

Abstract
Tumor infiltration by immunosuppressive myeloid cells, such as myeloid-derived suppressor cells (MDSCs), causes resistance to immunotherapy. Semaphorin4D, originally characterized for its axonal guidance properties, also contributes to endothelial cell migration and survival and modulates global immune cytokine profiles and myeloid cell polarization within the tumor microenvironment. Here, we show how a therapeutic murine Sema4D mAb improves responses to immune-checkpoint blockade (ICB) in two murine carcinoma models. Treatment of tumor-bearing mice with Sema4D mAb abrogated Ly6Ghi PMN-MDSC recruitment through reducing MAPK-dependent chemokine production by tumor cells in Murine oral cancer-1 (MOC1) tumors. PMN-MDSC suppressive capacity was reduced through inhibition of Sema4D-driven arginase expression. These changes led to enhanced tumor infiltration by CD8+ TIL and activation of tumor-draining lymph node T lymphocytes in response to tumor antigen. Sema4D mAb in combination with either CTLA-4 or PD-1 blockade enhanced rejection of tumors or tumor growth delay, resulting in prolonged survival with either treatment. This function of Sema4D mAb provides a rationale for its evaluation in combination with ICB to treat tumors with immunosuppressive myeloid infiltration.
Introduction
Despite advances in surgical techniques and standard anti-cancer therapeutics, outcomes for advanced head and neck carcinomas remain poor (1). A subset of patients with recurrent disease after standard treatment demonstrate durable antitumor responses to immune-checkpoint blockade (ICB) immunotherapy, though the majority of patients with recurrent or metastatic head and neck squamous cell carcinoma (HNSCC) do not respond to ICB (2, 3). Both extrinsic and intrinsic resistance to ICB immunotherapy occurs (4). One resistance mechanism appears to be due to immunosuppressive myeloid cells within the tumor microenvironment (TME; refs. 5–7). Many preclinical approaches to alter myeloid cell–driven immunosuppression within the TME, which may enhance effector immune cell activity and responses to ICB, have been investigated (5, 6, 8, 9).
Semaphorins are a family of transmembrane and soluble proteins that guide axonal sprouting (10, 11). Signaling through its cognate receptor Plexin B, Semaphorin4D (Sema4D, CD100), a class-4 sempahorin, regulates endothelial cell migration and survival and tumor vascularization (12, 13). A therapeutic murine Sema4D mAb shifts immunity toward a type I antitumor response with enhanced infiltration of CD11c+ myeloid cells and CD8+ tumor-infiltrating lymphocytes (TILs) into syngeneic murine Colon26 tumors (14). Tumor cell–derived Sema4D induces formation of myeloid-derived suppressor cells (MDSCs), a type of immunosuppressive myeloid cell, from PBMCs in an in vitro setting (15). Sema4D expression may drive carcinoma cell perineural invasion (16), and Sema4D expression correlates with poor outcome in multiple carcinoma types (17).
Given these data, we hypothesized that blockade of Sema4D with a therapeutic mAb could alter MDSCs within the TME and sensitize tumors to ICB. Murine oral cancer-1 (MOC1) is a carcinogen-induced, HPV-negative model of oral cavity carcinoma (18). The MOC1 TME is infiltrated with immunosuppressive Ly6G+ granulocytic MDSC (PMN-MDSC) but not FoxP3+ CD4+ regulatory T lymphocytes (Treg; refs. 5, 8). Herein, we describe effects of a murine Sema4D mAb on PMN-MDSC. Treatment with Sema4D mAb sensitized MOC1 tumors to rejection following CTLA-4 blockade and growth delay following PD-1 blockade. Enhanced sensitivity to ICB following Sema4D mAb treatment was separately validated in Lewis lung carcinomas (LLC). The ICB sensitization was not due to significant changes in tumor vascularity or direct inhibition of tumor cell proliferation or survival. Following Sema4D mAb treatment, TME infiltration of PMN-MDSCs was significantly reduced, due in part to reduced expression of the myeloid chemokines CXCL1, 2, and 5, but not due to inhibited PMN-MDSC expansion within the TME. The ability of PMN-MDSC to suppress T-cell proliferation and tumor antigen–specific killing was reduced due to blockade of Sema4D-induced PMN-MDSC arginase expression. These effects resulted in enhanced infiltration of CD8+ TIL in the MOC1 TME and enhanced tumor antigen–specific responses in CD8+ T lymphocytes from mice treated with Sema4D mAb. A phase I trial of a human Sema4D mAb (VX15) demonstrated a good safety profile in patients with advanced solid cancers (19). Thus, a treatment that combines Sema4D mAb with immune-checkpoint blockade may be useful against tumors infiltrated with MDSCs such as HNSCC.
Materials and Methods
Cell culture
MOC1 cells from the same stock that has been genomically characterized (20) were obtained from Ravindra Uppaluri (Washington University, St. Louis, Missouri) in 2014 and cultured as described (21). LLC cells were obtained from James Hodge (National Cancer Institute) in 2016 and cultured as described (22). Both cell lines were used for experiments within 20 passages and serially tested to ensure negativity for murine pathogens and mycoplasma. Cell lines were not reauthenticated within the past year. Recombinant Sema4D (rSema4D), Sema4D mAb, and isotype mAb were obtained from Vaccinex under a Cooperative Research and Drug Agreement. U0126 was purchased from Selleckchem.
Animal studies
All in vivo treatments were approved by the NIH animal care and use committee. MOC1 or LLC tumors were established via sub-cutaneous injection of MOC1 (5 × 106) or LLC (1.5 × 106) cells suspended in 30% matrigel (Corning) in the right flank of wild-type C57BL/6 (B6) mice (Taconic). Sema4D (clone 67–2, 200 μg/injection once every 7 days for 4 total doses, starting at day 4) or isotype control mAb treatments were performed via intraperitoneal injection (i.p.) and started on day 4. PD-1 (clone RMP1–14, 200 μg/injection once every 5 days for 4 total doses, starting at day 10) or CTLA-4 (clone 9H10, 100 μg/injection once every 5 days for 3 total doses, starting at day 10) mAb (Bio X Cell) injections were performed via i.p. and started on day 10. For tumor growth experiments, tumor measurements and weights were obtained 2 to 3 times per week, and volume was calculated as (length2 × width)/2. Tumors, draining lymph nodes, and spleens were harvested via standard dissection techniques. Digestion of tumors into single-cell suspensions was performed using a murine tumor dissociation kit and a gentleMACS dissociator (Miltenyi) per the manufacturer’s recommendations. When performed, leukocyte separation from whole tumor digests was achieved using a standard 80/40% percoll (Sigma-Aldrich) gradient. The >95% purity of CD45.2+ cells after gradient was verified by flow cytometry. For some experiments, positive selection of PMN-MDSCs (anti-Ly6G microbead kit) and negative selection of T lymphocytes (pan T-cell isolation kit II) was performed on an autoMACS (Miltenyi).
Flow cytometry
Only fresh cultured cells or tissues prepared into single-cell suspensions were analyzed. Nonspecific staining was minimized by staining with CD16/32 (FcR) blocking antibodies. Primary antibodies were applied for 30 minutes at concentrations titrated for each antibody. PlexinB1 antibody was purchased from Proteintech, phospho-AKT antibody was purchased from Cell Signaling Technology, phospho-ERK and phospho-STAT3 antibodies were from Thermo Fisher, and all other antibodies were from BioLegend or eBioscience. Dead cells were excluded via 7AAD uptake or a fixable viability dye and a “fluorescence-minus-one” technique was used to validate specific staining of all antibody combinations. Fixation and permeation for Ki67 and transcription factor staining was achieved with the transcription factor staining buffer set (eBioscience) per the manufacturer’s recommendations. Apoptosis was assessed using the annexin V apoptosis detection kit (eBioscience) per the manufacturer’s protocol. All analyses were performed on a BD FACSCanto analyzer running FACSDiva software and interpreted using FlowJo (vX10.0.7r2).
TIL culture
MOC1 tumors were harvested, minced into 1-mm fragments, and plated in RPMI1640-based media with rmIL2 (100 U/mL). After 72 hours, tumor fragments were removed. TILs were cultured in the presence of rmIL2 for 14 days, replenishing fresh media (50% of the volume) with rmIL2 every 48 hours. Flow-cytometric analysis revealed >90% of the cultured cells to be CD3+ lymphocytes.
Impedance analysis
To measure proliferation and viability of MOC1 cells, 1 × 104 cells were plated in the presence or absence of control or Sema4D mAb. In other experiments designed to measure cultured TIL-mediated antigen-specific cytotoxicity, MOC1 (1 × 104) cells were plated and allowed to gain impedance for 48 hours. Cultured TIL from day 14 MOC1 tumors were then added at a 10:1 effector:target (E:T) ratio in the presence or absence of tumor Ly6GhiLy6Clow myeloid cells (3:1 E:T ratio to the TIL), treated with control mAb or Sema4D mAb. TIL, Ly6GhiLy6Clow myeloid cells and mAbs were combined and incubated together for 2 hours before addition to the impedance plate. Impedance experiments were performed using 96-well E-Plates (ACEA Biosciences) and alteration of impedance was acquired using the xCELLigence Real-Time Cell Analysis (RTCA) platform per the manufacturer’s recommendations. For each plot, y-axis is cell index and x-axis is time in hours. Triton X-100 (0.2%) was added to some wells to verify complete loss of cell index with total cell lysis. Percent loss of the cell index for a given time point was calculated as: 1 – (experimental cell index/control cell index).
T-lymphocyte proliferation assay
Isolated tumor Ly6GhiLy6Clow myeloid cells were added to carboxyfluorescein succinimidyl ester (CFSE; Sigma) labeled T lymphocytes isolated from naïve B6 spleens. After 2 hours of Ly6GhiLy6Clow myeloid cells and T-lymphocyte coincubation at a 3:1 Ly6GhiLy6Clow myeloid cell:T lymphocyte ratio, T lymphocytes were stimulated with plate-bound CD3 (clone 145–2C11, eBioscience) and CD28 (clone 37.51, eBioscience) antibodies. Flow cytometry was used to measure CFSE dilution at 72 hours. Proliferation was quantified as the average number of divisions for all cells in the culture (division index) using FlowJo software (23). In some experiments, GM-CSF was added to prolong the viability of PMN-MDSCs (24).
Tumor-draining lymph node T-lymphocyte responses
From tumor-draining lymph nodes, T lymphocytes were isolated by negative magnetic selection on an auoMACS (Miltenyi) and combined with MOC1 tumor cells that had been irradiated (50 Gy was required to reverse tumor cell–mediated suppression of T-lymphocyte activation) and treated with IFN (20 ng/mL IFNγ × 24 hours to increase MHC class I expression). The cells were combined at a ratio of 3:1 T lymphocyte:MOC1. Supernatants were collected at 24 hours and analyzed for IFNγ concentration by ELISA. Other lymph node T lymphocytes were stimulated with CD3/28 dynabeads (Thermo Fisher) at a 1:1 T lymphocyte:bead ratio. Supernatant IFNγ concentration was measured by ELISA.
Immunofluorescence
Tumor sections (5 μm) were fixed for 7 minutes in ice-cold methanol at −20°C. Following several washes, samples were blocked with RTU normal goat serum (2.5%; Vector Laboratories) and Renaissance Antibody diluent (Biocare Medical). Anti-CD31 conjugated to APC (clone 390; eBioscience) was diluted 1:50 in Renaissance antibody diluent, and sections were stained for 1 hour on an orbital shaker (80 rpm). Slides were washed, mounted with DAPI VECTASHIELD mounting media, cover-slipped, and sealed. Images were obtained on a Zeiss LSM780 confocal microscope.
qRT-PCR
Cells were lysed or whole tumor lysates were generated using the Tissue Lyser II. RNA was purified using the RNEasy Mini Kit (Qiagen) per the manufacturer’s protocol. cDNA was synthesized utilizing a high-capacity cDNA reverse transcription kit with RNase inhibitor (Applied Biosystems). A TaqMan Universal PCR master mix was used to assess the relative expression of target genes compared with GAPDH on a Viia7 qPCR analyzer (Applied Biosystems). All primers were commercially available and purchased from Thermo Scientific.
Western analysis
Whole-cell lysates were obtained using NP40 lysis buffer, mixed with NuPAGE LDS sample buffer and reducing agent (Life Technologies), heated at 95°C for 5 minutes, and subjected to electrophoresis using Bis-Tris precast gels (Life Technologies) at 150 V for 100 minutes. The Invitrogen iBlot Dry Blotting System was used to transfer proteins onto a PVDF membrane. Primary antibodies were diluted in 5% BSA prepared from Tween 20-TBS (dilutions: pErk1/2 1:1,000; pAKT 1:1,000; pSTAT3 1:2000; ERK1/2 1:1,000; AKT1:1,000; STAT3 1:1,000; Actin 1:5,000). Primary antibodies were purchased from Cell Signaling Technologies. Blots were incubated with Chemiluminescent HRP Antibody Detection Reagent (Denville Scientific Inc.) and imaged using Image Studio software (LI-COR Biosciences).
ELISA
CXCL1 (R&D) and IFNγ (eBioscience) ELISAs were used per the manufacturer’s recommendations.
Statistical analysis
Tests of significance between pairs of data are reported as P values, derived using a Student t test with a two-tailed distribution and calculated at 95% confidence. Comparison of multiple sets of data was achieved with analysis of variance with Tukey multiple comparisons. Survival analysis was determined by log-rank (Mantel–Cox) analysis. All error bars indicate standard deviation. Statistical significance was set to P < 0.05. All analysis was performed using GraphPad Prism v7.
Results
Sema4D mAb sensitized carcinomas to CTLA-4 or PD-1 ICB
Sema4D mAb can polarize the TME toward antitumor activity (14) and carcinomas have an immunosuppressive microenvironment (5, 8). We therefore hypothesized that treatment of mice bearing MOC1 tumors with Sema4D mAb would sensitize them to growth control with CTLA-4 mAb. Although treatment of MOC1 tumor-bearing mice with Sema4D mAb alone had little effect, Sema4D mAb treatment enhanced the rate of tumor rejection after CTLA-4 mAb treatment from 50% to 90% (Fig. 1A), resulting in enhanced survival (Fig. 1B). Mice that rejected MOC1 tumors after combination treatment were rechallenged with MOC1 cells in the opposite flank. Mice that previously rejected MOC1 did not engraft tumors (Fig. 1C), suggesting the presence of immunologic memory.
Figure 1.
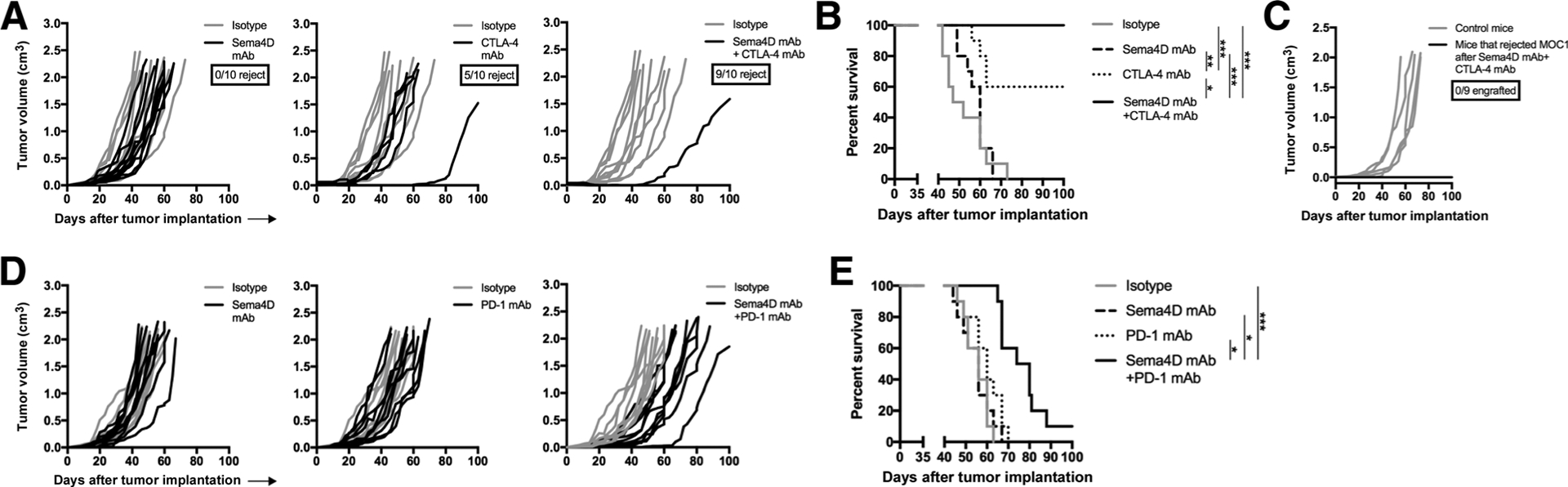
Sema4D mAb plus ICB enhanced rejection of MOC1 tumors. A, Mice bearing MOC1 tumors were treated with Sema4D mAb and CTLA-4 mAb alone or in combination (n = 10 mice/group). Individual tumor growth curves are shown. The ratio of mice that rejected tumors is shown below the graph legends. Representative results from two independent experiments. B, Survival analysis of mice treated with Sema4D mAb and CTLA-4 mAb alone or in combination. C, Mice that rejected MOC1 tumors following combination Sema4D and CTLA-4 mAb treatments were challenged with MOC1 (3 × 106 cells/mouse) in the contralateral flank and followed for tumor engraftment. Control mice were untreated, wild-type B6 mice. Ratio of mice that rejected tumor engraftment is shown below the graph legend. D, Mice bearing MOC1 tumors were treated with Sema4D mAb and PD-1 mAb alone or in combination (n = 10 mice/group). Individual tumor growth curves are shown. Representative results from two independent experiments. E, Survival analysis of mice treated with Sema4D mAb and PD-1 mAb alone or in combination. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We hypothesized that Sema4D mAb treatment could sensitize mice bearing MOC1 tumors to growth control with PD-1 mAb. Although neither treatment alone altered tumor growth, the combination of Sema4D mAb and PD-1 delayed primary tumor growth (Fig. 1D) and significantly prolonged survival of mice (Fig. 1E). Together, these data suggested that Sema4D mAb enhanced the rate of rejection of MOC1 tumors following CTLA-4 blockade, and sensitized resistant MOC1 tumors to growth delay following PD-1 blockade.
To validate these findings in a separate carcinoma model, these experiments were repeated in mice bearing LLC tumors. Although treatment with Sema4D or CTLA-4 mAb alone induced no tumor growth delay (Fig. 2A), combination treatment delayed tumor growth in 5 of 10 (50%) LLC tumors, leading to significantly prolonged survival (Fig. 2B). Treatment with PD-1 mAb alone initially delayed growth in some tumors, but these quickly rebounded. However, the combination of Sema4D and PD-1 mAb induced rejection of 2 of 10 (20%) established LLC tumors and slowed growth in the remaining tumors (Fig. 2C), leading to significantly prolonged survival (Fig. 2D). Thus, treatment with Sema4D mAb enhanced tumor control or tumor rejection following treatment with CTLA-4 or PD-1 ICB in two murine carcinoma models.
Figure 2.
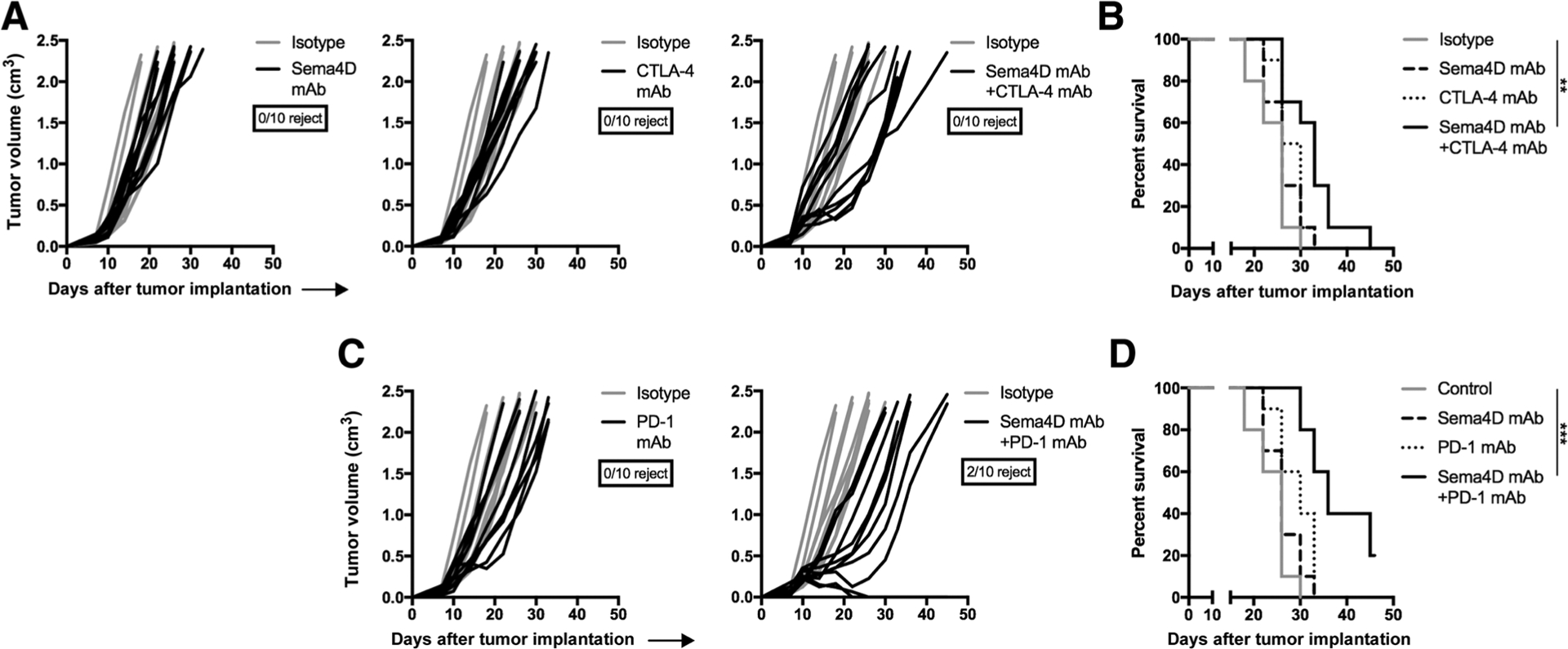
Sema4D mAb plus ICB enhanced rejection of LLC tumors. A, Mice bearing LLC tumors were treated with Sema4D mAb and CTLA-4 mAb alone or in combination (n = 10 mice/group). Individual tumor growth curves are shown. Ratio of mice that rejected tumors is shown below the graph legends. Representative results from two independent experiments. B, Survival analysis of mice treated with Sema4D mAb and CTLA-4 mAb alone or in combination. C, Mice bearing LLC tumors were treated with Sema4D mAb and PD-1 mAb alone or in combination (n = 10 mice/group). Individual tumor growth curves are shown. Representative results from two independent experiments. D, Survival analysis of mice treated with Sema4D mAb and PD-1 mAb alone or in combination. **, P < 0.01; ***, P < 0.001.
Sema4D mAb did not affect tumor cell growth or immunogenicity or tumor vascularity
To understand the mechanisms of enhanced responses to ICB following Sema4D mAb treatment, we explored the expression of Sema4D and its high-affinity receptor PlexinB1 in normal oral mucosa from B6 mice and MOC1 carcinomas from B6 mice (Fig. 3A). Compared with epithelial cells from the oral mucosa, MOC1 tumor cells expressed more cell-surface Sema4D and PlexinB1. Consistent with prior reports (12, 13), CD31+ endothelial cells expressed high levels of PlexinB1, and this expression was greater on tumor than oral mucosa endothelial cells. Although both CD11b+ myeloid cells and CD3+ T lymphocytes highly expressed Sema4D, Ly6GhiLy6CintF4/80− myeloid cells expressed more PlexinB1 than other leukocytes.
Figure 3.
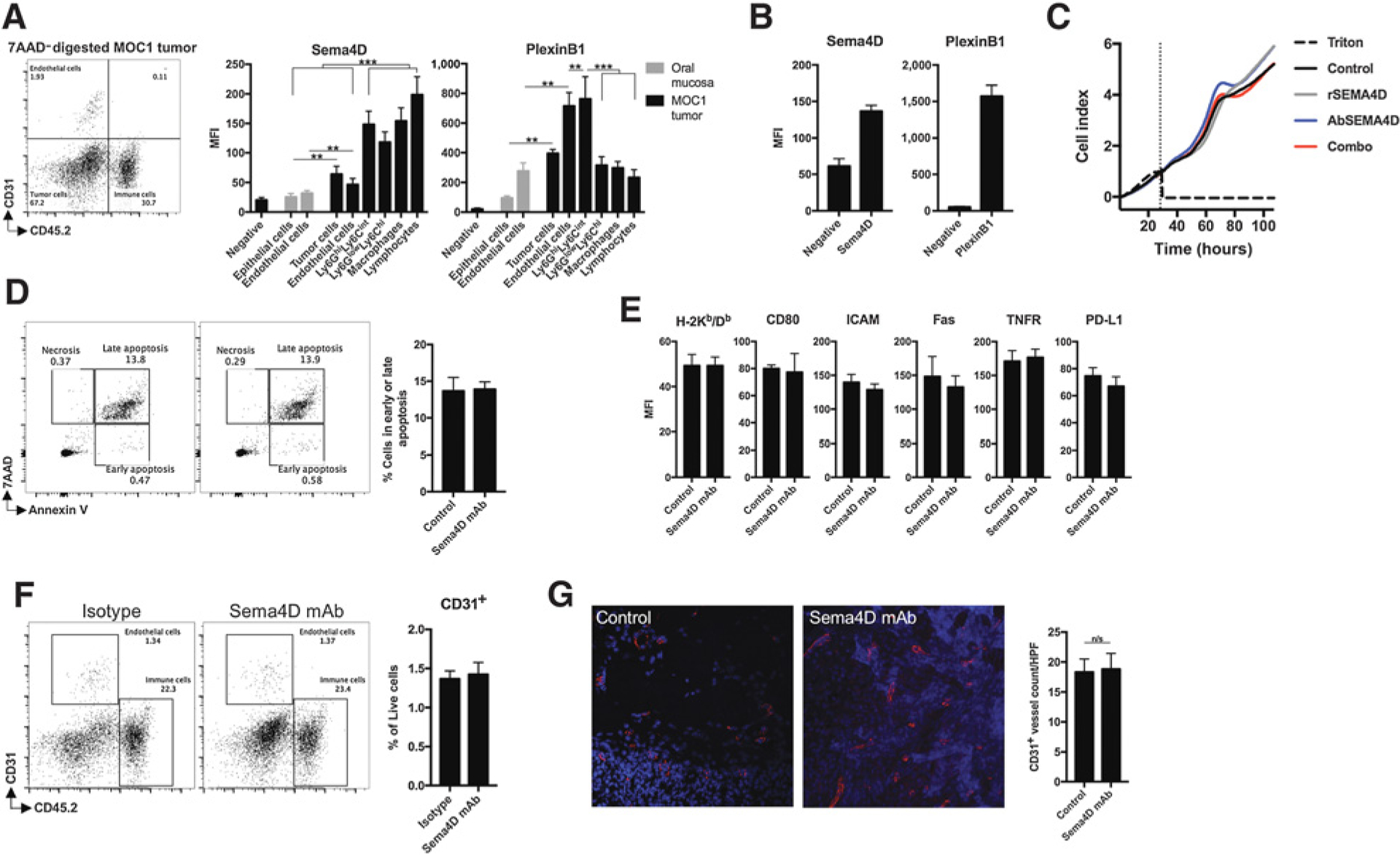
Sema4D mAbs did not alter MOC1 tumor vascularity in vivo or directly alter MOC1 tumor cells in vitro. A, MOC1 tumors (n = 5) or oral mucosa from the same mice were digested into single-cell suspensions, and cellular subsets were analyzed for Sema4D and PlexinB1 expression by flow cytometry. Representative dot plot of a digested MOC1 tumor is shown. Expression levels on CD45.2−CD31− epithelial or tumor cells, CD45.2−CD31+ endothelial cells, CD45.2+CD11b+Ly6GhiLy6CintF4/80− myeloid cells, CD45.2+CD11b+ Ly6GlowLy6ChiF4/80− myeloid cells, CD45.2+CD11b+F4/80+ macrophages, and CD45.2+CD11b−CD3+ lymphocytes are quantified on the right. Negative refers to isotype control for Sema4D or PlexinB1 mAbs used for staining. B, MOC1 cells in vitro were analyzed for Sema4D and PlexinB1 expression by flow cytometry. Negative refers to isotype control for Sema4D or PlexinB1 mAbs used for staining. C, Real-time in vitro impedance of MOC1 cells exposed to recombinant Sema4D protein (10 μg/mL), Sema4D mAb (10 μg/mL), or isotype (10 μg/mL) alone or in combination was measured to assess changes in cell adhesion and proliferation. Cells were plated at time 0 in recombinant protein or antibody. D, MOC1 cells in vitro were assessed for induction of apoptosis via annexin V/7AAD flow cytometry after exposure to Sema4D mAb for 24 hours. E, MOC1 cells in vitro were assessed for changes in cell-surface expression of components of immunogenicity by flow cytometry after exposure to Sema4D mAb for 24 hours. All in vitro experiments were repeated at least twice, each with at least three technical replicates. F, Mice bearing MOC1 tumors were treated with Sema4D mAb or isotype control (n = 5/group). Tumors harvested 1 day after the last Sema4D mAb treatment were analyzed via flow cytometry for endothelial cell infiltration. G, Tumors from mice treated as in F were assessed for CD31 vessel density by CD31 immunofluorescence. Representative photomicrographs on left, with quantification by vessel count per high-power field (10 fields/condition) on right. MFI, mean fluorescent intensity; rSema4D, recombinant Sema4D. **, P < 0.01; ***, P < 0.001.
We investigated how Sema4D mAb treatment altered MOC1 tumor cell growth or viability by assessing the effect of Sema4D mAb on the viability of MOC1 cells in vitro. Although MOC1 cells express both Sema4D and PlexinB1 (Fig. 3B), real-time impedance analysis suggested that neither recombinant Sema4D (rSema) nor Sema4D mAb significantly altered MOC1 tumor cell growth or proliferation (Fig. 3C). Similarly, treatment with Sema4D mAb did not induce MOC1 cell necrosis or apoptosis (Fig. 3D). To rule out the possibility that MOC1 tumor cells were made more susceptible to T-lymphocyte killing by Sema4D mAb, we assayed for expression of components of immunogenicity (Fig. 3E). Expression of MOC1 cell MHC class I, PD-L1, or related costimulatory factors or death receptors, such as ICAM, CD80, Fas, or TNFR, was not altered after Sema4D mAb treatment. Thus, despite expression of Sema4D and PlexinB1 on many cell types within MOC1 tumors, enhanced responses to ICB were not due to direct inhibition of proliferation or survival of MOC1 tumor cells or direct alteration of tumor cell immunogenicity that sensitizes cells to immune elimination.
Given expression of both Sema4D and PlexinB1 on endothelial cells, we next investigated alterations in CD31+ tumor endothelial accumulation within MOC1 tumors following Sema4D mAb treatment (Fig. 3F) and in CD31+ vessel density (Fig. 3G). Neither was significantly altered, suggesting that reduction in tumor vascularity was not the mechanism of Sema4D action in this model.
Sema4D mAb reduced myeloid cell infiltration by reducing chemokine production
MOC1 tumors recruit Ly6GhiLy6Cint myeloid cells into the TME between days 10 and 20 of tumor progression (5). Evaluation of MOC1 tumors after Sema4D mAb treatment revealed a significant reduction in tumor infiltration of Ly6GhiLy6Cint myeloid cells (Fig. 4A). Accumulation of either myeloid cell type in the periphery of MOC1 tumor–bearing mice was not significantly altered after Sema4D mAb treatment (Supplementary Fig. S1). Given reports that tumor cell–derived Sema4D can induce expansion of myeloid cell populations, we stained both tumor-infiltrating and peripheral myeloid cells with Ki67 to assess proliferation (Fig. 4B). Reduction in Ki67 positivity was not statistically significant following Sema4D treatment, suggesting that the reduced Ly6GhiLy6Cint myeloid cell accumulation in MOC1 tumors was not due to reduced expansion within the TME. We and others have reported the constitutive expression of the chemokine receptor CXCR2 on Ly6GhiLy6Cint myeloid cells (5). We next assessed expression of the ligands for CXCR2, myeloid chemokines CXCL1, 2, and 5, within the MOC1 TME (Fig. 4C). Given evidence that Ly6GlowLy6Chi myeloid cells were also reduced within the TME following Sema4D mAb treatment, we also assessed changes in expression of the CCR2 ligand and myeloid chemokine CCL2. PCR analysis of whole tumor digests demonstrated reductions in all three CXCR2 ligands but not in CCL2 following Sema4D mAb treatment. Analysis of the immune compartment alone demonstrated a reduction in CXCL1 but not in other myeloid chemokines, suggesting that reduced chemokine expression was primarily due to Sema4D mAb-induced alterations in the tumor and/or stromal cell compartments. Treatment of MOC1 cells in vitro with Sema4D mAb significantly reduced expression of CXCR2 ligands and, to a lesser extent, CCL2 (Fig. 4D), suggesting that signaling through Sema4D and PlexinB1 contributes to their expression. We next explored activation of signaling pathways that mediate alterations downstream of PlexinB1 (25–27). Although phosphorylation of STAT3 was low and alterations in AKT were variable, baseline phosphorylation of ERK1/2 increased with the addition of rSema4D and both baseline and rSema4D-induced phosphorylation was reduced by 30% to 40% with the addition of Sema4D mAb (Fig. 4E). We previously reported MAPK pathway-dependent expression of CXCL1 in MOC1 cells (21). To validate this finding in the current setting, we treated MOC1 cells with the MEK inhibitor U0126, which significantly reduced CXCL1 expression (Fig. 4F). These data suggested that Sema4D mAb treatment significantly inhibited MOC1 tumor Ly6GhiLy6Cint myeloid cell infiltration, and that this reduction was not due to reduced Ly6GhiLy6Cint myeloid cell expansion but rather due at least in part to reduced MOC1 tumor cell MAPK-dependent expression of the myeloid chemokines CXCL1, 2, and 5.
Figure 4.
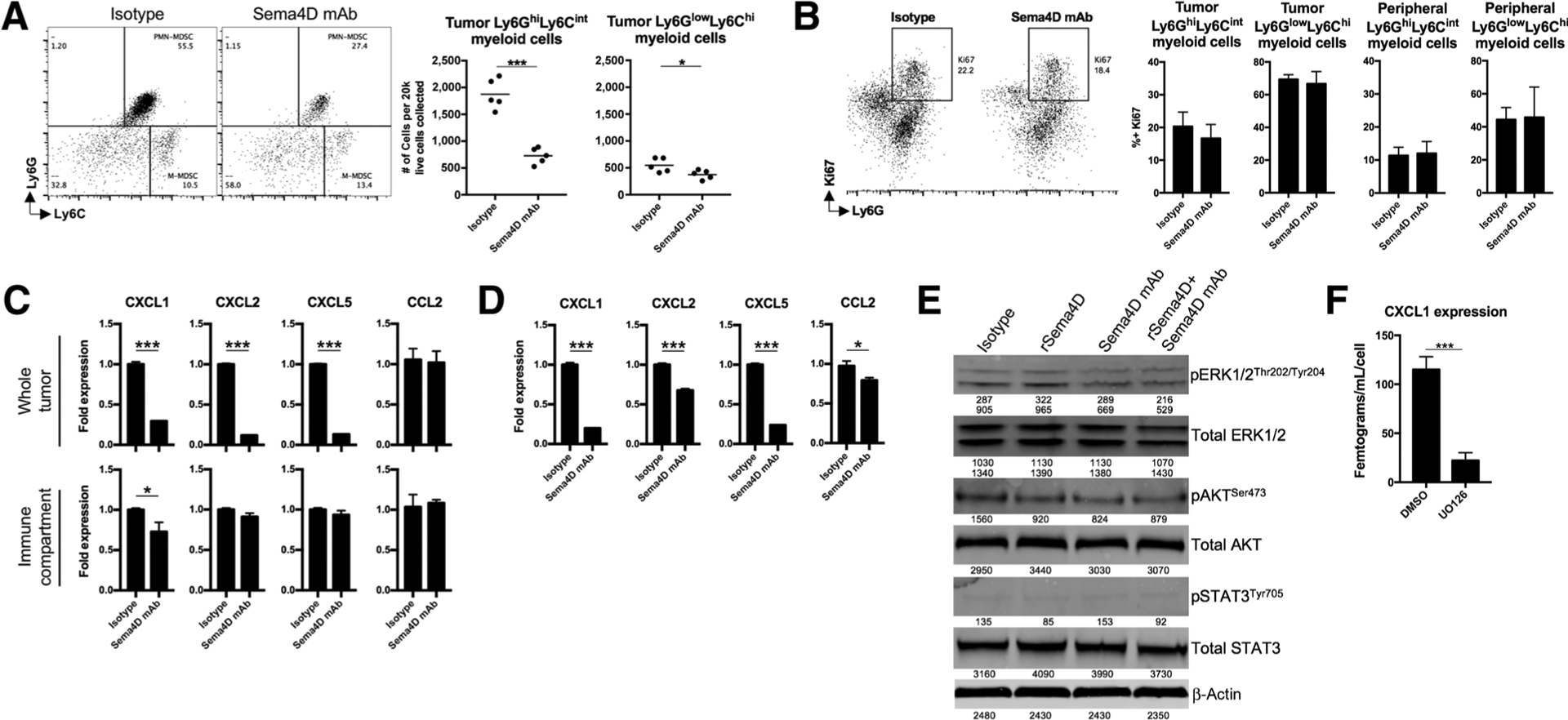
Sema4D mAb inhibited recruitment of myeloid cells into MOC1 tumors through reduction of myeloid chemokines. A, Mice bearing MOC1 tumors were treated with Sema4D mAb or isotype control (n = 5/group) and whole tumor digests harvested 1 day after the last Sema4D mAb treatment were analyzed via flow cytometry for Ly6GhiLy6Cint myeloid cell or Ly6GlowLy6Chi myeloid cell infiltration. Representative dot plots on the left, quantified on the right. B, Mice bearing MOC1 tumors were treated as above (n = 5/group), tumor digests subjected to a percoll gradient, and leukocytes were analyzed for myeloid cell markers and intracellular Ki67 by flow cytometry. Representative dot plots on the left, quantified on the right. C, Mice bearing MOC1 tumors were treated as above (n = 5/group). Some tumor digests were subjected to a percoll gradient (to obtain leukocyte fraction). Whole tumor or leukocyte fraction digests were analyzed for myeloid chemokine expression via qRT-PCR. D, MOC1 cells in vitro were exposed to Sema4D mAb (10 μg/mL) or isotype for 24 hours and analyzed for myeloid chemokine expression by qRT-PCR. E, MOC1 cells in vitro were exposed to recombinant Sema4D protein (10 μg/mL), Sema4D mAb (10 μg/mL), or isotype alone or in combination for 1 hour and subjected to Western blot analysis. Band densitometry quantified below each blot. F, MOC1 cells in vitro were exposed to the MEK inhibitor U0126 (5 μmol/L) for 24 hours, and cell culture supernatant was analyzed for CXCL1 concentration by ELISA. All in vitro experiments were repeated at least twice. rSema4D, recombinant Sema4D. *, P < 0.05; ***, P < 0.001.
Sema4D mAb reverses myeloid cell function through reduced arginase production
Younis and colleagues described Sema4D-induced arginase expression from human immunosuppressive myeloid cells generated in vitro (15). Ly6GhiLy6Cint myeloid cells infiltrating MOC1 tumors suppress T-lymphocyte activity at least through arginase-dependent mechanisms (8), suggesting they may represent PMN-MDSCs. This led us to the hypothesis that Sema4D mAb not only reduced Ly6GhiLy6Cint myeloid cell recruitment into MOC1 tumors, but also reversed their immune-suppressive capacity. Evaluation of the ability of Ly6GhiLy6Cint myeloid cells to suppress T-lymphocyte proliferation in an ex vivo T-lymphocyte proliferation assay demonstrated that these cells are immunosuppressive, and that both sorted peripheral and tumor-infiltrating Ly6GhiLy6Cint myeloid cells were less immunosuppressive following Sema4D mAb treatment (Fig. 5A). To be identified as PMN-MDSC, distinct from tumor-associated neutrophils, Ly6Ghi myeloid cells must suppress T-cell antigen–specific function (28, 29). We assessed the ability of Ly6GhiLy6Cint myeloid cells sorted form MOC1 tumors to be able to suppress MOC1 tumor cell killing by TIL cultured from MOC1 tumors (Fig. 5B). Ly6GhiLy6Cint myeloid cells at a 3:1 ratio to TIL (there are about three times as many Ly6GhiLy6Cint myeloid cells in MOC1 tumors as there are CD8+ T cells) suppressed killing of MOC1 cells, validating them as PMN-MDSC. PMN-MDSCs sorted from MOC1 tumors following Sema4D mAb treatment were less suppressive compared with control. Accordingly, PCR analysis of whole tumor digests revealed less arginase expression following Sema4D mAb treatment (Fig. 5C). To explore signaling alterations within PMN-MDSCs following Sema4D mAb treatment, whole leukocytes were sorted from tumors and analyzed for phosphorylation of AKT, ERK, and STAT3 by flow cytometry (Fig. 5D). Although phosphorylation of these signaling proteins was unaltered in CD3+ lymphocytes, phosphorylation of ERK and STAT3 was reduced in PMN-MDSCs following Sema4D mAb treatment. To validate these findings, we treated PMN-MDSCs sorted from treatment-naïve MOC1 tumors ex vivo with rSema4D or Sema4D mAb and assessed their suppressive capacity (Fig. 5E). Exposure of PMN-MDSCs to rSema4D enhanced suppressive capacity. Addition of Sema4D mAb reduced baseline and reversed rSema4D-induced suppressive capacity. Expression of arginase and phosphorylation of ERK and STAT3 followed a similar pattern (Fig. 5F and G), suggesting that arginase expression within PMN-MDSCs was dependent at least in part on ERK and STAT3 signaling downstream of PlexinB1. Cumulatively, these data suggest that in addition to reduced PMN-MDSC recruitment, Sema4D mAb treatment suppressed ERK- and STAT3-dependent arginase production downstream of PlexinB1 and PMN-MDSC suppressive capacity.
Figure 5.
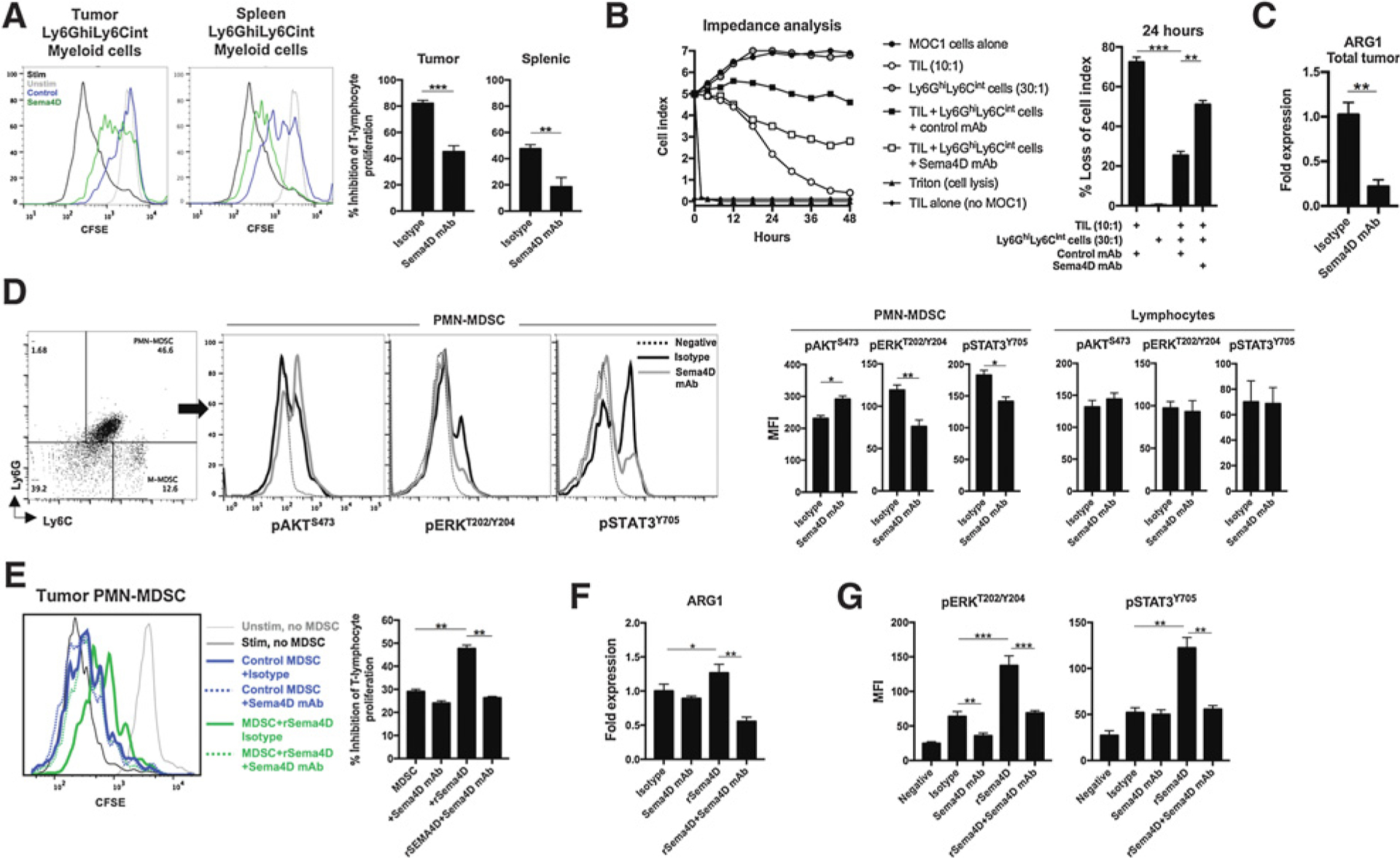
Sema4D mAb inhibited the T-lymphocyte-suppressive capacity of Ly6GhiLy6Clow myeloid cells within MOC1 tumors. A, Mice bearing MOC1 tumors were treated with Sema4D mAb or isotype control (n = 5/group), tumor digests harvested 1 day after the last Sema4D mAb treatment were subjected to a percoll gradient, and Ly6GhiLy6Clow myeloid cells were sorted via positive magnetic selection. Myeloid cell–suppressive capacity was assessed in a T-lymphocyte proliferation assay. Representative overlaid CFSE histograms on the left, quantified on the right. B, MOC1 TILs were cocultured with MOC1 tumor cells in the presence or absence of Ly6GhiLy6Clow myeloid cells, and changes in MOC1 cell impedance were assessed in real time. Representative impedance plot on the left, and loss of impedance at the 24-hour time point qualified on the right. C, Mice bearing MOC1 tumors were treated Sema4D mAb or isotype control (n = 5/group), and whole tumor digests were analyzed for ARG1 expression via qRT-PCR. D, Mice bearing MOC1 tumors were treated as in C (n = 5/group), tumor digests were subjected to a percoll gradient, and tumor leukocytes were analyzed for lineage markers and intracellular phosphotranscription factors by flow cytometry. Representative dot plots and overlaid histograms are on the left, quantified on the right. E, Tumor Ly6GhiLy6Clow myeloid cells were sorted from untreated MOC1 tumors and exposed ex vivo to recombinant Sema4D protein (10 μg/mL), Sema4D mAb (10 μg/mL), or isotype alone or in combination for 12 hours. Ly6GhiLy6Clow myeloid cell–suppressive capacity was assessed in a T-lymphocyte proliferation assay (n = 5/group). Representative overlaid histograms on the left, quantified on the right. F, PMN-MDSCs as in E were assessed for ARG1 expression via qRT-PCR after exposure to recombinant Sema4D protein or antibody for 3 hours (n = 5/group). G, PMN-MDSCs as in E were assessed for intracellular phosphotranscription factors by flow cytometry after exposure to recombinant Sema4D protein or antibody for 1 hour (n = 5/group). MFI, mean fluorescent intensity; rSema4D, recombinant Sema4D. **, P < 0.01; ***, P < 0.001.
Sema4D mAb enhanced tumor antigen–specific T-lymphocyte responses
To characterize the effect of reduced PMN-MDSC infiltration and suppressive capacity on effector immune cells, we assessed changes in infiltration of T lymphocytes and natural killer (NK) cells after Sema4D mAb treatment (Fig. 6A). Tumors were infiltrated with more CD8+ TILs after Sema4D mAb. Infiltrating CD8+ TIL did not display significant differences in PD-1 or CTLA-4 expression. Tumor infiltration of CD4+ TIL was also enhanced to a degree, but there was no change in infiltration of NK cells after Sema4D treatment (Supplementary Fig. S2). Ki67 expression was greater after Sema4D mAb treatment (Fig. 6B), indicating enhanced expansion of CD8+ TIL. Direct exposure of Sema4D mAb to T lymphocytes ex vivo did not alter proliferation (Supplementary Fig. S3), suggesting that the enhanced proliferation of T lymphocytes in vivo was not due to the antibody. To assess T-lymphocyte tumor antigen–specific responses, we combined T lymphocytes sorted from tumor-draining lymph nodes with irradiated MOC1 cells as an antigen source. T lymphocytes from mice treated with Sema4D mAb demonstrated greater IFNγ production upon exposure to MOC1 antigen (Fig. 6C). As an additional verification of enhanced T-lymphocyte responses, we stimulated sorted T lymphocytes nonspecifically with CD3/28 mAbs, and found similar increased responses in T lymphocytes from mice treated with Sema4D mAb. Lastly, we assessed MOC1 tumor cells for expression of known IFNγ-responsive proteins H-2Kb/Db and PD-L1 (Fig. 6D). Expression of both was elevated in mice treated with Sema4D mAb, indicating increased IFNγ production in the microenvironment of these tumors.
Figure 6.
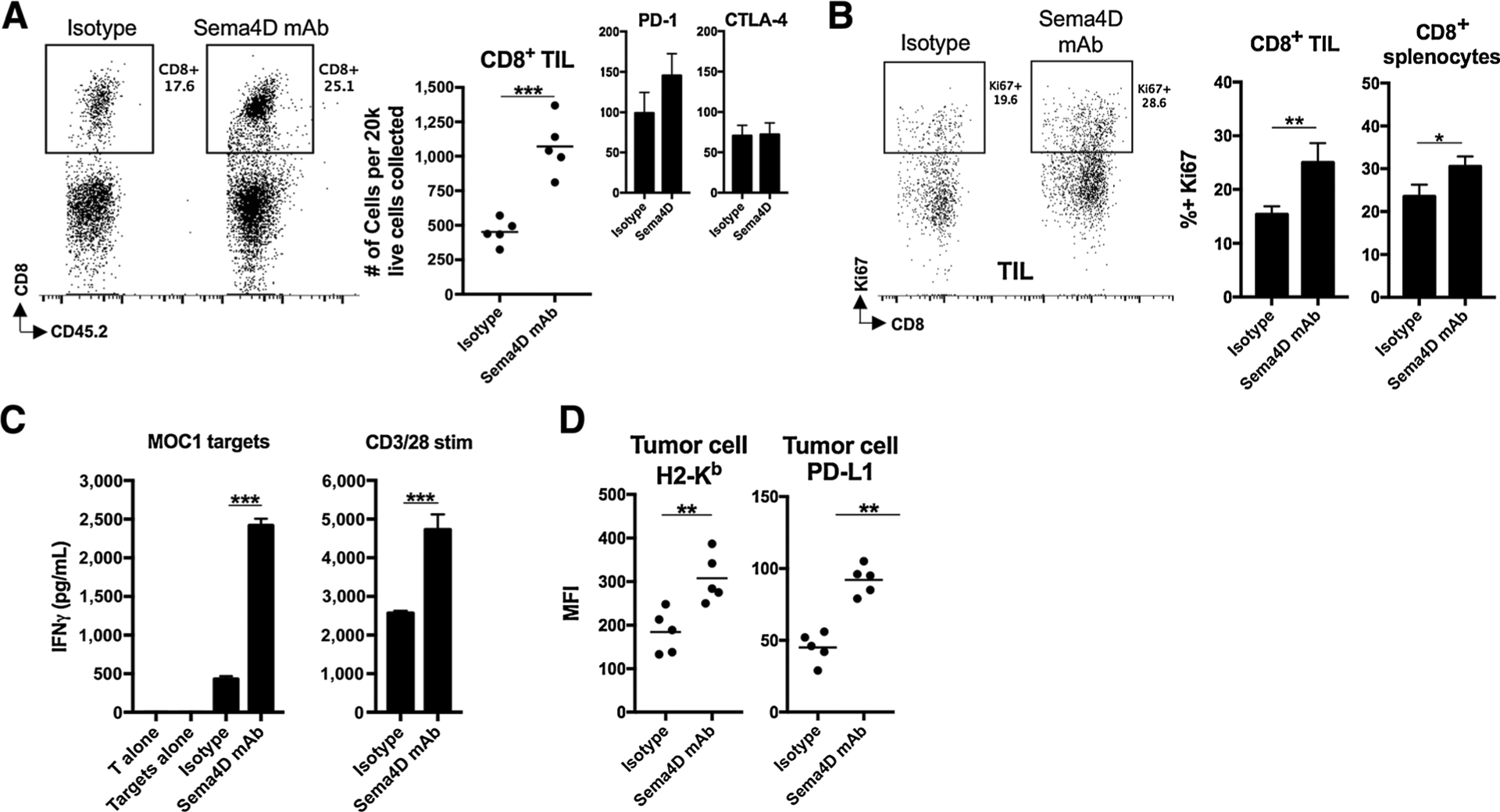
Sema4D mAb enhanced CD8+ TIL infiltration and expansion and draining lymph node T-lymphocyte tumor antigen–specific responses. A, Mice bearing MOC1 tumors were treated with Sema4D mAb or isotype control (n = 5/group), and whole tumor digests harvested 1 day after the last Sema4D mAb treatment were analyzed via flow cytometry for CD3+CD8+ T-lymphocyte infiltration. Representative dot plots are on the left, quantified on the right. Inset bar graphs show expression of immune checkpoints on CD8+ TIL. B, Mice bearing MOC1 tumors were treated as in A (n = 5/group), tumor digests subjected to a percoll gradient, and leukocytes were analyzed for T-lymphocyte markers and intracellular Ki67 by flow cytometry. Representative dot plots on the left, quantified on the right. C, Mice bearing MOC1 tumors were treated as in A (n = 5/group), tumor-draining lymph nodes were harvested 1 day after the last Sema4D mAb treatment, and T lymphocytes were assessed for IFNγ concentration after stimulation. D, Mice bearing MOC1 tumors were treated as in A (n = 5/group) and analyzed via flow cytometry for expression of H-2Kb/Db and PD-L1 on CD45.2−CD31− tumor cells. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Discussion
In this study, we describe mechanisms of action of a therapeutic Sema4D mAb in murine models of carcinoma. Blockade of Sema4D appeared to both reduce PMN-MDSC accumulation in MOC1 carcinomas through reduced myeloid chemokine expression from MOC1 tumor cells and decrease PMN-MDSC suppressive capacity through inhibition of PMN-MDSC arginase expression. These changes in PMN-MDSC accumulation and function resulted in enhanced T-lymphocyte tumor antigen–specific responses that enhanced responses to either CTLA-4 or PD-1 ICB. We assessed the performance of a Sema4D mAb in the MOC1 model given previous data that targeting Sema4D altered the myeloid immune compartment (14, 30, 31). Our previous work demonstrated the role of MDSCs in local immunosuppression within MOC1 tumors (5, 8). To ensure that these findings were not model specific, sensitization to ICB following Sema4D blockade was validated in mice bearing LLC tumors. Similar effects of Sema4D blockade have been demonstrated in models of colon and breast carcinoma (14).
This work enhances our understanding of how Sema4D blockade sensitizes carcinomas to ICB. Work by Evans and colleagues demonstrated enhanced tumor penetration of CD11c+ antigen-presenting myeloid cells, cytokine polarization to a type I antitumor profile, and enhanced infiltration of IFN-producing antigen-specific CD8+ TILs after Sema4D mAb treatment of Colon26 tumors (14). Our work suggests that, in addition to global alterations in immune profiles, disruption of Sema4D and PlexinB1 signaling within the TME affects tumor cells and PMN-MDSC.
Our finding that Sema4D mAb alters signaling through ERK in tumor cells validates the presence of MAPK pathway signaling downstream of PlexinB1 (25, 27). Although MAPK driven expression of CXCL1 in MOC1 cells was known, here we demonstrate regulation of a more diverse myeloid chemokine expression profile with interruption of signaling downstream of PlexinB1. PlexinB1 forms a signaling complex with the receptor tyrosine kinases c-Met or ErbB-2 (32, 33). Intracellular signaling networks downstream of c-Met regulate CXCL1 expression in murine cells (34), and work by Van Waes and colleagues in human HNSCC cells demonstrates the role of c-Met in the regulation of expression of IL8, the human homologue of murine CXCL1 (35, 36). Whether Sema4D mAb disrupts CXCL1 expression downstream of PlexinB1 or alters signaling downstream of c-Met or ErbB-2 due to disrupted receptor tyrosine kinase:plexinB1 interaction remains to be determined. Sema4D blockade and the resulting decreased chemokine expression seemed to slow the accumulation of PMN-MDSCs to a greater degree compared with Ly6Chi myeloid cells. This can be explained by more reduction in CXCR2 ligands CXCL1, 2, and 5 within the TME following Sema4D blockade, suggesting Sema4D-independent expression of CCL2 by multiple cell types. PMN-MDSCs are more susceptible to depletion given the high baseline infiltration present in MOC1 tumors. Among tumor-infiltrating myeloid cells, PlexinB1 expression was greater on PMN-MDSCs compared with Ly6GlowLy6Chi cells or macrophages. This may explain why Sema4D blockade appeared to have the greatest impact on PMN-MDSC function. Given the expression of PlexinB1 on PMN-MDSCs, we cannot rule out that Sema4D mAb blocked recruitment of PMN-MDSCs into MOC1 tumors independent of reduced myeloid chemokine expression. CXCL1 expression drives recruitment of CXCR2+ PMN-MDSCs in multiple tumor models (9, 37, 38), but control of migration of myeloid cells by semaphorin family members has also been demonstrated in other models (30, 31).
Sema4D-driven expression of arginase in PMN-MDSCs was first demonstrated by Younis and colleagues in vitro (15). Our work validates this finding in vivo. Reduced STAT3 phosphorylation and arginase expression in MOC1 tumor-infiltrating PMN-MDSCs after Sema4D treatment supports the work of Vasquez-Dunddel and colleagues demonstrating STAT3-dependent arginase expression in human MDSCs (6). Reduced ERK phosphorylation in MOC1 tumor-infiltrating PMN-MDSCs observed after Sema4D mAb treatment suggests that MAPK signaling may regulate expression of arginase as well. In addition to promoting chemotaxis, signaling through CXCR2 after CXCL1 ligation directly regulates arginase expression in MDSCs (39), offering the possibility that reduced PMN-MDSC suppressive capacity and arginase expression after Sema4D mAb treatment in vivo was due to reduced CXCL1 expression. This is unlikely, however, given that ex vivo rSema4D enhanced the suppressive capacity of sorted PMN-MDSCs, and that this suppressive capacity was abrogated in the presence of Sema4D mAb.
Previous work showed that tumor cell–derived or macrophage-derived Sema4D participates in tumor vascularization by recruiting and promoting the survival of PlexinB1+ endothelial cells (12, 13, 40). Induction of Sema4D expression by hypoxia inducible factor-1 is likely required (41). However, signaling through PlexinB1 could regulate independent processes in both myeloid and endothelial cells despite their common embryologic origin (42). Here, we demonstrate that accumulation of CD31+ endothelial cells and CD31+ vessels in MOC1 tumors was not altered by Sema4D mAb treatment.
In conclusion, we found reduced PMN-MDSC accumulation and suppressive capacity within the TME of carcinogen-induced, HPV-negative MOC1 oral cavity carcinomas. This adds mechanistic insight into how Sema4D alters the TME to enhance responses to ICB. With low overall response rates to ICB alone in patients with recurrent or metastatic HNSCC, adjuvant therapies are needed to improve outcomes in this patient cohort. These and previously described effects on tumor vasculature and myeloid cells within the TME provide a rationale for the combination of Sema4D mAb blockade and ICB in a clinical trial setting.
Supplementary Material
Acknowledgments
The authors thank Leslie Balch, Alan Howell, and Christine Reilly for production, purification, and characterization of reagents, recombinant SEMA4D and antibodies; and Rose Berman for technical assistance with immune assays. This work was supported by the Intramural Research Program of the NIH, NIDCD, project numbers ZIA-DC000087 and DC000074.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Disclosure of Potential Conflicts of Interest
E.S. Smith is Chief Scientific Officer at and has ownership interest in Vaccinex, Inc. M. Zauderer reports receiving commercial research funding from and has ownership interest in Vaccinex, Inc. No potential conflicts of interest were disclosed by the other authors.
Footnotes
Supplementary data for this article are available at Cancer Immunology Research Online (http://cancerimmunolres.aacrjournals.org/).
References
- 1.Cooper JS, Pajak TF, Forastiere AA, Jacobs J, Campbell BH, Saxman SB, et al. Postoperative concurrent radiotherapy and chemotherapy for high-risk squamous-cell carcinoma of the head and neck. N Engl J Med 2004; 350:1937–44. [DOI] [PubMed] [Google Scholar]
- 2.Ferris RL, Blumenschein G Jr, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med 2016;375:1856–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, Eder JP, et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol 2016;17:956–65. [DOI] [PubMed] [Google Scholar]
- 4.Davis RJ, Van Waes C, Allen CT. Overcoming barriers to effective immunotherapy: MDSCs, TAMs, and Tregs as mediators of the immunosuppressive microenvironment in head and neck cancer. Oral Oncol 2016;58: 59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clavijo PE, Moore EC, Chen J, Davis RJ, Friedman J, Kim Y, et al. Resistance to CTLA-4 checkpoint inhibition reversed through selective elimination of granulocytic myeloid cells. Oncotarget 2017;8:55804–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vasquez-Dunddel D, Pan F, Zeng Q, Gorbounov M, Albesiano E, Fu J, et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J Clin Invest 2013;123:1580–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Young MR, Petruzzelli GJ, Kolesiak K, Achille N, Lathers DM, Gabrilovich DI. Human squamous cell carcinomas of the head and neck chemoattract immune suppressive CD34(+) progenitor cells. Hum Immunol 2001;62: 332–41. [DOI] [PubMed] [Google Scholar]
- 8.Davis RJ, Moore EC, Clavijo PE, Friedman J, Cash H, Chen Z, et al. Anti-PD-L1 efficacy can be enhanced by inhibition of myeloid-derived suppressor cells with a selective inhibitor of PI3Kdelta/gamma. Cancer Res 2017;77:2607–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med 2014;6:237ra67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gurrapu S, Tamagnone L. Transmembrane semaphorins: Multimodal signaling cues in development and cancer. Cell Adh Migr 2016;10: 675–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hall KT, Boumsell L, Schultze JL, Boussiotis VA, Dorfman DM, Cardoso AA, et al. Human CD100, a novel leukocyte semaphorin that promotes B-cell aggregation and differentiation. Proc Natl Acad Sci USA 1996;93: 11780–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Basile JR, Castilho RM, Williams VP, Gutkind JS. Semaphorin 4D provides a link between axon guidance processes and tumor-induced angiogenesis. Proc Natl Acad Sci USA 2006;103:9017–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou H, Binmadi NO, Yang YH, Proia P, Basile JR. Semaphorin 4D cooperates with VEGF to promote angiogenesis and tumor progression. Angiogenesis 2012;15:391–407. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Evans EE, Jonason AS Jr, Bussler H, Torno S, Veeraraghavan J, Reilly C, et al. Antibody blockade of semaphorin 4D promotes immune infiltration into tumor and enhances response to other immunomodulatory therapies. Cancer Immunol Res 2015;3:689–701. [DOI] [PubMed] [Google Scholar]
- 15.Younis RH, Han KL, Webb TJ. Human head and neck squamous cell carcinoma-associated semaphorin 4D induces expansion of myeloid-derived suppressor cells. J Immunol 2016;196:1419–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Binmadi NO, Yang YH, Zhou H, Proia P, Lin YL, De Paula AM, et al. Plexin-B1 and semaphorin 4D cooperate to promote perineural invasion in a RhoA/ROK-dependent manner. Am J Pathol 2012;180: 1232–42. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Ch’ng ES, Kumanogoh A. Roles of Sema4D and Plexin-B1 in tumor progression. Mol Cancer 2010;9:251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Judd NP, Winkler AE, Murillo-Sauca O, Brotman JJ, Law JH, Lewis JS Jr, et al. ERK1/2 regulation of CD44 modulates oral cancer aggressiveness. Cancer Res 2012;72:365–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patnaik A, Weiss GJ, Leonard JE, Rasco DW, Sachdev JC, Fisher TL, et al. Safety, pharmacokinetics, and pharmacodynamics of a humanized anti-semaphorin 4D antibody, in a first-in-human study of patients with advanced solid tumors. Clin Cancer Res 2016;22: 827–36. [DOI] [PubMed] [Google Scholar]
- 20.Onken MD, Winkler AE, Kanchi KL, Chalivendra V, Law JH, Rickert CG, et al. A surprising cross-species conservation in the genomic landscape of mouse and human oral cancer identifies a transcriptional signature predicting metastatic disease. Clin Cancer Res 2014;20:2873–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cash H, Shah S, Moore E, Caruso A, Uppaluri R, Van Waes C, et al. mTOR and MEK1/2 inhibition differentially modulate tumor growth and the immune microenvironment in syngeneic models of oral cavity cancer. Oncotarget 2015;6:36400–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farsaci B, Sabzevari H, Higgins JP, Di Bari MG, Takai S, Schlom J, et al. Effect of a small molecule BCL-2 inhibitor on immune function and use with a recombinant vaccine. Int J Cancer 2010;127:1603–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roederer M Interpretation of cellular proliferation data: avoid the panglossian. Cytometry A 2011;79:95–101. [DOI] [PubMed] [Google Scholar]
- 24.Davis RJ, Silvin C, Allen CT. Avoiding phagocytosis-related artifact in myeloid derived suppressor cell T-lymphocyte suppression assays. J Immunol Methods 2017;440:12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aurandt J, Li W, Guan KL. Semaphorin 4D activates the MAPK pathway downstream of plexin-B1. Biochem J 2006;394(Pt 2):459–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang YH, Zhou H, Binmadi NO, Proia P, Basile JR. Plexin-B1 activates NF-kappaB and IL-8 to promote a pro-angiogenic response in endothelial cells. PLoS One 2011;6:e25826. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Basile JR, Gavard J, Gutkind JS. Plexin-B1 utilizes RhoA and Rho kinase to promote the integrin-dependent activation of Akt and ERK and endothelial cell motility. J Biol Chem 2007;282:34888–95. [DOI] [PubMed] [Google Scholar]
- 28.Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res 2017;5:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun 2016;7:12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Casazza A, Laoui D, Wenes M, Rizzolio S, Bassani N, Mambretti M, et al. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 2013;24:695–709. [DOI] [PubMed] [Google Scholar]
- 31.Takamatsu H, Takegahara N, Nakagawa Y, Tomura M, Taniguchi M, Friedel RH, et al. Semaphorins guide the entry of dendritic cells into the lymphatics by activating myosin II. Nat Immunol 2010;11:594–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giordano S, Corso S, Conrotto P, Artigiani S, Gilestro G, Barberis D, et al. The semaphorin 4D receptor controls invasive growth by coupling with Met. Nat Cell Biol 2002;4:720–4. [DOI] [PubMed] [Google Scholar]
- 33.Swiercz JM, Worzfeld T, Offermanns S. ErbB-2 and met reciprocally regulate cellular signaling via plexin-B1. J Biol Chem 2008;283:1893–901. [DOI] [PubMed] [Google Scholar]
- 34.Fang WB, Mafuvadze B, Yao M, Zou A, Portsche M, Cheng N. TGF-beta negatively regulates CXCL1 chemokine expression in mammary fibroblasts through enhancement of Smad2/3 and suppression of HGF/c-Met signaling mechanisms. PLoS One 2015;10:e0135063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dong G, Chen Z, Li ZY, Yeh NT, Bancroft CC, Van Waes C. Hepatocyte growth factor/scatter factor-induced activation of MEK and PI3K signal pathways contributes to expression of proangiogenic cytokines interleukin-8 and vascular endothelial growth factor in head and neck squamous cell carcinoma. Cancer Res 2001;61:5911–8. [PubMed] [Google Scholar]
- 36.Dong G, Lee TL, Yeh NT, Geoghegan J, Van Waes C, Chen Z. Metastatic squamous cell carcinoma cells that overexpress c-Met exhibit enhanced angiogenesis factor expression, scattering and metastasis in response to hepatocyte growth factor. Oncogene 2004;23:6199–208. [DOI] [PubMed] [Google Scholar]
- 37.Katoh H, Wang D, Daikoku T, Sun H, Dey SK, Dubois RN. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell 2013;24:631–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loukinova E, Dong G, Enamorado-Ayalya I, Thomas GR, Chen Z, Schreiber H, et al. Growth regulated oncogene-alpha expression by murine squamous cell carcinoma promotes tumor growth, metastasis, leukocyte infiltration and angiogenesis by a host CXC receptor-2 dependent mechanism. Oncogene 2000;19:3477–86. [DOI] [PubMed] [Google Scholar]
- 39.Kang X, Zhang X, Liu Z, Xu H, Wang T, He L, et al. CXCR2-mediated granulocytic myeloid-derived suppressor cells’ functional characterization and their role in maternal fetal interface. DNA Cell Biol 2016;35:358–65. [DOI] [PubMed] [Google Scholar]
- 40.Sierra JR, Corso S, Caione L, Cepero V, Conrotto P, Cignetti A, et al. Tumor angiogenesis and progression are enhanced by Sema4D produced by tumor-associated macrophages. J Exp Med 2008;205:1673–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun Q, Zhou H, Binmadi NO, Basile JR. Hypoxia-inducible factor-1-mediated regulation of semaphorin 4D affects tumor growth and vascularity. J Biol Chem 2009;284:32066–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bailey AS, Willenbring H, Jiang S, Anderson DA, Schroeder DA, Wong MH, et al. Myeloid lineage progenitors give rise to vascular endothelium. Proc Natl Acad Sci USA 2006;103:13156–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.