

Abstract
INTRODUCTION:
Studies of monogenic diseases have uncovered the importance of immune pathways in human tissue-specific immunity and antimicrobial defense. In particular, human inborn errors of the interleukin-17 (IL-17) receptor signaling pathway and corroborating mouse studies have established the critical contribution of type 17 responses inmucosa-specific fungal surveillance. The yeast Candida albicans is the signature pathogen in autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy (APECED), an inherited autoimmune disease caused by loss-of-function mutations in the autoimmune regulator (AIRE) gene. Fungal disease in APECED is limited to chronic mucocutaneous candidiasis (CMC) without dissemination, suggesting a central defect in barrier immunity.
RATIONALE:
AIRE deficiency impairs central immune tolerance, resulting in the generation of pathogenic autoreactive T cells and autoantibodies directed against many tissue-specific antigens and certain cytokines, including “type 17” cytokines. However, although a majority of APECED patients with CMC have type 17 cytokine–targeted autoantibodies, whether type 17 or other localmucosal immune responses are affected in AIRE deficiency has not been determined. Here, we broadly investigated oral mucosal immune responses both in a model of oropharyngeal candidiasis in Aire−/− mice and in a large cohort of APECED patients.
RESULTS:
Type 17 immune responses at the oralmucosa were unexpectedly intact in mice and humans with AIRE deficiency. To define alternative mechanisms of fungal susceptibility, we investigated Aire−/− mice, which exhibited oralmucosa-specific susceptibility to candidiasis without dissemination and controlled experimental challenges with viruses and bacteria normally, thereby phenocopying the infection predisposition observed in APECED patients. Notably, Aire−/− CD4+ and CD8+ T cells accumulated in increased numbers and displayed an activated and proliferative phenotype within the oral mucosa and were both necessary and sufficient to drive mucosal fungal infection in Aire deficiency. Enhanced production of interferon-γ (IFN-γ) by Aire−/− mucosal CD4+ and CD8+ T cells resulted in exacerbated IFN-γ/STAT1-mediated responses in the oral mucosa, which promoted IFN-γ–dependent epithelial barrier disruption and mucosal fungal susceptibility. Genetic and pharmacologic inhibition of IFN-γ or JAK-STAT signaling ameliorated mucosal fungal disease in Aire−/− mice. Aberrant type 1 responseswere also observed in the oral mucosa of APECED patients.
CONCLUSION:
We identify a T cell–dependent interferonopathy as a critical local mucosal mechanism underlying CMC in APECED. Although type 17 mucosal immunity is critical for host defense against barrier infection, mucosal type 17 responses were intact in patients with APECED and in a mouse model of the disease. These findings show that, in contrast to the known protective roles of T cells in antifungal host defense, aberrant type 1–associated T cell responses can be detrimental to antifungal mucosal immunity. They also support a paradigm by which exaggerated immunopathology may facilitate susceptibility to mucosal fungal infection by impairing the integrity of the epithelial barrier. Finally, they pave the way for investigating type 1 mucosal responses in other CMC-manifesting diseases and for the prevention and treatment of CMC in APECED patients using FDA-approved therapies that target IFN-γ or JAK-STAT signaling.
Human monogenic disorders have revealed the critical contribution of type 17 responses in mucosal fungal surveillance. We unexpectedly found that in certain settings, enhanced type 1 immunity rather than defective type 17 responses can promote mucosal fungal infection susceptibility. Notably, in mice and humans with AIRE deficiency, an autoimmune disease characterized by selective susceptibility to mucosal but not systemic fungal infection, mucosal type 17 responses are intact while type 1 responses are exacerbated. These responses promote aberrant interferon-γ (IFN-γ)– and signal transducer and activator of transcription 1 (STAT1)–dependent epithelial barrier defects as well as mucosal fungal infection susceptibility. Concordantly, genetic and pharmacologic inhibition of IFN-γ or Janus kinase (JAK)–STAT signaling ameliorates mucosal fungal disease. Thus, we identify aberrant T cell–dependent, type 1 mucosal inflammation as a critical tissue-specific pathogenic mechanism that promotes mucosal fungal infection susceptibility in mice and humans.
Graphical Abstract
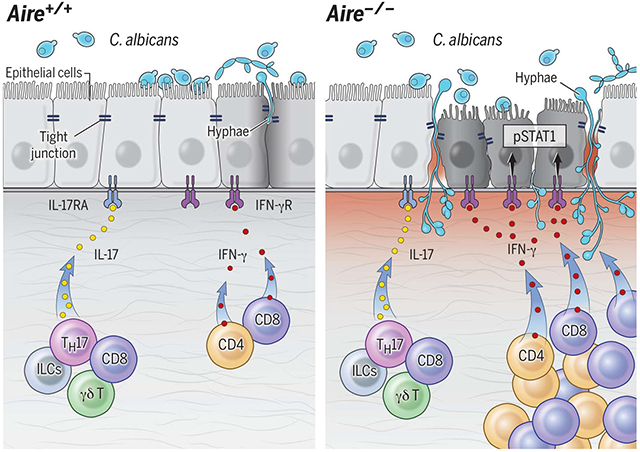
Aberrant type 1 mucosal immunity underlies mucosal fungal susceptibility. Left: Type 17 mucosal responses promote mucosal fungal clearance. Right: In AIRE deficiency, T cells drive enhanced IFN-γ/STAT1-dependent mucosal responses that disrupt the epithelial barrier and promote fungal susceptibility in the setting of intact type 17 mucosal responses. AIRE, autoimmune regulator; pSTAT1, phosphorylated signal transducer and activator of transcription 1; ILCs, innate lymphoid cells.
The study of inherited susceptibility to infectious disease has revealed important immunoregulatory pathways that are specialized for the tissue-specific control of particular pathogens. Recently, genetic deficiency of interleukin-17 receptor (IL-17R) signaling in humans has been shown to cause susceptibility to chronic mucocutaneous candidiasis (CMC), which is characterized by recurrent mucosal but not systemic infections with the commensal fungus Candida albicans (1–5).
Autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy (APECED), also known as autoimmune polyglandular syndrome (APS) type 1, is another inherited disease featuring selective susceptibility to CMC without systemic candidiasis. APECED is caused by mutations in the autoimmune regulator (AIRE) gene that disrupt central immune tolerance, resulting in the generation of pathogenic autoreactive T cells (6, 7). Neutralizing autoantibodies against T helper 17 (TH17) cell–derived cytokines have been reported in APECED patients, which suggests that CMC in APECED may be caused by impaired IL-17 signaling (7–9). However, not all APECED patients with CMC carry TH17 cytokine-targeted autoantibodies, indicating that other factors may also contribute to mucosal fungal infection susceptibility (8–10). In addition, immune responses at the mucosa, where candidiasis specifically develops in APECED, have not to date been thoroughly examined in the setting of AIRE deficiency. Here, we studied mucosal immune responses in APECED patients and in a model of oropharyngeal candidiasis (OPC) in Aire-deficient mice (11). We identify an aberrant pathogenic mechanism mediated by interferon-γ (IFN-γ) and signal transducer and activator of transcription 1 (STAT1) that promotes tissue-specific mucosal fungal infection susceptibility in the setting of intact IL-17R/IL-22–dependent mucosal responses.
Aire−/− mice exhibit specific infection susceptibility to OPC
The genetic background of Aire−/− mice determines the kinetics and severity of autoimmune tissue destruction. NOD Aire−/− mice, relative to those on a BALB/c background, present with disease that is more severe, has a more rapid onset, and clinically resembles that of APECED patients (12, 13). NOD Aire−/− mice showed severe, early-onset susceptibility to OPC, whereas BALB/c Aire−/− mice developed later-onset susceptibility and C57BL/6 Aire−/− mice exhibited late-onset susceptibility to mild disease (Fig. 1A and fig. S1, A and B).
Fig. 1. Aire deficiency causes mucosal fungalspecific infection susceptibility without a defect in mucosal IL-17R/IL-22–dependent immune responses.
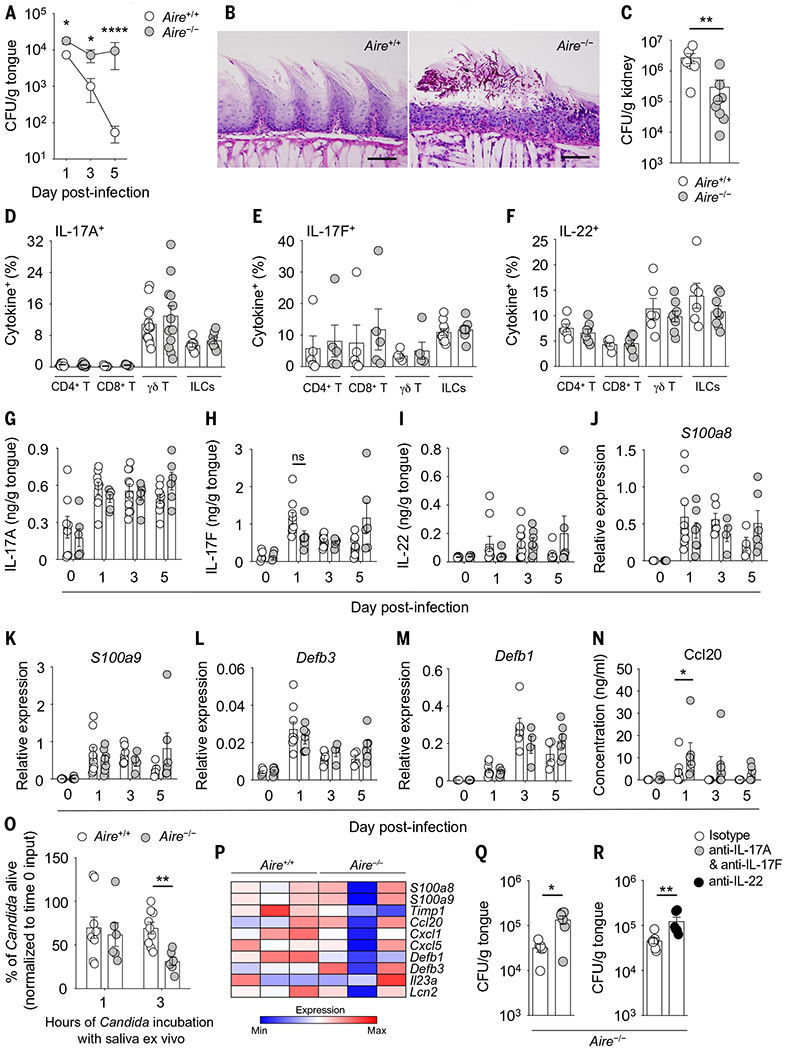
(A) Progression of fungal burden in wild-type and Aire−/− tongue tissue after oral C. albicans infection (N = 9 to 16 per group; three experiments). (B) Representative histological micrographs of periodic acid-Schiff staining in wild-type and Aire−/− tongue tissue at day 5 after infection. Scale bars, 100 μm (N = 8 per group; two experiments). (C) Fungal burden in wild-type and Aire−/− kidney tissue at day 4 after intravenous C. albicans challenge (N = 6 to 8 per group; two experiments). (D to F) Frequencies of indicated lymphoid cells with (D) IL-17A–, (E) IL-17F–, and (F) IL-22–producing potential in wild-type and Aire−/− oral gingival mucosal tissue at day 1 after infection (N = 5 to 13 per group; two to six experiments). ILCs, innate lymphoid cells. (G to I) Concentrations of IL-17A (G), IL-17F (H), and IL-22 (I) in wild-type and Aire−/− tongue tissue homogenates before and after infection (N = 6 to 10 per group; two to three experiments). (J to M) Relative mRNA expression of S100a8 (J), S100a9 (K), Defb3 (L), and Defb1 (M) in wild-type and Aire−/− tongue tissue before and after infection (N = 4 to 9 per group; two experiments). (N) Protein concentration of Ccl20 in wild-type and Aire−/− tongue tissue homogenates before and after infection (N = 6 to 10 per group; two experiments). (O) Saliva was harvested after pilocarpine administration in wild-type and Aire−/− mice over 20 min. The percentage of C. albicans that remained alive relative to the input inoculum after 1 hour or 3 hours of incubation ex vivo with salivary secretions harvested from wild-type and Aire−/− mice was calculated (N = 6 to 9 per group; two experiments). (P) The tongues of wild-type and Aire−/− mice were harvested at day 1 after infection. Epithelial cells were isolated by FACS. mRNA was extracted and RNA-seq was then performed. The heat map shows expression of selected IL-17R–regulated genes curated by Ingenuity Pathway Analysis (IPA) in RNA-seq of oral epithelial cells of Aire+/+ (N = 3) and Aire−/− (N = 3) mice with oral candidiasis. (Q and R) Fungal burden in Aire−/− tongue tissue at day 4 after infection following antibody-mediated depletion of IL-17A and IL-17F (Q) or IL-22 (R) relative to control antibody (N = 5 to 7 per group; two experiments). All quantitative data are means ± SEM. *P < 0.05, **P < 0.01, ****P < 0.0001 as calculated using unpaired t test [(A), day 1, (O), and (Q)] or Mann-Whitney U test [(A), days 3 and 5, (C), (N), and (R)]; ns, not significant.
To more closely model the CMC dynamics observed in APECED patients, we focused our studies on NOD Aire−/− mice. Early-onset susceptibility to OPC in Aire−/− mice was observed across tongue, buccal, and gingival mucosal surfaces, the sites corresponding to human oral candidiasis lesions (Fig. 1A and fig. S1C). This was evident from a panel of clinical isolates of C. albicans (Fig. 1A and fig. S1D). Furthermore, Aire−/− mice exhibited persistent epithelial fungal invasion and epithelial injury (Fig. 1B). As in APECED patients, fungal dissemination to systemic tissues was not observed after OPC (fig. S1E), and no susceptibility was observed after cutaneous infection with C. albicans (fig. S1F). Consistent with the segregation of host factors required for mucosal versus systemic anti-Candida immunity (5), Aire−/− mice did not have increased susceptibility to systemic C. albicans challenge (Fig. 1C). Moreover, Aire−/− mice controlled oral mucosal and systemic viral infections (fig. S2) and did not exhibit increased susceptibility to cutaneous infection with Staphylococcus aureus, gastrointestinal infection with Citrobacter rodentium, or ligature-induced experimental periodontitis (fig. S3), three models that rely on IL-17R/IL-22 immune responses for host defense (2, 14). Thus, Aire−/− mice display mucosa-specific susceptibility to C. albicans infection, thereby phenocopying the CMC observed in APECED patients.
Intact IL-17R/IL-22 mucosal responses in AIRE deficiency
Generation of neutralizing autoantibodies to TH17 cell–derived cytokines is posited as an underlying cause for CMC in patients with APECED (8, 9). Interestingly, <10% of NOD Aire−/− mice had detectable serum TH17 cell–derived cytokine autoantibodies (fig. S4, A and B), and transfer of Aire−/− serum into wild-type mice did not promote susceptibility to OPC (fig. S4, C and D). These data are consistent with previous observations that Aire−/− sera transferred to wild-type recipient mice do not precipitate autoimmune tissue destruction (15–17).
Given the established role of type 17 immunity in host defense against fungi (1–4, 18, 19), we evaluated IL-17R– and IL-22–dependent mucosal immune responses in Aire−/− mice during OPC. Oral mucosal CD4+ T cells, CD8+ T cells, γδ T cells, and innate lymphoid cells (ILCs) from Aire−/− mice displayed normal production of IL-17A, IL-17F, and IL-22 (Fig. 1, D to F, and fig. S5). Because IL-17A is commonly coproduced together with other inflammatory cytokines (20), we also assessed CD4+ T and γδ T cells for the generation of TNF-α, IFN-γ, and GM-CSF. Wild-type and Aire−/− IL-17A–producing CD4+ T and γδ T cells displayed similar frequencies of co-production of TNF-α, IFN-γ, and GM-CSF (fig. S6). Furthermore, levels of IL-17A, IL-17F, and IL-22 were comparable to those of wild-type mice in Aire−/− oral mucosal tissue during OPC (Fig. 1, G to I), and the induction of IL-17R– and IL-22–dependent proinflammatory cytokines, chemokines, and antimicrobial peptides (AMPs) (18, 19, 21) was maintained in Aire−/− oral mucosal tissue during OPC (Fig. 1, J to N, and figs. S7 and S8). The antifungal killing capacity and the production rate of Aire−/− salivary secretions were not impaired (Fig. 1O and fig. S9). Moreover, RNA sequencing (RNA-seq) analysis of fluorescence-activated cell sorter (FACS)–isolated oral epithelial cells from wild-type and Aire−/− mice revealed that IL-17R–dependent genes, as defined by a previous study (19), were not differentially expressed between wild-type and Aire−/− mice with oral candidiasis (fig. S10). Several genes curated as key IL-17R downstream gene targets by Ingenuity Pathway Analysis (IPA) (S100a8, S100a9, Timp1, Ccl20, Cxcl1, Cxcl5, Defb1, Defb3, Il23a, and Lcn2) were also not differentially expressed between wild-type and Aire−/− oral epithelial cells in this setting (Fig. 1P). Experimental antibody–mediated neutralization of IL-17A and IL-17F or IL-22 in Aire−/− mice further increased mucosal fungal burden relative to Aire−/− mice treated with control antibody (Fig. 1, Q and R), further supporting the notion that Aire deficiency does not disable IL-17R/IL-22–dependent oral mucosal immunity to C. albicans.
To corroborate these experimental findings, we obtained oral mucosal biopsies from APECED patients. We found no differences between APECED patients and healthy donors with respect to production of IL-17A by oral mucosal αβ T cells, γδ T cells, and ILCs (Fig. 2A and fig. S11); mRNA for IL17A, IL17F, and IL22 (Fig. 2B); and the induction of the IL-17R/IL-22–dependent AMPs S100A8 and S100A9 (22, 23) in oral mucosal tissue and saliva (Fig. 2, C and D). To further evaluate type 17 responses in the human oral mucosa, we performed RNA-seq analysis of oral mucosal tissue from uninfected healthy donors and patients with APECED and found no differences in the expression of the IL-17R–regulated genes defined by a previous study (19) (Fig. 2E and fig. S12). Thus, deficiency in type 17 immunity unexpectedly does not underlie the heightened susceptibility to mucosal candidiasis observed in AIRE-deficient mice and humans.
Fig. 2. APECED patients have intact mucosal type 17 immune responses.
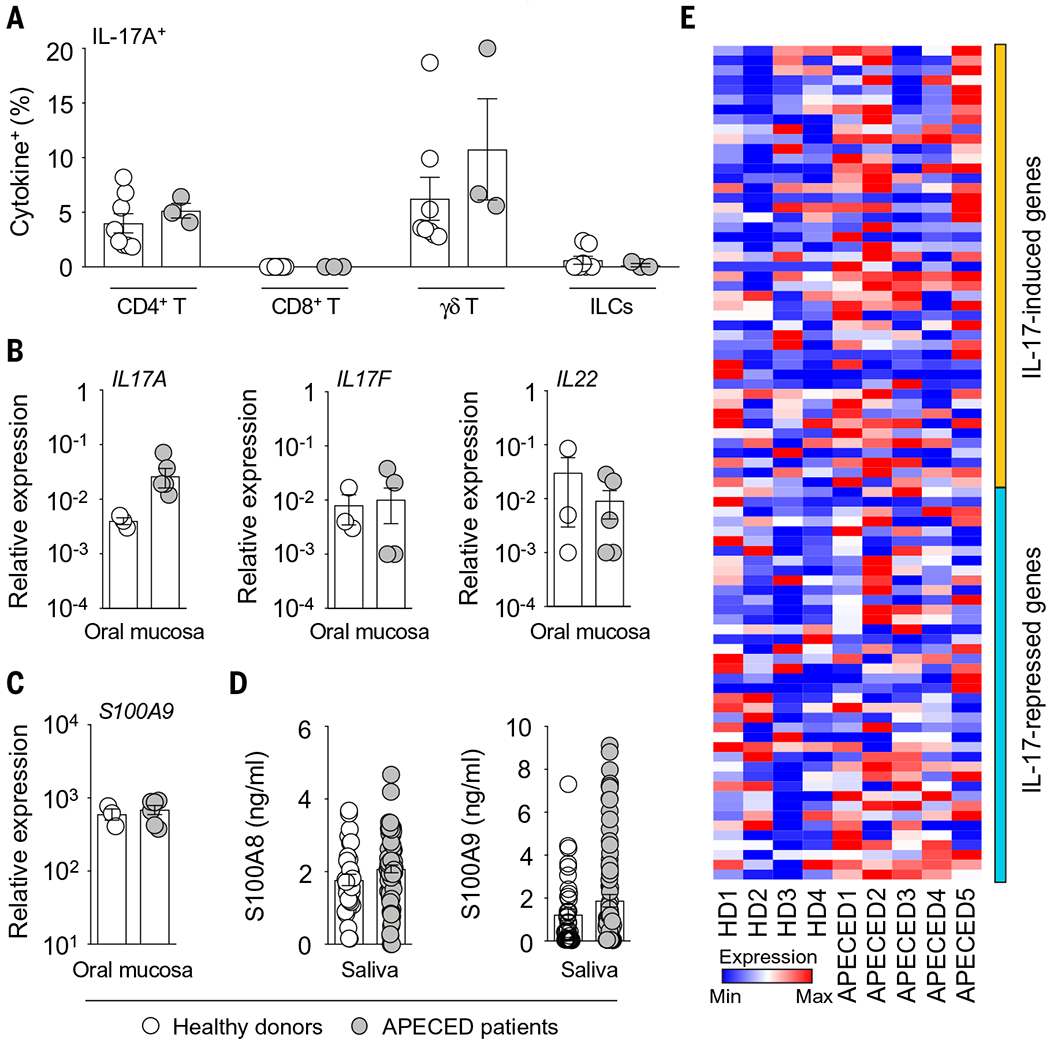
(A) Frequencies of indicated lymphoid cells with IL-17A–producing potential in healthy donor (N = 8) or APECED patient (N = 3) oral gingival mucosal tissue. Each dot represents an individual patient. ILCs, innate lymphoid cells. No IL-17A production by CD8+ T cells was observed in any of the healthy donors or APECED patients. (B and C) Relative mRNA expression of IL17A, IL17F, and IL22 (B) and S100A9 (C) in healthy donor (N = 3) and APECED patient (N = 6) oral gingival mucosal tissue. (D) S100A8 and S100A9 concentrations in saliva of healthy donors (N = 33 to 38) and APECED patients (N = 75 to 77). (E) Oral gingival mucosal tissue was obtained from uninfected healthy donors (HD; N = 4) and uninfected APECED patients (N = 5), mRNA was extracted, and RNA-seq was performed. The heat map shows expression of IL-17R–regulated genes, curated from an earlier study (19), in RNA-seq of oral gingival mucosal tissue from APECED patients versus healthy donors. All quantitative data are means ± SEM.
Accumulation of pathogenic T cells drives mucosal fungal infection susceptibility in Aire−/− mice
To determine alternative mechanisms that might account for mucosal fungal infection susceptibility in Aire−/− mice in the setting of intact IL-17R/IL-22–dependent mucosal responses, we assessed the temporal and spatial accumulation of myeloid and lymphoid cells in the oral mucosa following OPC. Neutrophils have been reported to promote normal fungal clearance in the oral mucosa, where they accumulate in an IL-17R– and IL-22–dependent manner during OPC (24, 25). We observed no defect in neutrophil accumulation or colocalization with fungal elements in the Aire−/− infected mucosa (fig. S13, A and B). Mucosal accumulation of other myeloid cell subsets in Aire−/− mice was equivalent to (or, in the case of monocytes and macrophages, higher than) that in wild-type mice (fig. S13, C to G). Notably, we also observed a persistent, significant increase in accumulation of T cells in the Aire−/− oral mucosa both before and after infection (fig. S13, H to M), which included both CD4+ and CD8+ T cells (Fig. 3, A and B). Both subsets exhibited an activated and proliferative (CD44hiCD69+Ki-67+) phenotype in the Aire−/− oral mucosa (Fig. 3, C and D, and fig. S14, A and B) and infiltrated the epithelial and submucosal layers of Aire−/− mice, a phenotype observed only sporadically within wild-type mucosal surfaces (Fig. 3, E and F, and fig. S14, C and D). A similar marked accumulation of CD4+ and CD8+ T cells was observed within the oral mucosa of APECED patients in the absence of oral candidiasis (Fig. 3G and fig. S15).
Fig. 3. Pathogenic TCRαβ+ cells drive mucosal fungal infection susceptibility in Aire deficiency.
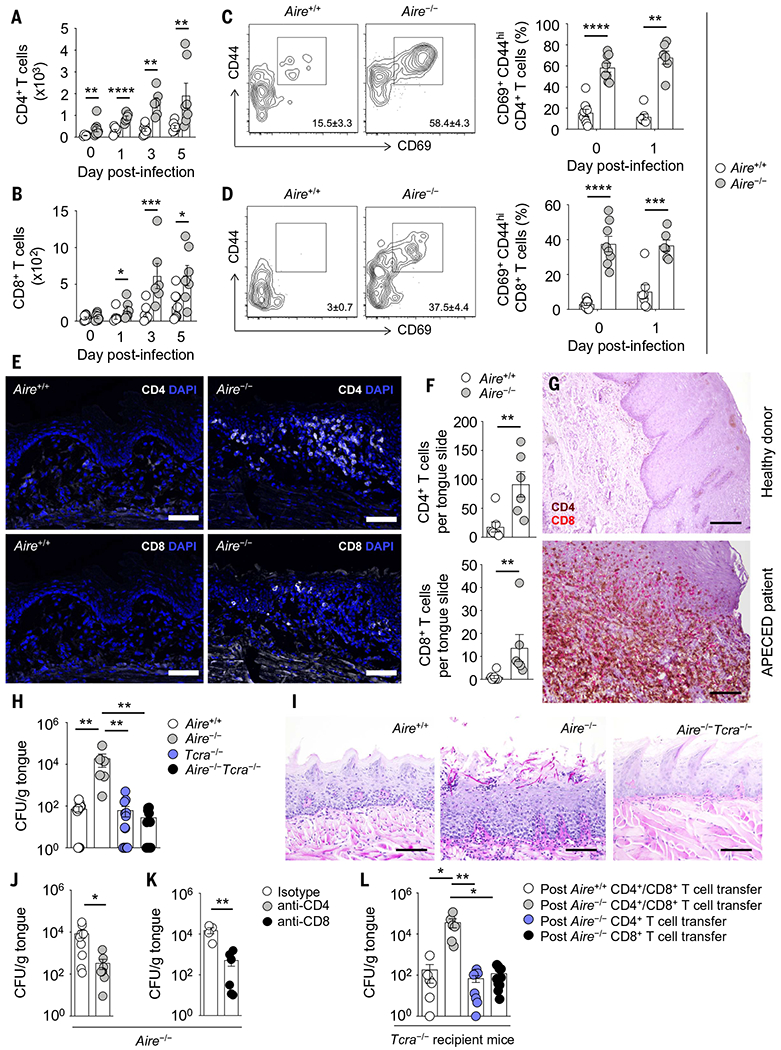
(A and B) Numbers of CD4+ (A) and CD8+ (B) T cells in wild-type and Aire−/− tonguetissue before and after oral C. albicans infection (N = 6 to 10 per group; two experiments). (C and D) Representative contour plots of CD44 and CD69 expression (left, day 0) and proportions of CD44hiCD69+ cells (right, days 0 and 1) within CD4+ (C) and CD8+ (D) T cells in wild-type and Aire−/− tongue tissue (N = 6 to 10 per group; two experiments). (E and F) Representative immunofluorescence images of CD4 and CD8 staining [(E), top and bottom, respectively] and quantification of CD4+ and CD8+ T cells (F) within epithelial and submucosal layers of wild-type and Aire−/− tongue tissue at day 1 after infection. Scale bars, 100 μm (N = 6 or 7 per group; two experiments). (G) Immunohistochemical analysis of CD4 (brown) and CD8 (red) in healthy donor and APECED patient oral gingival mucosal tissue. Scale bars, 1200 μm (top), 200 μm (bottom). Shown are images from one of two examined healthy donors and one of three examined APECED patients; images from all patients are shown in fig. S15. (H and I) Aire−/−Tcra−/− mice control mucosal fungal infection better than do Aire−/− mice, as indicated by fungal burden analysis in tongue tissue (H) and representative histological micrographs of periodic acid-Schiff staining in wild-type, Aire−/−, and Aire−/−Tcra−/− tongue tissue at day 5 after infection (I). Scale bars, 200 μm (N = 6 to 16 per group; three experiments). (J and K) In Aire−/− mice, antibody-mediated depletion of CD4+ T cells [(J); N = 8 or 9 per group, three experiments] or CD8+ T cells [(K); N = 4 to 6 per group, two experiments] results in decreased fungal burden in tongue tissue. (L) Fungal burden in tongue tissue of Tcra−/− recipient mice 10 weeks after adoptive transfer of the indicated T cells derived from wild-type or Aire−/− mice (N = 6 to 10 per group; three experiments). All quantitative data are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 as calculated using unpaired t test [(A), days 1 and 3, (B), days 0 and 5, (C), day 0, and (D)], Mann-Whitney U test [(A), days 0 and 5, (B), days 1 and 3, (C), day 1, (F), (J), and (K)], one-way ANOVA with Holm-Šidák multiple-comparisons test (H), or Kruskal-Wallis H test with Dunn’s multiple-comparisons test (L).
To determine whether the accumulation of activated T cells within the Aire−/− oral mucosa impeded host defense, we generated Aire−/−Tcra−/− mice, which lack αβ T cells, and infected them with C. clbiccns. Strikingly, Aire−/−Tcra−/− mice controlled fungal growth as well as wild-type mice (Fig. 3H) and did not develop the fungal epithelial invasion and epithelial injury observed in Aire−/− mice (Fig. 3I). Thus, pathogenic TCRαβ+ cells underlie the mucosal fungal infection susceptibility in Aire deficiency. Notably, the selective antibody-mediated depletion of CD4+ or CD8+ T cells in Aire−/− mice also decreased mucosal fungal growth (Fig. 3, J and K), revealing the contribution of both Aire−/− CD4+ and CD8+ T cells to mucosal fungal infection susceptibility in Aire−/− mice.
The adoptive transfer of CD4+ and CD8+ T cells derived from Aire−/− mice into Tcra−/− recipient mice promoted mucosal fungal growth, whereas the transfer of CD4+ or CD8+ T cells alone did not (Fig. 3L). These results show that Aire−/−-derived T cells are both necessary and sufficient to drive mucosal fungal infection susceptibility—in line with the pathogenic role of Aire−/−-derived TCRαβ+ cells in driving the multi-organ autoimmune phenotypes of Aire deficiency (15, 16)—and underscore the potential interplay between Aire−/−-derived CD4+ T cells and CD8+ T cells in vivo to promote mucosal fungal infection. Susceptibility to mucosal fungal infection in Aire−/− mice was not modulated by oral commensal bacterial communities (fig. S16), consistent with the previously reported non-essential role of microbiota in driving autoimmune tissue destruction in these mice (26). Thus, mucosal fungal infection susceptibility in Aire deficiency appears to be driven by pathogenic CD4+ and CD8+ T cells that infiltrate the epithelial and submucosal tissue layers. Strikingly, in contrast to the known protective roles of T cells in antifungal host defense (5), we show that aberrant T cell responses can exert detrimental effects on antifungal mucosal immunity.
Aberrant IFN-γ/STAT1 mucosal responses in AIRE deficiency
To investigate how Aire-deficient T cells promote Candida infection in the mucosa, we analyzed the production of TH1, TH2, and TH17 cell–derived cytokines. Although TH2 and TH17 cell–derived cytokines were comparable in wild-type and Aire−/− oral mucosal tissue (Fig. 1, G to I, and figs. S17A and S18), we identified a significant and persistent increase in Ifng expression in the Aire−/− mucosa both before and during OPC (Fig. 4A), which we confirmed at the protein level (Fig. 4B). Notably, both Aire−/− CD4+ and CD8+ mucosal T cells produced significantly more IFN-γ relative to wild-type cells both before and during OPC (Fig. 4, C and D). By contrast, Foxp3+ regulatory T cell frequencies were comparable in wild-type and Aire−/− mucosal tissue (fig. S17B). We also observed no decrease in Il10 induction in Aire−/− mice (fig. S17C) and no differences in the levels of other proinflammatory cytokines (fig. S18). Consistent with elevated IFN-γ production by mucosal T cells, Stat1 expression (Fig. 4E) and levels of the IFN-γ–inducible chemokines CXCL9 and CXCL10 were also elevated in the Aire−/− mucosa, both before and during OPC (Fig. 4, F and G). These increased type 1 responses were also observed in the mucosa of BALB/c Aire−/− mice, which also exhibited the intact type 17 mucosal responses seen in NOD Aire−/− mice (fig. S19).
Fig. 4. Aire deficiency results in IFN-γ–driven mucosal interferonopathy.
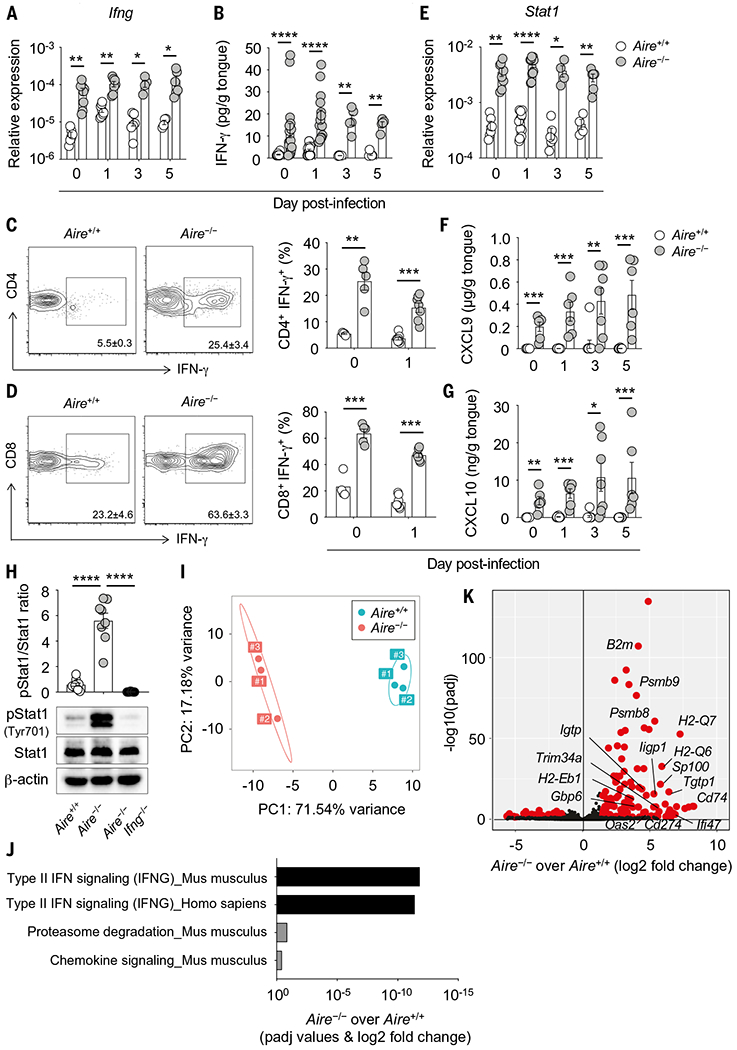
(A and B) Relative mRNA expression of Ifng [(A); N = 4 to 9 per group; two experiments] and concentration of IFN-γ [(B); N = 5 to 20 per group; two experiments] in wild-type and Aire−/− tongue tissue before and after infection. (C and D) Representative contour plots of IFN-γ expression (left; day 0) and frequencies of IFN-γ–producing cells (right; days 0 and 1) within CD4+ (C) and CD8+ (D) T cells in wild-type and Aire−/− oral gingival mucosal tissue (N = 4 to 8 per group; two experiments). (E) Relative mRNA expression of Stat1 in wild-type and Aire−/− tongue tissue homogenates before and after infection (N = 4 to 13 per group; two experiments). (F and G) Concentrations of IFN-γ–inducible CXCL9 (F) and CXCL10 (G) in wild-type and Aire−/− tongue tissue homogenates before and after infection (N = 6 to 10 per group; two to three experiments). (H) Epithelial layers of wild-type, Aire−/−, and Aire−/−Ifng−/− tongue tissues were harvested at day 1 after infection for immunoblot analysis of phospho-Stat1 (Tyr701), total Stat1, and β-actin as loading control. Top, quantification of protein immunoblot data; bottom, representative protein immunoblot images (N = 8 to 10 per group; three experiments). (I to K) Tongues of wild-type and Aire−/− mice were harvested at day 1 after infection. Epithelial cells were isolated by FACS. mRNA was extracted and RNA-seq was then performed. (I) Principal components analysis of RNA-seq in wild-type (N = 3) and Aire−/− (N = 3) oral epithelial cells at day 1 after infection. (J) Pathway analysis using differentially expressed genes between wild-type and Aire−/− oral epithelial cells was performed using Enrichr and graphed according to enrichment score for significant Reactome biological processes. (K) Volcano plot of RNA-seq demonstrating differential gene expression in wild-type and Aire−/− oral epithelial cells at day 1 after infection. Shown are IFN-γ–dependent genes. Each dot represents an individual mouse. All quantitative data are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 as calculated using unpaired t test [(A), (C), (D), day 0, and (G), day 0], unpaired t test with Welch’s correction [(B), day 3, and (E), days 0, 3, and 5], Mann-Whitney U test [(B), days 0, 1, and 5, (D), day 1, (E), day 1, (F), and (G), days 1, 3, and 5] or one-way ANOVA with Holm-Šidák multiple-comparisons test (H).
Attesting to the markedly elevated constitutive and Candida-induced levels of IFN-γ in the Aire−/− mucosa, oral epithelial cells, which express IFN-γR1 and IFN-γR2 (fig. S20, A and B), exhibited amplified IFN-γ–dependent STAT1 phosphorylation both before and after OPC challenge (Fig. 4H and fig. S20C). RNA-seq analysis of FACS-isolated oral epithelial cells from wild-type and Aire−/− mice verified a dominant IFN-γ/STAT1–dependent transcriptional signature of the oral epithelium in the context of Aire deficiency (Fig. 4, I to K, fig. S21, A and B, and tables S1 to S3).
To corroborate these experimental findings, we obtained oral mucosal biopsies and saliva from APECED patients and found increased production of IFN-γ by mucosal CD4+ and CD8+ T cells (Fig. 5, A to D), significantly increased levels of CXCL9 and CXCL10 in saliva (Fig. 5, E and F), and a prominent phospho-STAT1 signal in oral epithelial cells (Fig. 5G). We also performed RNA-seq analysis of oral mucosal tissue from uninfected healthy donors and patients with APECED and found a dominant IFN-γ/STAT1–dependent transcriptional signature in the AIRE-deficient human oral mucosa (Fig. 5, H to J, and tables S4 to S6). Notably, IFN-γ and STAT1 were the top two IPA-predicted upstream regulators of differentially expressed genes in both humans (APECED patients versus healthy donors) and mice (Aire−/− versus Aire+/+) (Fig. 5K). Thus, augmented IFN-γ production by mucosal CD4+ and CD8+ T cells drives a mucosal interferonopathy in AIRE-deficient mice and humans.
Fig. 5. APECED patients exhibit IFN-γ/STAT1–associated mucosal interferonopathy.
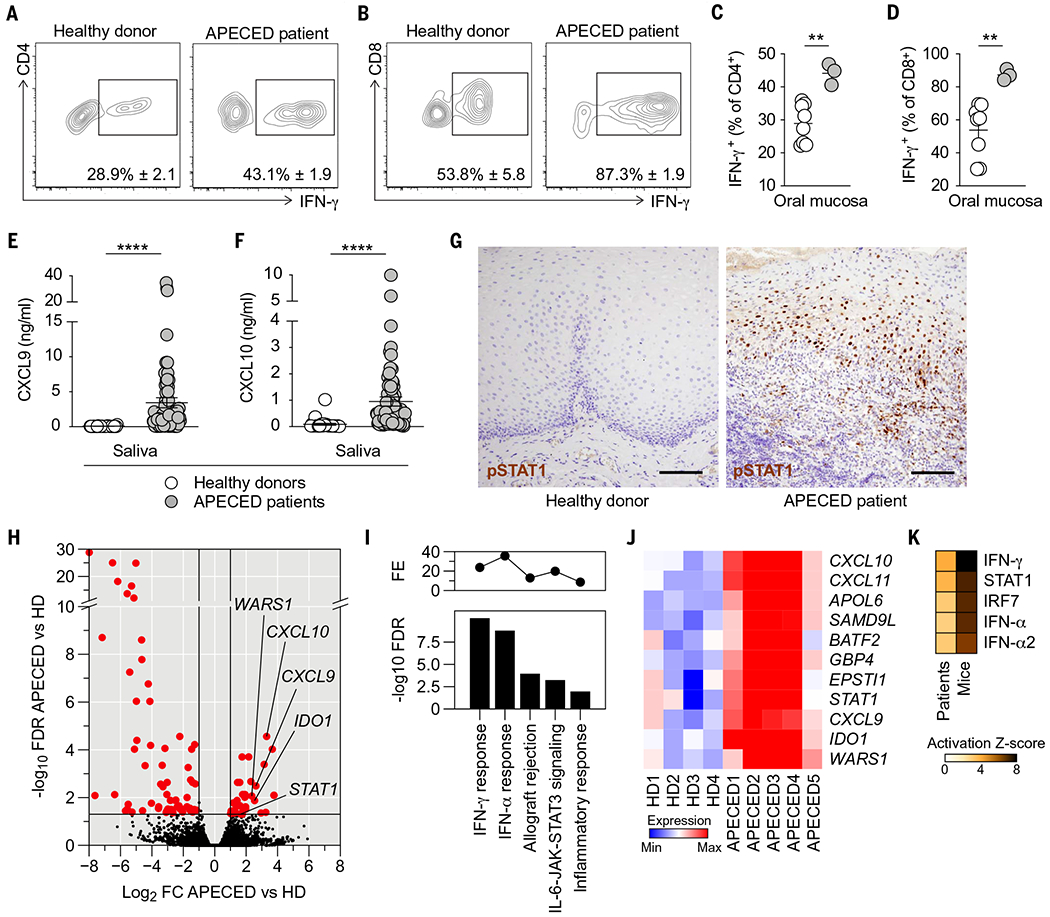
(A and B) Representative contour plots of IFN-γ expression within CD4+ (A) and CD8+ (B) T cells in healthy donor and APECED patient oral gingival mucosal tissue. (C and D) Frequencies of CD4+ (C) and CD8+ (D) T cells with IFN-γ–producing potential in healthy donor (N = 8) or APECED patient (N = 3) oral gingival mucosal tissue. (E and F) Concentrations of IFN-γ–inducible CXCL9 (E) and CXCL10 (F) in saliva of healthy donors (N = 28 to 31) and APECED patients (N = 73 to 79). (G) Immunohistochemical analysis of phospho-STAT1 in healthy donor and APECED patient oral gingival mucosal tissue. Scale bars, 400 μm. (H to K) Oral gingival mucosal tissue was obtained from uninfected healthy donors (N = 4) and uninfected APECED patients (N = 5), mRNA was extracted, and RNA-seq was performed. (H) Volcano plot comparing RNA-seq of oral gingival mucosal tissue from APECED patients (N = 5) versus healthy donors (HD; N = 4). Highlighted in red are differentially expressed genes [DEGs; fold change (FC) ≥ ±2 at FDR < 0.05]. Marked are select IFN-γ–regulated genes. (I) Top five enriched MSigDB hallmark gene sets in DEGs from (H). Shown are −log10 FDR q-value (bottom) and fold enrichment (FE; top). (J) Heat map showing expression of the 11 enriched IFN-γ response genes from (I) in each sample. (K) Ingenuity Pathway Analysis (IPA)–predicted upstream regulators of DEGs in humans (APECED patients versus HD) and mice (Aire−/− versus Aire+/+). The heat map shows activation Z-scores of the top five predicted cytokines or transcription factors. All quantitative data are means ± SEM. **P < 0.01, ****P < 0.0001 as calculated using unpaired t test [(C) and (D)] or Mann-Whitney U test [(E) and (F)].
Excessive IFN-γ/STAT1 responses in AIRE deficiency promote oral epithelial defects and drive mucosal fungal infection susceptibility
Excessive and prolonged exposure to IFN-γ has been shown to adversely affect survival and proliferation of intestinal epithelial cells and skin keratinocytes and to impair intestinal barrier permeability associated with decreased expression of tight junction proteins (27–29). As such, we evaluated the impact of the enhanced epithelial IFN-γ/STAT1–dependent responses of Aire−/− mice on oral epithelial cell and barrier integrity during oral candidiasis. Using flow cytometry, we observed decreased survival of EpCAM+ epithelial cells within the oral mucosa of Aire−/− mice (Fig. 6, A to C). Two-photon confocal microscopy confirmed, in situ, the increased frequency of dead epithelial cells in the Aire−/− oral mucosa (Fig. 6, D and E). Oral epithelial cells from Aire−/− mice exhibited elevated expression of phosphorylated histone H2AX, a surrogate marker of DNA damage, reflective of reduced cellular survival (Fig. 6F). In addition, we found decreased proliferation of Aire−/− epithelial cells by Ki-67 immunoblot and immunohistochemical analyses (Fig. 6, G to I). To examine oral mucosal barrier permeability, we measured the serum concentration of fluorescein isothiocyanate (FITC)–dextran after topical application of FITC-dextran onto the mouse tongue. We found increased oral mucosal barrier permeability in Aire−/− mice, associated with decreased expression of the tight junction protein claudin-1 in Aire−/− oral epithelial cells (Fig. 6, J and K). We then examined whether the epithelial defects of the Aire-deficient oral mucosa that we observed during OPC were also evident at the uninfected state. Indeed, we found increased frequency of dead epithelial cells in the Aire−/− oral mucosa in situ (fig. S22, A and B), elevated expression of phosphorylated histone H2A.X in Aire−/− oral epithelial cells (fig. S22C), increased oral mucosal barrier permeability in Aire−/− mice (fig. S22D), and decreased expression of claudin-1 in Aire−/− oral epithelial cells (fig. S22E) in uninfected mice. Thus, Aire deficiency leads to impaired oral epithelial cell survival and oral mucosal barrier disruption both before and during OPC.
Fig. 6. Excessive IFN-γ in Aire-deficient mice drives oral epithelial barrier defects.
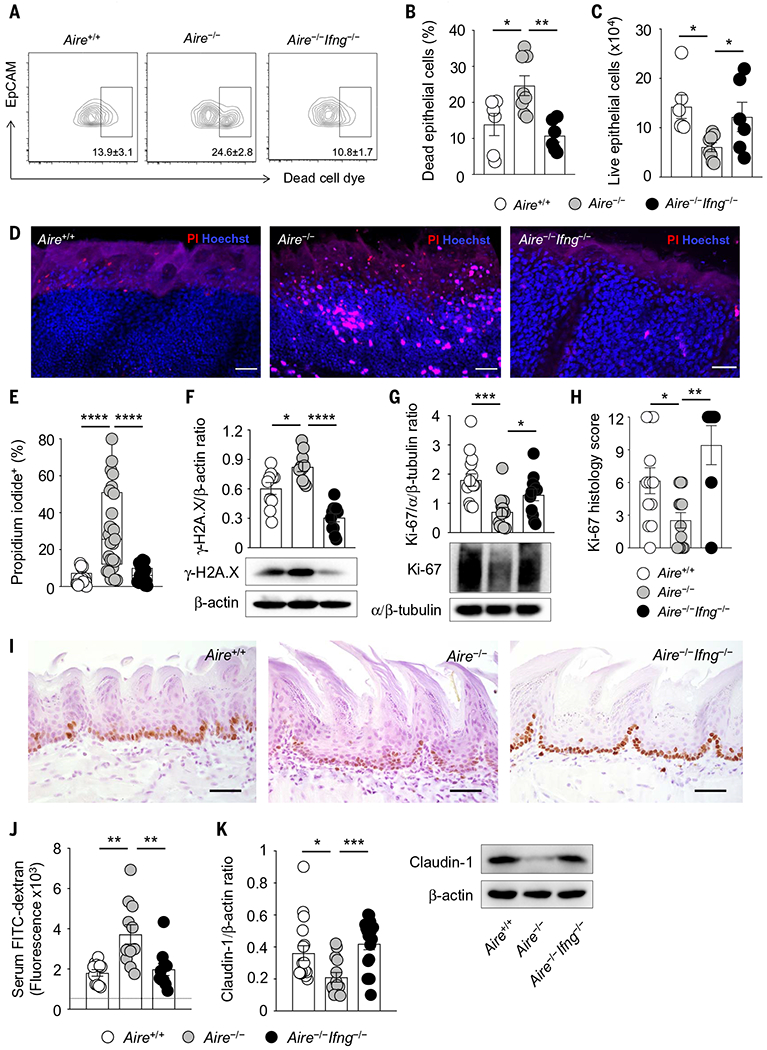
(A to E) Oral epithelial cells from tongue tissue of Aire−/− mice exhibit decreased survival relative to wild-type and Aire−/−Ifng−/− mice. (A) Representative contour plots of dead cell dye–positive oral epithelial cells; (B) frequencies of dead cell dye–positive oral epithelial cells; (C) numbers of live oral epithelial cells at day 1 after infection (N = 6 to 8 per group, two experiments); representative images (D) and frequencies (E) of propidium iodide (PI)–positive oral epithelial cells from wild-type, Aire−/−, and Aire−/−Ifng−/− mice as assessed by two-photon confocal imaging at day 1 after infection. Scale bars, 50 μm (N = 17 to 25 per group, three experiments). (F and G) Epithelial layers of wild-type, Aire−/−, and Aire−/−Ifng−/− tongue tissues were harvested at day 1 after infection for immunoblot analyses. Shown are quantification of protein immunoblot data and representative protein immunoblot images of γ-H2A.X (phosphorylated Ser139) [(F); N = 10 or 11 per group, three experiments] and Ki-67 [(G); N = 12 to 15 per group, three experiments]. β-Actin or α/β-tubulin were used as loading controls. (H and I) Immunohistochemical analysis of Ki-67 in the oral epithelium of tongue tissue of wild-type, Aire−/−, and Aire−/−Ifng−/− mice at day 1 after infection. (H) Summary histology score data; (I) representative immunohistochemistry images. Scale bars, 150 μm (N = 7 to 13 per group, three experiments). (J) Increased oral mucosal barrier permeability in Aire−/− mice, as shown by quantification of the amount of FITC-dextran measured in the serum of wild-type, Aire−/−, and Aire−/−Ifng−/− mice at day 1 after infection following topical application of FITC-dextran onto the mouse tongue (N = 10 or 11 per group, three experiments). The dotted horizontal line indicates the fluorescence values obtained in the serum of mice in which FITC-dextran was not applied onto the mouse tongue. (K) Epithelial layers of wild-type, Aire−/−, and Aire−/−Ifng−/− tongue tissues were harvested at day 1 after infection for immunoblot analyses. Left, quantification of protein immunoblot data; right, representative protein immunoblot images of claudin-1 with β-actin as loading control (N = 14 to 18 per group, four experiments). Each dot represents an individual mouse or an individual field of view of mouse tongue tissue imaged using two-photon confocal microscopy (E). All quantitative data are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 as calculated using one-way ANOVA with Newman-Keuls multiple comparisons test [(C) and (G)], one-way ANOVA with Holm-Šidák multiple comparisons test [(B), (E), (F), (H), and (J)], or Kruskal-Wallis H test with Dunn’s multiple-comparisons test (K).
On the basis of these findings, we next asked whether IFN-γ exerts direct detrimental effects on human oral epithelial cell survival and barrier integrity. We found that prolonged exposure of OKF6/TERT-2 human oral epithelial cells to IFN-γ resulted in dose-dependent oral epithelial cell death as measured by lactate dehydrogenase (LDH) release, an effect that was magnified in the presence of C. albicans (Fig. 7A). In addition, prolonged IFN-γ exposure caused a dose-dependent increase in oral epithelial cell permeability as measured by FITC-dextran fluorescence (Fig. 7B). This was associated with decreased expression of claudin-1 in oral epithelial cells upon IFN-γ exposure as assessed by immunoblot and immunofluorescence analyses (Fig. 7, C and D). Thus, excessive, prolonged exposure to IFN-γ impairs the survival and permeability of human oral epithelial cells.
Fig. 7. Prolonged IFN-γ exposure impairs the viability and barrier integrity of human oral epithelial cells.
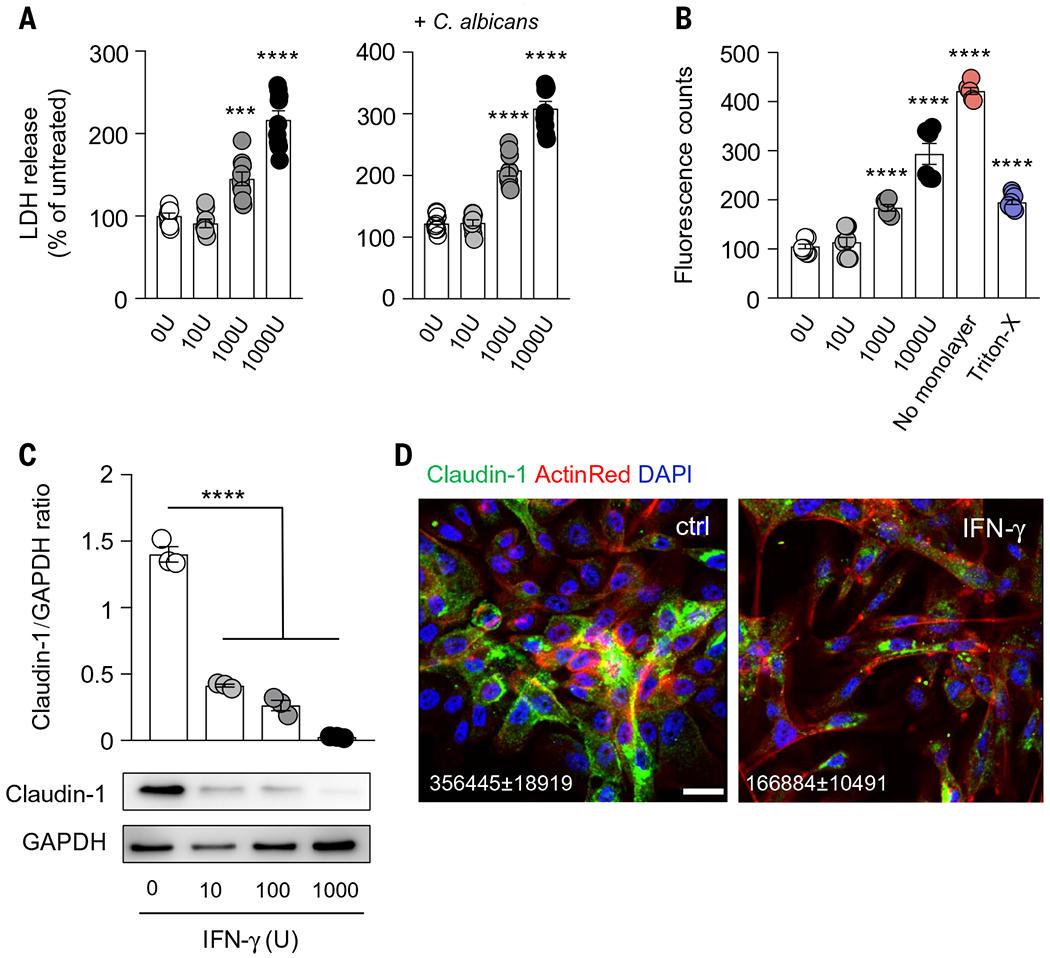
OKF6/TERT-2 human oral epithelial cells were exposed for 72 hours to the indicated concentrations of IFN-γ (U, units). (A) Lactate dehydrogenase (LDH) release was assessed after changing the culture media, 8 hours after C. albicans infection, with a starting point of similar confluency (N = 8 or 9 per group; three experiments). (B) Permeability of confluent cells assessed by measuring fluorescent counts of FITC-dextran in receiver wells (N = 6 to 9 per group; three experiments). (C) Quantification of protein immunoblot data (top) and representative protein immunoblot images (bottom) of claudin-1 with GAPDH as loading control (N = 3 per group; three experiments). (D) Representative images of claudin-1 expression and localization in human oral epithelial cells. Scale bar, 20 mm. Shown are mean (±SEM) fluorescent values of claudin-1 obtained from three randomly selected high-power fields per experiment (three independent experiments). All quantitative data are means ± SEM. ***P < 0.001, ****P < 0.0001 as calculated using one-way ANOVA with Dunnett’s post hoc multiple-comparisons test.
We hypothesized that the aberrant IFN-γ/STAT1-dependent responses of Aire−/− mice are detrimental to mucosal fungal host defense. Indeed, genetic deficiency of IFN-γ in Aire−/− mice ameliorated the oral epithelial cell and barrier defects both before and during OPC (Fig. 6, A to K, and fig. S22). Moreover, Aire−/−Ifng−/− mice had significantly reduced mucosal fungal growth relative to Aire−/− mice (Fig. 8A) and did not develop fungal epithelial invasion and epithelial injury (Fig. 8B). As a control, genetic deficiency of IFN-γ in wild-type mice did not affect mucosal fungal infection susceptibility (fig. S23), in agreement with prior reports (30). Notably, relative to Aire deficiency alone, double deficiency in Aire and Ifng did not alter the frequencies of IL-17A– and IL-17F–producing αβ T cells and γδ T cells in the oral mucosa (fig. S24). Adoptive transfer of CD4+ and CD8+ T cells derived from Aire−/−Ifng−/− mice into Tcrc−/− recipient mice did not promote susceptibility to mucosal fungal infection, as opposed to adoptive transfer of CD4+ and CD8+ T cells derived from Aire−/− mice (Fig. 8C). Thus, IFN-γ production by Aire−/−-derived T cells is sufficient to promote T cell–driven mucosal fungal infection in the setting of Aire deficiency. These results have translational implications, because antibody-mediated neutralization of IFN-γ or pharmacological inhibition of JAK-STAT signaling with ruxolitinib, both of which represent FDA-approved treatment modalities (31, 32), also protected Aire−/− mice from mucosal fungal infection (Fig. 8, D and E).
Fig. 8. Inhibition of IFN-γ or JAK-STAT signaling in Aire-deficient mice ameliorates mucosal fungal infection susceptibility.
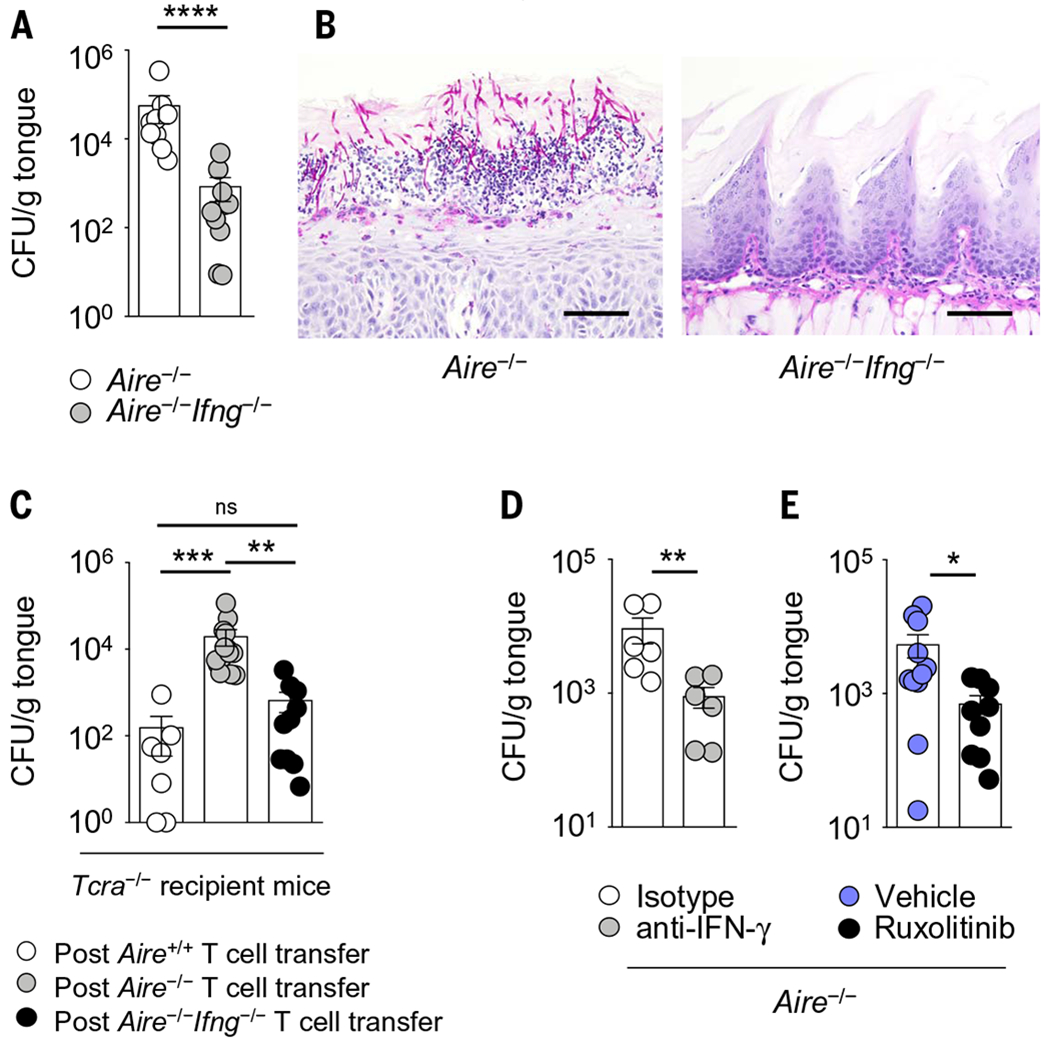
(A) Fungal burden in Aire−/− and Aire−/−Ifng−/− tongue tissue after oral C. albicans infection (N = 9 or 10 per group; three experiments). (B) Representative histological micrographs of periodic acid-Schiff staining in Aire−/− and Aire−/−Ifng−/− tongue tissue at day 5 after infection. Scale bars, 150 μm (N = 5 or 6 per group; two experiments). (C) Fungal burden in tongue tissue of Tcra−/− recipient mice 10 weeks after adoptive transfer of the indicated T cells derived from wild-type, Aire−/−, or Aire−/−Ifng−/− mice (N = 7 to 14 per group; five experiments). (D and E) Fungal burden in tongue tissue of Aire−/− mice at day 5 after infection following (D) antibody-mediated neutralization of IFN-γ (N = 6 per group; two experiments) or (E) ruxolitinib administration (N = 9 to 11 per group; three experiments). All quantitative data are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 as calculated using Mann-Whitney U test [(A), (D), and (E)] or Kruskal-Wallis H test with Dunn’s multiple-comparisons test (C).
Discussion
Our results show that Aire deficiency leads to CMC via an aberrant T cell response that causes a mucosal interferonopathy, which disrupts the oral epithelial barrier. Susceptibility to infection is typically thought of as being caused by a lack of appropriate surveillance responses that leads to impaired resistance against an invading pathogen; in the context of mucosal candidiasis, resistance is well recognized to be mediated by protective type 17 mucosal responses (fig. S25). Our findings support a paradigm by which susceptibility to mucosal fungal infection is caused by exacerbated type 1 mucosal inflammation that induces immunopathology, which in turn permits a fungal pathogen to favorably exploit the local niche. In this particular immunopathology, mucosal fungal infection develops without impaired type 17 mucosal immune responses (fig. S25). As such, our data unveil an IFN-γ/STAT1–mediated pathway that critically modulates tissue-specific mucosal antifungal host defense in mice and humans in the setting of intact IL-17R/IL-22–dependent mucosal responses (fig. S25).
These findings inform a mechanism-based targeted therapeutic intervention for CMC in APECED via the inhibition of IFN-γ or JAK-STAT with FDA-approved drugs. Our work expands on the critical roles of IFN-γ in mammalian host defense during infection. Deficiency of IFN-γ-dependent responses is well known to promote susceptibility to severe systemic infections by intramacrophagic pathogens such as mycobacteria and endemic dimorphic fungi (33,34). On the other hand, we show that excessive IFN-γ–dependent responses at the mucosal level lead to susceptibility to oral mucosal infection by the commensal fungus C. albicans, which expands on the recently reported pathogenic roles of excessive IFN-α/β– and IFN-λ–dependent responses in the lower respiratory epithelium during viral infection (35, 36).
Future studies will be required to determine the local mucosal factors that promote enhanced accumulation and aberrant IFN-γ production by mucosal T cells in Aire deficiency, to define the duration of prolonged IFN-γ exposure and the dose threshold of IFN-γ excess that are required at the oral mucosa to promote impaired barrier function and mucosal fungal infection susceptibility, to determine the makeup of T cell receptor repertoires of mucosal T cells in AIRE-deficient patients, and to examine the potential contribution of increased monocyte/macrophage accumulation in the Aire−/− mucosa to fungal infection susceptibility. Moreover, future studies will be needed to investigate whether aberrant T cell–dependent type 1 inflammation (15) may also be the pathogenic mechanism underlying the development of endocrine and nonendocrine autoimmune tissue destruction in AIRE-deficient mice and humans. If so, this could inform the development of targeted therapeutic interventions for autoimmune manifestations in APECED patients with the use of FDA-approved drugs.
In addition, ongoing work is aimed at determining whether similarly aberrant IFN-γ/STAT1–mediated mucosal responses contribute to susceptibility to mucosal fungal infection in other monogenic diseases that manifest with CMC and are associated with excessive inflammation. One such disease is STAT1 gain-of-function (37, 38), which features decreased numbers of TH17 cells in peripheral blood (37), and in which CMC is responsive to pharmacological inhibition of JAK-STAT signaling (39). However, no study has thus far examined immune responses at the mucosa where candidiasis specifically develops in STAT1 gain-of-function patients. Another such CMC-manifesting disease is Down syndrome, which features normal numbers of TH17 cells in peripheral blood, along with increased cellular expression of IFN-γR2 and IFN-γ–induced cellular responses associated with the presence of three copies of the IFNGR2 gene (40). Future studies will also be required to determine whether IFN-γ/STAT1–mediated immune responses are aberrant at the mucosa in interferonopathies in which CMC is not a prominent feature (41–44). Collectively, our findings identify enhanced type 1 immunity as a critical determinant of susceptibility to mucosal fungal infection and highlight the importance of tissue-specific immune responses, whether protective or detrimental, in the control of infectious disease.
Materials and methods
Mouse strains
Age- and sex-matched mice were used for all experiments and were kept under specific pathogen-free conditions. Aire+/− mice, on the NOD background, were rederived from embryos by Jackson Laboratories (JAX stock 006360) and bred as heterozygous breeder pairs to obtain wild-type (WT) and Aire−/− littermate mice, which were used for the majority of the experiments. NOD Aire−/− mice were rederived as heterozygous germ-free NOD Aire+/− by Taconic Biosciences and bred as heterozygous in flexible film isolators at the NIAID Microbiome Program Gnotobiotic Animal Facility. Aire+/− mice were crossed with Tcra−/− [provided by D. Mathis (Harvard Medical School)] or Ifng−/− mice [provided by D. Serreze (JAX Laboratories)] on the NOD background to generate Aire−/−Tcra−/− or Aire−/−Ifng−/− mice, respectively. BALB/c Aire+/− mice were provided by M. Anderson (UCSF) and bred in-house, whereas C57BL/6 Airegfp/gfp mice were provided by M. Matsumoto (Tokushima University, Japan) and bred in-house. IL-22Cre mice (JAX stock 027524), which lack IL-22 when bred to homozygosity, were provided by R. Caspi (National Eye Institute). All experiments were performed according to guidelines set forth by the Guide for the Care and Use of Laboratory Animals under a protocol approved by the NIAID Animal Care and Use Committee.
Fungal strains and oropharyngeal candidiasis (OPC) infection model
Candida albicans strain SC5314 was used for OPC infection, with the exception of the experiments in fig. S1, where C. albicans strains Y72 and Y42 were used. These strains were isolated from the oral mucosa of APECED patients with oral candidiasis and have been previously shown to cause infection in this model (45, 46).
For the OPC model of infection, C. albicans yeast was grown overnight in yeast extract, peptone, and dextrose (YPD) media with penicillin and streptomycin in a shaking incubator set at 30°C. To prepare C. albicans for infection, the yeast was centrifuged, washed in phosphate-buffered saline (PBS), and counted using a hemocytometer. C. albicans was then diluted to 107/ml in HBSS for infections. Cotton swabs were soaked in the C. albicans solution prior to insertion under the tongue for 90 min while the mice were anesthetized with ketamine and xylazine, as described (45–47). For intravenous infections, mice were infected with 2 × 104. C. albicans yeast cells via the lateral tail vein.
Fungal burdens
To determine the number of C. albicans colony-forming units (CFUs) in blood and organs of mice, blood was obtained via cardiac puncture and plated onto YPD plates, while tissues were weighed and homogenized with an Omni homogenizer (Omni International) in PBS. The homogenate was then left undiluted or diluted in PBS and plated onto YPD plates containing penicillin and streptomycin. The plates were incubated at 37°C for 24 to 48 hours and CFUs were counted. The CFU data were presented as the number of CFUs per gram of tissue or per milliliter of blood. To lower the limit of detection for CFUs in the tongue, buccal, or gingival tissues, the entire organ homogenate was plated onto YPD plates. When the entire organ was plated and no CFUs were counted, then a value of 1 was assigned.
Other infection models
Mouse cytomegalovirus (MCMV) purification, infection experiments, and determination of tissue viral load.
Mouse fibroblasts, NIH-3T3 and M2-10B4, were acquired from ATCC and maintained in DMEM and RPMI medium (Life Technologies), respectively, supplemented with 10% FBS. MCMV was recovered by transfection of NIH-3T3 cells with a pSM3fr-MCK-2fl bacmid (provided by B. Adler, Ludwig Maximilians University). This bacmid corrects the frameshift mutation found in the MCK-2 ORF of the original pSM3fr bacmid derived from MCMV strain Smith (48). MCMV was expanded by infection of M2-10B4 cells and purified from infected supernatants by centrifugation at 72,000g for 1 hour through a 15% sucrose cushion. Pelleted virus was resuspended in virus standard buffer (50 mM Tris-HCl pH 7.8, 12 mM KCl, and 5 mM EDTA), aliquoted, and stored at −80°C. The viral titer was determined by plaque assay on M2-10B4 cells as described (49). Parental WT and Aire−/− mice were infected intraperitoneally (i.p.) with 105 plaque-forming units (PFU) of tissue culture–derived MCMV. Lung, spleen, liver, and salivary glands were collected at days 4 and 14 after infection and homogenized using an Omni TH homogenizer (Omni International). Homogenates were clarified by centrifugation and viral titers were determined by plaque assay on M2-10B4 cells, as above.
Due to the observed high toxicity of liver homogenates on these cells, viral load in this organ was determined by quantitative PCR (qPCR) using a previously described protocol with some modifications (50). Briefly, genomic DNA was isolated from homogenates using the DNeasy Blood and Tissue kit (Qiagen). Viral load was assessed as the copy number ratio MCMV iE1/GAPDH detected by qPCR in a CFX96 Touch Real-Time System (Bio-Rad) using the SensiFast SYBR Hi-Rox kit (Bioline), following manufacturer instructions. MCMV iE1 was detected using the primers iE1MCMV2F (5′-CATCTCCTGTCCTGCAACCT-3′) and iE1MCMV2R (5′-CTTGGGCTGCTGTTGATTCT-3′). Mouse GAPDH was amplified with the primers mGAPDH F (5′-AACTTTGGCATTGTGGAAGG-3′) and mGAPDH R(5′-ACACATTGGGGGTAGGAACA-3′). The copy numbers of MCMV iE1 and GAPDH were calculated by interpolation of the corresponding Cq values in standard curves generated by 10-fold dilutions of pcDNA3.1-iE1 and pCMV-Sport6-mGAPDH plasmids, respectively. After the last extension cycle, a melting curve analysis was performed to control the amplification specificity.
Staphylococcus aureus infection and determination of tissue bacterial load.
S. aureus skin infections and CFU assessment were performed as described (51). In brief, the backs of mice were shaved, and 106 to 107 CFU of S. aureus (strain USA300-LAC, provided by F. DeLeo, NIAID) were injected intradermally in 100 μl of PBS, in a blinded manner to the mouse genotype. The resultant abscess size was measured daily. Skin tissue and kidneys were homogenized and plated onto blood agar plates (Remel) for CFU enumeration at day 5 after infection.
Vaccinia virus infection and determination of viral load.
Mice were anesthetized and infected in the lower lip with five pokes from a bifurcated needle dipped in thymidine kinase (TK)–negative recombinant vaccinia virus (VACV) expressing eGFP (stock titer, ~1 × 108 PFU/ml). Lips were gently wiped with sterile gauze to remove remaining viral inoculum before mice were allowed to recover from anesthesia. Lips were harvested at 7 days after infection, digested in collagenase for 40 min at 37°C, and disrupted by vigorous pipetting to generate single-cell suspensions. Suspensions were freeze-thawed three times, sonicated three times, serially diluted, and plated on TK-143B cells. Cells were incubated at 37°C for 2 days and stained with crystal violet. Plaques were then counted.
Candida albicans footpad infection and CFU determination.
C. albicans was grown in YPD broth overnight and washed with PBS, and 5 × 106 blastospores (in a 50-μl volume of PBS) were injected in the left hind paw. Footpad swelling was measured every 2 days until day 10 after infection, at the widest area of the foot just above the toes, using Vernier calipers. Uninfected footpad thickness was also measured and the percent swelling of the infected footpad was recorded relative to the uninfected footpad. For determination of footpad CFUs at day 10 after infection, the infected footpads were harvested using a scalpel, weighed, and homogenized in 0.5 ml of PBS using an Omni tip homogenizer (Omni International). The homogenates were plated on YPD agar plates and incubated at 37°C for 48 hours for CFU enumeration.
Citrobacter rodentium infection and bacterial burden quantification.
Citrobacter rodentium (nalidixic acid–resistant strain ICC169, a kind gift from G. Frankel, Imperial College London) was cultured for 18 hours in Luria-Bertani (LB) broth containing nalidixic acid (50 mg/ml) at 37°C as described (52). Mice were infected orally by gavage with 108 CFUs resuspended in sterile PBS. For quantification of bacterial burden, cecum or colon were collected, weighed, placed in tubes containing 2.8-mm stainless steel beads (Sigma-Aldrich), and homogenized using a Precellys 24 tissue homogenizer (Bertin Instruments), followed by serial dilution and plating on LB agar plates containing nalidixic acid (52). Colonies were then counted after 16 hours of incubation at 37°C, and CFUs were reported per milligram of tissue.
Ligature-induced periodontitis and periodontal bone loss.
Experimental periodontitis was induced by a silk suture placed around the left maxillary second molar for 7 days. The contralateral second molar was used as a negative control. After micro–computed tomography scanning (Scanco μCT 50, 7-μm voxel size, 70kV/114 μA), bone loss was measured at six predetermined anatomical landmarks as described (53). The bacterial load was then determined (54).
Antibody and drug administrations
In some experiments, Aire−/− mice at 6 to 7 weeks of age were administered depleting or neutralizing antibodies to determine their impact on fungal clearance. For IL-17 neutralization, anti-IL17A (BioXCell, clone 17F3) and anti-IL-17F (eBioscience, clone RN17) or isotype control (BioXCell, clone MOPC-21) were administered i.p. at a dose of 500 μg or 300 μg, respectively, on days −1, 1, and 2 after infection and CFUs were determined at day 4 after infection, according to a dosing scheme that has been used previously and was shown to increase fungal susceptibility in the OPC model of infection (55). For IL-22 neutralization experiments, Aire−/− mice were administered 150 μg of anti–IL-22 (Genentech, clone 8E11) or the isotype control (BioXCell, clone MOPC-21) once daily starting at day −1 through 3 after infection for five total injections. CFUs were determined on day 4 alter infection, according to a dosing scheme that has been used previously and was shown to increase fungal susceptibility in the OPC model of infection (25).
In some experiments, a ~3-week treatment was administered to Aire−/− mice starting at 3 to 4 weeks of age to determine the impact of the corresponding cellular or molecular immune factors in the OPC model. For IFN-γ neutralization, 3- to 4-week-old Aire−/− mice were administered 500 μg of anti–IFN-γ (BioXCell, clone R4-6A2) or its isotype control (BioXCell, clone HRPN) twice a week for a total of eight injections, including one at day 1 after infection, and CFUs were determined at day 5 after infection. To determine the impact of enhanced JAK/STAT pathway on fungal clearance, 3- to 4-week-old Aire−/− mice were administered the JAK1/2 inhibitor ruxolitinib (50 mg/kg; Chemietek) or vehicle (1% carboxymethylcellulose; Sigma) once daily via oral gavage for 3 weeks. Mice were then infected with C. albicans and CFUs were determined at day 5 after infection, with ruxolitinib administration continuing through day 4 after infection. To determine the impact of CD4+ and CD8+ T cells on fungal clearance, 3- to 4-week-old Aire−/− mice were administered 500 μg of anti-CD4 (BioXCell, clone GK1.5), anti-CD8 (BioXCell, clone YTS169.4), or isotype control (BioXCell, clone LTF-2) for three consecutive daily doses i.p., followed by twice-weekly administration for a total of eight injections, according to previously described methods that have been shown to effect T cell depletion in vivo (56, 57). We confirmed 86 to 100% depletion of CD4+ or CD8+ T cells in blood by flow cytometry, using different clones for staining (clone RM4-5 for CD4+ T cells; clone 53-6.7 for CD8+ T cells).
Serum transfers
To determine the impact of serum-derived factors (including autoantibodies) on susceptibility to oral candidiasis, we performed serum transfers, as previously described for the assessment of the role of serum-derived factors in the development of autoimmune tissue destruction in Aire deficiency (15–17). WT mice were administered 200 μl of serum i.p. from either WT or Aire−/− mice twice weekly for 7 weeks. One day before the last serum injection, recipient mice were orally infected with C. albicans. At day 5 after infection, tongue CFUs were determined.
Autoantibody determination
Particle-based multiplex assay.
Serum from WT or Aire−/− mice was screened for TH17 cell–derived autoantibodies using a particle-based approach as described (58). Briefly, 2.5 μg of recombinant mouse IL-17A, IL-17F, or IL-22 (eBioscience) were coupled to differentially fluorescing magnetic beads (Bio-Rad). These beads were incubated with 1:10-diluted serum for 30 min. The beads were washed and incubated with PE-labeled goat anti-mouse IgG (1 μg/ml, eBioscience) for 30 min. Beads were washed again and run as a multiplex on the Bio-Plex 200 (Bio-Rad) instrument. The fluorescence intensity for each bead was plotted as a function of autoantibody titer (GraphPad Prism version 8). The seropositivity of these autoantibodies in Aire−/− mice was calculated based on the means plus three standard deviations of the WT group.
Radioligand binding assay (RLBA).
Full-length cDNAs for mouse IL-17A, IL-17F, and IL-22 were used as templates for in vitro transcription and translation and labeled with [35S]methionine using the TNT system kit (Promega). Radio-labeled proteins were immunoprecipitated in duplicate or triplicate with serum or control antibodies in 96-well PVDF filtration plates (Millipore). In each well, 20,000 counts per min (cpm) of 35S-labeled proteins and 2.5 μl of WT and Aire−/− serum were used for immunoprecipitation. The radioactivity of the immunoprecipitated material was quantified with the use of a liquid scintillation counter. For each assay, a polyclonal rabbit anti-mouse IL-17A antibody (Sigma-Aldrich, prs4887), a polyclonal rabbit anti-mouse IL-17F antibody (Abnova, pab16929), or a polyclonal goat anti-mouse IL-22 antibody (Santa Cruz Biotechnology, sc-14436) were used as positive controls. Serum from NOD WT mice served as negative standards. The autoantibody index was calculated as follows: [(cpm in the unknown sample) – (cpm in the negative standard)] / [(cpm in the positive standard) – (cpm in the negative standard)] × 100. RLBAs were performed in two independent laboratories using the same antigen construct. The upper limit of the normal range for each assay was defined as the mean value obtained for the WT mice tested plus three standard deviations.
Oral microbiome characterization using 16S rRNA gene sequencing
Oral murine microbial samples were obtained from uninfected WT and Aire−/− mice as described (59) . DNA was extracted using the DNeasy Blood and Tissue kit (Qiagen) with modifications as described (59). Library preparation for sequencing of the 16S rRNA V4 region by PCR used primers 515F and 806R and the amplicon libraries were sequenced based on the 2 × 250 bp paired-end protocol (Illumina).
Bioinformatics analysis was done as described (60). Briefly, the reads were denoised with DADA2 (61) in QIIME 2 2019.4 (62). The taxonomy was assigned to amplicon sequence variant (ASV) using naive Bayes classifier trained with the SILVA database (v 132) (63) at 99% similarity. The number of reads per sample was rarefied to 8500. Alpha-diversity was calculated using Chaol (64) and Shannon. Beta-diversity was calculated using Jaccard distance. The distances were used for principal coordinate analysis and visualized in 3D plots using EMPeror (65), and analysis of similarities (Anosim) was used to check for significant differences at the community level. Individual bacterial features that showed differential abundance due to Aire deficiency were identified using the linear discriminant analysis (LDA) effect size (LEfSe) (66) with default settings.
Histological, immunofluorescence, and immunohistochemical evaluations of mucosal tissue
For hematoxylin and eosin (H&E) and periodic acid-Schiff (PAS) staining, tongues were harvested and placed in 10% formalin for 24 to 48 hours. Formalin was then replaced with 70% ethanol to store until paraffin embedding. Longitudinal sections of the tongue were prepared and stained with H&E and/or PAS (Histoserv Inc.).
For immunofluorescence, tongues from WT and Aire−/− mice were placed in 2% paraformaldehyde at 4°C for 2 hours in the dark, followed by incubation in 30% sucrose in the dark until the tongues sank (~6 to 8 hours). Tongues were then placed in OCT media, snap-frozen on dry ice, and stored at −80°C until ready for sectioning. Tongues were sectioned into 15- to 20-μm-thick sections onto Superfrost Plus slides (Fisher), using a Cryostat (Leica). Frozen sections were fixed with acetone for 10 min, blocked with Fc block (anti-CD16/32) and 5% goat serum for 1 hour at room temperature, and incubated overnight at 4°C with Alexa Fluor 647-conjugated anti-mouse CD4 (clone GK1.5; Biolegend; 5 mg/ml) and Alexa Fluor 488-conjugated anti-mouse CD8a (clone 53-6.7; R&D Systems; 2 μg/ml). Slides were mounted with ProLong Gold Antifade Reagent with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; Life Technologies), and microscopy was performed using a Leica SP8 Dual MP microscope. The fluorophores were excited using 405-nm, 488-nm, and 633-nm laser lines for DAPI, AF488, and AF647, respectively. Emission was collected via sequential scanning, whereby emission from DAPI was collected in one sequence and emission for AF488 and AF647 was collected in another to minimize signal bleed-through. To acquire a large area across the tissue surface, tile scanning was performed using the “Mark & Find Tile Scan” function, where tiled fields of view (FOV) of 18-μm total thickness per sample were imaged (seven image slices, 3-μm apart) using a 40×1.3 NA oil-immersion objective lens. Fluorescence from DAPI was detected on a standard PMT, whereas fluorescence from AF488 and AF647 was detected on HyD detectors (Leica). The tiles were stitched using the “Mosaic Merge” function of Leica’s LAS X. For quantification of CD4+ and CD8+ T cells, the images were exported to Bitplane Imaris 9.0 and FOVs of equal areas were extracted for quantifying cell numbers. The fluorophore-positive cells were classified using the “Spot” creation algorithm of Imaris.
Ki-67 immunohistochemical staining was performed on formalin-fixed and paraffin-embedded tongue tissue slides from WT, Aire−/−, and Aire−/−Ifng−/− mice. Slides were deparaffinized in xylene and rehydrated in graded alcohol and distilled water. Heat-induced antigen retrieval was performed using 1:10 Dako Target Retrieval Solution, Citrate pH 6.1 (10×) (S1699) in a pressure cooker for 6 min, and the slides were rinsed in Tris buffer solution. The slides were immersed in TBS/3% normal goat serum to block nonspecific binding. For mouse tissue, the primary antibody, Ki-67 (D3B5) rabbit mAb from Cell Signaling, was added at a 1:400 dilution and incubated for 1 hour at room temperature followed by a 30-min incubation of anti-rabbit secondary antibody. Dako Liquid DAB+ Substrate Chromogen System Code (K3468) was used as the detection chromogen, and the slides were counterstained with hematoxylin. The histology score for Ki-67 was completed in a blinded manner to the mouse genotype by two viewers scanning the entire mucosal surface at 10× to determine the percent of positive cells by quartiles (range, 0 to 4) and intensity of positivity (range, 1+ to 3+). These values were multiplied to calculate the final score (range, 0 to 12).
Two-photon confocal microscopy
To visualize and quantify cell death in oral epithelial cells of mouse tongues, 100 μl of nuclear stain solution containing 6.7 μg of propidium iodide (Sigma Aldrich) and 0.5 mg of Hoechst 33258 (Thermo Fisher Scientific) in PBS was injected retro-orbitally in anesthetized WT, Aire−/−, and Aire−/−Ifng−/− mice on day 0 and on day 1 after C. albicans infection. Ten minutes after injection of nuclear stain solution, the mice were killed via cervical dislocation and tongues were excised. The excised tongues were washed briefly in PBS and placed on a cover glass. The cover glass was then mounted on a microscope stage insert using an adhesive tape for subsequent imaging. A PBS-dipped, moist Kimwipe (Kimberly-Clark) was placed atop the tongues to avoid tissue dehydration. Microscopy was performed immediately using a Leica SP8 Dual MP microscope. The fluorophores were excited using a Mai Tai laser (Spectra-Physics), tuned at 820 nm. For each mouse tongue, 3 to 5 independent FOVs were acquired using a 25× 0.95 NA water immersion objective. At each FOV, a volumetric image stack was acquired with dimensions of 465 μm across the x and y axes and 60 mm across the z axis (xyz voxel size of 0.455 μm, 0.455 μm, and 1.999 μm, respectively). Fluorescence for Hoechst 33258 and propidium iodide were collected using external HyD detectors (Leica). For quantification of Hoechst 33258 and propidium iodide–positive cells, the images were exported to Bitplane Imaris 9.0. The fluorophore-positive cells were classified using the “Spot” creation algorithm of Imaris. Statistics were exported to Microsoft Excel to compute “percent propidium iodide positive cells” relative to the total Hoechst-positive cells. For display purposes, the image stacks were exported to FIJI (67), and “Maximum Intensity Projections” were computed for display.
Determination of concentrations of cytokines and chemokines in mouse mucosal tissue homogenates
To assess the relative abundance of cytokines and chemokines in the tongue, tongues from mice that were uninfected or sublingually infected with C. albicans were collected on days 1, 3, and 5 alter infection. Tongues were homogenized with an Omni Tissue Homogenizer (Omni International) in PBS containing 0.5% Tween 20 and a protease inhibitor cocktail (Roche). The homogenate was centrifuged at ~16,000g for 10 min at 4°C. Supernatants were filtered through a 22-μm filter and snap-frozen on dry ice. Homogenates were stored at −80°C until analysis. Proteins were then analyzed either via enzyme-linked immunosorbent assay (ELISA) or Luminex-based detection methods. For ELISA, supernatants were spun down at 13,200g for 10 min at 4°C, and proteins were determined using to the manufacturers’ protocols. The following ELISA kits were used for the corresponding cytokines: IL-17A (R&D), IL-17F (R&D), IL-23p19 (R&D), TNF-α (R&D), and IFN-γ (Thermo Fisher Scientific, Extra Sensitive), according to the manufacturer’s instructions. Luminex-based detection of mouse IL-1b, IL-2, IL-4, IL-6, IL-22, GM-CSF, CCL20, CXCL1, CXCL5, CXCL9, and CXCL10 was performed as described (47).
Gene expression in mouse mucosal tissue
Tongues were homogenized in Trizol with an Omni Tissue Homogenizer (Omni International) and mRNA was isolated with the RNeasy Kit (Qiagen), according to the manufacturer’s protocol. mRNA was then converted to cDNA using the qScript cDNA Supermix Kit (Quanta Biosciences). qPCR was performed using TaqMan detection (PerfeCTa qPCR FastMix ROX, Quanta Biosciences), Taqman primers/probes predesigned by Applied Biosystems, and the 7500 Real-Time PCR system (Applied Biosystems). To determine relative mRNA levels for S100a8, S100a9, Defb1, Defb3, Lcn2, Il4, Il6, Il10, Il17a, Il17f, Il22, Il23p19, Ifng, Cxcl1, Cxcl5, Cxcl9, Cxcl10, Ccl20, and Stat1, Ct values were determined and compared against Gapdh. All qPCR assays were completed in duplicate wells and normalized to Gapdh using the ΔΔCT method.
Single-cell suspensions from mouse mucosal tissues
Single-cell suspensions were obtained from whole tongue tissue, based on a previously described technique (68). Briefly, the tongue was minced and placed in a digestion solution containing collagenase type 4 (385 U/ml; Worthington), dispase II (2 U/ml; Gibco), and DNase I (50 mg/ml; Roche) in 10 ml of RPMI in a shaking water bath set at 37°C. After 45 min, PBS + 5 mM EDTA (Quality Biological) was added and the solution placed on ice to stop the digestion. The cells were then filtered through a 70-μm filter, washed, filtered through a 40-μm filter, and washed again. To form a Percoll gradient, 40% and 70% Percoll solutions were made from 100% Percoll (10× PBS diluted 1:10 in Percoll). The cells were resuspended in 40% Percoll and carefully overlaid onto 70% Percoll. The gradient was then centrifuged at 872g for 30 min at room temperature to separate out the leukocytes. The cells at the interface between the 40% and 70% layers were collected for flow cytometric analysis.
A single-cell suspension was obtained from the tongue epithelium, using a previously established protocol (69). Briefly, the tongue was harvested and turned upside down. The ventral epithelium (tongue underside) was split down the middle and the underlying muscle tissue was carefully removed with a scalpel. The tissue that remained was finely minced using a scalpel. The tongue epithelium tissue was digested in a PBS solution containing trypsin (1 mg/ml), DNase I (200 mg/ml), and collagenase IV (2.4 mg/ml) in a 37°C water bath for 45 min. The cell suspension was then centrifuged, resuspended in ice-cold PBS + 1% FBS and 2 mM EDTA, and passed through a 70-μm filter to remove large particles.
A single-cell suspension was obtained from the oral gingival mucosal tissue of mice according to a previously established method (69). Briefly, mice were killed, and the body and head were immobilized on a pad with the stomach facing upward. The maxillary and mandibular molar areas and the surrounding gingiva were incubated in 5 ml of a collagenase/DNase mixture for 1 hour at 37°C in a shaking incubator. During the last 5 min of the incubation, 50 μl of 0.5 M EDTA was added to the mixture. The mixture was quenched with 5 ml of DNase-containing RPMI media. The gingiva was separated from the teeth using a scalpel blade and cut into small pieces in a petri dish. The tissue was filtered through a 70-μm filter and washed with DNase-containing RPMI. The cells were then spun at 314g for 6 min and resuspended in complete RPMI media.
Flow cytometry of mouse samples
Unless otherwise stated, single-cell suspensions from the tongue (in PBS) were stained with a LIVE/DEAD fluorescent dye (Invitrogen) at 4°C for 10 min. The cells were then incubated for 10 min at 4°C with rat anti-mouse CD16/32 (eBioscience) to block Fc receptors. Cell surface markers were then stained for 30 min at 4°C.
To stain for intracellular cytokines in mouse oral mucosal gingival cells, cells were stimulated with PMA (50 ng/ml) and ionomycin (2.5 μg/ml) for 3.5 hours at 37°C + 5% CO2 in the presence of Brefeldin A (1 mg/ml). Cells were stained with live/dead and cell surface markers as mentioned above, and were then fixed and permeabilized using the Foxp3 intracellular staining kit (eBioscience). Cells were stained for IFN-γ (clone XMG1.2, eBioscience), IL-17A (clone TC11-18H10.1, BioLegend), IL-17F (clone eBio18F10, eBioscience), TNF-α (clone MP6-XT22, BioLegend), GM-CSF (clone MP1-22E9, BD Biosciences), and/or IL-22 (clone IL-22JOP, eBioscience) and analyzed on a BD Fortessa. For Foxp3 and Ki-67 staining, gingival mucosal cells were left unstimulated or were stimulated with anti-CD3/C28 (BD Pharmingen) in Dulbecco’s minimum essential medium (Life Technologies) for 5 hours at 37°C + 5% CO2, in the presence of Brefeldin A (1 μg/ml), respectively, and stained for cell surface markers and live/dead as mentioned above. Cells were then resuspended in Fix/Perm buffer overnight and intracellular staining was performed for Foxp3 (clone FJK-16s) or Ki-67 (clone B56). Samples were acquired on a LSRII flow cytometer (BD Biosciences) and analyzed with FlowJo software.
The fluorophores used for flow cytometry included eFluor450, AmCyan, BV421, BV510, BV570, BV605, BV711, BV785, FITC, PE, eVolve 605, PE/Texas Red, PE-eFluor 610, PerCP-eFluor 710, PE-Cy7, PerCP-Cy5.5, APC, AF700, APC-Cy7, and APC-eFluor 780. The antibodies used for cell surface staining were as follows: From eBioscience, CD3 (clone 145-2C11), CD5 (clone 53-7.3), CD44 (clone IM7), FcεR1 alpha (clone MAR-1), CD90.2 (clone 30-H12), CD117 (clone 2B8), CD11b (clone M1/70), Gr-1 (clone RB6-8C5), Sca-1 (clone D7), NKp46 (clone 29A1.4), B220 (clone RA3-6B2), CD14 (clone Sa2-8), CD11c (clone N418), CD102 (clone 3C4), CD45 (clone 30-F11); from Biolegend: CD8a (clone 53-6.7), CD4 (clone RM4-5), NKp46 (clone 29A1.4), CD103 (clone 2E7), MHC-II RT1B (clone OX-6), F4/80 (clone BM8), CD11b (clone M1/70), CD119 (IFNγR1) (clone 2E2), Armenian Hamster IgG isotype control (clone HTK888), CD31 (clone 390), EpCAM (clone G8.8), CD69 (clone H1.2F3), CD3ε (clone 145-2C11), TCRβ (clone H57-597), CD19 (clone 1D3/CD19), CD90.2 (clone 30-H12), and CD44 (clone IM7); from BD: TCRγd (clone GL3), CD3 (clone 17A2), Ly6G (clone 1A8), CD45 (clone 30-F11), and CD4 (clone GK1.5) (table S7).
Different leukocyte populations were first gated as live CD45+ cells and were defined as follows: Neutrophils were defined as Ly6G+Ly6CintCD11b+CD3−B220−NKp46− cells, monocytes were defined as Ly6C+CD11b+Ly6G−MHCII−F4/80lo-intCD3−B220−NKp46− cells, macrophages were defined as MHCIIhiCD11b+F4/80hiLy6G−CD3−B220−NKp46− cells, CD11b+ dendritic cells were defined as MHCIIhiF4/80− CD11chiCD3−B220−NKp46− cells, eosinophils were defined as MHCII−F4/80lo-intLy6CloLy6G−SSChiCD3−B220−NKp46− cells, mast cells were defined as c-kit+ FcεR1+ cells, innate lymphoid cells were defined as lineage-negative (Gr-1−CD5−CD3−CD19−CD11b−) CD90.2hiCD127lo-int cells, CD4+ T cells were defined as SSCloCD3+CD4+CD8−gdTCR−NKp46− cells, CD8+ T cells were defined as SSCloCD3+CD4−CD8+gdTCR−NKp46− cells, B cells were defined as SSCloCD19+ cells, NK cells were defined as SSCloNKp46+CD3− cells, NKT cells were defined as SSCloNKp46+CD3+ cells, and γδ T cells were defined as SSCloCD3+ gdTCR+ cells.
Isolation of epithelial cells by FACS
To sort tongue epithelial cells, a single-cell suspension was obtained from the tongue epithelium as described above using sterile technique. Cells were then stained with EpCAM, CD31, CD45, CD102, and a live/dead cell marker. Live CD45−CD31−CD102−EpCAM+ tongue epithelial cells were sorted with a FACS ARIA cell sorter (>95% purity) and resuspended in Trizol (Thermo Fisher Scientific).
Isolation of T cells by FACS and adoptive transfer experiments
CD4+ and CD8+ T cells were harvested for adoptive transfer into recipient Tcra−/− mice. The spleens and submandibular lymph nodes were harvested from WT, Aire−/−, and Aire−/−Ifng−/− mice and passed through a 70-μm filter twice. Single-cell suspensions were surface-stained with eFluor450-conjugated CD45, PerCP-Cy5.5-conjugated CD3, APC-Cy7-conjugated CD4, and PE-Cy7–conjugated CD8, passed through a 35-μm filter, and sorted on a BD FACS Aria II Cell Sorter. Post-sorting purity of CD4+ T cells and CD8+ T cells was 98.2 to 99.5% and 96 to 99.8%, respectively. CD4+ (4 × 106 T cells), CD8+ (4 × 106 T cells), or a 1:1 ratio of CD4+ and CD8+ T cells (4 × 106 CD4+ T cells and 4 × 106 CD8+ T cells) were injected intravenously into ~5-week-old NOD Tcra−/− recipient mice. After 10 weeks, the mice were sublingually infected with C. albicans and CFUs were determined at day 5 after infection.
RNA-seq of mouse oral epithelial cells
WT and Aire−/− mice (N = 3 per group) were infected with C. albicans and tongue epithelial cells were sorted at day 1 after infection, as described above, and lysed in Trizol. RNA was isolated from the purified epithelial cells according to the manufacturer’s guidelines for the kit (Norgen Biotek RNA Clean-Up and Concentration Micro-Elute Kit, cat# 61000). Sequencing libraries were prepared by the Illumina Nextera XT method, following the manufacturer’s recommendations. The multiplexed libraries were sequenced in a NextSeq500 instrument in 150-bp paired end mode. Demultiplexed samples were mapped and transcripts quantified to GRCm38.v11 mouse genome using the STAR v2.5.2 aligner. Gene-level counts were filtered to remove low-expression genes (keeping genes with ≥5 counts in at least one sample). Differential gene expression was evaluated using DESeq2. Genes differentially expressed (twofold, Padj < 0.05) between infected WT and Aire−/− samples were subjected to pathway analysis using Enrichr and graphed based on enrichment score [−log10(adjusted P value)]. Enrichment of hallmark genes in differentially expressed genes of Aire−/− mice versus WT mice was also determined using MSigDB. Upstream regulators of differentially expressed genes were identified using Ingenuity Pathway Analysis (IPA, Qiagen). IL-17 receptor–induced and repressed genes in tongue tissue were previously determined in a mouse model of oral candidiasis (19). Heat maps were drawn using Morpheus (Broad Institute).
Protein immunoblots
Tongue epithelium was homogenized in 1× RIPA buffer (Thermo Fisher Scientific) containing protease and phosphatase inhibitors (Thermo Fisher Scientific). Protein concentrations were determined using the Bradford Protein Assay (Bio-Rad), according to the manufacturer’s protocol. The proteins (20 mg) were resolved in 10% or 12% SDS-PAGE and electrotransferred onto 0.2-μm PVDF or nitrocellulose membranes. Equivalently loaded proteins were resolved by NuPAGE 3 to 8% Tris-Acetate (Invitrogen). Membranes were blocked in 5% nonfat dry milk and incubated with primary antibodies against pStat1 (Tyr701) (clone 58D6; Cell Signaling; 0.2 μg/ml), total Stat1 (clone D4Y6Z; Cell Signaling; 0.2 μg/ml), gamma H2AX (phosphoS139) [clone EP854(2)Y; Abcam; 0.15 μg/ml], Ki-67 (clone SP6; Abcam; 1 μg/ml), IFNγR2 (Proteintech; cat# 10266-1-AP; 0.15 μg/ml), claudin-1 (Invitrogen; cat# 51-9000; 0.5 μg/ml), β-actin (clone D6A8; Cell Signaling; 0.2 μg/ml), and α/β tubulin (Cell Signaling; cat# 2148; 0.2 μg/ml), followed by secondary anti-rabbit IgG, HRP-linked antibodies (Cell Signaling). Chemiluminescence detection was performed with Clarity Western ECL Blotting Substrate (Bio-Rad) or Radiance ECL Substrate (Azure Biosystems), using the ChemiDoc MP Imaging System (Bio-Rad). Quantification was obtained by densitometry image analysis using Image Lab 5.2 Software (Bio-Rad).
Assessment of oral mucosal permeability in mice
The paracellular permeability of the oral mucosa of the tongue of WT, Aire−/−, and Aire−/−Ifng−/− mice was evaluated by the passage of 4-kDa FITC-dextran (Sigma-Aldrich) as measured in the serum. Specifically, uninfected mice or mice infected with C. albicans on day 1 after infection were fasted for 4 hours before the assay. The mice were then anesthetized with ketamine and xylazine, and 100 ml of 4-kDa FITC-dextran dissolved in PBS (40 mg/ml) was applied topically on the anterior surface of the tongue. Serum was collected 4 hours after FITC-dextran topical tongue application and transferred to a black 96-well plate (Greiner). Fluorescence of the serum FITC-dextran was determined at an excitation wavelength of 485 nm and an emission wavelength 535 nm using a Synergy H1 multimode microplate reader (BioTek).
Saliva collection and measurement of salivary antifungal killing capacity
To collect saliva, WT and Aire−/− mice were anesthetized with ketamine and xylazine and placed on an elevated position to allow for saliva collection. Pilocarpine (20 μl of a 0.5 mg/ml solution in PBS) was injected subcutaneously in the scruff of the neck and one end of a hematocrit tube was placed in the corner of the mouth, with the other end connected with a 1.5-ml microcentrifuge collection tube. Saliva was collected for 20 min and the total volume was measured. After collection, saliva was mixed 1:1 with a protease inhibitor cocktail (Roche) in 1× PBS, according to the manufacturer’s instructions. Saliva was snap-frozen on dry ice and stored at −80°C until testing.
For assessment of salivary antifungal killing capacity, C. albicans was cultured as previously described, washed three times using 50 mM sodium phosphate buffer (pH 7.4), and resuspended in the same buffer at a concentration of 105 CFU/ml. Then, 100 μl of saliva was incubated with 100 μl of the C. albicans suspension in a water bath at 37°C. At different time points (0, 1, and 3 hours), 10 μl of the suspension was with-drawn, diluted in 90 μl of PBS, and plated onto YPD agar plates. The plates were incubated at 37°C for 24 to 48 hours and the C. albicans CFUs were counted. The percentage of remaining live C. albicans was calculated to assess the killing capacity of the saliva.
Antibiotic treatment of mice
To determine the impact of antibiotic treatment on mucosal fungal clearance in Aire−/− mice, the drinking water for mice was substituted with autoclaved water containing a mixture of ampicillin, metronidazole, neomycin, and vancomycin (AMNV) or autoclaved water without antibiotics (control) and provided ad libitum for 4 weeks. Mice were then sublingually infected with C. albicans and fungal CFUs were determined in tongue homogenates at day 5 after infection.
Human subjects
Thirty-eight healthy volunteers and 79 APECED patients enrolled from 2013 to 2018 in NIH IRB-approved protocols (NCT01386437, NCT01568697). Healthy volunteers were 18 to 75 years of age who did not meet the following exclusion criteria: (i) smoking history of ≥10 pack-years, a current smoker, or tobacco-free <1 year; (ii) positive HIV status by HIV PCR; (iii) acute or chronic hepatitis based on viral hepatitis serologies; (iv) pregnant or breastfeeding; (v) history of pulmonary disorders; (vi) history of bleeding disorders or significant bruising or bleeding with intramuscular injections or blood draws; (vii) anticoagulant use; and (viii) taking immunosuppressive medications, cytotoxic medications, or inhaled corticosteroids within the past 3 months.
Inclusion criteria for APECED patient enrollment consisted of either a clinical APECED diagnosis based on developing any two manifestations within the classic triad of CMC, adrenal insufficiency, and hypoparathyroidism (N = 70), or a genetic APECED diagnosis (i.e., biallelic AIRE mutations and/or deletions) without developing a classic diagnostic dyad (N = 9). Forty-nine subjects were female (62%) and 30 were male. Thirty-five subjects were children (44%) with a mean age of 11.5 years (range 3 to 17 years). Among the classic triad manifestations, 65 (82%) had hypoparathyroidism, 62 (78%) had adrenal insufficiency, and 69 (87%) had CMC. Sixty-three subjects were from North America, 11 were from Europe, three were from South America, and two were from Asia. Seventy-seven subjects self-identified as Caucasian, among whom nine co-identified as Hispanic, and two identified as Asian. All study participants provided written informed consent in accordance with the Declaration of Helsinki.
Human saliva collection
APECED patients and healthy volunteers abstained from eating or drinking anything other than water for 90 min prior to saliva collection. Over a 5-min time period, APECED patients or healthy volunteers spit into a 50-ml conical tube. After collection, the saliva was mixed 1:1 with a protease inhibitor cocktail (Roche) in 1× PBS, according to the manufacturer’s instructions. Saliva was snap-frozen on dry ice and stored at −80°C for analysis.
Determination of concentrations of chemokines and antimicrobial peptides in human saliva
Duoset ELISA kits (R&D) were used to determine the concentration of CXCL9, CXCL10, S100A8, and S100A9 in human saliva according to the manufacturer’s instructions.
Human mucosal biopsies
Standardized 4-mm-long, 2-mm-wide gingival collar biopsies were obtained under local anesthesia. For all experiments, each biopsy was analyzed separately and not pooled (table S8). APECED patients and healthy donors had no signs of mucosal candidiasis, inflammation, or infection at the time of tissue harvesting.
immunohistochemical evaluations of human oral mucosal tissue
For CD4+ and CD8+ T cell staining of human oral mucosal tissue, immunohistochemical CD4/CD8 double staining was performed on formalin-fixed and paraffin-embedded tissue slides using Roche-Ventana BenchMark ULTRA standard manufacturer protocol. Antigen retrieval was conducted in ULTRA Cell Conditioning (ULTRA CC1) (950-224) buffer for 6 min. The slides were incubated with Ventana prediluted anti-CD4 (SP35; Roche; pre-diluted) rabbit monoclonal primary antibody for 32 min. Signal was detected using ultraView Universal DAB Detection Kit (760-500). The slides were then incubated with Ventana prediluted anti-CD8 (SP57; Roche; pre-diluted) rabbit monoclonal primary antibody for 32 min. Signal was detected using ultraView Universal Alkaline Phosphatase Red Detection Kit (760-501). Hematoxylin was used to counterstain.
For phospho-STAT1 immunohistochemical staining of human oral mucosal tissue, formalin-fixed and paraffin-embedded tissue slides were processed using a standard labeled strep-tavidin biotin protocol. Antigen retrieval was conducted in Agilent Citrate pH 6.1 (RTU) (51699) buffer at 85°C for 20 min. The slides were incubated with pSTAT1 (Tyr701) (58D6; Cell Signaling; 1:200) rabbit monoclonal primary antibody overnight. Signal was detected using DAB from Sigma (D5905). Hematoxylin was used to counterstain.
Gene expression in human oral mucosal tissue
Total cellular RNA was isolated from human oral mucosal tissues using Trizol (Life Technologies). For real-time PCR, 1 μg of total RNA was reverse-transcribed using oligodeoxythy-midylic acid primers (Invitrogen) and cDNA was amplified by PCR using the 7500 Real-Time PCR system (Thermo Fisher Scientific). Amplification was performed using the TaqMan expression assay for IL17A (Hs00174383_m1), IL17F (Hs00369400_m1), IL22 (Hs00220924_ m1), S100A9 (Hs00610058_m1), and HPRT (Hs99999909_m1) as a normalization control (Thermo Fisher Scientific). Data were examined by the 2-ΔCt method and results shown as relative expression.
Single-cell suspensions from human oral mucosal tissues
Human oral mucosal gingival tissues were minced and digested for 50 min at 37°C with collagenase IV (Gibco) and DNase (Sigma). A single-cell suspension was then generated by mashing digested samples through a 70-μm filter (Falcon, Corning), as described (70).
Flow cytometry of human oral mucosal tissue samples
Single-cell suspensions from oral mucosal gingival tissues were incubated with mouse serum (Jackson ImmunoResearch Lab) and fluorochrome-conjugated antibodies to surface markers in PBS with 2.5% FBS, for 20 min at 4°C in the dark, and then washed. Dead cells were excluded with Live/Dead fixable dye (Molecular Probes by Life Technologies). Anti-human antibodies for IL-17A (clone eBio64-DEC17; 1:20), CD3 (clone SK7; 1:20), TCRγd (clone B1.1; 1:20), and CD8α (clone RPA-T8; 1:20) were purchased from Thermo Fisher Scientific-eBioscience. Anti-human CD4 (clone OKT4; 1:20), CD19 (clone HIB19; 1:20), CD56 (clone HCD56; 1:20), and CD45 (clone H130; 1:50) antibodies were purchased from Biolegend, and anti-human antibody for IFN-γ (clone 45-15; 1:10) was obtained from Miltenyi Biotec. For examination of IL-17A and IFN-γ production, human gingival cells were stimulated with PMA (50 ng/ml; Sigma-Aldrich) and ionomycin (5 μg/ml; Sigma-Aldrich) in the presence of GolgiPlug (Brefeldin A; BD Biosciences). After 3.5 to 4 hours, cells were stained for surface markers, then washed and fixed in a solution of 2% paraformaldehyde (Electron Microscopy Sciences). Cells were stained intracellularly with fluorochrome-conjugated antibodies against IL-17A and IFN-γ for 40 min in BD Perm/Wash buffer (BD Bioscience). Innate lymphoid cells were defined as live single SSCloCD45+CD3− CD19−CD56− cells. Cell acquisition was performed on a BD LSRFortessa machine using FACSDiVa software (BD Biosciences). Data were analyzed with FlowJo software (TreeStar).
RNA-seq of human oral mucosal tissue
Oral mucosal biopsies were obtained from healthy donors (N = 4) and APECED patients (N = 5) as described above. APECED patients and healthy donors had no signs of mucosal candidiasis, inflammation, or infection at the time of tissue harvesting. Oral tissue was homogenized using Miltenyi Gentlemacs M tube (cat# 130-093-236). Total RNA was extracted from whole tissue using Trizol Reagent using the manufacturer’s protocol (Thermo Fisher). Sequencing libraries were prepared using the Truseq stranded total RNA kit. The multiplexed libraries were sequenced in a NextSeq500 instrument in 150-bp paired-end mode. Demultiplexed samples were mapped and transcripts quantified to GRCh38.v25 genome using the STAR v2.5.2 aligner. Gene-level counts were filtered to remove low-expression genes (keeping genes with >5 counts in at least one sample). Differential gene expression was evaluated using edgeR. Differentially expressed genes were defined as genes with a fold change of at least ±2 in either direction at false discovery rate (FDR) less than 0.05. Enrichment of hallmark genes in differentially expressed genes of APECED patients versus healthy donors was determined using MSigDB (71, 72). Upstream regulators of differentially expressed genes were identified using IPA (Qiagen). IL-17 receptor–induced and repressed genes were previously determined in a mouse model of oral candidiasis (19). Enrichment of IL-17 receptor–induced and repressed genes, previously defined in (19), was determined using gene set enrichment analysis (GSEA) (72). Heat maps were drawn using Morpheus (Broad Institute).
Assessment of viability and barrier integrity in human oral epithelial cells
The OKF6/TERT-2 human oral epithelial cell line (provided by J. Rheinwald, Dana-Farber/Harvard Cancer Center, Boston, MA) was grown as described (73). OKF6/TERT-2 cells have been authenticated by RNA-seq (18) and have been tested for mycoplasma contamination. Recombinant IFN-γ (PeproTech) was reconstituted in Dulbecco’s PBS containing 0.1% bovine serum albumin (BSA) (Sigma). To determine the effects of prolonged IFN-γ exposure in human oral epithelial cells, the cells were incubated with 10, 100, or 1000 U of IFN-γ for 72 hours or incubated without IFN-γ as control. To correct for IFN-γ–induced epithelial cell death, control untreated and 10 U of IFN-γ–treated OKF6/TERT-2 cells were seeded at 4 × 104/ml, 100 U of IFN-γ–treated OKF6/TERT-2 cells were seeded at 6 × 104/ml, and 1000 U of IFN-γ–treated OKF6/TERT-2 cells were seeded at 8 × 104/ml for cells. All experiments were performed at >98% confluency.
The extent of host cell damage caused by IFN-γ in the absence or presence of C. albicans was measured using the LDH release assay. Briefly, OKF6/TERT-2 cells were grown to confluence in 96-well tissue culture plates in the presence or absence of IFN-γ (10, 100, or 1000 U), and then infected with 4 × 105 cells of the C. albicans strain SC5314. Uninfected epithelial cells were processed in parallel as a negative control. After an 8-hour incubation, the amount of LDH that was released into the culture medium was quantified using the CytoTox 96 assay (Promega). Each experiment was performed three independent times in triplicate.
To determine the effects of prolonged IFN-γ exposure on human oral epithelial permeability we performed an In Vitro Permeability Assay (ECM644, Millipore). In brief, OKF6/TERT-2 cells were seeded in cell culture inserts and treated with IFN-γ (10, 100, or 1000 U) for 72 hours. After treatment, FITC-dextran (1:20 in growth media) was added on top of the confluent monolayer. The extent of permeability was determined after 1 hour by measuring the fluorescence of the receiver plate well solution with a BioTek Synergy 2 Multi-Mode Microplate Reader. Each experiment was performed three independent times in duplicate.
OKF6/TERT-2 cells in 24-well tissue culture plates were incubated for 72 hours with different concentrations of IFN-γ (10, 100, or 1000 U). Next, the cells were rinsed with cold HBSS containing protease and phosphatase inhibitors and removed from the plate with a cell scraper. After collecting the cells by centrifugation, they were boiled in sample buffer. The lysates were separated by SDS-PAGE, and the proteins were detected by immunoblotting with specific antibodies, including anti–claudin-1 (D5H1D; Cell Signaling; 0.6 μg/ml) and anti-GAPDH (D16H11; Cell Signaling; 0.2 μg/ml). The blots were developed using enhanced chemiluminescence and imaged with the C400 digital imager (Azure Biosystems). Each experiment was performed three independent times.
The localization of claudin-1 was visualized by confocal microscopy using a slight modification of our previously described method (74). Oral epithelial OKF6/TERT-2 cells were incubated with 1000 U of IFN-γ for 72 hours. The cells were fixed in 3% paraformaldehyde (wt/vol), blocked with 10% BSA (vol/vol), and incubated with antibodies to claudin-1 (D5H1D; Cell Signaling; 0.1 μg/ml) followed by an AlexaFluor 488-labeled goat anti-rabbit antibody (cat# A-11008; Thermo Fisher, 1 μg/ml). Actin was visualized using ActinRed 555 (Thermo Fisher, 3 U/ml) and nuclei using DAPI. The cells were then imaged with the Leica TCS SP8 confocal system, and the final images were generated by stacking optical sections along the z axis. To enable comparison of fluorescence intensity between slides, the same image acquisition settings were used for each experiment. Each experiment was performed three independent times.
Statistical analyses
To determine statistical significance, unpaired t tests (with or without Welch’s correction, as appropriate), Mann-Whitney U tests, one-way analysis of variance (ANOVA) with Newman-Keuls or Holm-Šidák or Tukey’s or Dunnett’s post hoc multiple-comparison test, two-way ANOVA test, or Kruskal-Wallis H test with Dunn’s multiple-comparisons test were used, as appropriate, using GraphPad Prism 8 software. Quantitative data are presented as means ± SEM, and a P value of ≤0.05 was considered significant.
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Cohen, T. Hawley, T. Moyer, and I. Douagi (NIAID Research Technologies Branch, Flow Cytometry Section) for assistance with FACS sorting; D. Trageser-Cesler, C. Acevedo, and E. Lewis (NIAID Microbiome Program Gnotobiotic Animal Facility) for assistance with generation of germ-free wild-type and Aire−/− mice; and W. Yuan, R. R. Rodrigues, and V. Thovarai (Frederick National Laboratory for Cancer Research and Microbiome and Genetics Core, Laboratory of Integrative Cancer Immunology, Center for Cancer Research, NCI) for assistance with assistance with 16S rRNA sequencing, data analysis, and data deposition, respectively. This work used the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov). We thank the NIAID animal facility staff.
Funding: Supported by the NIAID Division of Intramural Research (M.S.L., Y.B., S.M.H., P.M.M., H.D.H., L.D.N., D.L.B., and I.A.M.); the NIDCR Division of Intramural Research (N.M.M.); the NCI Division of Intramural Research (S.P. and C.-C.R.L.); the NIDDK Division of Intramural Research (B.A.); NIDCD Division of Intramural Research grant ZIC DC000086 to the Genomics and Computational Biology Core; NIH grants R01DE022600 and R01AI124566 (S.G.F.); NIH grant R00DE026856 (Ma.S.); Deutsche Forschungsgemeinschaft research fellowship FR 3851/2-1 (T.F.); Juan Rodes program grant JR 18/00042 (P.O.); Instituto de Salud Carlos III, Madrid; Consejería de Salud y Familias de Andalucia grant EF-0038-2016; Estancias Formativas de Larga Duración; and the Asociación de Pediatría (Beca para Estancias de Larga Duración), Madrid.
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability: Anti–IL-22 (clone 8E-11) is available under a material agreement with Genentech. The mouse and human RNA-seq datasets are available from the NCBI’s GEO accession GSE133603. Raw sequence data from all 16S sequencing experiments are deposited in the NCBI Sequence Read Archive under BioProject accession number PRJNA640597. All other data needed to evaluate the conclusions in this paper are present either in the main text or the supplementary materials.
SUPPLEMENTARY MATERIALS
science.sciencemag.org/content/371/6526/eaay5731/suppl/DC1
List of Genomics and Computational Biology Core Members MDAR Reproducibility Checklist View/request a protocol for this paper from Bio-protocol.
REFERENCES AND NOTES
- 1.Puel A et al. , Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 332, 65–68 (2011). doi: 10.1126/science.1200439; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lévy R et al. , Genetic, immunological, and clinical features of patients with bacterial and fungal infections due to inherited IL-17RA deficiency. Proc. Natl. Acad. Sci. U.S.A 113, E8277–E8285 (2016). doi: 10.1073/pnas.1618300114; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boisson B et al. , An ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity 39, 676–686 (2013). doi: 10.1016/j.immuni.2013.09.002; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ling Y et al. , Inherited IL-17RC deficiency in patients with chronic mucocutaneous candidiasis. J. Exp. Med. 212, 619–631 (2015). doi: 10.1084/jem.20141065; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lionakis MS, Levitz SM, Host Control of Fungal Infections: Lessons from Basic Studies and Human Cohorts. Annu. Rev. Immunol. 36, 157–191 (2018). doi: 10.1146/annurev-immunol-042617-053318; [DOI] [PubMed] [Google Scholar]
- 6.Anderson MS et al. , Projection of an immunological self shadow within the thymus by the aire protein. Science 298, 1395–1401 (2002). doi: 10.1126/science.1075958; [DOI] [PubMed] [Google Scholar]
- 7.Constantine GM, Lionakis MS, Lessons from primary immunodeficiencies: Autoimmune regulator and autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Immunol. Rev. 287, 103–120 (2019). doi: 10.1111/imr.12714; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Puel A et al. , Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J. Exp. Med. 207, 291–297 (2010). doi: 10.1084/jem.20091983; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kisand K et al. , Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J. Exp. Med. 207, 299–308 (2010). doi: 10.1084/jem.20091669; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferre EM et al. , Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. JCI Insight 1, e88782 (2016). doi: 10.1172/jci.insight.88782; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Solis NV, Filler SG, Mouse model of oropharyngeal candidiasis. Nat. Protoc. 7, 637–642 (2012). doi: 10.1038/nprot.2012.011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hubert FX et al. , Aire-deficient C57BL/6 mice mimicking the common human 13-base pair deletion mutation present with only a mild autoimmune phenotype. J. Immunol. 182, 3902–3918 (2009). doi: 10.4049/jimmunol.0802124; [DOI] [PubMed] [Google Scholar]
- 13.Jiang W, Anderson MS, Bronson R, Mathis D, Benoist C, Modifier loci condition autoimmunity provoked by Aire deficiency. J. Exp. Med. 202, 805–815 (2005). doi: 10.1084/jem.20050693; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cho JS et al. , IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J. Clin. Invest. 120, 1762–1773 (2010). doi: 10.1172/JCI40891; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeVoss JJ et al. , Effector mechanisms of the autoimmune syndrome in the murine model of autoimmune polyglandular syndrome type 1. J. Immunol. 181, 4072–4079 (2008). doi: 10.4049/jimmunol.181.6.4072; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gavanescu I, Benoist C, Mathis D, B cells are required for Aire-deficient mice to develop multi-organ autoinflammation: A therapeutic approach for APECED patients. Proc. Natl. Acad. Sci. U.S.A. 105, 13009–13014 (2008). doi: 10.1073/pnas.0806874105; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Niki S et al. , Alteration of intra-pancreatic target-organ specificity by abrogation of Aire in NOD mice. J. Clin. Invest. 116, 1292–1301 (2006). doi: 10.1172/JCI26971; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conti HR et al. , IL-17 Receptor Signaling in Oral Epithelial Cells Is Critical for Protection against Oropharyngeal Candidiasis. Cell Host Microbe 20, 606–617 (2016). doi: 10.1016/j.chom.2016.10.001; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conti HR et al. , Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J. Exp. Med. 206, 299–311 (2009). doi: 10.1084/jem.20081463; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirota K et al. , Fate mapping of IL-17-producing T cells in inflammatory responses. Nat. Immunol. 12, 255–263 (2011). doi: 10.1038/ni.1993; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trautwein-Weidner K, Gladiator A, Nur S, Diethelm P, LeibundGut-Landmann S, IL-17-mediated antifungal defense in the oral mucosa is independent of neutrophils. Mucosal Immunol. 8, 221–231 (2015). doi: 10.1038/mi.2014.57; [DOI] [PubMed] [Google Scholar]
- 22.Krueger JG et al. , IL-17A is essential for cell activation and inflammatory gene circuits in subjects with psoriasis. J. Allergy Clin. Immunol. 130, 145–54.e9 (2012). doi: 10.1016/j.jaci.2012.04.024; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krueger JG et al. , IL-17A inhibition by secukinumab induces early clinical, histopathologic, and molecular resolution of psoriasis. J. Allergy Clin. Immunol. 144, 750–763 (2019). doi: 10.1016/j.jaci.2019.04.029; [DOI] [PubMed] [Google Scholar]
- 24.Huppler AR et al. , Role of neutrophils in IL-17-dependent immunity to mucosal candidiasis. J. Immunol. 192, 1745–1752 (2014). doi: 10.4049/jimmunol.1302265; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aggor FEY et al. , Oral epithelial IL-22/STAT3 signaling licenses IL-17-mediated immunity to oral mucosal candidiasis. Sci. Immunol. 5, eaba0570 (2020). doi: 10.1126/sciimmunol.aba0570; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gray DH, Gavanescu I, Benoist C, Mathis D, Danger-free autoimmune disease in Aire-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 104, 18193–18198 (2007). doi: 10.1073/pnas.0709160104; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nava P et al. , Interferon-γ regulates intestinal epithelial homeostasis through converging beta-catenin signaling pathways. Immunity 32, 392–402 (2010). doi: 10.1016/j.immuni.2010.03.001; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bruewer M et al. , Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J. Immunol. 171, 6164–6172 (2003). doi: 10.4049/jimmunol.171.11.6164; [DOI] [PubMed] [Google Scholar]
- 29.Niehues H et al. , STAT1 gain-of-function compromises skin host defense in the context of IFN-γ signaling. J. Allergy Clin. Immunol. 143, 1626–1629.e5 (2019). doi: 10.1016/j.jaci.2018.11.033; [DOI] [PubMed] [Google Scholar]
- 30.Farah CS, Hu Y, Riminton S, Ashman RB, Distinct roles for interleukin-12p40 and tumour necrosis factor in resistance to oral candidiasis defined by gene-targeting. Oral Microbiol. Immunol. 21, 252–255 (2006). doi: 10.1111/j.1399-302X.2006.00288.x; [DOI] [PubMed] [Google Scholar]
- 31.Al-Salama ZT, Emapalumab: First Global Approval. Drugs 79, 99–103 (2019). doi: 10.1007/s40265-018-1046-8; [DOI] [PubMed] [Google Scholar]
- 32.Verstovsek S et al. , A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 366, 799–807 (2012). doi: 10.1056/NEJMoa1110557; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kerner G et al. , Inherited human IFN-γ deficiency underlies mycobacterial disease. J. Clin. Invest. 130, 3158–3171 (2020). doi: 10.1172/JCI135460; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu UI, Holland SM, Host susceptibility to non-tuberculous mycobacterial infections. Lancet Infect. Dis. 15, 968–980 (2015). doi: 10.1016/S1473-3099(15)00089-4; [DOI] [PubMed] [Google Scholar]
- 35.Broggi A et al. , Type III interferons disrupt the lung epithelial barrier upon viral recognition. Science 369, 706–712 (2020). doi: 10.1126/science.abc3545; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Major J et al. , Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science 369, 712–717 (2020). doi: 10.1126/science.abc2061; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu L et al. , Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J. Exp. Med. 208,1635–1648 (2011). doi: 10.1084/jem.20110958; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van de Veerdonk FL et al. , STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N. Engl. J. Med. 365, 54–61 (2011). doi: 10.1056/NEJMoa1100102; [DOI] [PubMed] [Google Scholar]
- 39.Higgins E et al. , Use of ruxolitinib to successfully treat chronic mucocutaneous candidiasis caused by gain-of-function signal transducer and activator of transcription 1 (STAT1) mutation. J. Allergy Clin. Immunol. 135, 551–553 (2015). doi: 10.1016/j.jaci.2014.12.1867; [DOI] [PubMed] [Google Scholar]
- 40.Kong XF et al. , Three Copies of Four Interferon Receptor Genes Underlie a Mild Type I Interferonopathy in Down Syndrome. J. Clin. Immunol. 40, 807–819 (2020). doi: 10.1007/s10875-020-00803-9; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Del Bel KL et al. , JAK1 gain-of-function causes an autosomal dominant immune dysregulatory and hypereosinophilic syndrome. J. Allergy Clin. Immunol. 139, 2016–2020.e5 (2017). doi: 10.1016/j.jaci.2016.12.957; [DOI] [PubMed] [Google Scholar]
- 42.Gruber CN et al. , Complex Autoinflammatory Syndrome Unveils Fundamental Principles of JAK1 Kinase Transcriptional and Biochemical Function. Immunity 53, 672–684.e11 (2020). doi: 10.1016/j.immuni.2020.07.006; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uggenti C, Lepelley A, Crow YJ, Self-Awareness: Nucleic Acid-Driven Inflammation and the Type I Interferonopathies. Annu. Rev. Immunol. 37, 247–267 (2019). doi: 10.1146/annurev-immunol-042718-041257; [DOI] [PubMed] [Google Scholar]
- 44.de Jesus AA, Canna SW, Liu Y, Goldbach-Mansky R, Molecular mechanisms in genetically defined autoinflammatory diseases: Disorders of amplified danger signaling. Annu. Rev. Immunol. 33, 823–874 (2015). doi: 10.1146/annurev-immunol-032414-112227; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Break TJ et al. , VT-1598 inhibits the in vitro growth of mucosal Candida strains and protects against fluconazole-susceptible and -resistant oral candidiasis in IL-17 signalling-deficient mice. J. Antimicrob. Chemother. 73, 2089–2094 (2018). doi: 10.1093/jac/dky170; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Break TJ et al. , VT-1161 protects mice against oropharyngeal candidiasis caused by fluconazole-susceptible and -resistant Candida albicans. J. Antimicrob. Chemother. 73, 151–155 (2018). doi: 10.1093/jac/dkx352; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Break TJ et al. , CX3CR1 is dispensable for control of mucosal Candida albicans infections in mice and humans. Infect. Immun. 83, 958–965 (2015). doi: 10.1128/IAI.02604-14; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jordan S et al. , Virus progeny of murine cytomegalovirus bacterial artificial chromosome pSM3fr show reduced growth in salivary Glands due to a fixed mutation of MCK-2. J. Virol. 85, 10346–10353 (2011). doi: 10.1128/JVI.00545-11; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zurbach KA, Moghbeli T, Snyder CM, Resolving the titer of murine cytomegalovirus by plaque assay using the M2-10B4 cell line and a low viscosity overlay. Virol. J. 11, 71 (2014). doi: 10.1186/1743-422X-11-71; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wheat RL, Clark PY, Brown MG, Quantitative measurement of infectious murine cytomegalovirus genomes in real-time PCR. J. Virol. Methods 112, 107–113 (2003). doi: 10.1016/S0166-0934(03)00197-6; [DOI] [PubMed] [Google Scholar]
- 51.Myles IA et al. , TNF overproduction impairs epithelial staphylococcal response in hyper IgE syndrome. J. Clin. Invest. 128, 3595–3604 (2018). doi: 10.1172/JCI121486; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bouladoux N, Harrison OJ, Belkaid Y, The Mouse Model of Infection with Citrobacter rodentium. Curr. Protoc. Immunol 119, 19 15 11-19 15 25 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abe T, Hajishengallis G, Optimization of the ligature-induced periodontitis model in mice. J. Immunol. Methods 394, 49–54 (2013). doi: 10.1016/j.jim.2013.05.002; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Williams DW et al. , Indigenous Microbiota Protects against Inflammation-Induced Osteonecrosis. J. Dent. Res. 99, 676–684 (2020). doi: 10.1177/0022034520908594; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Whibley N et al. , Antibody blockade of IL-17 family cytokines in immunity to acute murine oral mucosal candidiasis. J. Leukoc. Biol. 99, 1153–1164 (2016). doi: 10.1189/jlb.4A0915-428R; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kruisbeek AM, In vivo depletion of CD4- and CD8-specific T cells. Curr. Protoc. Immunol Chapter 4, Unit 4 1 (2001). [DOI] [PubMed] [Google Scholar]
- 57.Vanpouille-Box C et al. , TGFβ Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity. Cancer Res. 75, 2232–2242 (2015). doi: 10.1158/0008-5472.CAN-14-3511; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ding L et al. , Determination of human anticytokine autoantibody profiles using a particle-based approach. J. Clin. Immunol. 32, 238–245 (2012). doi: 10.1007/s10875-011-9621-8; [DOI] [PubMed] [Google Scholar]
- 59.Dutzan N et al. , A dysbiotic microbiome triggers TH17 cells to mediate oral mucosal immunopathology in mice and humans. Sci. Transl. Med. 10, eaat0797 (2018). doi: 10.1126/scitranslmed.aat0797; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bae HR et al. , Multi-omics: Differential expression of IFN-γ results in distinctive mechanistic features linking chronic inflammation, gut dysbiosis, and autoimmune diseases. J. Autoimmun. 111, 102436 (2020). doi: 10.1016/j.jaut.2020.102436; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Callahan BJ et al. , DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016). doi: 10.1038/nmeth.3869; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bolyen E et al. , Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 37, 852–857 (2019). doi: 10.1038/s41587-019-0209-9; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Quast C et al. , The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013). doi: 10.1093/nar/gks1219; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hill TC, Walsh KA, Harris JA, Moffett BF, Using ecological diversity measures with bacterial communities. FEMS Microbiol. Ecol. 43, 1–11 (2003). doi: 10.1111/j.1574-6941.2003.tb01040.x; [DOI] [PubMed] [Google Scholar]
- 65.Vázquez-Baeza Y, Pirrung M, Gonzalez A, Knight R, EMPeror: A tool for visualizing high-throughput microbial community data. Gigascience 2, 16 (2013). doi: 10.1186/2047-217X-2-16; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Segata N et al. , Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60 (2011). doi: 10.1186/gb-2011-12-6-r60; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schindelin J et al. , Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). doi: 10.1038/nmeth.2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Trautwein-Weidner K et al. , Antigen-Specific Th17 Cells Are Primed by Distinct and Complementary Dendritic Cell Subsets in Oropharyngeal Candidiasis. PLOS Pathog. 11, e1005164 (2015). doi: 10.1371/journal.ppat.1005164; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sparber F et al. , Langerin+ DCs regulate innate IL-17 production in the oral mucosa during Candida albicans-mediated infection. PLOS Pathog. 14, e1007069 (2018). doi: 10.1371/journal.ppat.1007069; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dutzan N, Abusleme L, Konkel JE, Moutsopoulos NM, Isolation, Characterization and Functional Examination of the Gingival Immune Cell Network. J. Vis. Exp. 53736 (2016). doi: 10.3791/53736; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liberzon A et al. , The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015). doi: 10.1016/j.cels.2015.12.004; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Subramanian A et al. , Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). doi: 10.1073/pnas.0506580102; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Swidergall M, Solis NV, Lionakis MS, Filler SG, EphA2 is an epithelial cell pattern recognition receptor for fungal β-glucans. Nat. Microbiol. 3, 53–61 (2018). doi: 10.1038/s41564-017-0059-5; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Solis NV, Swidergall M, Bruno VM, Gaffen SL, Filler SG, The Aryl Hydrocarbon Receptor Governs Epithelial Cell Invasion during Oropharyngeal Candidiasis. mBio 8, e00025–17 (2017). doi: 10.1128/mBio.00025-17; [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.