

Abstract
We report on a fetus with cardiomegaly and increased middle cerebral artery-peak systolic velocity at 25 weeks of gestation. Severe fetal anemia (hemoglobin (Hb) level 37 g/L) was confirmed by cordocentesis. Hb analysis showed that Hb Bart’s was 9% in cord blood. Molecular analysis of the proband’s family found that the mother was a carrier of Hb Quong Sze (Hb QS, HBA2:c.377T>C), the father was a carrier of Hb Zurich-Albisrieden (Hb ZA, HBA2:c.178G>C), and the fetus was a compound heterozygote for Hb ZA and Hb QA. Despite intrauterine blood transfusions, the fetus experienced problems including oligohydramnios, growth retardation, placental thickening, and heart enlargement in the third trimester. The couple chose to terminate the pregnancy, and fetal autopsy confirmed the above diagnosis. This is the first report of a case of Hb ZA compounded with Hb QS, and provides a reference for genetic counselling and prenatal diagnosis in the Chinese population.
Keywords: Hemoglobin Zurich-Albisrieden, hemoglobin Quong Sze, fetal anemia, hydrops fetalis, cardiomegaly, prenatal diagnosis
Introduction
Fetal anemia is a serious condition with various possible causes, including maternal alloimmunization, virus infection, fetal aneuploidy, vascular tumors, arteriovenous malformations of the fetus or placenta, and inherited conditions such as hemoglobinopathies or genetic metabolic disorders.1,2 Hemoglobinopathies are the main cause of non-immune fetal anemia and hydrops fetalis (NIHF) in Southeast Asia. 1 Hemoglobin (Hb) Bart’s disease and some homozygous or compound heterozygote Hb variants, such as Hb Constant Spring (Hb CS) and Hb Quong Sze (Hb QS), which are relatively common in Asia, can also cause severe fetal anemia and hydrops fetalis.3–5 Hb Zurich-Albisrieden (Hb ZA) (α2 59(E8)Gly>Arg; HBA2:c.178G>C) was first reported in an adult male in 2004 and has been confirmed as a highly unstable Hb variant resulting in α-thalassemia. 6 Compound heterozygosity for Hb ZA and the Southeast Asian (--SEA) deletion has also recently been reported to cause hydrops fetalis, 7 and a Brazilian child with thalassemia major phenotype homozygous for the Hb ZA mutation (ZA/ZA) has also been described. 8 However, there have been no reports of compound heterozygosity for Hb ZA and Hb QS. Here, we report a rare case of severe fetal anemia and hydrops fetalis caused by compound heterozygosity for Hb ZA and Hb QS in a fetus in a Chinese woman.
Case report
A 31-year-old pregnant woman from Guangzhou, Guangdong Province, and her 32-year-old husband from Zhangzhou, Fujian Province, presented at our center at 25+3 weeks of gestation. Prenatal ultrasound examination at 25 weeks of gestation indicated fetal anemia with cardiomegaly (cardiothoracic area ratio 0.45, cardiothoracic diameter ratio 0.67) (Figure 1a), an enlarged placenta (3.3 cm), and increased fetal middle cerebral artery peak systolic velocity (MCA-PSV) (49.7 cm/s) (Figure 1b). There were no other structural anomalies. This was their first pregnancy. The couple agreed to participate in our study and signed informed consent for treatment and for publication. This study was approved by the Ethics Committee of Guangdong Women and Children Hospital (approval no.: 201901085) and the study was reported in accordance with the CARE guidelines. 9
Figure 1.

Prenatal ultrasound at 25 weeks’ gestation showing cardiomegaly (a) and increased fetal middle cerebral artery peak systolic velocity (b).
The mother’s blood group was A, Rhesus-positive. Tests for cytomegalovirus, rubella virus, parvovirus, and syphilis infections, glucose-6-phosphate dehydrogenase deficiency, red blood cell antibodies, and feto-maternal hemorrhage using the Kleihauer–Betke test were all negative. Thalassemia testing showed that the mother was heterozygous for Hb QS(αQSα/αα). Her husband had decreased mean corpuscular volume (70.7 fL), mean corpuscular Hb (25.1 pg), and HbA2 (2.6%), but was normal for both α- and β-thalassemia according to suspension-array system analysis, which detected the 23 most frequent deletions and mutations in southern China. Sanger sequencing and multiplex ligation-dependent probe amplification were carried out to check the husband’s thalassemia status. Sanger sequencing identified a heterozygous G > C mutation at the first base of codon 59 of the HBA2 gene, previously reported as Hb ZA, in the husband, following polymerase chain reaction amplification of the HBA1 and HBA2 genes using the following primers: HBA1 forward, 5′-TGGAGGGTGGAGACGTCCTG-3′ and reverse 5′-TCCATCCCCTCCTCCCGCCCCTGCCTTTTC-3′; HBA2 forward 5′-TGGAGGGTGGAGACGTCCTG-3′ and reverse 5′-CCATTGTTGGCACATTCCGG-3′. Multiplex ligation-dependent probe amplification analysis found no deletions or duplications in the α-globin genes.
After genetic counseling, the couple decided to undergo umbilical cord blood puncture to determine the cause of the fetal anemia and prepared for fetal intrauterine transfusion (IUT). Routine fetal blood examination showed severe fetal anemia (Hb 29 g/L) and Hb electrophoresis showed significantly elevated Hb Bart’s (9.4%) (Figure 2). Suspension-array analysis and Sanger sequencing indicated that the fetus was compound heterozygous for Hb ZA and Hb QS. The hematological data and genotypes of the family are summarized in Table 1 and Figure 3. Fetal aneuploidies were excluded by chromosome karyotyping and chromosome microarray analysis. Screening for rubella virus and cytomegalovirus in the amniotic fluid and IgM antibody detection of rubella virus, cytomegalovirus, and parvovirus in umbilical cord blood were all negative. Whole-exome sequencing (WES) revealed no other genetic causes of fetal anemia. The fetal Hb pre-IUT was 29 g/L and the MCA-PSV pre-IUT was 49.7 cm/s. Infusion of 57 mL of concentrated washed red blood cells via the umbilical vein increased the post-IUT Hb and MCA-PSV to 91 g/L and 26.6 cm/s, respectively. Subsequent ultrasonography was performed every 2 weeks to monitor fetal development and anemia. Ultrasound examination at 35 weeks of pregnancy showed oligohydramnios (amniotic fluid index 42 mm), fetal growth retardation, cardiomegaly with cardiothoracic diameter ratio of 0.65 and cardiothoracic area ratio of 0.43, an enlarged placenta (48 mm), and increased MCA-PSV (84 cm/s). Fetal hemoglobin was 65 g/L, confirmed by cordocentesis. After counseling, the decision was made to terminate the pregnancy. Postmortem autopsy indicated fetal hydrops with cardiomegaly, ascites, bilateral pleural effusion, and right foot varus.
Figure 2.

Hemoglobin analysis of fetal cord blood.
Table 1.
Hematological and genotypic data of the proband’s family.
| Parameter | Father | Mother | Proband pre IUT | Proband post IUT | Proband TOP |
|---|---|---|---|---|---|
| Age (years) | 32 | 31 | 25 weeks’ gestation | 25 weeks’ gestation | 25 weeks’ gestation |
| Hb (g/L) | 149 | 119 | 29 | 91 | 65 |
| RBC (1012/L) | 5.95 | 4.88 | 1.26 | 3.15 | 2.84 |
| HCT (%) | 46.5 | 36.9 | 10.6 | 28.0 | 21.1 |
| MCV (fL) | 78.2 | 75.1 | 84.1 | 88.9 | 75 |
| MCH (pg) | 25.0 | 24.2 | 23.0 | 28.9 | 23.8 |
| Hb A (%) | 97.4 | 97.3 | 6.8 | NA | NA |
| Hb A2 (%) | 2.6 | 2.7 | / | NA | NA |
| Hb F (%) | / | / | 83.8 | NA | NA |
| Hb Bart’s (%) | / | / | 9.4 | NA | NA |
| Serum ferritin | 278.3 | 28.0 | / | NA | NA |
| MCA-PSV (cm/s) | NA | NA | 49.7 | 26.6 | NA |
| HBA genotype | αZAα/αα | αQSα/αα | αZAα/αQSα | αZAα/αQSα | αZAα/αQSα |
| HBB genotype | βN/βN | βN/βN | βN/βN | βN/βN | βN/βN |
TOP, pregnancy termination; Hb, hemoglobin; RBC, red blood cell count; HCT, hematocrit; MCV, mean corpuscular volume; MCH, mean corpuscular Hb; ZA, Zurich-Albisrieden; NA, not available
Figure 3.
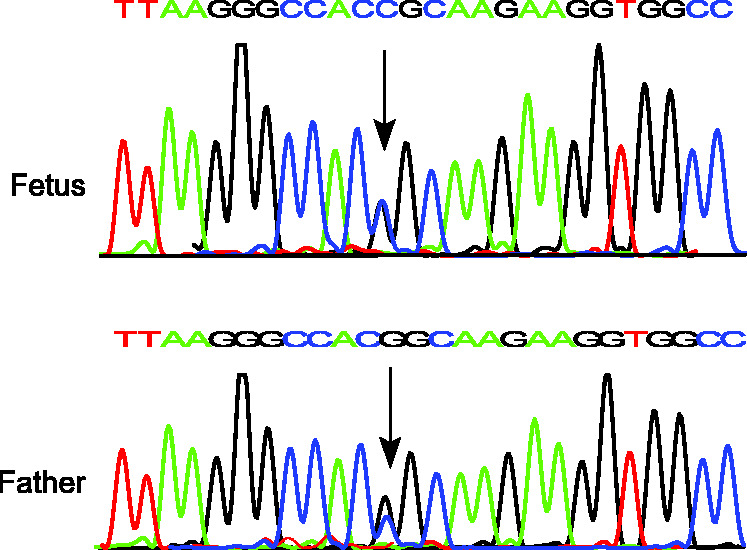
DNA sequencing of the HBA2 gene showing heterozygous Hb ZA mutation (HBA2: c.178G>C) in the fetus and father. Arrow indicates mutation.
Discussion
Hemoglobinopathies are the most common inherited disorders worldwide. Hb variants caused by mutations in globin genes leading to structural changes in the Hb molecule are responsible for a group of hereditary Hb diseases, ranging from asymptomatic to severe clinical manifestations. Hb ZA is a highly unstable Hb variant, in which arginine replaces glycine at codon 59 in the HBA1 or HBA2 gene. It was first recorded in 2004, 6 in a proband heterozygous for Hb ZA, with normal Hb and α-thalassemia trait manifestation. Two children with transfusion-dependent Hb H disease were subsequently reported, both of whom had Hb ZA compounded with the Southeast Asian (--SEA) deletion.10,11 A Brazilian child with thalassemia major phenotype homozygous for the Hb ZA mutation, who was dependent on blood transfusions and was being prepared for bone marrow transplantation, has also been reported. 8 A case of Hb H hydrops fetalis associated with compound heterozygosity for Hb ZA and the --SEA deletion, with Hb 55 g/L, was reported at 25 weeks of gestation. 7 However, to the best of our knowledge, the current report is the first case of severe fetal anemia and hydrops fetalis associated with compound heterozygosity for Hb ZA and Hb QS.
The common causes of fetal anemia and hydrops fetalis include maternal alloimmunization, parvovirus B19 infection, fetal aneuploidy, vascular tumors, arteriovenous malformations of the fetus or placenta, and inherited conditions such as α-thalassemia or genetic metabolic disorders. No aneuploidy was detected in the current fetus. Several monogenic disorders are also associated with fetal anemia, including dehydrated hereditary stomatocytosis, glucose-6-phosphate dehydrogenase deficiency, congenital erythrocyte membrane disorders, pyruvate kinase deficiency, lysosomal storage diseases, and Fanconi’s anemia. Further studies are needed to determine if high-throughput sequencing approaches such as WES are useful for the clinical management of hydrops fetalis. Given the large number of genes associated with NIHF, we propose that WES is necessary for the diagnosis of monogenic hydrops fetalis and may be required for the timely and efficient identification of monogenic NIHF.
Footnotes
Declaration of conflicting interest: The authors declare that there is no conflict of interest.
Funding: This study was funded by the Science and Technology Program of Guangzhou, China (grant no. 202002030390).
ORCID iD: Xiuqin Bao https://orcid.org/0000-0002-9935-9851
References
- 1.Prefumo F, Fichera A, Fratelli N, et al. Fetal anemia: Diagnosis and management. Best Pract Res Clin Obstet Gynaecol 2019; 58: 2–14. [DOI] [PubMed] [Google Scholar]
- 2.Abbasi N, Johnson JA, Ryan G. Fetal anemia. Ultrasound Obstet Gynecol 2017; 50: 145–153. [DOI] [PubMed] [Google Scholar]
- 3.Liao C, Li J, Li DZ. Fetal anemia and hydrops associated with homozygosity for hemoglobin Quong Sze. Prenat Diagn 2008; 28: 862–864. [DOI] [PubMed] [Google Scholar]
- 4.He Y, Zhao Y, Lou JW, et al. Fetal anemia and hydrops fetalis associated with homozygous Hb Constant Spring (HBA2: c.427T > C). Hemoglobin 2016; 40: 97–101. [DOI] [PubMed] [Google Scholar]
- 5.Komvilaisak P, Komvilaisak R, Jetsrisuparb A, et al. Fetal anemia causing hydrops fetalis from an alpha-globin variant: homozygous hemoglobin Constant Spring. J Pediatr Hematol Oncol 2018; 40: 405–408. [DOI] [PubMed] [Google Scholar]
- 6.Dutly F, Fehr J, Goede JS, et al. A new highly unstable alpha chain variant causing alpha(+)-thalassemia: Hb Zurich Albisrieden [alpha59(E8)Gly–>Arg (alpha2)]. Hemoglobin 2004; 28: 347–351. [DOI] [PubMed] [Google Scholar]
- 7.Yang X, Yan JM, Li J, et al. Hydrops fetalis associated with compound heterozygosity for Hb Zurich-Albisrieden (HBA2: C.178G > C) and the Southeast Asian (--(SEA)/) Deletion. Hemoglobin 2016; 40: 353–355. [DOI] [PubMed] [Google Scholar]
- 8.Pedroso GA, Kimura EM, Santos MNN, et al. Thalassemia major phenotype caused by HB Zurich-Albisrieden [alpha2 59(E8) Gly > Arg (HBA2: C.178G > C)] in a Brazilian child. Pediatr Blood Cancer 2018; 65: e27413. [DOI] [PubMed] [Google Scholar]
- 9.Gagnier JJ, Kienle G, Altman DG, et al. The CARE guidelines: consensus-based clinical case reporting guideline development. Headache 2013; 53: 1541–1547. [DOI] [PubMed] [Google Scholar]
- 10.Fang J, Chen L, Zeng R, et al. The Hb H disease genotypes in Southern China. Hemoglobin 2014; 38: 76–78. [DOI] [PubMed] [Google Scholar]
- 11.Jiang H, Huang LY, Zhen L, et al. Two alpha1-globin gene point mutations causing severe Hb H disease. Hemoglobin 2017; 41: 293–296. [DOI] [PubMed] [Google Scholar]