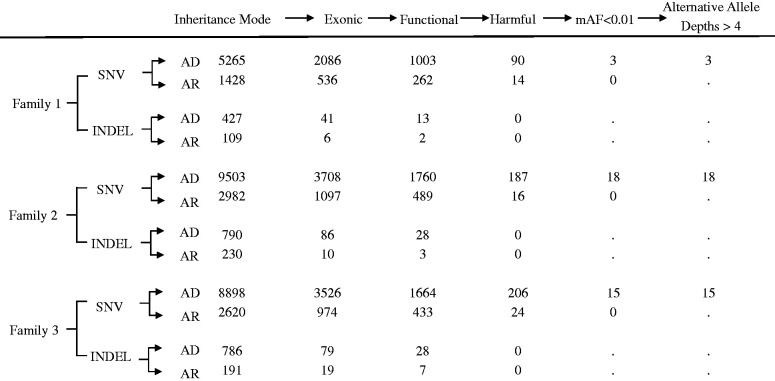
|
The column headings represent the work flow of the filtering for candidate mutations, which is described in details in the section of “Identification of Potential Pathogenic Variants” in Materials and Methods. The numbers of remaining SNV(s) or indel(s) in the family after each filtering step are shown. AD: autosomal dominant inheritance; AR: autosomal recessive inheritance.
Table 3.
The 36 SNVs in the exon regions meeting the screening criteria.
| Family | Carrier | SNV | Chr | Pos(GRCh37) | Ref | Alt | Gene | ChinaMap | dbSNP | ExAC | GnomAD | SIFT | Polyphen2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | I-2 II-1 II-2 | rs375155430 | 4 | 8613781 | G | A | CPZ | 1.42E-04 | 1.03E-03 | 5.E-04 | 3.E-04 | 0.001 | 1 |
| rs200622924 | 5 | 40958328 | C | T | C7 | 8.50E-04 | 3.42E-04 | 5.E-04 | 6.E-04 | 0 | 1 | ||
| rs12210538 | 6 | 110760008 | A | G | SLC22A16 | 1.98E-03 | 3.42E-04 | 2.E-04 | 3.E-04 | 0.004 | 0.959 | ||
| 2 | I-2 II-1 | . | 1 | 93682299 | T | G | CCDC18 | . | . | . | . | 0 | 1 |
| . | 1 | 180065192 | A | C | CEP350 | . | . | . | . | 0.013 | 0.999 | ||
| rs144715956 | 2 | 99013615 | G | A | CNGA3 | 9.44E-05 | 7.20E-05 | 2.E-04 | 2.E-04 | 0 | 1 | ||
| . | 2 | 179482520 | A | C | TTN | . | . | . | . | 0.001 | 0.989 | ||
| rs116608946 | 2 | 238258801 | G | A | COL6A3 | 4.25E-04 | 2.95E-04 | 2.E-04 | 1.E-04 | 0 | 1 | ||
| . | 5 | 43659448 | G | C | NNT | . | . | . | . | 0 | 1 | ||
| rs145644461 | 5 | 145499963 | C | T | LARS | . | 1.09E-03 | 2.E-04 | 1.E-04 | 0.027 | 1 | ||
| rs749833988 | 7 | 99022765 | G | A | ATP5J2-PTCD1 | 9.44E-05 | 1.60E-05 | 2.E-04 | 1.E-04 | 0.001 | 1 | ||
| rs1381527067 | 8 | 11406537 | G | C | BLK | 4.72E-05 | 2.40E-05 | . | . | 0 | 1 | ||
| rs1224964414 | 9 | 1052118 | C | G | DMRT2 | . | . | . | . | 0 | 1 | ||
| rs767474343 | 9 | 77567319 | G | T | C9orf40 | 5.19E-04 | 4.00E-05 | 3.E-04 | 6.E-04 | 0.001 | 1 | ||
| rs1374393461 | 10 | 88421115 | T | A | OPN4 | . | . | . | . | 0 | 1 | ||
| rs748523810 | 11 | 5345338 | G | T | OR51B2 | . | . | . | . | 0 | 1 | ||
| rs1272574331 | 11 | 5529289 | C | A | UBQLN3 | 1.89E-04 | . | . | . | 0.011 | 0.996 | ||
| . | 14 | 45658252 | A | G | FANCM | . | . | . | . | 0 | 1 | ||
| rs775421550 | 16 | 8890128 | C | A | TMEM186 | 1.23E-03 | 2.40E-03 | 8.E-04 | 6.E-04 | 0.004 | 1 | ||
| . | 16 | 66430084 | C | T | CDH5 | 4.72E-05 | . | . | . | 0 | 1 | ||
| rs1177611736 | 16 | 70900211 | G | T | HYDIN | . | 1.60E-05 | . | 2.E-04 | 0 | 0.999 | ||
| 3 | I-2 II-1 | rs758732580 | 1 | 11775244 | G | A | DRAXIN | 9.44E-04 | 3.42E-04 | 1.E-04 | 5.E-04 | 0 | 1 |
| rs768428875 | 1 | 177909759 | G | A | SEC16B | 1.42E-04 | 3.50E-04 | 1.E-04 | 1.E-04 | 0 | 1 | ||
| rs200878036 | 2 | 15613385 | C | G | NBAS | 1.42E-04 | . | . | . | 0.003 | 0.999 | ||
| rs374591393 | 2 | 175289331 | T | A | SCRN3 | . | . | . | . | 0 | 1 | ||
| . | 5 | 179155613 | T | A | CANX | . | . | . | . | 0.012 | 0.982 | ||
| rs762123072 | 6 | 110062680 | C | T | FIG4 | 8.03E-04 | 6.83E-04 | 7.E-04 | 7.E-04 | 0.015 | 0.998 | ||
| rs760314999 | 6 | 158924417 | C | T | TULP4 | 3.31E-04 | 6.80E-05 | 8.E-04 | 9.E-04 | 0 | 1 | ||
| rs764960008 | 9 | 74365207 | A | G | TMEM2 | 2.83E-04 | 2.80E-05 | 3.E-04 | 4.E-04 | 0 | 0.997 | ||
| rs1316987134 | 17 | 4045943 | A | T | ZZEF1 | . | . | . | . | 0 | 1 | ||
| . | 17 | 5086381 | G | C | ZNF594 | . | . | . | . | 0.002 | 0.994 | ||
| rs1006825019 | 17 | 73836605 | C | T | UNC13D | 9.44E-05 | 8.00E-06 | . | . | 0.009 | 0.994 | ||
| rs140002610 | 21 | 44486410 | G | A | CBS | 7.56E-04 | 1.71E-03 | 7.E-04 | 5.E-04 | 0.003 | 0.999 | ||
| rs376115617 | 22 | 25599848 | C | T | CRYBB3 | 2.83E-04 | 6.00E-05 | 2.E-04 | 2.E-04 | 0 | 1 | ||
| . | 22 | 36902126 | G | A | FOXRED2 | . | . | . | . | 0.001 | 1 | ||
| rs764088622 | X | 132161219 | G | A | USP26 | . | 6.84E-04 | 8.E-04 | 5.E-04 | 0.039 | 0.99 |
All mutations are nonsynonymous SNV. None of the above mutations have been reported in the East Asian population of the 1000G database.
Family: family number that inherited this mutation; Carrier: family members with this mutation; SNV: rsID of the single nucleotide variant in dbSNP database; Chr: chromosome which the SNV located in; Pos(GRCh37): GRCh37 coordinate position of the SNV; Ref: reference allele of the SNV; Alt: alternative allele of the SNV; Gene: gene which the SNV is located in or nearest to; ChinaMap: allele frequency of the SNV in ChinaMap database; dbSNP: allele frequency of the SNV in KOREAN population from KRGDB in dbSNP database; ExAC: allele frequency of the SNV in East Asian population from ExAC database; GnomAD: allele frequency of the SNV in East Asian population from exome data of GnomAD database; SIFT: harmfulness score of the SNV given by SIFT, the smaller the score, the stronger the harmfulness; Polyphen2: harmfulness score of the SNV given by Polyphen2 HDIV, the greater the score, the stronger the harmfulness.
Among the candidate genes with the identified SNVs, genes involved in bone development and digital anomalies deserve particular attention. In Family 2, three functional candidates were highlighted, i.e. the titin gene (TTN), the collagen type VI alpha 3 chain gene (COL6A3), and the leucyl-tRNA synthetase gene (LARS). In Family 3, two genes were highlighted, i.e. the FIG4 phosphoinositide 5-phosphatase gene (FIG4), and the cystathionine beta-synthase gene (CBS). Furthermore, the complement C7 gene (C7) was highlighted in Family 1, which suggested a different mechanism. These abovementioned mutation sites have been verified by Sanger sequencing. Sanger sequencing results were consistent with WES results (Figure 2).
Figure 2.

Sanger sequencing results of highlighted variants. The chromatograms show the sanger sequencing results of some of the mutations that this study focused on. The coordinates of the mutation (GRCh37) are marked below the graph group, and the gray vertical shading in the chromatogram indicates its location. The subject number corresponding to each chromatogram is marked on the upper left. The color of each base peak corresponds to the color of the base letter above. The height of the peak represents the relative signal strength of this base.
Discussion
This study identified functional candidate SNVs from three HV families. The candidate genes identified in each family bring insights into the mechanisms of HV occurrence and development.
Among the candidate genes identified in Family 2, TTN, COL6A3, and LARS have been suggested of their roles in the anatomical development of long toe and long fingers by previous studies.32–34 TTN contains as many as 363 exons and encodes titin which is the largest known protein. 35 Titin plays an important role in maintaining the physiological position of myosin molecules and passive muscle tension. It connects the Z-line and M-line of the sarcomere and is especially important in the contraction of striated muscle.36,37 COL6A3 is necessary for the generation of type VI collagen. Type VI collagen can be located in the extracellular matrix of skeletal muscle cells, thereby affecting the movement of the muscles; it can also be located around the cells in the connective tissue to provide strength and flexibility to the joints.38–41 LARS encodes the cytosolic leucine-tRNA synthetase which belongs to the class I aminoacyl-tRNA synthetase family, while its mutation causes infantile liver failure syndrome 1 with the phenotypes of long fingers and long toe. 42 Among Family 3, FIG4 and CBS related to long fingers were highlighted.43,44 FIG4 encodes phosphatidylinositol 3,5-bisphosphate 5-phosphatase. It was proved that FIG4 mutations can cause Yunis-Varón syndrome which is an autosomal-recessive disorder with clinical signs of cleidocranial dysplasia and digital anomalies. 43 CBS encodes Cystathionine-β-synthase which catalyzes the first step of the transsulfuration pathway from homocysteine to cystathionine. Mutations in CBS may relate to pyridoxine-responsive homocystinuria patients with clinical signs including slender limbs, spidery slender fingers and toes, weak muscles, and arched feet. 44 Combining our findings in the above two families and the reported physiological functions related to the differentiation of osteoblasts, as well as the phenotypic associations of these five genes, this study suggests that HV may be a consequence of abnormal digital and bone development, and related to the maintenance of muscle tension and the performance of joint functions. The local bone stress tolerance changes caused by genetic variants may thus be related to the occurrence and development of HV.
In contrast, a different mechanism is suggested by the finding in Family 1. Complement component 7 is an important component in the complement system and plays an essential role in innate immune system. C7 is a component of the membrane attack complex which can mediate cell lysis and death. 45 C7 deficiency is related to ankylosing spondylitis 46 and rheumatoid arthritis. 47 Our findings suggest that C7 may trigger the pathological changes similar to inflammatory joint diseases such as rheumatoid arthritis and ankylosing spondylitis by regulating the immune response including innate immunity, leading to the deformity of the bone structure. Therefore, different from the above five genes related to digital abnormalities and bone development, the SNV of C7 suggests that HV could be related to (and a consequence of) chronic inflammation.
Compared with GWAS catalog 48 and the previous studies,10,49,50 all the SNVs identified in this study have not been reported to be associated with HV before. The reason lies in two aspects. First, the approach of WES used in our study examined the coding region of the human genome. However, the vast majority of the SNPs surveyed by the GWAS approach are located in the non-coding region. 11 Second, our study focused on SNVs with low-mAF ( < 0.01), 51 while GWAS was not sensitive to these low-frequency variants and instead focus on SNPs with mAF > 0.01. 11 In summary, the research target of WES and GWAS are distinct and complementary to each other.
Whole-genome sequencing is a more comprehensive sequencing method, especially for mutations in introns, regulatory regions, and repetitive DNA. As mentioned in the “Introduction” section, although the exome accounts for only ∼1% of the whole human genome, 85% of the pathogenic mutations related to Mendelian diseases were harbored in this region.12,13 Therefore, compared with whole genome sequencing, WES is more cost-efficient. However, the disadvantages of WES must also be acknowledged. First, it is sometimes less accurate than first-generation sequencing. Therefore, Sanger sequencing is used to validate important findings from WES. Secondly, the significance of a considerable part of the variants discovered by next-generation sequencing was not thoroughly clear.52,53 Therefore, it is still challenging to determine whether certain variants are related to patients’ disease, phenotype, etc. To get a better interpretation of the results obtained from WES sequencing, this study jointly applied the 1000 Genomes Project (1000G), the Exome Aggregation Consortium (ExAC), the Genome Aggregation Database (GnomAD), ChinaMAP, KRGDB, SIFT, Polyphen2, the HGMD, and other databases for mutation annotations.
Through WES, our study give additional insight into the genetic basis underlying the development of HV. So far, orthomorphia was still the main treatment for HV. If non-invasive therapeutic approaches can be used to intervene HV at an early stage, some patients may have the opportunity to be exempt from surgery. This study laid the foundation for a deeper understanding of the molecular mechanism of HV, and it helps to provide targets to facilitate the development of novel therapeutic approaches.
Conclusions
This study acquires critical insights into the physiological foundations of HV, represented by genes involved in the anatomical development of long toe, long fingers, and other digital anomalies, as well as a gene related to chronic arthritis. These discoveries may have important clinical implication, e.g. by enabling the early identification of patients with high risk of HV (e.g. with long toe or chronic arthritis) for prevention and early intervention. More importantly, chronic arthritis underlying HV should not be overlooked. At the same time, more than one functional candidate SNVs identified in the affected families may suggest that some family-types of HV may be digenic or oligogenic, instead of a monogenic dominant-inherited disease. However, we have to admit that this study has limitations, mainly due to the small sample size. Further study by recruiting more patients is warranted to confirm the findings of this study.
Supplemental Material
Supplemental material, sj-pdf-1-ebm-10.1177_15353702211008641 for New insights into hallux valgus by whole exome sequencing study by Jun Jia, Junyi Li, Huiqi Qu, Mengyu Li, Sipeng Zhang, Jun Hao, Xinyi Gao, Xinyi Meng, Yan Sun, Hakon Hakonarson, Xiantie Zeng, Qianghua Xia and Jin Li in Experimental Biology and Medicine
Footnotes
AUTHORS’ CONTRIBUTIONS: Conceptualization: XZ, QX, and JL; methodology: QX and JL; software: JYL and XM; validation: SZ; formal analysis: JYL and HQ; investigation: JJ, JYL, HQ, ML, and XG; resources: JJ, JH, and XZ; data curation: QX and JL; writing—original draft preparation: JJ, JYL, and HQ; writing—review and editing: HQ, XM, YS, HH, XZ, QX, and JL; visualization: JYL, HQ, and SZ; supervision: HH and JL; project administration: QX and JL; funding acquisition: JL. JJ, JYL, and HQ contributed equally to this paper. All authors have read and agreed to the published version of the manuscript.
DECLARATION OF CONFLICTING INTERESTS: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
ETHICAL APPROVAL: The study was approved by the institutional review board of Tianjin Hospital, and the informed consents were obtained from all the participants.
FUNDING: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by National Natural Science Foundation of China (grant number 81771769) and Natural Science Foundation of Tianjin City (grant number 18JCYBJC42700).
DATA AVAILABILITY: The scripts used for the analysis are available at GitHub for open access (https://github.com/JunyiLi-TMU/New-Insights-into-Hallux-Valgus-by-Whole-Exome-Sequencing-Study).
ORCID iD: Huiqi Qu https://orcid.org/0000-0001-9317-4488
SUPPLEMENTAL MATERIAL: Supplemental material for this article is available online.
References
- 1.Wülker N, Mittag F. The treatment of hallux valgus. Dtsch Arztebl Int 2012; 109:857–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nix S, Smith M, Vicenzino B. Prevalence of hallux valgus in the general population: a systematic review and meta-analysis. J Foot Ankle Res 2010; 3:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hecht PJ, Lin TJ. Hallux valgus. Med Clin North Am 2014; 98:227–32 [DOI] [PubMed] [Google Scholar]
- 4.Perera A, Mason L, Stephens M. The pathogenesis of hallux valgus. J Bone Joint Surg Am 2011; 93:1650–61 [DOI] [PubMed] [Google Scholar]
- 5.Zirngibl B, Grifka J, Baier C, Gotz J. Hallux valgus: etiology, diagnosis, and therapeutic principles. Orthopade 2017; 46:283–96 [DOI] [PubMed] [Google Scholar]
- 6.Mann RA, Coughlin MJ. Hallux valgus–etiology, anatomy, treatment and surgical considerations. Clin Orthop Relat Res 1981;(157):31–41 [PubMed] [Google Scholar]
- 7.Pique-Vidal C, Sole MT, Antich J. Hallux valgus inheritance: pedigree research in 350 patients with bunion deformity. J Foot Ankle Surg 2007; 46:149–54 [DOI] [PubMed] [Google Scholar]
- 8.Hannan MT, Menz HB, Jordan JM, Cupples LA, Cheng C-H, Hsu Y-H. High heritability of hallux valgus and lesser toe deformities in adult men and women. Arthritis Care Res 2013; 65:1515–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee CH, Lee S, Kang H, Jung DE, Song YM, Lee K, Lee K, Hwang J, Sung J. Genetic influences on hallux valgus in Koreans: the healthy twin study. Twin Res Hum Genet 2014; 17:121–26 [DOI] [PubMed] [Google Scholar]
- 10.Hsu YH, Liu Y, Hannan MT, Maixner W, Smith SB, Diatchenko L, Golightly YM, Menz HB, Kraus VB, Doherty M, Wilson AG, Jordan JM. Genome-wide association meta-analyses to identify common genetic variants associated with hallux valgus in Caucasian and African Americans. J Med Genet 2015; 52:762–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, Yang J. 10 Years of GWAS discovery: biology, function, and translation. Am J Hum Genet 2017; 101:5–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, Shaffer T, Wong M, Bhattacharjee A, Eichler EE, Bamshad M, Nickerson DA, Shendure J. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009; 461:272–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, Nayir A, Bakkaloglu A, Ozen S, Sanjad S, Nelson-Williams C, Farhi A, Mane S, Lifton RP. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A 2009; 106:19096–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 2011; 17:10–12 [Google Scholar]
- 15.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20:1297–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, Gabriel S, DePristo MA. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 2013; 43:11.10.1–11.10.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011; 43:491–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics 2009; 25:2078–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jun G, Flickinger M, Hetrick Kurt N, Romm Jane M, Doheny Kimberly F, Abecasis Gonçalo R, Boehnke M, Kang Hyun M. Detecting and estimating contamination of human DNA samples in sequencing and array-based genotype data. Am J Hum Genet 2012; 91:839–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R. The variant call format and VCFtools. Bioinformatics 2011; 27:2156–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin ) 2012; 6:80–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016; 32:3047–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Auton A, Abecasis GR, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, Donnelly P, Eichler EE, Flicek P, Gabriel SB, Gibbs RA, Green ED, Hurles ME, Knoppers BM, Korbel JO, Lander ES, Lee C, Lehrach H, Mardis ER, Marth GT, McVean GA, Nickerson DA, Schmidt JP, Sherry ST, Wang J, Wilson RK, Gibbs RA, Boerwinkle E, Doddapaneni H, Han Y, Korchina V, Kovar C, Lee S, Muzny D, Reid JG, Zhu Y, Wang J, Chang Y, Feng Q, Fang X, Guo X, Jian M, Jiang H, Jin X, Lan T, Li G, Li J, Li Y, Liu S, Liu X, Lu Y, Ma X, Tang M, Wang B, Wang G, Wu H, Wu R, Xu X, Yin Y, Zhang D, Zhang W, Zhao J, Zhao M, Zheng X, Lander ES, Altshuler DM, Gabriel SB, Gupta N, Gharani N, Toji LH, Gerry NP, Resch AM, Flicek P, Barker J, Clarke L, Gil L, Hunt SE, Kelman G, Kulesha E, Leinonen R, McLaren WM, Radhakrishnan R, Roa A, Smirnov D, Smith RE, Streeter I, Thormann A, Toneva I, Vaughan B, Zheng-Bradley X, Bentley DR, Grocock R, Humphray S, James T, Kingsbury Z, Lehrach H, Sudbrak R, Albrecht MW, Amstislavskiy VS, Borodina TA, Lienhard M, Mertes F, Sultan M, Timmermann B, Yaspo M-L, Mardis ER, Wilson RK, Fulton L, Fulton R, Sherry ST, Ananiev V, Belaia Z, Beloslyudtsev D, Bouk N, Chen C, Church D, Cohen R, Cook C, Garner J, Hefferon T, Kimelman M, Liu C, Lopez J, Meric P, O’Sullivan C, Ostapchuk Y, Phan L, Ponomarov S, Schneider V, Shekhtman E, Sirotkin K, Slotta D, Zhang H, McVean GA, Durbin RM, Balasubramaniam S, Burton J, Danecek P, Keane TM, Kolb-Kokocinski A, McCarthy S, Stalker J, Quail M, Schmidt JP, Davies CJ, Gollub J, Webster T, Wong B, Zhan Y, Auton A, Campbell CL, Kong Y, Marcketta A, Gibbs RA, Yu F, Antunes L, Bainbridge M, Muzny D, Sabo A, Huang Z, Wang J, Coin LJM, Fang L, Guo X, Jin X, Li G, Li Q, Li Y, Li Z, Lin H, Liu B, Luo R, Shao H, Xie Y, Ye C, Yu C, Zhang F, Zheng H, Zhu H, Alkan C, Dal E, Kahveci F, Marth GT, Garrison EP, Kural D, Lee W-P, Fung Leong W, Stromberg M, Ward AN, Wu J, Zhang M, Daly MJ, DePristo MA, Handsaker RE, Altshuler DM, Banks E, Bhatia G, del Angel G, Gabriel SB, Genovese G, Gupta N, Li H, Kashin S, Lander ES, McCarroll SA, Nemesh JC, Poplin RE, Yoon SC, Lihm J, Makarov V, Clark AG, Gottipati S, Keinan A, Rodriguez-Flores JL, Korbel JO, Rausch T, Fritz MH, Stütz AM, Flicek P, Beal K, Clarke L, Datta A, Herrero J, McLaren WM, Ritchie GRS, Smith RE, Zerbino D, Zheng-Bradley X, Sabeti PC, Shlyakhter I, Schaffner SF, Vitti J, Cooper DN, Ball EV, Stenson PD, Bentley DR, Barnes B, Bauer M, Keira Cheetham R, Cox A, Eberle M, Humphray S, Kahn S, Murray L, Peden J, Shaw R, Kenny EE, Batzer MA, Konkel MK, Walker JA, MacArthur DG, Lek M, Sudbrak R, Amstislavskiy VS, Herwig R, Mardis ER, Ding L, Koboldt DC, Larson D, Ye K, Gravel S, The Genomes Project C, Corresponding a, Steering c, Production g, Baylor College of M, Shenzhen BGI, Broad Institute of MIT, Harvard, Coriell Institute for Medical R, European Molecular Biology Laboratory EBI, Illumina, Max Planck Institute for Molecular G, McDonnell Genome Institute at Washington U, Health USNIo, University of O, Wellcome Trust Sanger I, Analysis g, Affymetrix, Albert Einstein College of M, Bilkent U, Boston C, Cold Spring Harbor L, Cornell U, European Molecular Biology L, Harvard U, Human Gene Mutation D, Icahn School of Medicine at Mount S, Louisiana State U, Massachusetts General H, McGill U, National Eye Institute NIH. A global reference for human genetic variation . Nature 2015; 526:68–7426432245 [Google Scholar]
- 25.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won H-H, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG, Exome Aggregation C. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016; 536:285–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O’Donnell-Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, Aguilar Salinas CA, Ahmad T, Albert CM, Ardissino D, Atzmon G, Barnard J, Beaugerie L, Benjamin EJ, Boehnke M, Bonnycastle LL, Bottinger EP, Bowden DW, Bown MJ, Chambers JC, Chan JC, Chasman D, Cho J, Chung MK, Cohen B, Correa A, Dabelea D, Daly MJ, Darbar D, Duggirala R, Dupuis J, Ellinor PT, Elosua R, Erdmann J, Esko T, Färkkilä M, Florez J, Franke A, Getz G, Glaser B, Glatt SJ, Goldstein D, Gonzalez C, Groop L, Haiman C, Hanis C, Harms M, Hiltunen M, Holi MM, Hultman CM, Kallela M, Kaprio J, Kathiresan S, Kim B-J, Kim YJ, Kirov G, Kooner J, Koskinen S, Krumholz HM, Kugathasan S, Kwak SH, Laakso M, Lehtimäki T, Loos RJF, Lubitz SA, Ma RCW, MacArthur DG, Marrugat J, Mattila KM, McCarroll S, McCarthy MI, McGovern D, McPherson R, Meigs JB, Melander O, Metspalu A, Neale BM, Nilsson PM, O’Donovan MC, Ongur D, Orozco L, Owen MJ, Palmer CNA, Palotie A, Park KS, Pato C, Pulver AE, Rahman N, Remes AM, Rioux JD, Ripatti S, Roden DM, Saleheen D, Salomaa V, Samani NJ, Scharf J, Schunkert H, Shoemaker MB, Sklar P, Soininen H, Sokol H, Spector T, Sullivan PF, Suvisaari J, Tai ES, Teo YY, Tiinamaija T, Tsuang M, Turner D, Tusie- Luna T, Vartiainen E, Vawter MP, Ware JS, Watkins H, Weersma RK, Wessman M, Wilson JG, Xavier RJ, Neale BM, Daly MJ, MacArthur DG, Genome Aggregation Database C. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020; 581:434–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sim N-L, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res 2012; 40:W452–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7:248–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, Cooper DN. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet 2017; 136:665–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao Y, Li L, Xu M, Feng Z, Sun X, Lu J, Xu Y, Du P, Wang T, Hu R, Ye Z, Shi L, Tang X, Yan L, Gao Z, Chen G, Zhang Y, Chen L, Ning G, Bi Y, Wang W. The ChinaMAP analytics of deep whole genome sequences in 10,588 individuals. Cell Res 2020; 30:717–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jung KS, Hong KW, Jo HY, Choi J, Ban HJ, Cho SB, Chung M. KRGDB: the large-scale variant database of 1722 Koreans based on whole genome sequencing. Database: The Journal of Biological Databases and Curation 2020;2020:baz146 [DOI] [PMC free article] [PubMed]
- 32.Demir E, Sabatelli P, Allamand V, Ferreiro A, Moghadaszadeh B, Makrelouf M, Topaloglu H, Echenne B, Merlini L, Guicheney P. Mutations in COL6A3 cause severe and mild phenotypes of Ullrich congenital muscular dystrophy. Am J Hum Genet 2002; 70:1446–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Casey JP, McGettigan P, Lynam-Lennon N, McDermott M, Regan R, Conroy J, Bourke B, O'Sullivan J, Crushell E, Lynch S, Ennis S. Identification of a mutation in LARS as a novel cause of infantile hepatopathy. Mol Genet Metab 2012; 106:351–8 [DOI] [PubMed] [Google Scholar]
- 34.Hackman P, Vihola A, Haravuori H, Marchand S, Sarparanta J, De Seze J, Labeit S, Witt C, Peltonen L, Richard I, Udd B. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am J Hum Genet 2002; 71:492–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bang ML, Centner T, Fornoff F, Geach AJ, Gotthardt M, McNabb M, Witt CC, Labeit D, Gregorio CC, Granzier H, Labeit S. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res 2001; 89:1065–72 [DOI] [PubMed] [Google Scholar]
- 36.Itoh-Satoh M, Hayashi T, Nishi H, Koga Y, Arimura T, Koyanagi T, Takahashi M, Hohda S, Ueda K, Nouchi T, Hiroe M, Marumo F, Imaizumi T, Yasunami M, Kimura A. Titin mutations as the molecular basis for dilated cardiomyopathy. Biochem Biophys Res Commun 2002; 291:385–93 [DOI] [PubMed] [Google Scholar]
- 37.Opitz CA, Kulke M, Leake MC, Neagoe C, Hinssen H, Hajjar RJ, Linke WA. Damped elastic recoil of the titin spring in myofibrils of human myocardium. Proc Natl Acad Sci U S A 2003; 100:12688–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baker NL, Mörgelin M, Peat R, Goemans N, North KN, Bateman JF, Lamandé SR. Dominant collagen VI mutations are a common cause of Ullrich congenital muscular dystrophy. Hum Mol Genet 2005; 14:279–93 [DOI] [PubMed] [Google Scholar]
- 39.Bönnemann CG. The collagen VI-related myopathies: muscle meets its matrix. Nat Rev Neurol 2011; 7:379–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bushby KM, Collins J, Hicks D. Collagen type VI myopathies. Adv Exp Med Biol 2014; 802:185–99 [DOI] [PubMed] [Google Scholar]
- 41.Baker NL, Mörgelin M, Pace RA, Peat RA, Adams NE, Gardner RJ, Rowland LP, Miller G, De Jonghe P, Ceulemans B, Hannibal MC, Edwards M, Thompson EM, Jacobson R, Quinlivan RC, Aftimos S, Kornberg AJ, North KN, Bateman JF, Lamandé SR. Molecular consequences of dominant Bethlem myopathy collagen VI mutations. Ann Neurol 2007; 62:390–405 [DOI] [PubMed] [Google Scholar]
- 42.Liu RJ, Long T, Li H, Zhao J, Li J, Wang M, Palencia A, Lin J, Cusack S, Wang ED. Molecular basis of the multifaceted functions of human leucyl-tRNA synthetase in protein synthesis and beyond. Nucl Acids Res 2020; 48:4946–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Campeau PM, Lenk GM, Lu JT, Bae Y, Burrage L, Turnpenny P, Román Corona-Rivera J, Morandi L, Mora M, Reutter H, Vulto-van Silfhout AT, Faivre L, Haan E, Gibbs RA, Meisler MH, Lee BH. Yunis-Varón syndrome is caused by mutations in FIG4, encoding a phosphoinositide phosphatase. Am J Hum Genet 2013; 92:781–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aral B, Coudé M, London J, Aupetit J, Chassé JF, Zabot MT, Chadefaux-Vekemans B, Kamoun P. Two novel mutations (K384E and L539S) in the C-terminal moiety of the cystathionine beta-synthase protein in two French pyridoxine-responsive homocystinuria patients. Hum Mutat 1997; 9:81–82 [DOI] [PubMed] [Google Scholar]
- 45.Morgan BP, Boyd C, Bubeck D. Molecular cell biology of complement membrane attack. Semin Cell Dev Biol 2017; 72:124–32 [DOI] [PubMed] [Google Scholar]
- 46.Jiménez FB, Rico GR, Bravo CG, Mintz GS. Functional abnormalities of complement in familial and sporadic ankylosing spondylitis. Archivos de investigación médica 1989; 20:79–86 [PubMed] [Google Scholar]
- 47.Alcalay M, Bontoux D, Peltier A, Vial MC, Vilde JM, Wautier JL. C7 deficiency, abnormal platelet aggregation, and rheumatoid arthritis. Arthritis Rheum 1981; 24:102–3 [DOI] [PubMed] [Google Scholar]
- 48.Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, Suveges D, Vrousgou O, Whetzel PL, Amode R, Guillen JA, Riat HS, Trevanion SJ, Hall P, Junkins H, Flicek P, Burdett T, Hindorff LA, Cunningham F, Parkinson H. The NHGRI-EBI GWAS catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucl Acids Res 2019; 47:D1005–D1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang J, Wang J, Liang X, Zhao H, Lu J, Ma Q, Tian F. Relationship between genetic polymorphisms of the TNF gene and hallux valgus susceptibility. Genet Test Mol Biomark 2019; 23:380–86 [DOI] [PubMed] [Google Scholar]
- 50.Arbeeva L, Yau M, Mitchell BD, Jackson RD, Ryan K, Golightly YM, Hannan MT, Nelson A, Jordan JM, Hochberg MC. Genome-wide meta-analysis identified novel variant associated with hallux valgus in Caucasians. J Foot Ankle Res 2020; 13:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17:405–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nykamp K, Anderson M, Powers M, Garcia J, Herrera B, Ho YY, Kobayashi Y, Patil N, Thusberg J, Westbrook M, Topper S. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med 2017; 19:1105–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lappalainen T, Scott AJ, Brandt M, Hall IM. Genomic analysis in the age of human genome sequencing. Cell 2019; 177:70–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-pdf-1-ebm-10.1177_15353702211008641 for New insights into hallux valgus by whole exome sequencing study by Jun Jia, Junyi Li, Huiqi Qu, Mengyu Li, Sipeng Zhang, Jun Hao, Xinyi Gao, Xinyi Meng, Yan Sun, Hakon Hakonarson, Xiantie Zeng, Qianghua Xia and Jin Li in Experimental Biology and Medicine