

Abstract
Objectives:
The highly heterogeneous clinical presentation of lupus is characterized by the unpredictable appearance of flares of disease activity and important organ damage. Attempts to stratify lupus patients have been limited to clinical information, leading to unsuccessful clinical trials and controversial research results. Our aim was to develop and validate a robust method to stratify patients with lupus according to longitudinal disease activity and whole-genome gene expression data in order to establish subgroups of patients who share disease progression mechanisms.
Methods:
We applied a clustering-based approach to stratify SLE patients based on the correlation between disease activity scores and longitudinal gene expression information. Clustering robustness was evaluated by bootstrapping and the clusters were characterized in terms of clinical and functional features.
Results:
Using two independent sets of patients, one pediatric and another adult, our results show a clear partition into three different disease clusters not influenced by treatment, race or other source of bias. Two of the clusters differentiate into a neutrophil correlated disease group and a lymphocyte correlated disease group, while the third that correlated to a lesser extent with neutrophils, was functionally more heterogeneous. The neutrophil-driven clusters were associated with increased development towards proliferative nephritis.
Conclusions:
We found three subgroups of patients that show different mechanisms of disease progression and are clinically differentiated. Our results have important implications for treatment options, the design of clinical trials, the etiology of the disease, and the prediction of severe glomerulonephritis.
Keywords: Systemic lupus erythematosus, stratification, clustering, gene expression, activity scores, longitudinal
Introduction
SLE disease activity varies unpredictably over time and this variation is heterogeneous between patients and within patients. Patients go through periods of flaring, with both disease activity and treatment with corticosteroids resulting in organ damage and premature death (1). The Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) is the most used scoring system for disease activity(2). However, patients with similar disease activity by SLEDAI may have different prognosis and treatment responses (3). Therefore, there is urgent need to establish new lupus patient stratification. The clinical heterogeneity of lupus manifests itself also in the diversity of abnormalities that have been described at cellular, serological, and other levels (4). Not all patients share the same abnormalities suggesting that specific pathways leading to active disease are different from patient to patient.
In gene expression studies, clustering analyses have been broadly used to discover sets of samples that share similarities in their gene expression programs. These have resulted in successful results establishing new disease classifications in cancer (5,6).
Nevertheless, most patient stratification algorithms are designed to cluster samples from independent measurements. Several clustering algorithms that deal with longitudinal data or time series are mainly used for defining gene clusters. For example, TSclust R package (7) implements 20 different metrics and approaches to time-series clustering. But unlike most diseases, autoimmune diseases have no known progression patterns over time and hence, defined stages of disease cannot be established. So, we cannot assume a similar time-dependent modulation for different patients. Therefore, the classical methods of patient stratification based on disease progression over time are not valid.
Using two independent datasets, a previously reported and publicly available dataset of pediatric lupus patients (8) and a new adult dataset that we generated with the follow-up of the cohort reported in (9), we established three groups of patients from gene expression profiles correlated with disease activity progression in time. The clusters we report are robust and highly reproducible in both datasets differentiated by patterns of lymphocyte and neutrophil composition that occur with disease activity, the progression to proliferative nephritis, and presence of other clinical manifestations.
Materials and Methods
Population cohorts and design
We used two independent sets of SLE patients. As training set we used the public longitudinal data from Banchereau, et al (8). That study contained a unique set of pediatric SLE patients traced over time. Clinical variables and genome-wide gene expression levels were measured at different time points for every patient. Gene expression data was downloaded from NCBI GEO (ID GSE65391). Patients with less than three visits and whose SLEDAI magnitude does not change with time were discarded from analysis. A total of 80 patients were selected for further analysis, each one with a variable number of visits, continuous and categorical clinical variables, and gene expression data.
An independent dataset was generated from adult patients obtained from the SPARE (Study of biological Pathways, disease Activity and Response markers in patients with systemic lupus Erythematosus) (9) study protocol approved by Johns Hopkins University School of Medicine Institutional Review Board. Patients were enrolled from the Hopkins Lupus Cohort following informed consent. Adult patients were eligible if aged 18–75 years old and met the definition of SLE by the revised American College of Rheumatology classification criteria from 1997. At entry (baseline), patient’s medical history was reviewed, and information on current medications recorded. Visits were scheduled quarterly or more often if required for disease activity over a 2-year period. All patients were evaluated at entry and at all subsequent cohort visits (MP) by the same physician. A total of 306 SLE patients were enrolled. The demographics were 58.9% Caucasian, 33.9% African–American, 91.1% female, mean age 46.0±11.9 years. The number of visits per patient the following year ranged from 1 to 9. Six patients had 1 visit, 46 had 2–3 visits, 159 had 4 visits, and 81 had more than 4 visits.
Patients were treated according to standard clinical practice. To assess disease activity, the Safety of Estrogens in Lupus Erythematosus: National Assessment (SELENA) version of the SLEDAI as well as physician global assessment (PGA), were completed at each visit. C3, C4, anti-dsDNA (Crithidia), complete blood cell count and urinalysis were performed at every visit. Affymetrix GeneChip HT HG-U133+ arrays were used to measure gene expression profiles. The experimental protocol and gene expression data processing methods were reported (9).
Sixty-five adult patients with over three visits varying in their SLEDAI values were selected for analysis. We used this novel, unpublished longitudinal dataset to validate the results obtained with the pediatric dataset.
Processing of the data
Each set was processed independently. Transcripts with flat expression profiles and standard deviation below 0.1 across samples were removed. Retained transcripts were annotated to a gene symbol. Duplicated genes were merged assigning them their mean expression value. This yielded 15344 genes from743 samples (80 pediatric patients) as the final gene expression training dataset and 20741 genes from 288 samples (65 adult patients) as replication set. Healthy samples were left out from stratification analysis.
Stratification method
Because SLE is a disease with unknown progression, we cannot assume a temporal relationship between patients. For each patient we had gene expression data across a sparse and asynchronous number of visits (ranging from 3 to 22), with an associated SLEDAI score and clinical variables measured at each visit. Therefore, to stratify patients based on similarities in gene expression profiles and global disease progression, rather than to cluster individual time points, we calculated a gene by patient correlation matrix computing a stringent Pearson correlation coefficient between gene expression data and SLEDAI scores across each patients’ visits (Table S1). Correlation values summarize behavior of expression of each gene in each patient in relation with disease activity, with positive and negative correlation values. Genes with highest absolute correlation values across samples were selected by applying the Rank Sum method (10). Briefly, genes were ranked by absolute correlation values and the sum of ranks from all patients was computed obtaining a unique score value per gene. Finally, gene scores were sorted and we calculated an empirical p-value for each gene against the probability to obtain genes with higher scores by chance. For this we randomly created 1000 bi-dimensional inter-patient matrices by altering rows and columns and comparing the new score obtained for each gene with their original score. The cut-off p-value corrected by false discovery rate was <0.05.With this approach, the possible alterations in gene expression in individual patients are not included thus selecting only genes that are highly correlated with SLEDAI. In this way the differential effect of treatment on gene expression would not influence stratification.
To scale the correlation values these were normalized for each patient from 1 to −1 as maximum and minimum values, respectively. This normalization removes the effect of the different visit numbers for different patients, as the probability to obtain higher correlations is increased when fewer points are compared. The process is summarized in Figure 1A.
Figure 1. Summary of the clustering process.
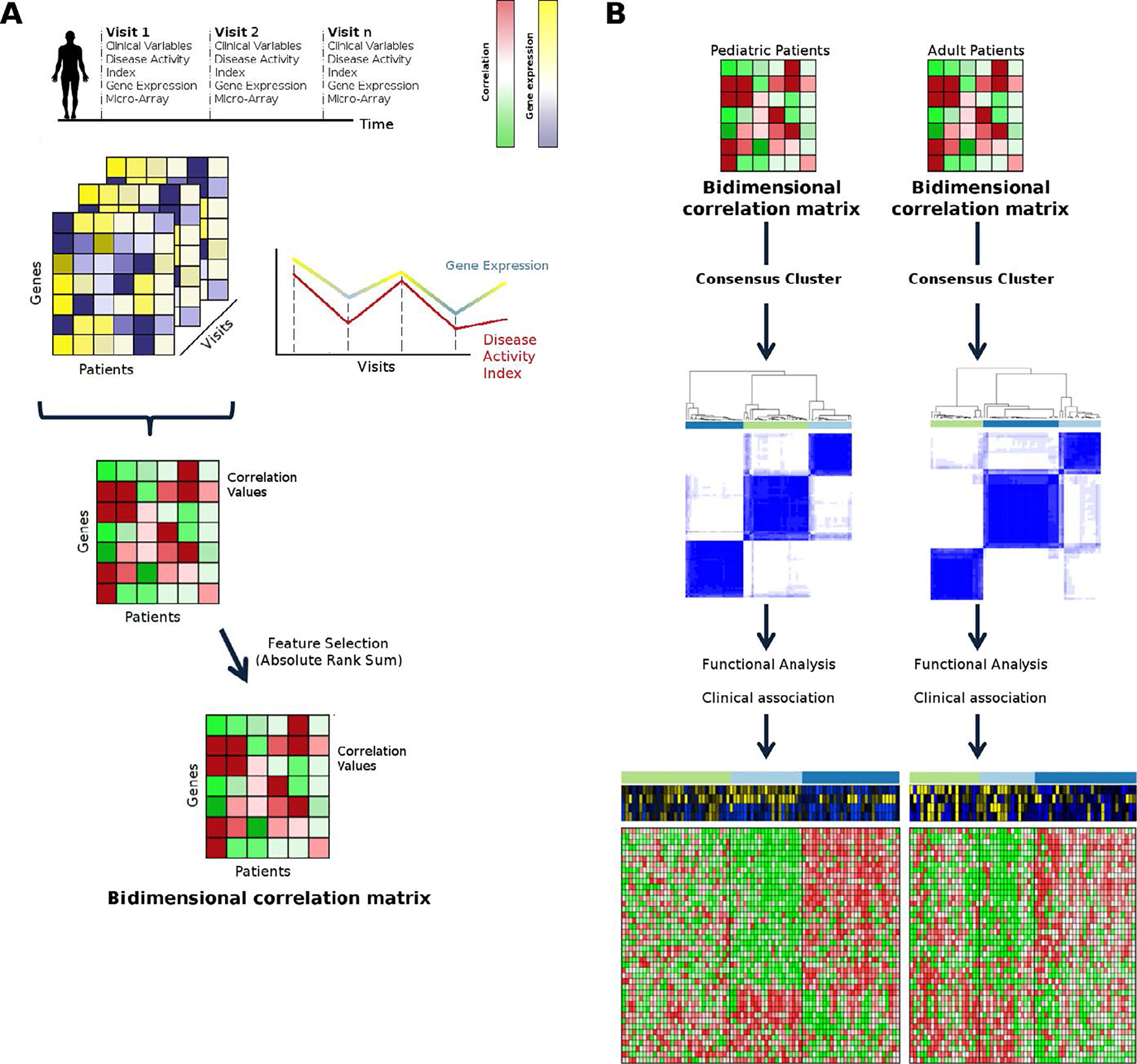
A) Obtaining the bi-dimensional correlation matrix. We part from a dataset with genes in rows, patients in columns and different visitsfor which we have gene expression and clinical variables for every patient. A correlation value for each gene and each patient is calculated throughout visits and a bi-dimensional matrix of genes, patients and correlation values is created. Feature selection is applied to select the best gene candidates to stratify and filter out the rest of genes. B) We performed consensus clustering on pediatric and adult bi-dimensional correlation matrices independently and then a functional and clinical characterization is performed.
Clustering analysis was performed using the normalized patient correlation matrix gene set. First, Bayesian Criterion Information (BIC) from mclust R package (11) was used to evaluate the optimal number of clusters and k-means based consensus clustering was used to stratify patients based on the optimal number of K. This analysis was performed using the ConsensusClusterPlus R package (12)(Figure 1B).
Cluster stability
Cluster robustness was evaluated by a bootstrap-based approach. Subsets of 75, 62 and 50 percent of samples were randomly selected from the original dataset and clustering analysis repeated. We calculated the stability of the clusters using the Jaccard coefficient by comparing our original clusters with the new clusters obtained in each subset and mean Jaccard coefficient across the cluster permutations. This analysis was performed using clusteval R package. We evaluated the robustness of the feature selection and clustering results with respect to the number of visits. For this, we randomly selected 3, 4 or 5visits for each patient and re-calculated the correlation values. We constructed the bi-dimensional matrix resulting from the genes selected in the original feature selection but with new correlation values for each number of visits, and repeated the clustering procedure. We performed 1000 permutations and estimated the stability with Jaccard.
Functional analysis of the clusters
To characterize the main biological processes associated to each cluster we performed functional enrichment analysis of Gene Ontology terms (http://www.geneontology.org/)in the set of genes selected in the feature selection using enrichR (13). To evaluate broader groups of biological processes GO terms were re-annotated and grouped with terms from the common superior levels of GO hierarchy, and compared correlation values of the genes of each category between the different clusters obtained. This analysis determines the biological functions that are represented by genes increasing or decreasing in their transcription with respect to disease activity in the clusters, allowing us to discern differential biological mechanisms between clusters.
Tests for association of clinical variables
Fisher’s exact test was used to evaluate if categorical variables (gender, race, and treatment) were enriched in a cluster with respect to the others. For continuous variables, we measured their correlation with SLEDAI scores for each patient and ANOVA test was applied to evaluate if clusters were enriched in samples with variables highly correlated with disease activity scores in pair-wise comparisons. With this analysis, we could measure not only the clinical variables that determine disease activity in each cluster, but also those differentially correlated with disease activity between clusters. Parameters measured on pediatric patients were downloaded from http://websle.com along with expression data.
Analysis of Treatment effects
To analyze if our selected stratification genes were modulated by treatment use we correlated gene expression of each patient with treatment doses for acetylsalicylic acid, cytotoxic drugs and prednisone, the treatments for which we had dose information in adults. No data was available for the pediatric set. We performed the rank sum method to select the top 100 positively and 100 negatively most drug-correlated genes for each treatment, that is, genes modified by treatment doses. We compared these three lists of drug-correlated genes with the genes selected for stratification.
Finally, we analyzed if treatments were differentially affecting cell proportions between clusters. We measured the tendency of neutrophil proportions of each patient at the time point when treatment was applied and at the next visit and compared the results between clusters.
Imputation of Cell Proportions
Twelve adult patients had no cell proportions available. Missing neutrophil cell proportions were imputed using CiberSort (14). We used real cell proportions from the pediatric set as control measuring the correlation of real and imputed data. Missing lymphocyte proportions were not imputed because CiberSort has different lymphocyte subtypes obtained through different methods so comparison with whole lymphocyte data is not direct.
Modular Functional Analysis
To assess differences between clusters in terms of biological pathways we defined the set of genes differentially expressed in each cluster using limma [15] selecting those genes with corrected p-values < 0.05. Tmod R package [16] was used to determine functional pathways differentially over and under-represented in each cluster. This analysis was performed separately in samples with low and high SLEDAI categories (scores<3 or >8, respectively).
Results
Gene expression correlation with activity results in three clusters or subgroups of lupus patients
The gene selection process of the pediatric patient set yielded777 significant genes (Table S1). Not unexpectedly, a large number of genes belonged to the type I interferon signature (17, 18).We found k=3as the optimal number of clusters (Figure 2A and 2B). We named the clusters P1 (P: pediatric), with 31, P2, with 21 and P3 with 28 patients (Table S2).
Figure 2. Evaluation of the best number of clusters.
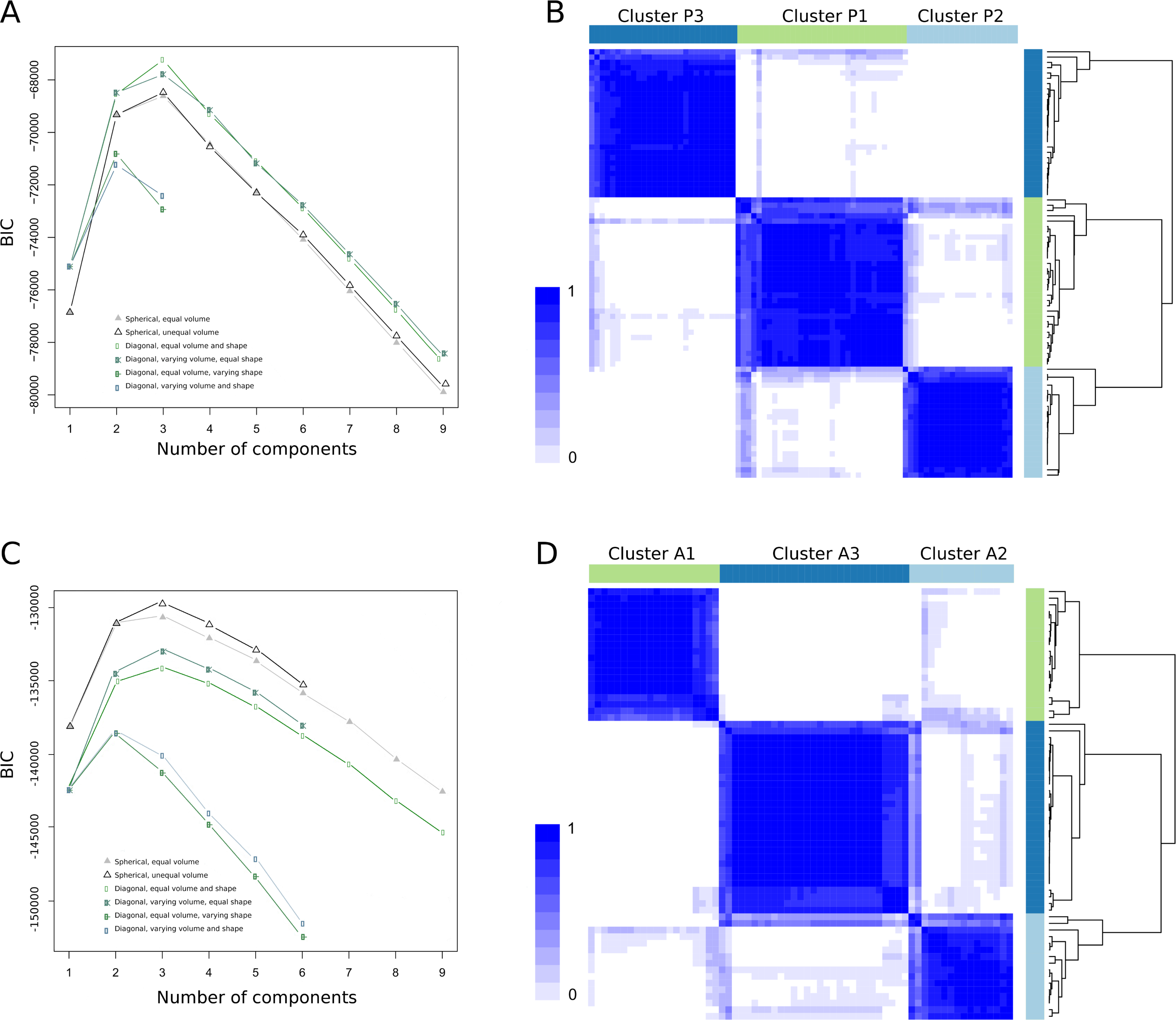
A) The plot shows the BIC values in y-axis and number of clusters in x-axis. The optimal number of clusters was three, value at which we found the highest BIC value. B) Stratification of pediatric patients using ConsensusClusterPlus R package. Rows and columns represent patients in the same order and color intensity represent the probability of that two patients clustering together. C) Estimation of the optimal number of clusters for the adult patient set, resulting in 3as the best number of clusters. D) Stratification of the adult patients.
For the adult set, 1051 significant genes were selected (Table S1). Again, k=3 was obtained as best cluster number (Figure 2C and Figure 2D). The three clusters, A1 (A: adult), A2 and A3 grouped 20, 16, and 29 patients, respectively (Table S2).
The clusters are highly stable and not biased by demography or treatment
Cluster stability measurements are summarized in Table S3.Of the two methods used for cluster validation, only when sample size was halved did bootstrapping give low stability retaining still a high Jaccard coefficient value in the pediatric (0.77), and adult set (0.7). In the feature selection stability test, no individuals were miss-classified, demonstrating high reproducibility of the clusters. These results showed that the clusters are highly stable and the genes selected for stratification are maintained independently of the number of visits (for correlation, at least three visits must be measured).
Table S4 shows results comparing demographic or treatment variables and Table S5 numbers of patients for each variable between clusters. In neither the three pediatric nor the three adult clusters did we observe statistical association with race, gender, or treatment, excluding these as drivers for stratification.
Behavior of numbers and proportions of neutrophils and lymphocytes in time differentiate the clusters
Figure 3A shows continuous variables significantly enriched in the pediatric clusters. As can be noted, there is differential distribution of neutrophil and lymphocyte proportions in the obtained clusters that correlate with disease activity, that is, increase or decrease with SLEDAI. The sharpest differences were observed in proportions of neutrophils and lymphocytes between clusters P2 and P3. The cellular proportion mean was not biased between clusters (Table S6) and SLEDAI values of patients from each cluster were within the same ranges. This means that the percentage of neutrophils increased with activity in clusters P2 and P1, and decreased with activity in P3 (Figures S1A–H). Percentage of lymphocytes showed an opposite trend. Thus, proportions of these cellular populations had a completely different behavior between clusters despite having in average similar disease activity (see below). Other interesting differences between clusters were observed withC3 and C4 complement levels. The correlation values were strongly negative in patients from clusters P2 and P3 and less negatively correlated in P1,having this cluster, enrichment in patients that develop proliferative nephritis(see below). We found also correlation with increased aspartate aminotransferase, a hepatic function enzyme (19) in P3 and a somewhat higher erythrocyte sedimentation rate (ESR) in P1.
Figure 3. Clinical characterization of the clusters.
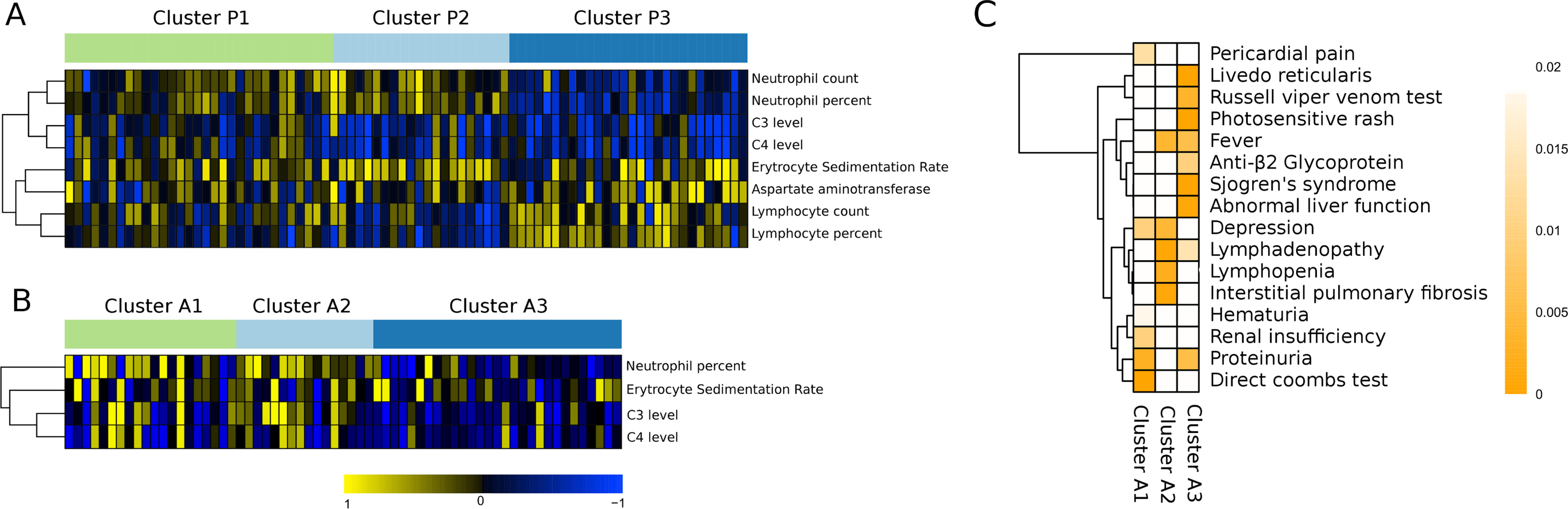
A) Heatmap showing continuous significant clinical variables found in the pediatric set. Columns and rows represent the different patients and clinical variables, respectively. Summarized by the color scale is the correlation between SLEDAI and each clinical variable. The significance was obtained using ANOVA when comparing the correlation values of each cluster with respect to the others. B) Continuous significant variables found in adult set. C) Categorical clinical variables significantly enriched in adult set. The enrichment was calculated by Exact Fisher’s test. Color represents the p-value of the enrichment.
Figure 3B shows the significant continuous clinical variables of the adult set. We used pediatric set as control of the cell proportions imputation for the missing data (Figure S2). Neutrophil proportions decreased in cluster A3 when the SLEDAI increased and increased in clusters A2 and A1, with increased activity, and the lymphocytes go in the opposite direction as in the pediatric clusters. For the 12 patients whose lymphocyte data was missing, the correlation data with SLEDAI was set to zero. C3, C4 and ESR conserved the same pattern in both sets, but differences between clusters A1 and A2 were less marked. So, in two independent analyses applied to different datasets we found a similar partition of SLE patients associated with different cell behavior following disease activity, differentiating A3 and P3 as lymphocyte-driven clusters, and the remaining groups as neutrophil-driven clusters.
Treatment did not influence cluster formation
Treatment can modulate gene expression and blood cell proportions (20, 21). We tested whether expression of the selected genes could be modulated by doses of cytotoxic drugs, acetylsalicylic acid or by prednisone. The presence of treatment-modulated genes did not exceed 2% of our gene selection (Figure S3).Using trajectory analyses of the pediatric set we observed that treatment did not affect neutrophil proportions between clusters (Figure S4).Thus our stratification approach is not influenced by treatment, and treatment is not differentially influencing cell proportions between clusters.
Clusters are not influenced by disease activity
We then analyzed if there was differential distribution of disease activity scores between clusters. Pediatric clusters had mean SLEDAI scores of 6.45, 6.44 and 6.65, for P1, P2 and P3, respectively. Adult SLEDAI scores were lower and less variable, and had mean values of 3.29, 2.31, and 2.80, excluding such bias. Also, there was no difference in the overall magnitude of the change in SLEDAI across visits when comparing the clusters (Table S6). We also independently evaluated the clinical variables that compose the SLEDAI score. Differences were found only when the SLEDAI score was between 8–11 in the pediatric clusters (Figure S5). Specifically, the only SLEDAI related variables showing significant differences were pyuria and hematuria for cluster P2 (P = 0.0064 and P = 0.0028, respectively), and pyuria for P3 (P = 0.0449), as compared to the other clusters. Therefore the clusters were similar and clinically indistinguishable by SLEDAI parameters.
We analyzed the trajectory of severe proliferative nephritis development, the most common and serious organ affectation in pediatric lupus. We found that 65% of patients of cluster P1 developed nephritis with time compared to 53% and 45% of patients from clusters P2 and P3, respectively. A tendency towards nephritis for P1 and P1-P2 combination compared with P3 (Fisher’s exact test P=0.12 and p=0.16 respectively) was observed. In the adult set we observed the same pattern, where 45%, 42% and 13% of patients in clusters A1, A2 and A3, respectively, developed proliferative nephritis. Nephritis was significantly enriched in A1, A2 and A1-A2 combination compared with A3 (p=0.022, p=0.035, and p=0.014, respectively). So, clusters P1 and A1 show a high nephritis incidence followed by clusters P2 and A2, both with neutrophil-driven disease activity, suggesting a direct relationship between the neutrophil-driven clusters and risk to develop severe nephritis.
We had additional clinical variables for the adult set. Figure 3C shows those significantly enriched in each cluster. Cluster A1 was enriched in renal damage-related manifestations, A2 in lymphopenia, similar to the lymphocyte decrease observed for P2, lymphadenopathy and interstitial pulmonary fibrosis, and A3 was enriched in patients with secondary Sjögren’s syndrome, photosensitive rash, and signs of anti-phospholipid syndrome, among others. Increased levels of aspartate aminotransferase activity correlated with cluster P3, showing that both adult and pediatric cluster 3 is related to abnormal hepatic function (Figure 3C).
Clusters P2/A2 and P3/A3 showed opposite correlation of activity with biological pathways, while P1 and A1 were heterogeneous
Figure 4A and 4B show the fifteen top biological pathways represented by the selected genes stratifying pediatric and adult patients, respectively.
Figure 4. Functional analysis of the genes selected to stratify.
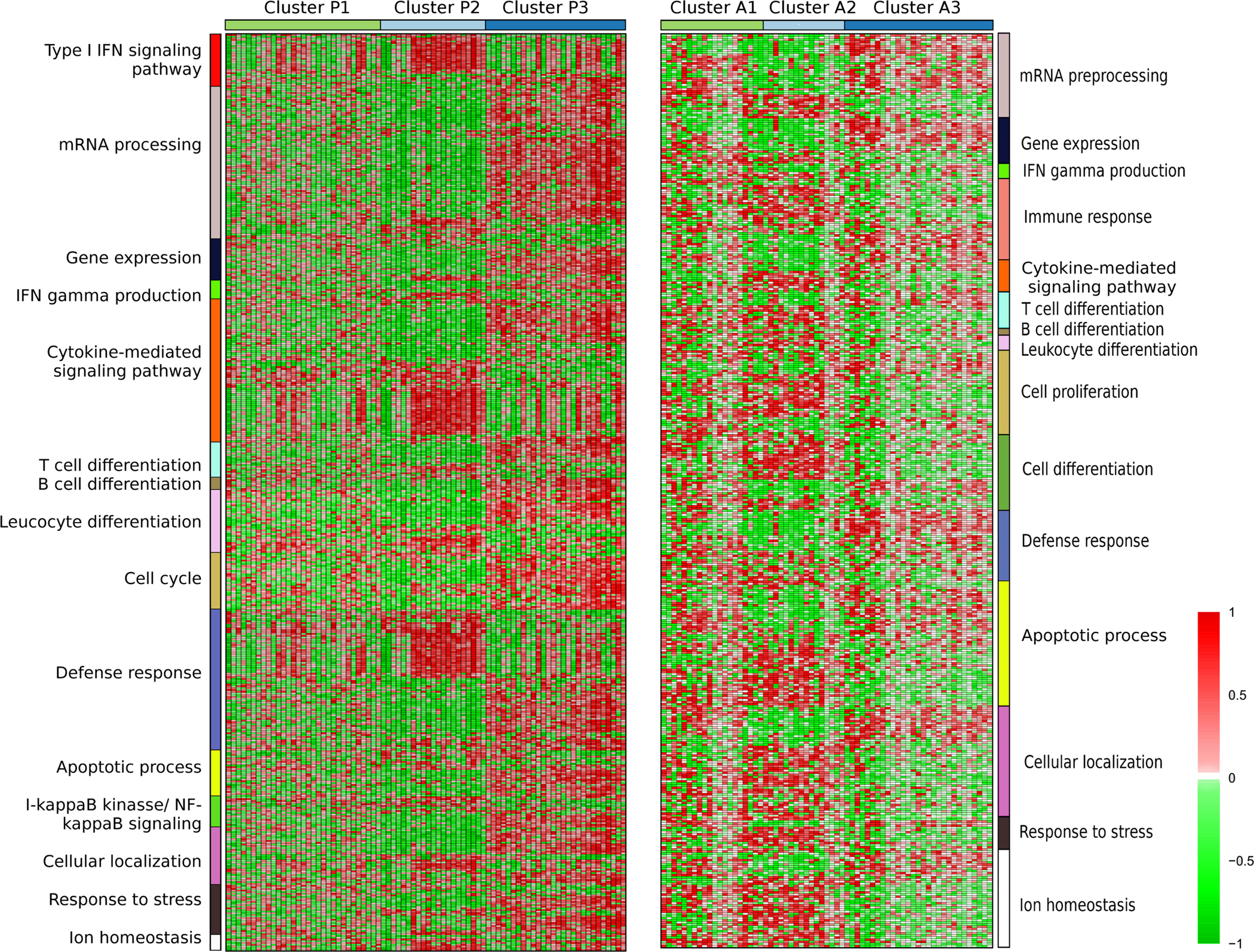
The color scale represents the correlation between gene expression and SLEDAI across visits. Significant Gene Ontology pathways were defined by EnrichR web tool and GO pathways were grouped according to the highest common hierarchy level (see methods).
Interestingly, the genes forming the clusters and their biological pathways in P2 and A2 correlated with SLEDAI in a pattern opposite to that ofP3 and A3. For example, cluster P2 had a strong positive correlation with type I interferon, infection and cytokine-mediated signaling pathways, found also in A2, while in P3 this correlation was strongly negative. The same differences were observed in other pathways. In clustersP1 and A1, the biological pathways correlated in both directions. The genes in these clusters were heterogeneous in their response to disease activity. If we focus on individual pathways, some patients of clustersP1 and A1could be classified intoP2 or P3 and A2 or A3, respectively, but if we consider all pathways and correlated genes, the functional profiles of patients in P1 and A1 were totally different from the profiles of the other two clusters. So, we can differentiate 3 groups by their differential biological function and the behavior of the correlated genes.
Modular functional pathways are different for the different clusters
In order to specifically address functional differences between clusters, we performed a functional modular analysis comparing clusters at low and high SLEDAI scores and differential gene expression. Figure 5A and 5B summarize the modular pathways with differential gene expression between clusters. A comprehensive modular analysis is shown in Figure S6. During high activity (>8) clusters P3 and A3 were over-represented with T lymphocyte-related functions and under-represented of neutrophil-related functions, while clusters P2 and A2 showed the opposite pattern. Interestingly, type I IFN-related pathways were over-expressed in clusters P3 and A3 at low SLEDAI values (Figure S6). Therefore, the clusters are differentially correlated in neutrophil and lymphocyte cell proportions, but also, the functions related with these cell types are differentially expressed between them.
Figure 5. Modular functional analysis according to disease activity.
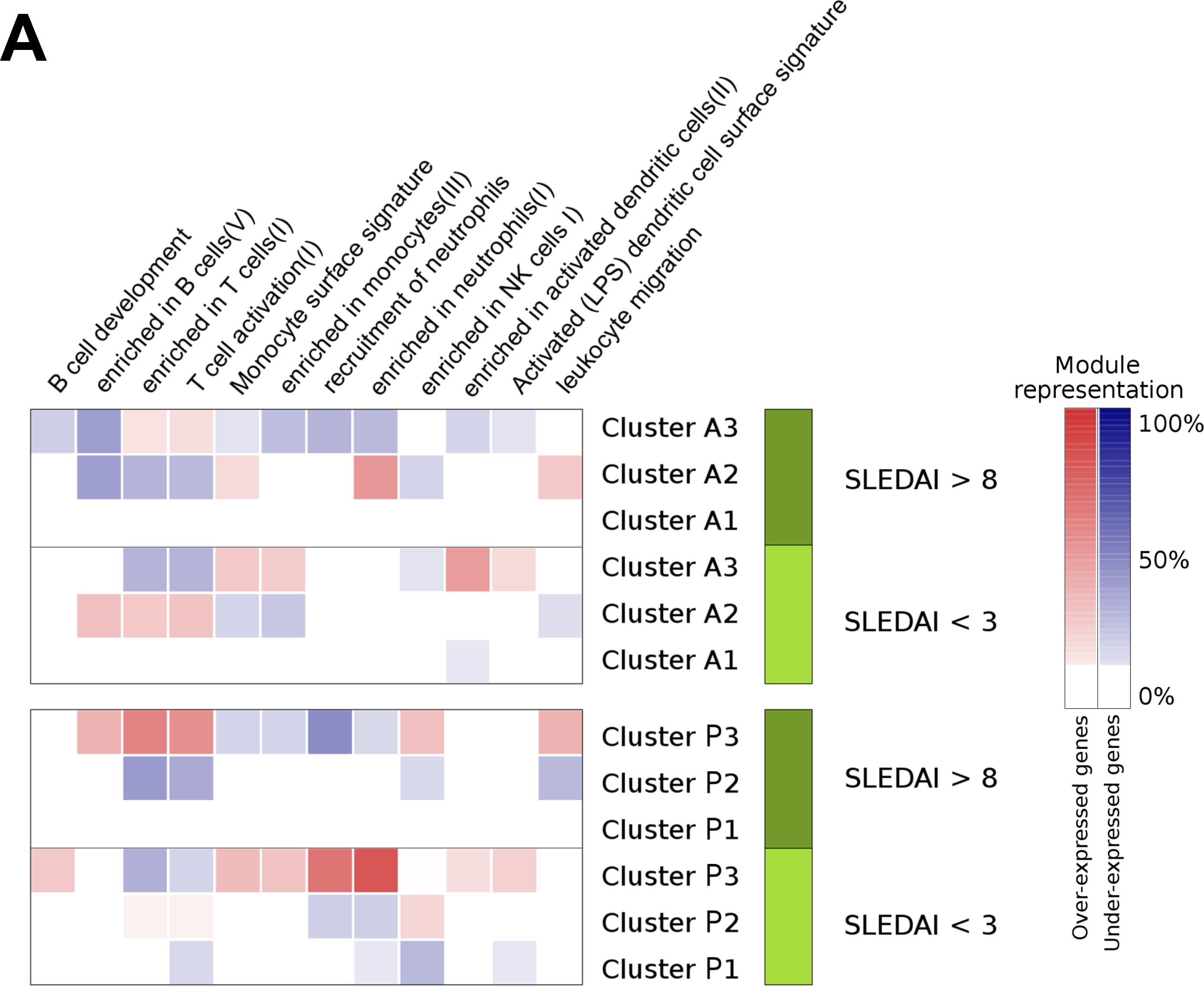
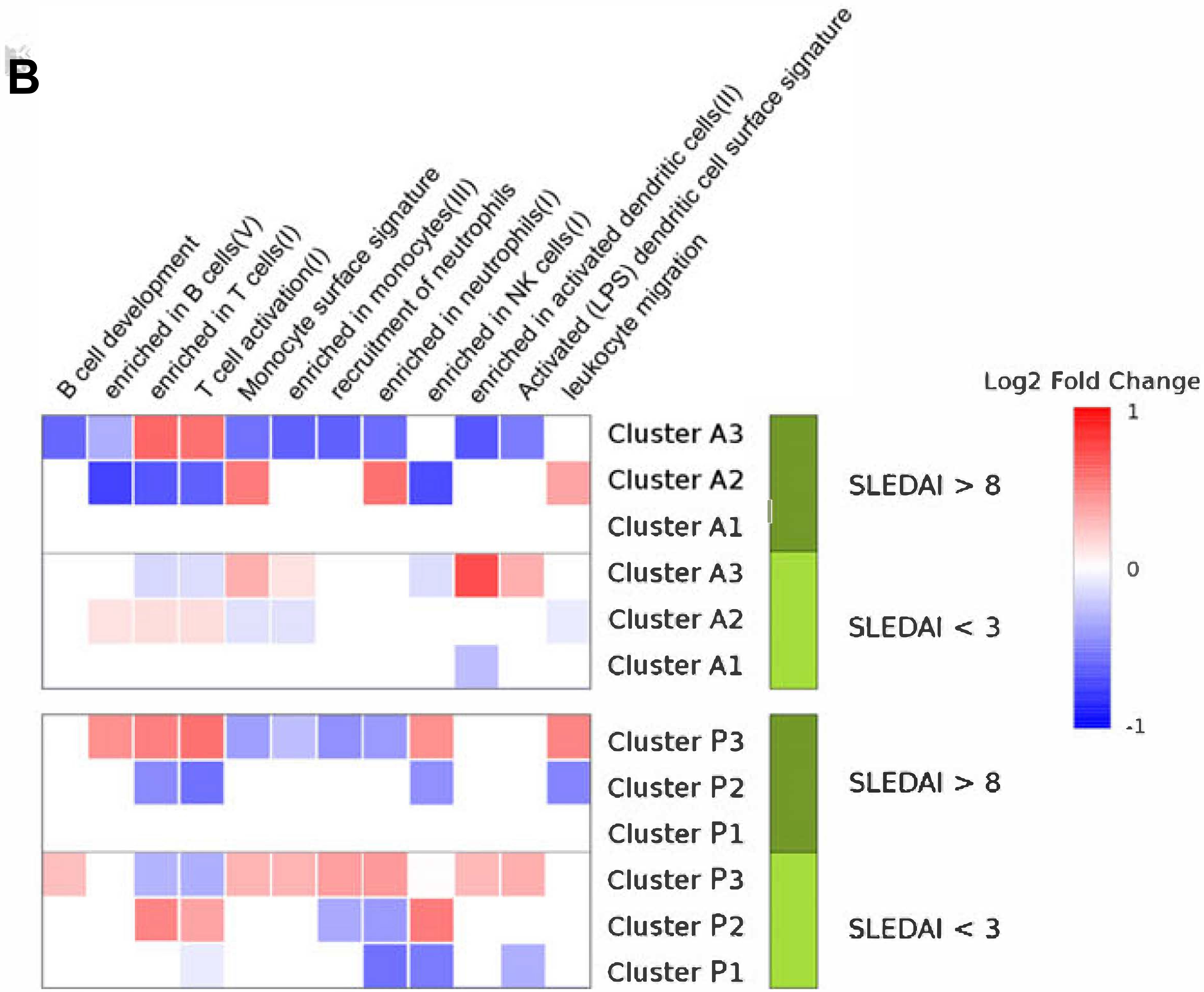
Summary of the significant modular pathways related with cluster’s progression across sets. We selected patients from each set with SLEDAI higher than 8 and performed a differential gene expression analysis between clusters. The same analysis was performed selecting patients with SLEDAI less than 3. With the lists of significant genes resulting in each differential gene expression comparison, we performed functional modular analyses. A)Red and blue color intensity represents the percentage of the genes from each modular pathway that is significantly over or under-expressed in a cluster respect to the other clusters at the same SLEDAI range, respectively. All significant pathways are shown in Figure S5. B) Red and blue color intensity represents the mean of the log2 fold change ofall significant genes that appears in each module for each subset.
Discussion
We propose a lupus stratification based on longitudinal gene expression data that robustly correlates with disease activity and shows clear clinical, functional, and cellular differences. This stratification reveals parameters that may be used in predictive models of disease progression. Our results suggest that different immune system mechanisms occur during disease activity that may determine the predisposition towards developing different clinical manifestations.
Banchereau, et al., (8) established patient stratification applying a weighted gene co-expression network analysis (WGCNA) (22). For each patient they selected agene module that best correlated with the SLEDAI score over time (called SLEDAI WGCNA module). Although useful, this strategy has drawbacks. The number of genes selected for each moduleshows large variation between patients (ranging from 31 to 3434 genes), which can bias patient comparison. The selected genes were different between patients, implying a lack of a common gene space for clustering patients. Thus, Banchereau proposed to stratify patients by comparing behavior of different genes between patients indirectly, in a functional common space. This space represented by the WGCNA modules selected for each patient projected by correlation into predefined functional and cellular gene modules obtained from blood (23) is from where seven groups were obtained. Therefore, patient stratification was subjected to a set of predefined modules, which could underestimate relevant relationships not found in the pre-established modules. By selecting one module for every patient, genes were discarded that might correlate with disease activity. Having selected the genes most correlated with the SLEDAI profile in each patient individually the global behavior of these genes was not evaluated in all patients and therefore, gene expression alterations caused by external factors such as treatment may have influenced their analysis.
Our approach considers a common gene correlation space for all patients. The correlation space is constructed calculating the correlation of each gene and a continuous clinical variable for every patient (SLEDAI). The method is useful for complex diseases where samples have been taken at different times or disease states for a variable number of visits. In addition, data does not need to be corrected for treatment or other confounders that affect gene expression, as our feature selection approach considers as significant genes having a strong correlation value across all patients or homogeneous groups of patients, removing possible individual alterations. We validated the stability of our clusters.
We established three clearly differentiated clusters of SLE patients replicated in two independent sets. We obtained largely the same cellular behavior, clinical and functional patterns across pediatric and adult sets in spite of widely reported differences between pediatric and adult patients. Clinically the groups show interesting similarities, such as hepatic disease in cluster P3 and A3, and increased risk of proliferative nephritis in neutrophil-driven clusters.
The completely opposite behavior of neutrophils and lymphocytes between clusters 2 and 3leadsus to conclude that the involvement of specific cell types is key to differentiating SLE patients during disease activity and suggests a fundamental difference in the mechanisms driving disease activity. Those driven by lymphocytes had functional pathways related to all lymphocytic populations: B cells, T cells, and NK cells, while those driven by neutrophils also have monocyte related biological functions.ClustersA1 and P1 were, however heterogeneous, representing the most severe renal disease cases. Intuitively in these patients the disease appears to be driven mainly by neutrophils, but both cell types appear to play functional roles, with variable patterns of gene expression and cellular responses during disease activity. Why this is so, remains unexplained.
The SLEDAI is an activity index that detects disease activity with an overrepresentation towards nephritis, so it might be perceived that there is a bias when clusters separate nephritis cases in our data. However this is highly improbable for several reasons. The mean SLEDAI value between clusters and the magnitude of its components were very similar with small differences between clusters 2 and 3 only at indexes between 8–11. At this point we do not have a set of patients with other activity index, such as BILAG, which is broader in the components of activity it detects. However, differences as substantial and dependent on the activity-driving cell types suggests that most probably we would observe the same pattern as we do using SLEDAI. Our result therefore supports the role of neutrophils in severe nephritis (8) but not in other manifestations, such as Sjögren’s syndrome (24, 25).
That three gene expression samples with varying SLEDAIs per patient are required to estimate the clusters makes it clinically unfeasible to use this method to classify new patients. A classifier on a single time is necessary.
The molecular mechanisms behind the clusters are unknown. Evaluation of the cytokine signaling group provides insight (Figure S7). P2 showed increased expression of a precursor of LL-37, CAMP (26), and the necroptosis gene RIPK3 (27). STAT4 a T cell transcription factor and SLE susceptibility gene (28), CARD11, a B lymphocyte differentiation gene (29), and LAG3, a T and NK cell differentiation gene (30) were increased in P3, among others.
From the point of view of a disease like lupus, the clusters we identified or the parameters strongly associated with them could be used in future studies to improve therapy used and achieve greater efficacy. It may be possible to prevent severe nephritis having a selection of genes and cellular counts at hand to define the cluster to which they belong.
In short, we suggest three mechanisms of lupus progression influenced by cell proportions and their expressed gene having different behavior in time.
Supplementary Material
Acknowledgements:
The data for analysis of the pediatric lupus patients was downloaded from the NCBI GEO database (https://www.ncbi.nlm.nih.gov/geo/) with the identifier GSE65391.
The authors would like to thank Ann Ranger, Normand Allaire, Chris Roberts and Huo Li, who contributed to the SPARE study at Biogen and produced the gene expression data for the adult SLE patients.
Funding:
Daniel Toro-Domínguez is supported through the grant GA#115565 from the Innovative Medicines Initiative Joint Undertaking of the European Union.
Footnotes
Competing interests: None
Data and materials availability: Pediatric data is publicly available in GEO through ID: GSE65391. Adult dataset is available at (GEO number to be added) including treatment doses.
References
- 1.Pons-Estel G, Alarcón GS, Scofield S, Reinlib L, Cooper GS. Understanding the epidemiology and progression of SLE. Semin Arthritis Rheum 2010; 39: 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bombardier C, Gladman DD, Urowitz MB, Caron D, Chang CH. Derivation of the SLEDAI.A disease activity index for lupus patients.The Committee on Prognosis Studies in SLE. Arthritis Rheum 1992; 35: 630–640. [DOI] [PubMed] [Google Scholar]
- 3.Chambers SA, Rahman A, Isenberg DA. Treatment adherence and clinical outcome in systemic lupus erythematosus. Rheumatology 2007; 46: 895–898. [DOI] [PubMed] [Google Scholar]
- 4.Mohan C, Putterman C. Genetics and pathogenesis of systemic lupus erythematosus and lupus nephritis. Nat Rev Nephrol 2015; 11: 329–341. [DOI] [PubMed] [Google Scholar]
- 5.Sorlie T Molecular portraits of breast cancer: tumour subtypes as distinct disease entities. Eur J Can 2004; 40: 2667–2675. [DOI] [PubMed] [Google Scholar]
- 6.Wang C, Machiraju R, Huanga K. Breast cancer patient stratification using a molecular regularized consensus clustering method. Methods 2014; 67: 304–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Montero P, Vilar JA. TSclust: an R package for time series clustering. J Statistic Soft 2014; 62: 1–43. [Google Scholar]
- 8.Banchereau R, Hong S, Cantarel B, Baldwin N, Baisch J, Edens M et al. Personalized immunomonitoring uncovers molecular networks that stratify lupus patients. Cell 2016; 165: 551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zollars E, Courtney S, Wolf B, Allaire N, Ranger A, Hardiman G et al. Clinical application of a modular genomics technique in systemic lupus erythematosus. Progress towards precision medicine. Int J Genomics 2016; ID:7862962, 7 pages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Breitling R, Herzyk P. Rank-based methods as a non-parametric alternative of the T-statistic for the analysis of biological microarray data. J Bioinform Comput Biol 2005; 3: 1171–1189. [DOI] [PubMed] [Google Scholar]
- 11.Fraley C, Raftery AE, Murphy TB, Scrucca L. Mclust version 4 for R: normal mixture modeling for model-based clustering, classification, and density Estimation. J Am Stat Assoc 2012; 97: 611–631. [Google Scholar]
- 12.Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 2010; 26: 1572–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016; 44: 90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W and Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Research 2015; 201543: pp. e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiner J 3rd, Domaszewska T tmod: an R package for general and multivariate enrichment analysis. PeerJ Preprints 2016; 4: 22420v1. [Google Scholar]
- 17.Crow MK. Type I interferon in the pathogenesis of lupus. J Immunol 2014; 192: 5459–5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Toro-Domínguez D, Carmona-Sáez P, Alarcón-Riquelme ME. Shared signatures between rheumatoid arthritis, systemic lupus erythematosus and Sjögren’s syndrome uncovered through gene expression meta-analysis. Arthritis ResTher 2014;16: 489–x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y, Yu J, Oaks Z, Marchena-Mendez I, Francis L, Bonilla E et al. Liver injury correlates with biomarkers of autoimmunity and disease activity and represents an organ system involvement in patients with systemic lupus erythematosus. Clin Immunol 2015; 160: 319–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tchetina EV, Pivanova AN, Markova GA, Lukina GV, Aleksandrova EN, Aleksankin AP et al. Rituximab Downregulates Gene Expression Associated with Cell Proliferation, Survival, and Proteolysis in the Peripheral Blood from Rheumatoid Arthritis Patients: A Link between High Baseline Autophagy-Related ULK1 Expression and Improved Pain Control. Arthritis 2016; 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salmon JH, Cacoub P, Combe B, Sibilia J, Pallot-Prades B, Fain O et al. Late-onset neutropenia after treatment with rituximab for rheumatoid arthritis and other autoimmune diseases: data from the AutoImmunity and Rituximab registry. RMD Open 2015; 1:e000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008; 9: 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chaussabel D, Quin C, Shen J, Patel P, Glaser C, Baldwin N et al. A modular analysis framework for blood genomics studies: application to systemic lupus erythematosus. Immunity 2008; 29: 150–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hochberg MC, Boyd RE, Ahearn JM, Arnett FC, Bias WB, Provost TT et al. Systemic lupus erythematosus: a review of clinico-laboratory features and immunogenetic markers in 150 patients with emphasis on demographic subsets. Medicine (Baltimore) 1985; 64:285–95. [PubMed] [Google Scholar]
- 25.Gustafsson JT, Herlitz Lindberg M, Gunnarsson I, Pettersson S, Elvin K, et al. Excess atherosclerosis in systemic lupus erythematosus,-A matter of renal involvement: Case control study of 281 SLE patients and 281 individually matched population controls. PlosONE 2017; 12: e0174572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Trans Med 2011; 73: 73ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun L, Wang H, Wang Z, He S, Chen S, Liao D et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012; 148: 213–27. [DOI] [PubMed] [Google Scholar]
- 28.Hagberg N, Joelsson M, Leonard D, Reid S, Eloranta ML, Mo J, et al. The STAT4 SLE risk allele rs7574865[T] is associated with increased IL-12-induced IFN-γ production in T cells from patients with SLE. Ann Rheum Dis 2018; Epub Ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brohl AS, Stinson JR, Su HC, Badgett T, Jennings CD, Sukumar G et al. Germline CARD11 Mutation in a Patient with Severe Congenital B Cell Lymphocytosis. J Clin Immunol 2015; 35:32–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Triebel F, Jitsukawa S, Baixeras E, Genevee C, Viegas-Pequignot E et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med 1990; 171:1393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.